

Abstract
Introduction.
Clinical standard of care for newborn screening (NBS) is acylcarnitine metabolites quantitation by tandem mass spectrometry (MS/MS) from dried blood spots. Follow up sequencing often results in identification of one or more variants of uncertain significance (VUS). Isovaleric acidemia (IVA) is an autosomal recessive inborn error of metabolism caused by deficiency of isovaleryl-CoA dehydrogenase (IVDH) in the Leu catabolism pathway. Many IVD mutations are characterized as VUS complicating IVA clinical diagnoses and treatment. We present a testing platform approach to confirm the functional implication of VUS identified in newborns with IVA applicable to multiple inborn errors of metabolism identified by NBS.
Methods.
An IVD null HEK293T cell culture model was generated by using a dual sgRNA CRISPR/Cas9 genome-editing strategy targeting IVD exons 2-3. Clonal cell lines were confirmed by a combination of genomic breakpoint sequencing and droplet digital PCR. The IVD null model had no IVDH antigen signal and 96% reduction in IVDH enzyme activity. The IVD null model was transfected with vectors containing control or variant IVD and functional assays were performed to determine variant pathogenicity.
Results.
c.149G>C (p.Arg50Pro; precursor numbering), c.986T>C (p.Met329Thr), and c.1010G>A (p.Arg337Gln), c.1179del394 fs mutant proteins had reduced IVDH protein and activity. c.932C>T (p.Ala311Val), c.707C>T (p.Thr236Ile), and c.1232G>A (p.Arg411Gln) had stable IVDH protein, but no enzyme activity. c.521T>G (p.Val174Gly) had normal IVDH protein and activity. IVD variant transfection results confirmed results from IVA fibroblasts containing the same variants.
Conclusions.
We have developed an IVD null HEK293T cell line to rapidly allow determination of VUS pathogenicity following identification of novel alleles by clinical sequencing following positive NBS results for suspected IVA. We suggest similar models can be generated via genome-editing for high throughput assessment of VUS function for a multitude of inborn errors of metabolism and can ideally supplement NBS programs.
Keywords: Organic acidemia, isovaleric acidemia, isovaleryl-CoA dehydrogenase, newborn screening, variants of uncertain significance
Introduction
The clinical standard of care for newborn screening (NBS) for many inborn errors of metabolism is quantitation of acylcarnitine metabolites by tandem mass spectrometry (MS/MS) from dried blood spots on Guthrie cards. Secondary testing by sequential liquid chromatography with MS/MS further improves NBS specificity for many disorders [1]. Follow up genetic diagnosis is recommended by molecular analysis and functional studies. Sequencing-based methods often result in identification of variants of uncertain significance (VUS) in at least one allele, though the frequency varies among disorders [2]. When VUS are present, a firm genetic diagnosis of an inborn errors of metabolism remains in question, as specific functional testing for the novel variants is often unavailable or difficult to obtain, and can lead to a significant delay of final diagnoses and treatment implementation [2, 3].
Isovaleric acidemia (IVA, OMIM #243500) is an autosomal recessive inborn error of metabolism of the leucine (Leu) catabolic pathway [4-6]. It results from biallelic mutations in the gene encoding isovaleryl-CoA dehydrogenase (IVDH, EC 1.3.8.4), leading to the accumulation of isovaleryl-CoA and its metabolites. Symptoms of untreated IVA can first appear during infancy, childhood or adolescence; and include characteristic sweaty foot odor, poor feeding, vomiting, seizures, and mental disabilities [4, 7, 8]. IVA is typically detected via MS/MS-based NBS and is marked by a characteristic increase of C5-carnitine [9, 10]. However, the C5-carnitine can represent isovaleryl- or 2-methylbutyryl-carnitine and genetic confirmation of the diagnosis is required. IVA patients are typically treated with a Leu restricted diet, and L-glycine and/or L-carnitine supplementation to replenish exhausted endogenous supply [8, 11-15]. In a study of IVA patients identified by NBS in Germany and the United States of primarily Caucasian and Arabic ethnicities, a common IVD mutation 932C>T (p.Ala311Val) was identified in nearly half of the individuals, and predicts a mild clinical presentation and no need for therapy [8, 16, 17]. However, there are a growing number of IVD variants across diverse ethnicities being reported with unknown functional effects. As of this writing, 107 VUS in the IVD gene are reported in ClinVar, leading to uncertainty in the diagnosis and treatment in identified infants. Classification of these VUS as pathogenic or benign is critical to appropriately define the need for treatment following NBS.
In this study, we describe a scalable approach to determine the functional significance of VUS identified by NBS in a timely manner to facilitate better diagnostic and therapeutic outcomes. We present proof-of-concept of this approach by examining the functional implication of VUS identified in newborns with elevated C5-carnitine indicative of IVA. To implement the study of IVD variants, an IVD null HEK293T cell line was generated using CRISPR/Cas9 genome-editing. Expression of control or variant IVD cDNAs within the IVD null HEK293T line then allowed characterization of IVDH enzyme activity in cellular extracts. This model enables determining pathogenicity of individual IVD VUS and is amendable for development as a high throughput platforms for screening VUS in other inborn errors of metabolism.
Materials & Methods
Experiments were performed in accordance with the approved guidelines and regulations. Experimental human protocols were approved by the Institutional Review Board at the University of Pittsburgh, protocol 19030195.
Subjects
Patients were detected after abnormal NBS of elevated C5-carnitine consistent with IVA (Supplementary Table 1). Skin biopsies for fibroblast culture were performed on a clinical basis from infants and subsequent analysis was performed written informed consent from parents and/or legal guardians. Sequencing of IVD was performed on a clinical basis in a CLIA certified laboratory on IVA patients and parents to determine IVD mutation presence in mutations were in trans. Control fibroblast cells were obtained from the American Type Culture Collection (ATCC.org).
