

Abstract
In oncology, comprehensive omics and functional enrichment studies have led to an extensive profiling of (epi)genetic and neurobiological alterations that can be mapped onto a single tumor’s clinical phenotype and divergent clinical phenotypes expressing common pathophysiological pathways. Consequently, molecular pathway-based therapeutic interventions for different cancer typologies, namely tumor type- and site-agnostic treatments, have been developed, encouraging the real-world implementation of a paradigm shift in medicine.
Given the breakthrough nature of the new-generation translational research and drug development in oncology, there is an increasing rationale to transfertilize this blueprint to other medical fields, including psychiatry and neurology. In order to illustrate the emerging paradigm shift in neuroscience, we provide a state-of-the-art review of translational studies on the β-site amyloid precursor protein cleaving enzyme (BACE) and its most studied downstream effector, neuregulin, which are molecular orchestrators of distinct biological pathways involved in several neurological and psychiatric diseases. This body of data aligns with the evidence of a shared genetic/biological architecture among Alzheimer’s disease, schizoaffective disorder, and autism spectrum disorders.
To facilitate a forward-looking discussion about a potential first step towards the adoption of biological pathway-based, clinical symptom-agnostic, categorization models in clinical neurology and psychiatry for precision medicine solutions, we engage in a speculative intellectual exercise gravitating around BACE-related science, which is used as a paradigmatic case here.
We draw a perspective whereby pathway-based therapeutic strategies could be catalyzed by high-throughput techniques embedded in systems-scaled biology, neuroscience, and pharmacology approaches that will help overcome the constraints of traditional descriptive clinical symptom and syndrome-focused constructs in neurology and psychiatry.
Keywords: β-site amyloid precursor protein cleaving enzyme (BACE), systems biology, precision medicine, systems pharmacology, neurology, psychiatry
1. INTRODUCTION
Translational and in-human studies indicate shared pathophysiological commonalities between complex multi-factorial neurological and psychiatric diseases such as brain proteinopathies and neurodegenerative diseases, i.e., Alzheimer’s disease (AD), schizoaffective disorder, autism spectrum disorders, and mood disorders [1, 2]. A multi-system overlap, from genetic to molecular, cellular, synaptic, and large-scale neural networks, coupled with comorbidities and brain structural and functional commonalities, led to inferring common etiologic and neurobiological determinants among clinically and phenotypically divergent neurological and psychiatric (neuropsychiatric) diseases and disorders [1, 2].
Therefore, it is conceivable that one single compound targeting specific aberrant molecular pathways shared by a wider set of neuropsychiatric conditions could be effectively developed for clinical phenotypic disease spanning disease-modifying therapeutic approaches. This theoretical research and development framework in neuroscience is becoming a reality in some medical specialties, such as oncology, where tumor- and site-agnostic treatments have already been approved or are under late-stage development [3-5].
In the last 3 years, the U.S. Food and Drug Administration (FDA) has authorized tissue-and tumor-agnostic solutions, whereby the patient’s tumor biomarker-based profiling, reflecting genetic factors and primary aberrant molecular pathways, rather than “traditional” assessments (histology, site, and clinical manifestation) play a critical role in the therapeutic decision-making process [3-5].
Here, we propose the concept of a similar clinical syndrome-agnostic therapeutic paradigm that could be explored and applied in neuroscience and neuropharmacology, taking advantage of operating models adopted in oncology experimental and clinical research.
The first objective of this approach involves identifying molecular factors that increase vulnerability to complex neuropsychiatric diseases and are responsible for maladaptive biological responses and/or ineffective compensatory mechanisms to incipient pathophysiological alterations [6, 7]. The second goal is to map out critical molecular pathways that account for adaptive and compensatory capacity and ensure improved thresholds of resilience under initiated pathophysiological dynamics, which seems a viable therapeutic avenue supported by methodological advances, including high-throughput techniques and omics sciences [8, 9]. Achieving these two objectives would represent a crucial step for time-sensitive and effective disease-modifying therapies capable of fostering brain resilience to incipient pathophysiological alterations as well as prolonging healthspan.