Cell lines & culture
HEK293T (obtained from ATCC.org) and fibroblast cell lines were grown in Dulbecco’s Modified Eagle Medium (DMEM; Corning Life Sciences, Manassas, VA) containing 4.5 g/l glucose and supplemented with 10% fetal bovine serum, 4 mM glutamine and 100 IU penicillin and 100 μg/ml streptomycin (Corning Life Sciences) at 37 °C in a 5% CO2 humidified atmosphere.
CRISPR-Cas9 genome-editing
CRISPR sgRNAs targeting IVD repeat-masked intron 1 and intron 3 (Supplementary Table 2) were designed using the Crispor.org web application [18] with optimal residues at the protospacer adjacent motif (PAM) (5’-NGG-3’) +1 and −4 positions and cloned into the Bbs I site of the pSpCas9(BB)-2A-GFP vector (PX458; Addgene #48138) [19]. HEK293T cells were seeded into 6-well plates and co-transfected with 1 μg of each PX458 plasmid complexed with Lipofectamine 3000 (Invitrogen, Waltham, MA) at a 1:3 plasmid to reagent ratio for 24 hours. Cells were disaggregated after 48 hours and flow sorted for GFP(+) single cells into 96-well plates. Colonies were grown for 2-3 weeks, scaled up to 24-well plates and DNA extracted using the DNeasy Blood and Tissue kit (Qiagen). PCR amplification of deletion or inversion breakpoints (Primers available in Supplemental Table 2) was performed was performed using Q5 high-fidelity polymerase (New England Biolabs). PCR products were gel extracted using a Zymoclean Gel DNA Recovery kit (Zymo Research, Irvine, CA) and either directly Sanger sequenced or cloned into pJet2.1 (Thermo Fisher Scientific, Waltham, MA) and sequenced.
IVD cDNA analysis
IVA patient fibroblast and IVD variant plasmid transfected IVD KO HEK293T cell mRNA was isolated using a RNeasy Mini kit (Qiagen, Valencia, CA) with on column DNase I digestion. First strand synthesis of complementary DNA (cDNA) from 500 ng of mRNA was performed using the Superscript Vilo IV Master Mix (Qiagen). Reverse transcription PCR (RT-PCR) of full-length and partial IVD cDNA regions was performed with Q5 DNA polymerase (New England Biolabs, Ipswich, MA) and directly Sanger sequenced to identify sequence variants (Primers available in Supplementary Table 4).
Western blot
Fibroblasts and HEK293T cells were grown in T175 flasks to 90% confluence, harvested by trypsinization, pelleted, and stored at −80°C for western blot. Frozen pellets were treated with 50 μL of radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific) and 1X Protease Inhibitor Cocktail (PI) (Roche, St Louis, MO) for 30 minutes on ice and centrifuged at 14,000 x g for 15 minutes at 4°C. Supernatants were collected and 25 μg of protein was loaded onto a 4 to 15% gradient Criterion precast SDS-PAGE gel (Biorad, Hercules, CA). Following electrophoresis, the gel was blotted onto a nitrocellulose membrane and incubated with mouse anti-IVDH antibody (1:2000; Origene, Rockville, MD), then incubated with secondary goat anti-mouse-HRP antibody (1:3000, Biorad). Pierce ECL Western Blotting Substrate kit (Thermo Fisher Scientific) was used to visualize bands. Membranes were stripped and re-probed with mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:25,000) monoclonal antibody (Abcam, Cambridge, MA) to verify equal loading.
ETF fluorescence reduction assay
The electron transfer flavoprotein (ETF) fluorescence reduction assay was performed using a Jasco FP-6300 spectrofluorometer (Jasco, Inc., Easton, MD) with cuvette holder heated with circulating water at 32 °C, as previously described [20, 21]. ETF was diluted 1200-fold into a buffer containing 50 mM Tris, pH 8.0, 5 mM EDTA and 50% glycerol, and 10 μl were used for each assay. The ETF concentration in the reaction mixture was 2 μM. 30 μM of isovaleryl-CoA lithium salt hydrate (iC5-CoA; Sigma-Aldrich Co., St. Louis, MO) or octanoyl-CoA lithium salt hydrate (C8-CoA; Sigma Aldrich Co.) were used to measure IVDH and medium chain acyl-CoA dehydrogenase (MCAD) activity, respectively. Spectra Manager 2 software (Jasco, Inc.) was used to collect data and calculate reaction rate and Microsoft Excel was used to calculate kinetic parameters.
Digital droplet polymerase chain reaction (ddPCR)
Genomic DNA from HEK293T parental, control and IVD null clonal lines, and from patient fibroblasts was restriction digested with XbaI and diluted to a concentration of 18 ng/μl with 2 μl used as input per reaction. Multiplex probe-based ddPCR assays (Supplementary Table 3) were setup with FAM labeled Taqman probe PCR assays for IVD exons (1, 2, 3, 4, 9 or 12) and either HEX labeled Taqman probe PCR assays for reference loci either external (RPP30, XIST) or internal (IVD exon 4). The ratio of the target IVD exon to expected diploid reference gene was calculated based on the concentration of each (copies per μl) within a single reaction with a 95% confidence interval indicated by Poisson statistics [22].
IVD variant vector design and isolation
Control and variant IVD gene pcDNA3.1(+) mammalian expression vectors were constructed by BioMatik (Willmington, DE). IVD pcDNA3.1(+) vectors were transformed into XL1-Blue supercompetent Escherichia coli (Agilent Technologies, Santa Clara, CA) and grown in Luria-Bertani (LB) broth and 100 μg/mL ampicillin. Supercoiled plasmid DNA was prepared using a midi prep kit (Zymo Research).
Transfection of IVD mutant vectors
HEK293T IVD KO cells were seeded into 6-well plates or 10 cm2 dishes and cotransfected with 2.5 or 15 μg of plasmid DNA, respectively, at 60% confluency using TransIT X2 (Mirus Bio LLC, Madison, WI). Cells were incubated for 48 hours and then harvested for western blotting and ETF fluorescence reduction assay for IVDH protein presence and enzyme activity.
Computational Molecular Modeling
To examine the environment of residues and their interactions with others, computational computer model of IVDH was visualized using a Silicon Graphics Fuel workstation (Mountain View, CA) and the Insight II 2005 software package, which included Homology/Modeler, and Discover modules and the atomic coordinates of monomers A and B of human IVDH (PDB: 1IVH) in the dimer form, as reference molecule [23]. An IVDH:ETF ternary complex was modeled using the MCAD complexed with the ETF (PDB: 1T9G), [24]. Using Homology, the known IVDH and MCAD structures were “Superimposed” by matching their secondary structures backbone to examine the IVDH:ETF complex interactions and possible effect on IVDH ETF interaction.
Statistics
Data are presented as mean ± standard deviation (SD) for replicates and analyzed using unpaired Students t test (Graphpad Version 7, graphpad.com). Statistical significance was considered p < 0.05.
Results
CRISPR/Cas9 IVD gene editing
The IVD gene on human chromosome 15 contains 12 exons and encodes a 423 amino acid polypeptide [25]. Traditionally, the 30 amino acid mitochondrial targeting leader sequence has been removed from protein variant nomenclature [8, 21, 26, 27] but the full-length protein annotation is used in ClinVar. An in-frame ATG that is 9-nt upstream is annotated in alternate GENCODE transcript annotation gives the potential for an alternative translational initiator with a slightly larger product, but is poorly conserved and deviates from the consensus Kozak sequence [25, 27], and is not reflected in the ClinVar SNP annotation. In addition, alternative spliced products with either exon 2 skipping or an extended exon 2 associated with an alternate downstream UTR are annotated in the NCBI RefSeq database. Notably, IVD intron 1 splice acceptor mutations leading to constitutive exon 2 skipping is associated with IVA diagnoses [26].
To evaluate the function of VUS identified through clinical sequencing we sought to create a robust method that would ultimately be amenable to analysis of variants in multiple genes as well as high throughput techniques. We first generated functional null mutations in IVD in HEK293T cells by targeting IVD exons 2-3 to recapitulate the murine Ivdtm1b(EUCOMM)Hmgu homozygous lethal null allele [28]. A dual CRISPR single guide (sgRNA) strategy [29] was implemented with a pair of sgRNAs designed to induce Cas9-nuclease mediated double-stranded DNA breaks (DSBs) in cis at IVD introns 1 and 3 to generate deletion or inversions of the intervening 847 bp sequence containing exons 2-3 (Fig. 1A). Genomic breakpoint PCR assays using primers flanking the sgRNA sites enabled the identification of genome-edited alleles (Figs. 1A-B; S1A-B). Vector-based CRISPR/Cas9 reagents for the expression of sgRNAs and spCas9-P2A-GFP [19] were transfected into HEK293T cells and clonal lines were derived by isolation and expansion of single GFP-positive cells. We initially tested the efficiency of dual sgRNA targeting with four permutations of IVD sgRNAs in bulk transfected cells before selecting the most efficient pair for clonal derivatization. In our IVD CRISPR-Cas9 genome-editing screen 13 out of 21 HEK293T clonal lines amplified a deletion breakpoint PCR band (Figs. 1B and S1B, top panel). A similar, high frequency of inversion alleles was detected by proximal (11 of 21) and distal (12 of 21) inversion breakpoint PCR assays (Figs 1B and S1B, second panel). Further, as evidenced by the lack of amplification of intact sgRNA sites we identified potential homozygous genome-edited lines (Figs. 1B and S1B, bottom panel).
Figure 1. Targeted deletion of IVD exons 2-3 by using CRISPR/Cas9 genome-editing.

(A) Location of sgRNA and genotyping primers used to generate and screen for IVD null alleles. Key: Blue box, IVD exons with intervening introns; pink rectangles, sgRNA intron 1 and 3 sites; directional arrows, genotyping primers. (B) Genotyping PCR assays identifying 3 IVD null clonal HEK293T lines through presence of deletion (top) and/or inversion (second from top) breakpoint-PCR bands and lack of intact sgRNA sites (bottom). * indicates non-specific band. (C) Genomic copy number of IVD as ascribed by the ratio of IVD to RPP30 by ddPCR. Note that HEK293T is likely pseodotriploid for the RPP30 locus and that technical variability led to lower-than-expected exon 1 estimates. Bars represent absolute copy number ratio of IVD exons across the locus with Poisson distribution 95% confidence intervals.
Confirmation of genomic deletion, inversion and sgRNA site PCRs by Sanger sequencing revealed the exact molecular composition of genome-edited alleles in 3 homozygous IVD null lines (4-3, 4-13 and 4-16) and two controls (4-11 and 4-20) (Figs. S2, S3 and S4). Sanger sequencing identified a single deletion-breakpoint and distal inversion allele in 4-3 (Figs. S1A, S2B) and subsequently the proximal inversion breakpoint was identified that contained a partial deletion of intron 1 (Fig. S3B). Clone 4-13 likewise had a single deletion-breakpoint (Fig. S2A) but on further PCR analysis also contained a second larger deletion allele spanning from exon1 into intron 3 (Fig. S4A). In contrast, clone 4-16 yielded a dual chromatograph for the deletion-breakpoint PCR indicating heterozygosity, and two alleles distinguished by a single nt were subsequently cloned and sequenced (Supplemental Figs. 2A and 2A). Sequencing of control line 4-11 revealed heterozygosity of alleles with small indels at each intronic CRISPR sgRNA site indicating two unique intact alleles (Figs. S2C-D and S2B-C). In contrast line 4-20 contained a single intact allele (Figs S2C-D).