The β-secretase, a membrane-bound aspartic protease termed β-site amyloid precursor protein (APP) cleaving enzyme (BACE), is widely known for the isoform 1 (BACE1) and its role in AD pathophysiology [10, 11]. BACE1 molecular pathways are expressed across different pathophysiological conditions, spanning neurodevelopment and neurodegeneration as well as childhood, adulthood, and aging. BACE1 (and isoform 2) is a crucial enzyme required not only for the processing of APP into amyloid-β (Aβ) but also for other potential substrates that are implicated in neuronal/glia functions and synaptic homeostasis [11, 12]. Translational and large-scale genetic studies show that both BACE1 and BACE2 and related downstream pathways are involved in a broad set of neuropsychiatric diseases with clinically divergent phenotypes such as age-related cognitive diseases, including AD [11, 12], AD-Down Syndrome (DS) complex [13, 14], schizoaffective disorder [15-18], and autism spectrum disorders (ASD) [19-21]. To date, the most relevant support of this hypothesis is that germline BACE knockout (KO) mice exhibit complex neurological and behavioral phenotypes resembling clinical and neurobiological features lying in the spectrum of several human cerebral illnesses [22-24].
In the present article, we revise the most prominent literature on the association between BACE biology (with a focus on the BACE-neuregulin axis for which more extensive experimental and in-human evidence is available) and neuropsychiatric diseases. The intent of our somewhat speculative intellectual endeavor, gravitating around BACE as a mere paradigmatic model, is to stimulate the debate regarding the opportunity of extending and planning future clinical research in the direction of biological pathway-based and symptoms-agnostic therapies in the field of neurology and psychiatry. While we are utterly aware that several knowledge gaps still exist in the field of BACE biology, and therefore we do not claim that clinical trials should be put in place, we also point at an existing body of experimental and in-human evidence that could support further research in the direction mentioned above. In this perspective, capitalizing on the success of oncology operating models and blueprints may represent the doorway for accomplishing precision medicine in neuroscience.
1.1. BACE1 and BACE2 Biology: A Concise Overview
BACE1 is a type I transmembrane aspartyl protease belonging to the pepsin family. The identification of BACE1 [10, 25], encoded by a chromosome 11 gene, dates back to more than 20 years ago, and it shares 59% of its amino acid sequence with the homolog BACE2 [26, 27], residing in the obligate DS region of chromosome 21 [25, 28]. The two proteases have an identical structural domain and localize on the plasma membrane, endoplasmic reticulum, Golgi apparatus, and in the endosomal compartments as well as in healthy synaptic terminals [25, 26, 28].
BACE1 is widely expressed in the brain, especially in neurons, oligodendrocytes, and astrocytes, with particular abundance in various neuronal cell types [10]. Unlike BACE1, BACE2 mRNA expression levels are very low or undetectable in human adult and fetal brains. In contrast, its expression is higher in peripheral tissues, including melanocytes, colon, kidney, pancreatic beta cells, placenta, prostate, stomach, endothelial cells, and trachea [26, 27].
1.2. BACE 1 and 2 Physiological Functions: The Amyloidogenic Pathway
The Aβ pathway is a physiological, molecular pathway that serves several physiological brain functions and is primarily known for its dysregulation and pathophysiological role in AD and DS [12].
BACE1 is the β-secretase enzyme that cleaves the transmembrane APP and, together with γ-secretase, generates the 42-amino acid-long amyloid β peptide (Aβ42), the likely toxic initiator of AD. BACE1 proteolytic processing of APP represents the rate-limiting step for Aβ production (Fig. 1). APP is a type I transmembrane protein and is highly expressed in neurons and at synapses. Experimental evidence indicates that APP is involved in synaptic remodeling, plasticity, and excitatory/inhibitory balance maintenance [29,30]. Soluble APP is a γ-aminobutyric acid type B receptor (GABABR)-ligand that regulates neurotransmission [31]. Moreover, Aβ monomers derived from the APP cleavage may have intrinsic physiological properties since there are experimental data showing Aβ-related stimulation of intracellular signaling essential for hippocampal synaptic and neuronal bioenergetic homeostasis [32-35]. Moreover, some truncated APP forms can initiate CREB-mediated neuroprotective pathways involved in hippocampal neurogenesis, a critical mechanism for adult long-term potentiation (LTP), which represents the neural substrate of synaptic plasticity, memory, and learning [32-35].
Fig. (1).
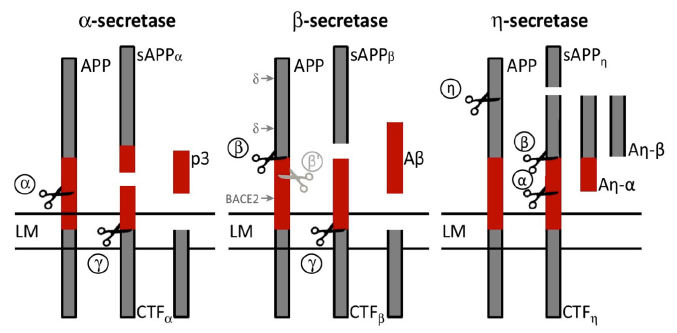
Schematic representation of amyloid precursor protein (APP) processing pathways. BACE1 functions as the β-secretase enzyme by cleaving the transmembrane APP to release the β-stubs. BACE1 cleavage of APP represents the rate-limiting step for Aβ production. Cleavage of APP by BACE1 liberates the soluble N-terminus of APP, while the C-terminal fragment (CTF-β or C99) remains bound to the membrane. To produce Aβ, the fragment CTF-β is cleaved by β-secretase, which finally releases Aβ into the extracellular space and the APP intracellular domain into the cytoplasm. In a parallel competing non-amyloidogenic pathway, APP is cleaved either by α-secretase or η-secretase to release two additional variants of the APP ectodomain, namely sAPP-α and sAPP-η
Abbreviations: Aβ = amyloid-β; APP = amyloid precursor protein; BACE1 = β-site amyloid precursor protein cleaving enzyme 1; CTF-β = β-C-terminal fragment; sAPP = soluble amyloid precursor protein.
Note: Adapted from The β-Secretase BACE1 in Alzheimer's Disease. Biological Psychiatry, 2020, S0006-3223(20)30063-9. https://doi.org/10.1016/j.biopsych.2020.02.001
As observed for BACE1, BACE2 can cleave APP at the β-site level and compete for the substrate with its most common homolog [26, 36]. In addition to its pro-amyloidogenic role as an auxiliary β-secretase, BACE2 exerts two other additional activities with regards to APP processing: I) it acts at the θ-site (Phe20) as a θ-secretase degrading the β-C-terminal fragment (CTFβ) and producing C80, thus preventing the formation of Aβ [37, 38]; II) it serves as an Aβ-degrading protease (AβDP) cutting, at overly acidic pH, synthetic Aβ peptides after aa20 and aa34, to generate the 1-20 and 1-34 peptide products. BACE2 can also degrade Aβ with an efficiency second only to the insulin-degrading enzyme (IDE) [26, 39].
1.3. BACE 1 and 2 Physiological Functions: Neuronal and Synaptic Homeostasis
Consistent with high BACE1 expression and presynaptic localization in neurons, KO of the BACE1 gene in the germline of mice leads to toxic phenotypes, including impaired neurogenesis and astrogenesis, reduced spine density, axon targeting errors, defective myelination, neurochemical abnormalities, increased vulnerability to neurodegeneration, retinal pathology, endophenotypes of schizophrenia (e.g., neuregulin), memory and learning deficits, and seizures (Table 1) [10, 22, 23, 40]. When BACE1 is deleted in adult mouse brains, an impaired synaptic function is reported, as manifested in LTP reduction. Moreover, a decrease in the hippocampal mossy fiber length infrapyramidal bundle is observed [10, 22, 23, 40]. Moreover, there are subtle alterations in the steady-state inactivation of voltage-gated sodium channels (NaV) in cortical neurons, leading to increased excitability [10, 22, 23, 40].
Table 1.
BACE1 null phenotypes in the Central Nervous System.
| Phenotype | Substrate |
|---|---|
| Astrogenesis increase, neurogenesis decrease | Jag1 |
| Axon guidance defects | CHL1 |
| Hyperactivity | NRG1 |
| Hypomyelination | NRG1 |
| Memory deficits | - |
| Neurochemical deficits | - |
| Neurodegeneration w/ age | NaVβ2 |
| Post-natal lethality, growth retardation | - |
| Retinal pathology | VEGFR1 |
| Schizophrenia endophenotypes | NRG1 |
| Seizures | NaVβ2 |
| Spine density reduction | NRG1 |
Abbreviations: CHL1 = neural cell adhesion molecule L1; Jag1 = Jagged-1; NaVβ2 = Voltage-gated sodium channels beta 2; NRG1 = neuregulin 1; SEZ6 = seizure-related protein 6; VEGFR1 = Vascular endothelial growth factor receptor 1.