Quantitative genomic copy number analysis across the IVD locus by droplet digital PCR (ddPCR) provided definitive proof of clonal IVD null HEK293T cell line genotypes (Fig. 1C). Parental HEK293T showed a ratio of IVD exons 1-12 to RPP30, an unlinked single copy locus on chr10 that was close to 0.8 reflective of a mixed population of pseudotriploid lineages as has been reported by cytogenetic studies [30, 31]. In contrast, the clonal lines had a ratio closer to 0.65 at intact exons suggesting a stable ratio of 2:3 indicative of diploid chr15 (IVD) and triploid chr10 (RPP30). Exons 2 and 3 show a clear reduction to heterozygous levels in 4-3 as the inversion allele is still detected. In contrast the copy number of exons 2-3 in 4-13 and 4-16 are completely ablated, evidence of homozygous deletion. Heterozygosity is also observed in 4-13 exon 1 reflective of the larger deletion allele and across all exons in 4-20 indicative of aneuploidy. Similar results were observed in genomic copy number ddPCR assays using either XIST (known triploid locus in HEK293T), or by using IVD exon 4 as an internal reference (Fig S5). Thus, we have identified and fully characterized genome editing events in three independent IVD null HEK293T clonal lines as well as a normal and haploid control.
Functional characterization of IVD null HEK293T cells
Western blotting was performed to determine the amount of IVDH protein present in the deleted HEK293 cell lines (Fig. 2A). Intact clonal line 4-11 had equivalent IVDH protein abundance compared to the parental lineage control, whereas aneuploid clone 4-20 had reduced IVDH protein levels. As expected, IVD null HEK293T clones 4-13, 4-16, and 4-3 had no detectable IVDH protein. IVDH and MCAD enzyme activity was assessed was assessed using the ETF fluorescence reduction assay (Fig. 2B-C). While 4-11 had the same level of activity as the parental HEK293T, clone 4-20 had partial activity and the three IVD null clones 4-3, 4-11 and 4-16 had minimal activity (Fig. 2B). There was no statistical difference in MCAD enzyme activity in control and all IVD deletion clones (Fig. 2C).
Figure 2. Protein and enzymatic activity of control and IVD null HEK293T lines.

(A) Western blot for IVDH confirms its absence in clonal lines 4-13, 4-16 and 4-3 and reduction in line 4-20. (B) Enzymatic activity of IVDH (isovaleryl-CoA as substrate) and (C) and MCAD (octanoyl-CoA as substrate). IVDH activity assays were done in triplicates and octanoyl-CoA assays were done in duplicates. Means and standard deviations were calculated. Data were analyzed using paired T-test. ****p<0.0001, *** p<0.001, **p<0.01, ns = no statistical difference.
Genetic and functional validation of variants of uncertain significance in IVA patient fibroblasts.
Fibroblasts from four IVA patients containing clinically determined mutations and VUS were analyzed for IVDH activity using the ETF fluorescent reduction assay (Table 1). Genetic mutations were confirmed at the mRNA level through Sanger sequencing of RT-PCR products (Fig. S6). All variants were detected at heterozygous levels in the sequence chromatograph except for c.1232G>A (p.Arg411Gln) in FB909, which was homozygous for a c.1232G>A (p.Arg411Gln) mutation. To rule out changes in IVD copy number in the IVA patients we utilized ddPCR and found the locus to be diploid across the entire gene (Fig S7).
Table 1.
Identified IVD mutations and their corresponding pcDNA3.1(+) plasmid designations and corresponding fibroblast line origin.
| Cell Line Designation Origin |
Mutation | Functional Protein Seq# /location |
Plasmid ID | Key Interacting Residue(s)**/ Ligand |
Experimental Finding |
Current ClinVar Status |
Proposed ClinVar Status*** |
|---|---|---|---|---|---|---|---|
| FB826 | Control | N/A | IVD-C | N/A | IVDH protein: normal Enzyme activity: normal |
— | — |
| FB825 | C.9320T*, p.Ala311Val | Ala282Val α-helices G-H loop |
IVD-932 | Adenine of the C5-CoA | IVDH protein: normal Enzyme activity: reduced |
Pathogenic | Likely Pathogenic |
| FB825 | C.707C>T, p.Thr236Ile | Thr207Ile β-strand 6 |
IVD-707 | Phe205 | IVDH protein: normal Enzyme activity: reduced |
VUS | Pathogenic/Likely Pathogenic |
| FB118 | c.149G>C*. p.Arg50Pro | Arg21Pro | IVD-149 | Anchors Asp7 to Tyr312, Asn11, Leu81 | IVDH protein: reduced Enzyme activity: reduced |
Pathogenic/Likely Pathogenic | Pathogenic/Likely Pathogenic |
| FB827 | c.986T>C, p.Met329Thr |
Met300Thr α-Helix H |
IVD-986 | Thr273, Val342, Cys349 | IVDH protein: reduced Enzyme activity: reduced |
VUS, Not in ClinVar | Pathogenic/Likely Pathogenic |
| FB827 | c.1010G>A, p.Arg337Gln |
Arg308Gln α-Helix H |
IVD-1010 | Asp7, Gln267, Leu304 | IVDH protein: reduced Enzyme activity: reduced |
VUS | Pathogenic/Likely Pathogenic |
| FB909 | c.1232G>A*, p.Arg411Gln |
Arg382Gln α-Helix K |
IVD-1232 | Asp299", Glu337, Glu379 | IVDH protein: normal Enzyme activity: reduced |
VUS, Conflicting Interpretations | Pathogenic/Likely Pathogenic |
| FB925 | c.521T>G, p.Val174Gly | Val145Gly | IVD-521 | Met248, Ser190 | IVDH protein: normal Enzyme activity: normal |
VUS | Benign/Likely Benign |
| FB925 | c.1179del > fs, p.Leu394Phe | Leu365Phe, 7 nonsense residues, and truncating α-Helix J and K |
IVD-1179 | -- | IVDH protein: absent Enzyme activity: absent |
Pathogenic/Likely pathogenic | Pathogenic |
See detailed molecular study [32]
In bold, invariant residues.
Pathogenicity increases exponentially under stress conditions.
Additionally, functional analysis revealed that all four patient fibroblast lines had reduced IVDH protein presence in cell lysates (Fig. 3A) and reduced IVDH enzyme activity compared to control (Fig 3B), consistent with clinically defined IVA. While FB825 and FB827 also had reduced MCAD activity compared to the concurrent control (Fig. 3C); it was within the range that we have observed in other control fibroblast lines.