Neuregulin 1 (NRG1) is the most widely investigated synaptic substrate of BACE1 in experimental models and human individuals and patients across different neuropsychiatric diseases. NRG1 interacts with the epidermal growth factor receptor (EGFR) family of receptors to exert signaling cascades crucial for central nervous system (CNS) development and synaptic plasticity [15, 41]. BACE1 cleavage of NRG1 promotes myelination in the central/peripheral nervous systems (CNS/PNS) and regulates muscle spindle formation and maintenance [15, 42]. BACE1-dependent NRG1/receptor tyrosine-protein kinase erbB-4 (ErbB4) signaling is also implicated in neuronal excitability, synaptic plasticity, learning and memory processes, and neuronal survival (Fig. 2), mainly by the modulation of group 1 metabotropic glutamatergic receptors (mGluRI) activity [15, 42].
Fig. (2).
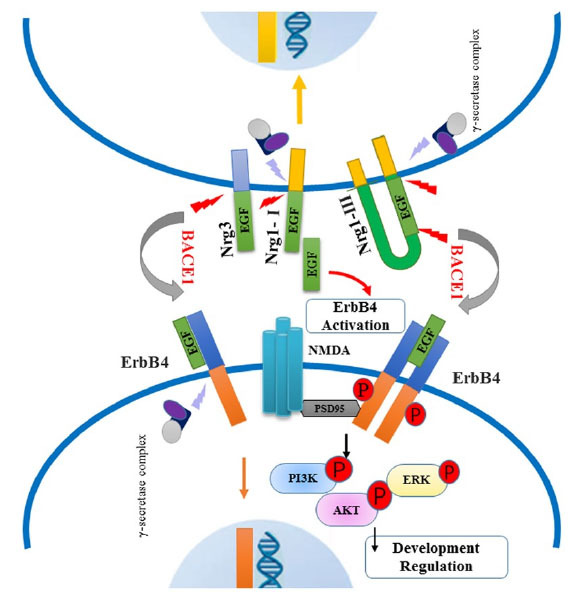
The BACE1-NRG1 axis. NRG1 is a transmembrane protein expressed mainly in the frontal cortex, hippocampus, and cerebellum, as well as in the peripheral nervous system (PNS) dopaminergic neurons. The NRG1-type I and NRG1-type III isoforms (the latter is the primary variant expressed in the brain) are validated substrates of BACE1. NRG1-type III is a hairpin-shaped protein precursor with two membrane-spanning domains. Its cleavage performed by BACE1 leads to two separate fragments anchored to the membrane: the N-terminal fragment (NTF) (NRG1-NTF) and the C-terminal fragment (NRG1-CTF), on the external and internal (cytosolic) sides of the membrane, respectively. NRG1-induced cellular responses are primarily mediated by binding to tyrosine kinase receptors of the ErbB family, especially to ErbB2, ErbB3, and ErbB4.
BACE1-cleaved NRG1-NTF fragment contains an epidermal growth factor (EGF)-like domain that binds and activates the ErbB receptors involved in the NRG1/ErbB signaling pathway. This increases the phosphorylation of downstream signaling molecules: the extracellular signal-regulated kinase (ERK) and the serine/threonine kinase (Akt). The activation of the NRG1/ErbB signaling pathway is necessary to modulate cell survival, synaptic development, glutamatergic transmission (through regulation of NMDA receptor expression and turn-over), neuronal migration, and myelination, and control the internalization of mGluR1 on dopaminergic neurons.
Abbreviations: BACE1 = β-site amyloid precursor protein cleaving enzyme 1; EGF = epidermal growth factor; ERK = extracellular signal-regulated kinase; mGLUR1 = group 1 metabotropic glutamatergic receptors; NMDA = N-methyl-D-aspartate; NRG1 = neuregulin-1; NRG1-CTF = NRG1 C-terminal fragment; NRG1-NGF = NRG1 N-terminal fragment; PNS = peripheral nervous system.
Note: Adapted from BACE1-Dependent Neuregulin-1 Signaling: An Implication for Schizophrenia. Front Mol Neurosci, 2017, Sep 25;10:302. doi: 10.3389/fnmol.2017.00302.
BACE1 is essential for producing soluble seizure-related protein 6 (SEZ6), whose suppression by either gene KO or pharmacological inhibition in mouse models of brain development is associated with typical phenotypes exhibiting abnormal dendritic spines density, impaired hippocampal LTP, and loss of synaptic plasticity [43, 44]. BACE1 cleavage of the close homolog of neural cell adhesion molecule L1 (CHL1) and/or its homolog L1 cell adhesion molecule (L1CAM) generates an intracellular membrane-bound C-terminal peptide that is active in the regulation of axon guidance (cytoskeleton remodeling and turn-over) upon presentation of the semaphorin 3A (Sema3A) cue [45, 46]. Conditional BACE1 KO mice show axon guidance abnormalities in the hippocampus, which may be related to a lack of CHL1 functions [45, 46].
Despite close homology, the synaptic substrate profiles of BACE1 and BACE2 do not entirely overlap. Neuregulin 2 (NRG2), a homolog of NRG1 equally expressed in different brain regions, including the hippocampus, is cleaved by BACE2, thus generating a C-terminal fragment that serves as a substrate for γ-secretase [47, 48]. Vascular cell adhesion molecule 1 (VCAM1) is an exclusive BACE2 substrate, expressed in astrocytes and microglia and cleaved in a pro-inflammatory microenvironment induced by the tumor necrosis factor alfa (TNF-α) [48].
2. PHARMACOLOGICAL MODULATION OF BACE PATHWAYS IN ALZHEIMER’S DISEASE PATHOLOGY
2.1. BACE1 in Alzheimer’s Disease
In AD, the evidence of the role of BACE1 in the dysregulation of the Aβ pathway and disease pathophysiological progression has been extensively reported and documented. Briefly, the discovery of the Icelandic genetic variant of APP (a point, missense protective variant for AD) provided one of the first demonstrations of the role of BACE1 in the aberrant Aβ pathway characterizing AD pathophysiology. The Icelandic variant (A673T mutation) is associated with a different BACE1 recognition motif at P2 (A673T), resulting in 30% lower levels of Aβ40 and Aβ42 in plasma of individuals carrying the mutation compared to control individuals [49]. By contrast, the E682K and the Swedish mutations in APP, located at the P1’ and P2-P1 β’ subsites, are characterized by a shift of BACE1 cleavage toward the β-site, thus increasing Aβ production [50, 51].
Neuropathological studies show that AD brain neurons have higher BACE1 enzymatic activity compared to cognitively healthy individuals [25, 52]. Moreover, mouse models of AD and human pathological studies report a relatively large accumulation of BACE1 in neuritic dystrophies in close vicinity of Aβ fibrillary deposits and plaques [53, 54].
When taken together, all these human studies provide a robust proof-of-principle for inhibition of the β-cleavage of APP as a viable therapeutic strategy for AD. Despite the strong scientific rationale, all BACE1 inhibitors investigated so far were discontinued for futility or safety reasons. Such a dramatic rate of failures has raised theoretical concerns around the validity of BACE1 as a molecular target for AD. Potential critical factors accounting for trial failures are under debate within the scientific community and may lie in the lack of biomarker-driven trial decision making (including proof of target engagement and treatment effect/safety) coupled with an incomplete understanding of BACE1 homeostasis/biology that may have led to over-inhibition or scarce substrate selectivity (see the Discussion section for more extensive and granular argumentation).
2.2. BACE1 in the Alzheimer’s-Down Syndrome Complex
Adult individuals carrying the 21-chromosome trisomy and manifesting DS clinical phenotypes have a higher risk of developing AD pathology and cognitive decline in an age-dependent fashion [13, 55, 56]. DS adults in their fourth decade of age have a likelihood of up to 50% to manifest overt cognitive impairment, and a lifetime prevalence of 70% of AD pathophysiological hallmarks including Aβ and tau pathology and dementia has been reported [13, 55, 56]. Besides in-human neuropathological findings showing co-occurrence of AD pathology in the DS brain, preliminary experimental models support the involvement of BACE1 in AD-like pathological changes observed in DS. In particular, the first and only study conducted so far in the mouse model of DS-AD suggests a potential therapeutic role of BACE1 inhibitors in DS patients. Individuals with DS are at increased risk of developing AD compared to age-matched healthy subjects, as a consequence of trisomy 21 with subsequent over-production of the APP and of its amyloidogenic peptide products (Aβ40 and Aβ42), although not all DS patients invariably develop AD symptoms during adult age [14, 57]. According to this scenario, BACE1 inhibition might prevent Aβ accumulation and degeneration of cholinergic neurons in the DS brain. Along this line, the deletion of one BACE1 allele (BACE1+/-) is associated with halting of AD-like Aβ pathway dysregulation and cholinergic disruption in the trisomic mouse line (Ts2), a model of DS generated by a spontaneous Robertsonian fusion. In particular, the reduction of BACE1 by-products, such as APP-βCTF, whose related signaling in endosomes initiates the pathological activation of rab5 that downstream impairs the neurotrophin signaling, which is essential for cholinergic neurons homeostasis and whose downregulation is reported in DS and AD experimental models [14]. The authors concluded, “the therapeutic effects of partial BACE1 inhibition in our DS model are consistent with those in β-amyloidosis models.”