Figure 3. Protein expression and enzymatic activity assays for IVA patient-derived fibroblasts.

(A) Western blot of IVA patient and control fibroblasts for detection of IVDH and GAPDH (B) Enzymatic activity assay for IVDH (isovaleryl-CoA as substrate). (C) Enzymatic ETF assay for MCAD (octanoyl-CoA substrate). IVDH activity assays were done in triplicates and octanoyl-CoA assays were done in duplicates. Means and standard deviations were calculated. Data were analyzed using unpaired T-test. ***p<0.001, ns = no statistical difference.
Functional analysis of individual VUS alleles in HEK293T IVD null lines
To evaluate the effect of IVD VUS alleles we evaluated protein expression and enzymatic activity in IVD null HEK293T cells that were transfected with vectors driving expression of normal consensus IVD cDNA or IVD cDNA containing individual mutations from variants identified in IVA patients (Table 1, Fig. 4). Abundant IVD protein levels were observed with expression of control, c.521T>G (p.Val174Gly), c.932C>T (p.Ala311Val), c.707C>T (p.Thr236Ile), and c,1232G>A (p.Arg411Gln) cDNAs (Fig. 4A). In contrast, IVDH protein was hardly observed with expression of c.149G>C (p.Arg50Pro), c.986T>C (p.Met329Thr), c.1010G>A (p.Arg337Gln) and c.1179del (p.Leu394 fs) cDNAs (Fig. 4A). To confirm that the lack of IVDH was due to unstable protein and not lack of expression from the plasmid, RT-PCR was performed on transfected IVD KO HEK293T cells. All transfected cells had IVD mRNA confirming the expression plasmid was expressing the IVD gene properly (Fig. 4D). GPI was used as an internal control and had no change in mRNA expression in untransfected or transfected cells (Fig. 4D). Expression of the control cDNA in IVD null HEK293T cells led to dramatic increase in IVDH activity (Fig. 4B). While expression of c.521T>G (p.Val174Gly) IVD cDNA partially rescued enzymatic activity all other tested mutations showed little or no enzyme activity. Expression of the common c.932C>T (p.Ala311Val) variant had 12% of the control plasmid, consistent with previous expression studies of this variant [32]. MCAD activity was unperturbed by transfection of either control or variant IVD cDNAs (Fig. 4C).
Figure 4. Expression and enzymatic activity of IVD VUS in an IVD null HEK293T cell line.
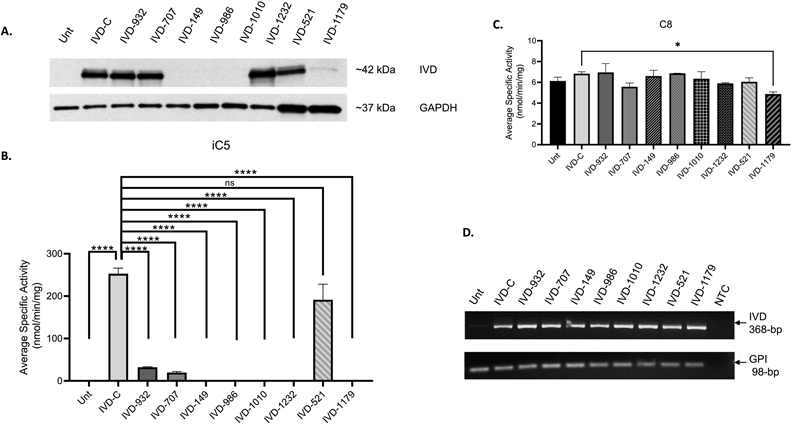
(A) Detection of IVDH protein levels by western blot to evaluate protein stability of control and variants expressed from cDNA vectors with GAPDH an internal loading standard. (B) Activity of IVDH and (C) MCAD using the ETF fluorescence reduction assay. IVDH activity assays were done in triplicates and octanoyl-CoA assays were done in duplicates. Means and standard deviations were calculated. Data were analyzed using unpaired t test showing significant difference, ****p<0.0001, ns = no statistical difference.
Computational prediction of mutations’ adverse structure function effect
To assess the potential adverse structure/function changes caused by these mutations, two tools are utilized: in silico molecular modeling using the crystal structure of IVDH, a dimer of dimers, and a modeled IVDH:ETF mutant ternary complex and homology alignment of IVDH from various eukaryotic species (Fig. 5). Arg21 (mature numbering), an invariant across species, is located at the middle of α-helix A and is a structurally important residue as it functions synergistically with Arg308 (mature numbering), an invariant, to anchor the N-terminus α-helix A region to the rest of the monomer through α-helices G and C [23] (Fig. 5). Both arginine residues interact with Asp7, highly conserved, with the Arg21 also interacting with the invariant Tyr312 (mature numbering) and the Arg308 interacting with the invariant Gln267 (mature numbering). The ridged kink introduced by the Arg21Pro mutation affects the secondary and supersecondary structure significantly of the monomer but also tetramer assembly as the N-terminus α-helix A region appears to be involved in the final process of monomer folding as the 29-amino acid-precursor peptide is removed (Fig. 5). Moreover, proper positioning of residues in this region affects tetramer assembly as Ile10 residue, monomer A, would lose key hydrophobic interaction with the invariants Val314' and Ala317' (mature numbering) from the opposite fourth monomer D (Fig. 5). On the other hand, the Arg308Gln replacement is predicted as a milder change, albeit weakening the same region proper side chains alignments of key residues. Unlike patients with the Arg21Pro replacement, patients with Arg308Gln replacement may potentially respond to treatments that promote stability of the monomer, including substrate analogs and riboflavin (Fig. 5).
Figure 5. Ribbon representation of the three dimensional structure of an IVDH monomer using published atomic coordinates, PDB: 1IVH [23].