In summary, experimental evidence, albeit preliminary, encourages the investigation of the therapeutic effects of partial BACE1 inhibition in the DS model, which seem consistent with those in β-amyloidosis models and may open up therapeutic solutions for the AD-DS complex [14].
2.3. The Emerging Role of BACE2 in Alzheimer’s Disease and Alzheimer’s-Down Syndrome Complex: A New Pharmacological Target?
Genetic studies indicate the polymorphisms of the BACE2 gene as risk factors for I) late-life dementia in individuals with no familial history of AD [27, 58, 59] and II) early-onset AD [59, 60].
An increased BACE2 expression and activity have been reported in human AD neurons derived from brains of asymptomatic subjects, suggesting a tight biological connection between BACE homeostasis and AD pathophysiology [61]. A recent study performed in two independent datasets showed an association between several single nucleotide polymorphisms (SNPs) in BACE2 and AD in ε4 allele of the gene encoding for the apolipoprotein E (APOE ε4) non-carriers, with the SNP variants hypothesized to alter BACE2 expression-mediated Aβ generation and clearance [27].
The molecular mechanisms accounting for BACE2 involvement in AD pathophysiology are under investigation. A defective neuronal proteasome and lysosome systems activity is hypothesized to be one of the most upstream AD pathophysiology mechanisms and may induce downstream loss of BACE2 homeostasis with accumulation [62, 63].
BACE2 co-overexpression with APP (TgBACE2-APP), Flemish, and Arctic mutations on the APP gene indicate that BACE2 and downstream pathways overexpressed in the AD brain, including clusterin, may play a role in the loss of Aβ homeostasis [59, 64]. Recent studies also suggest an emerging role of BACE2 in type 2 diabetes mellitus (T2DM) and insulin resistance [65, 66], two well-known risk and precipitant factors for AD and other neurodegenerative diseases [67, 68]. Different neurobiological links have been identified between T2DM and AD, such as insulin resistance, low-grade inflammation, increased oxidative stress, and accumulation of advanced glycation end-products [67, 68]; second-generation antidiabetic drugs are currently studied as disease-modifying drugs in AD (Fig. 3) [67, 68].
Fig. (3).
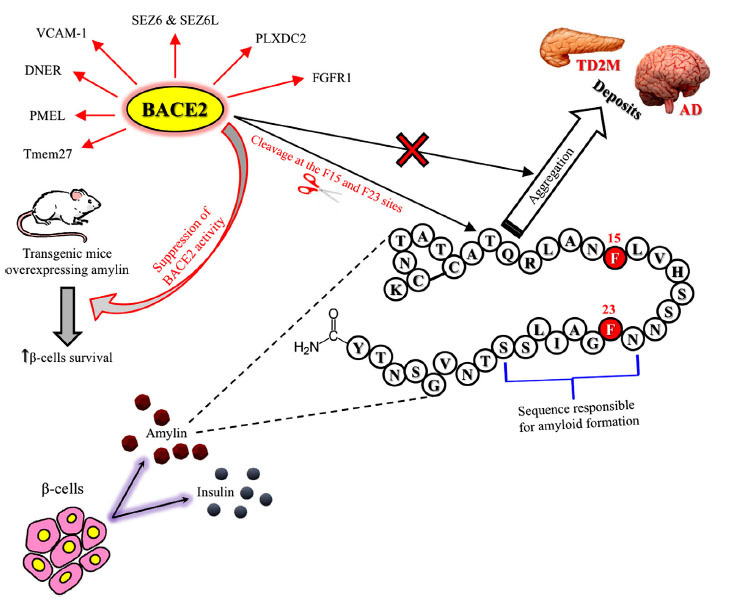
BACE2 as a common pharmacological target for type 2 diabetes mellitus and Alzheimer’s disease. The evidence observed in animal models of T2DM suggests that BACE2 might represent a novel pharmacological target for the treatment of T2DM [1]. Mice with an in-frame deletion of exon 6 of BACE2 on both alleles (Bace2ΔE6/ΔE6) and ob/ob mice, representing a model of obesity-related insulin resistance, treated with a BACE2 inhibitor (CpdJ), displayed reduced blood glucose levels, improved glucose tolerance, and increased β-cell mass and function. BACE2 suppression is known to promote beta-cell survival and function in an animal model of T2DM induced by human amylin over-expression. These new data have therefore stimulated the development of highly selective and potent human BACE2 inhibitors (Ki < 2nM and selectivity over BACE1 over 500-fold) [2] whose preclinical efficacy in animal models of T2DM remains to be evaluated. These selective BACE 2 inhibitors will represent new pharmacological tools to better validate the role of BACE 2 as a new pharmacological target for the treatment of AD in the future. The one-letter code was used to describe the amylin peptide amino acid sequence.
Abbreviations: AD = Alzheimer’s disease; BACE2 = β-site amyloid precursor protein cleaving enzyme 2; DNER = delta and notch-like epidermal growth factor-related receptor; FGFR1 = fibroblast growth factor receptor 1; PLXDC2 = plexin domain-containing protein 2; PMEL = pigment cell-specific melanocyte protein; SEZ6 = seizure-related protein 6; SEZ6L = seizure-related 6 homolog-like; Tmem27 = transmembrane protein 27; T2DM = type 2 diabetes mellitus; VCAM1 = vascular cell adhesion molecule 1.
Note: Finally, BACE2 cleaves SEZ6 and its homolog seizure-related 6 homolog-like (SEZ6L) in pancreatic β-cells but not in neurons. BACE2, by acting on its transmembrane protein 27, is implicated in the regulation of pancreatic β-cell function and mass, two parameters that, when impaired, can contribute to type 2 diabetes mellitus (T2DM) pathogenesis. An additional substrate of BACE2 is amylin (also known as human islet amyloid polypeptide), a peptide co-secreted with insulin by β-cells, directly associated with T2DM, whose aggregates and deposits have also been observed in blood vessels and brain parenchyma of late-onset AD patients. As shown in vitro, BACE2 lowers the human amylin aggregation rate through the cleavage at the F15 and F23 sites, reducing its intracellular concentration in β-cells. In contrast, in transgenic mice overexpressing amylin, BACE2 suppression promotes β-cells survival, counteracting the adverse outcomes induced by amylin. BACE2 is also responsible for the proteolytic processing of the pigment cell-specific melanocyte protein required to form functional amyloid fibrils during melanogenesis, explaining the loss of pigmentation observed in preclinical studies of BACE1/2 inhibition. Table 2 illustrates more information about BACE2 substrates, substrate physiological functions, and the effects related to enzyme-substrate interaction.
Note: references: [1] Alcarraz-Vizan, G., et al. (2017) Cellular and molecular life sciences: CMLS 74, 2827-2838; [2] Ghosh, A.K., et al. (2019) ChemMedChem 14, 545-560
Specific BACE2 SNP variants are associated with AD cognitive decline in clinically defined AD-DS complex patients [59,69]. However, experimental evidence counteracts such risk association linkage reported in humans [56, 70]. In particular, a recent study conducted in cerebral organoids grown from induced pluripotent stem cells (iPSC) generated by non-integrational reprogramming of primary cells donated by people with DS showed that the BACE2-trisomy might have a protective mechanism, partially cross-antagonized through pharmacological manipulation with BACE1 inhibitors [56]. These findings suggest that BACE2 may exert a dose-sensitive AD pathology-suppressor gene, potentially accounting for the discrepancy between the monogenic forms of APP mutations (penetrance 100%) and AD dementia in DS individuals, around 70% during lifespan.
3. BACE1 AND BACE2 IN NEUROPSYCHIATRIC DISEASES
3.1. Schizophrenia
Schizophrenia is a major psychiatric disorder whose diagnosis is entirely based on clinical symptomatology, although experimental models of the disease indicate multi-factorial and multi-faceted neurobiology alongside complex spatial-temporal pathophysiological dynamics [101-103].