iC5-CoA is modeled in the active site in place of the CoA-persulfide published. The A and B views, at near right angle, depict the position of the backbone carbon atom, in blue, of amino acid residues replaced due to missense mutations in the IVD gene in patients with IVA listed in Table 1. The c.1179del cause a frameshift and a Leu365Phe mutation, position marked in red, and premature termination and leading to the loss of most of the a-helix J and a-helix K and undetectable IVDH antigen band, see Figure 4.
While Thr207 residue is located on β-strand 6, an important component of the β-sheet domain critical for FAD binding [23], the residue is structurally dispensable as it is conserved among higher eukaryotes but not so in lower eukaryotes (it is a valine in Drosophila, glutamine in C. elegcins, and Arg in mosquito). Since the Thr207 methyl moiety is within interacting distance, 3.9 Å to, Phe205, which is an invariant, this implies that other than threonine side chain has to fit in a specific conformation. While the side chain of a valine would nearly fit spatially in the space occupied by a threonine side chain, but in silico replacement predicts that a glutamine side chain is less accommodated unless it projects out towards solvent. Interestingly, in silico replacement with an Arg at this position, and Pro at position 209, has significantly lower energy as the side chain, the guandinium moiety, projects out towards the solvent. As discussed previously Ala282Val adversely affect binding of the substrate bound to the second subunit in the dimer set. However, the replacement also has some impact on protein stability [32, 33]. The IVDH Ala282Val instability is exhibited by the recombinant purified form lower melting temperature that does not improve in the presence of substrate or substrate analogues [33], providing evidence that fever is a risk factor for IVA patients with this mutation.
The c.986T>C nucleotide mutation cause an Met300Thr (mature numbering) change. Met300 residue located near the middle of α-helix H is an invariant residue located in a hydrophobic pocket formed by Val269, Thr273, Leu304, Val342 (invariant), and others (Fig. 5). The hydrophobic pocket stabilizes the supersecondary interactions of α-helices H and G. Although replacement of Met300 with a Thr should disrupt the supersecondary “glue” stabilizing function of the hydrophobic pocket, it could be tolerated to some extent provided folding is completed to the functionally active protein. Similar to the Arg308Gln mutation discussed above, this mutation may also respond to substrate analogue treatment.
The effect of c.1232G>A mutation leading to the Arg382Gln (mature numbering) change on protein stability has been examined previously [32], errored as an Arg382Leu change). The guanidinium group of Arg382 in monomer A plays a role in tetramer assembly and maintain the protein quaternary structure as both its η-N nitrogens are locked in their spatial position by the carboxylates of the invariants Glu337 and Glu379 from monomer A while its ε-N nitrogen interact with the carboxylate of the invariant Asp299" of monomer D [23]. The Arg382Gln should thus disrupt the efficiency of tetramer assembly and cause instability when formed. Because of the contribution the Gln291" residue (an invariant) of monomer D in stabilizing binding of the adenine of the FAD bound to monomer A, enzyme kinetic mechanism is also likely affected assuming cooperativity between the two subunits is a valid proposition [34, 35]. In addition, it is also reasonable to assume with multiple points of contact between the adenine of the FAD bound to monomer A and monomer D that the FAD adenine contributes to the quaternary stability of the tetramer, and therefore maximum amounts of free FAD in mitochondria may be helpful in stabilizing this IVDH Arg382Gln mutant and hence riboflavin (vitamin B2) treatment may be beneficial in treating patients with this mutation.
The c.521T>G nucleotide mutation cause a Val145Gly change. Val145 (mature numbering) is located at the end of the loop connecting β-strands 1 and 2 and juxtaposed to the ribose moiety of the CoA substrate and the loop connecting β-strands 4 and 5, which is involved in binding of the ribose moiety 2'-phosphate of the CoA substrate through the highly conserved Arg191 (mature numbering) (Fig. 5). While binding affinity and Km of the substrate is expected to be affected, folding and/or stability is also expected to be affected, but to a lesser extent compared to the IVDH Ala311Val (Ala282Val) mutant.
Discussion
Except in specific disorders, most patients with inborn errors of metabolism are identified with rare unique mutations in the presumed affected gene, many of which are formally classified as VUS. Indeed, new guidance by ClinVar following the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) standards and guidelines for variant calling, requires specific demonstration of interrupted function of individual variant alleles to be classified as pathogenic [36-38]. To meet this goal, it will be necessary to develop high throughput methods to assess the functional effects of mutant alleles on enzyme activity for most of the inborn errors of metabolism identified by NBS. We have previously used both prokaryotic and eukaryotic expression systems (including in HEK293 deletion cell lines) to demonstrate the effect of variants in most of the acyl-CoA dehydrogenases including mutations identified in ACADVL, ACADM, ACADS, IVD, SBCADH, ACAD8, and ACAD9 [21, 32, 39-44]. However, our previous techniques were not particularly well suited to high throughput analysis. In this study, we describe the generation of a deletion in the IVD gene in HEK293T cells using CRISPR/Cas9 technology that can readily be applied to essentially any gene of interest. HEK293T is a well-established cell line to study expression recombinant proteins [45] and though it is aneuploid at multiple loci [30, 31] we were able to obtain a high frequency of CRISPR/Cas9 genome-edited IVD null lines. By using a commercial gene synthesis service to rapidly generate IVD variants for expression in our IVD null HEK293T cells, we have been able to confirm pathogenicity of 5 previously uncharacterized variants, and confirmed the partial retention of activity of the common IVD variant identified through NBS. These studies were in keeping with confirmation of IVDH-deficiency in cell lines from four patients identified by NBS as likely having IVA. Of note, one IVA patient (FB909) was of Ashkenazi Jewish origin and had a homozygous c.1232G>A (p.Arg411Gln) VUS with minimal enzymatic activity suggesting this mutation may be an IVD variant specific and of importance to this population. Molecular modeling results are consistent with our in vitro findings and provide additional insight into the molecular pathophysiology of the individual mutants. results are consistent with our in vitro findings and provide additional insight into the molecular pathophysiology of the individual mutants.
Based on the results above, we propose that the VUS examined here be reclassified in ClinVar. Variants c.707T>C, (p.Thr236Ile), c.986T>C (p.Met329Thr), c.1010G>A (p.Arg337Gln), and c.1232G>A (p.Arg411Gln) should be reclassified from VUS to pathogenic/likely pathogenic; and c.521T>G (p.Val174Gly) from VUS to benign/likely benign. Variants c.932C>T (p.Ala311Val), c.149G>C (p.Arg50Pro), and c.1179del (p.Leu394 fs) should maintain their pathogenic/likely pathogenic status.
While this study used a traditional cuvette based fluorometric assay to measure IVDH activity, we have recently published a microplate version of this assay that is more amenable to high throughput analysis and can be applied to any of the acyl-CoA dehydrogenases [46]. For enzymes relevant to metabolic disease identified by NBS but without an assay amendable to high throughput analysis, an indirect assay can still lead to high throughput confirmation of pathogenicity. In the context of most disorders identified by tandem mass spectrometry, an adaptation of the standard whole cell acylcarnitine profiling should be possible with appropriate gene deleted HEK293T or other cell lines. Ultimately, development of a panel of such cell lines should allow relatively rapid confirmation of pathogenicity of variants identified in infants through NBS without the need for an invasive procedure such as a skin biopsy.
In summary, we have developed an IVD null HEK293T cell line with no IVDH protein or activity that allowed determination of pathogenicity of several previous VUS found in patients with suspected IVA identified by NBS. This model will allow for high throughput assessment of the function of VUS in multiple genes for disorders identified by NBS and would be an ideal supplement to existing NBS programs to provide greater clarity of screening results.
Supplementary Material
Acknowledgements
JV was supported in part by NIH grant R01 DK109907. We thank the staff from the Core Flow Cytometry Laboratory from the University of Pittsburgh, Children’s Hospital of UPMC for help with flow sorting of GFP positive cells. We thank Yuxun Zhang, PhD from Eric Goetzman, PhD Laboratory, University of Pittsburgh, Children’s Hospital of UPMC for ETF supply for the ETF fluorescence reduction assays.
Footnotes
Conflict of interest
The authors certify that they have no affiliations with or involvement in any organization or entity with any financial or non-financial interest in the subject matter or materials discussed in this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Oglesbee D, et al. , Second-tier test for quantification of alloisoleucine and branched-chain amino acids in dried blood spots to improve newborn screening for maple syrup urine disease (MSUD). Clin Chem, 2008. 54(3): p. 542–9. [DOI] [PubMed] [Google Scholar]
- 2.Narravula A, et al. , Variants of uncertain significance in newborn screening disorders: implications for large-scale genomic sequencing. Genet Med, 2017. 19(1): p. 77–82. [DOI] [PubMed] [Google Scholar]
- 3.Wilcken B, Medicine. Newborn screening: gaps in the evidence. Science, 2013. 342(6155): p. 197–8. [DOI] [PubMed] [Google Scholar]
- 4.Tanaka K, et al. , Isovaleric acidemia: a new genetic defect of leucine metabolism. Proc Natl Acad Sci USA, 1966. 56(1): p. 236–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Online, M.I.o.M., OMIM Isovaleric Acidemia, IVA #243500. OMIM, Mendelian Inheritance of Man, OMIM. [Google Scholar]
- 6.Vockley J, et al. , Branched Chain Organic Acidurias, in The Online Metabolic and Molecular Bases of Inherited Disease, Valle D, et al. , Editors. 2019, McGraw-Hill Education: New York, NY. [Google Scholar]
- 7.Tanaka K, Isovaleric acidemia: personal history, clinical survey and study of the molecular basis. Prog Clin Biol Res, 1990. 321: p. 273–90. [PubMed] [Google Scholar]
- 8.Vockley J and Ensenauer R, Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity. Am J Med Genet C Semin Med Genet, 2006. 142C(2): p. 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McHugh D, et al. , Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet Med, 2011. 13(3): p. 230–54. [DOI] [PubMed] [Google Scholar]
- 10.Fingerhut R and Olgemoller B, Newborn screening for inborn errors of metabolism and endocrinopathies: an update. Anal Bioanal Chem, 2009. 393(5): p. 1481–97. [DOI] [PubMed] [Google Scholar]
- 11.Berry GT, Yudkoff M, and Segal S, Isovaleric acidemia: medical and neurodevelopmental effects of long-term therapy. J Pediatr, 1988. 113(1 Pt 1): p. 58–64. [DOI] [PubMed] [Google Scholar]
- 12.Chinen Y, et al. , Isovaleric acidemia: Therapeutic response to supplementation with glycine, l-carnitine, or both in combination and a 10-year follow-up case study. Mol Genet Metab Rep, 2017. 11: p. 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krieger I and Tanaka K, Therapeutic effects of glycine in isovaleric acidemia. Pediatr Res, 1976. 10(1): p. 25–9. [DOI] [PubMed] [Google Scholar]
- 14.Mayatepek E, Kurczynski TW, and Hoppel CL, Long-term L-carnitine treatment in isovaleric acidemia. Pediatr Neurol, 1991. 7(2): p. 137–40. [DOI] [PubMed] [Google Scholar]
- 15.Roe CR, et al. , L-carnitine therapy in isovaleric acidemia. J Clin Invest, 1984. 74(6): p. 2290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ensenauer R, et al. , A common mutation is associated with a mild, potentially asymptomatic phenotype in patients with isovaleric acidemia diagnosed by newborn screening. Am J Hum Genet, 2004. 75(6): p. 1136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ensenauer R, et al. , Newborn screening for isovaleric acidemia using tandem mass spectrometry: data from 1.6 million newborns. Clin Chem, 2011. 57(4): p. 623–6. [DOI] [PubMed] [Google Scholar]
- 18.Concordet JP and Haeussler M, CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res, 2018. 46(W1): p. W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ran FA, et al. , Genome engineering using the CRISPR-Cas9 system. Nat Protoc, 2013. 8(11): p. 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frerman FE and Goodman SI, Fluorometric assay of acyl-CoA dehydrogenases in normal and mutant human fibroblasts. Biochem Med, 1985. 33(1): p. 38–44. [DOI] [PubMed] [Google Scholar]
- 21.Vockley J, et al. , Mammalian branched-chain acyl-CoA dehydrogenases: molecular cloning and characterization of recombinant enzymes. Methods Enzymol, 2000. 324: p. 241–58. [DOI] [PubMed] [Google Scholar]
- 22.Dube S, Qin J, and Ramakrishnan R, Mathematical analysis of copy number variation in a DNA sample using digital PCR on a nanofluidic device. PLoS One, 2008. 3(8): p. e2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiffany KA, et al. , Structure of human isovaleryl-CoA dehydrogenase at 2.6 A resolution: structural basis for substrate specificity. Biochemistry, 1997. 36(28): p. 8455–64. [DOI] [PubMed] [Google Scholar]
- 24.Toogood HS, et al. , Extensive domain motion and electron transfer in the human electron transferring flavoprotein.medium chain Acyl-CoA dehydrogenase complex. J Biol Chem, 2004. 279(31): p. 32904–12. [DOI] [PubMed] [Google Scholar]
- 25.Parimoo B and Tanaka K, Structural organization of the human isovaleryl-CoA dehydrogenase gene. Genomics, 1993. 15(3): p. 582–90. [DOI] [PubMed] [Google Scholar]
- 26.Vockley J, et al. , Exon skipping in IVD RNA processing in isovaleric acidemia caused by point mutations in the coding region of the IVD gene. Am J Hum Genet, 2000. 66(2): p. 356–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsubara Y, et al. , Molecular cloning and nucleotide sequence of cDNAs encoding the precursors of rat long chain acyl-coenzyme A, short chain acyl-coenzyme A, and isovaleryl-coenzyme A dehydrogenases. Sequence homology of four enzymes of the acyl-CoA dehydrogenase family. J Biol Chem, 1989. 264(27): p. 16321–31. [PubMed] [Google Scholar]
- 28.Dickinson ME, et al. , High-throughput discovery of novel developmental phenotypes. Nature, 2016. 537(7621): p. 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraft K, et al. , Deletions, Inversions, Duplications: Engineering of Structural Variants using CRISPR/Cas in Mice. Cell Rep, 2015. 10(5): p. 833–839. [DOI] [PubMed] [Google Scholar]
- 30.Bylund L, et al. , Analysis of the cytogenetic stability of the human embryonal kidney cell line 293 by cytogenetic and STR profiling approaches. Cytogenet Genome Res, 2004. 106(1): p. 28–32. [DOI] [PubMed] [Google Scholar]
- 31.Lin YC, et al. , Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat Commun, 2014. 5: p. 4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohsen A-W, et al. , Characterization of molecular defects in isovaleryl-CoA dehydrogenase in patients with isovaleric acidemia. Biochemistry, 1998. 37(28): p. 10325–35. [DOI] [PubMed] [Google Scholar]
- 33.Nasser I, et al. , Thermal unfolding of medium-chain acyl-CoA dehydrogenase and iso(3)valeryl-CoA dehydrogenase: study of the effect of genetic defects on enzyme stability. Biochim Biophys Acta, 2004. 1690(1): p. 22–32. [DOI] [PubMed] [Google Scholar]
- 34.Mohsen A-W, Navarette B, and Vockley J, Identification of Caenorhabditis elegans isovaleryl-CoA dehydrogenase and structural comparison with other acyl-CoA dehydrogenases. Mol Genet Metab, 2001. 73(2): p. 126–37. [DOI] [PubMed] [Google Scholar]
- 35.Kormanik K, Identification and characterization of the biochemical and physiological functions of acyl-coa dehydrogenase 10, in Human Genetics. 2014, University of Pittsburgh: d-scholarship.pitt.edu. p. 376. [Google Scholar]
- 36.Richards S, et al. , Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015. 17(5): p. 405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landrum MJ, et al. , ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res, 2018. 46(D1): p. D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landrum MJ, et al. , ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res, 2014. 42(Database issue): p. D980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schiff M, et al. , Complex I assembly function and fatty acid oxidation enzyme activity of ACAD9 both contribute to disease severity in ACAD9 deficiency. Hum Mol Genet, 2015. 24(11): p. 3238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goetzman ES, et al. , Expression and characterization of mutations in human very long-chain acyl-CoA dehydrogenase using a prokaryotic system. Mol Genet Metab, 2007. 91(2): p. 138–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mohsen A-W and Vockley J, High-level expression of an altered cDNA encoding human isovaleryl-CoA dehydrogenase in Escherichia coli. Gene, 1995. 160(2): p. 263–7. [DOI] [PubMed] [Google Scholar]
- 42.Pedersen CB, et al. , Misfolding, degradation, and aggregation of variant proteins. The molecular pathogenesis of short chain acyl-CoA dehydrogenase (SCAD) deficiency. J Biol Chem, 2003. 278(48): p. 47449–58. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen TV, et al. , Identification of isobutyryl-CoA dehydrogenase and its deficiency in humans. Mol Genet Metab, 2002. 77(1-2): p. 68–79. [DOI] [PubMed] [Google Scholar]
- 44.Schowalter DB, Matern D, and Vockley J, In vitro correction of medium chain acyl CoA dehydrogenase deficiency with a recombinant adenoviral vector. Mol Genet Metab, 2005. 85(2): p. 88–95. [DOI] [PubMed] [Google Scholar]
- 45.Thomas P and Smart TG, HEK293 cell line: a vehicle for the expression of recombinant proteins. J Pharmacol Toxicol Methods, 2005. 51(3): p. 187–200. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, et al. , An acyl-CoA dehydrogenase microplate activity assay using recombinant porcine electron transfer flavoprotein. Anal Biochem, 2019. 581: p. 113332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.