The lack of validated and multi-dimensional biomarkers for the in-vivo, in-human investigation of schizophrenia molecular mechanisms hinders the comprehensive understanding of its neurobiological substrates [101, 102, 104]. The current construct of schizophrenia and schizoaffective disorders as clinically defined diseases appears insufficient to capture the entire complexity of the condition as indicated by the considerable inter-individual phenotypical and treatment-response heterogeneity and by the lack of compounds with a long-lasting biological effect [101-103].
Although the roadmap to reconceptualize schizophrenia according to a clinical-biological construct is steep, some in-human evidence enriched by functional annotation in animal models may represent the starting point. In this context, the molecular axis BACE1 - NRG1 has stood out.
Linkage analysis studies, performed in large multiplex families and extensive fine-mapping of the 8p locus, show that NRG1, a critical substrate of BACE1 (Fig. 2), is a candidate gene for schizophrenia [41, 102, 105]. This potential link is also supported by the observation of SNPs - the Val-to-Leu exchange at Val322 amino acid in the C-terminal transmembrane domain of NRG1-type III (NRG1-CTF) - associated with schizophrenia symptoms responsible for schizophrenia-like phenotypes in BACE1-/- mice [106-108].
3.1.1. BACE1 and BACE1-dependent NRG1 Signal in Schizophrenia: Evidence from Human Genetic and Transcriptomic Studies
The physiological roles of NRG1 are consistent with the glutamatergic hypothesis of schizophrenia since NRG1 regulates the expression and activation of neurotransmitter receptors (N-methyl-D-aspartate [NMDA] and mGluRI).
The glutamatergic hypothesis of schizophrenia is also backed by experimental models of the disease, indicating that 1) alteration NRG1-dependent signaling pathways contribute to the disease pathophysiology [109, 110] and 2) BACE1-dependent NRG1 cleavage may play an upstream critical role in the pathogenesis of schizophrenia [111-113].
In particular, NRG1 stimulation suppresses NMDA receptor activation in the human (and the rodent) prefrontal cortex with a significantly more pronounced effect in schizophrenia patients than healthy control individuals, thus indicating that enhanced NRG1 signaling may contribute to NMDA hypofunction in schizophrenia [15, 114]. Moreover, NRG1 controls glutamatergic LTP/long-term depression (LTD) and GABAergic LTD at hippocampal CA3-CA1 synapses, as well as glutamatergic LTD in midbrain DA neurons [15, 114]. The BACE1-dependent NRG1 signaling is involved in several biological pathways altered in schizophrenia and spanning synaptic homeostasis and development, neuronal migration, and myelination [115-117].
Aberrant cleavage of NRG in schizophrenia has been suggested by postmortem studies contributing to the understanding of the neurobiology of the disease. In fact, a positive correlation between BACE1 and full-length NRG1 precursor is reported in the Brodmann area 6 (BA6) brain region of healthy individuals, but not in the schizophrenia group [118]. The same study shows that a marked decrease in NRG1-CTF expression was documented in the same region of schizophrenia patients [118].
The same working group found brain region-specific alterations of NRG1 cleavage in postmortem brains of schizophrenic patients versus controls. In particular, both NRG1-NTF expression and NRG1-NTF/full-length NRG1 ratio were significantly increased in the prefrontal cortex BA9 region. In contrast, two other studies reported no significant alterations of BACE1 expression levels in schizophrenic patients than healthy control individuals [119].
The expression of a 50-kDa NRG1 fragment was decreased in the disease [120], which is consistent with previous results, where lower levels of 50-kDa NRG1 were observed in the BA6 region of schizophrenic patients [118].
Aberrant gene expression and epigenetic dysregulation in schizophrenia may also encompass ErbB3, one of the NRG1 receptors, as observed in the examination of prefrontal cortex slices derived from schizophrenic patients [117].
3.1.2. BACE1-dependent NRG1 Signal in Schizophrenia: The Roadmap to Biomarkers-guided Clinical Research
Different studies, employing different methodological approaches, attempted to quantify the dysregulation of BACE1 and NRG1 protein concentrations (supposed to reflect gene expression levels) and rates of activity in schizophrenic patients.
The first in-vivo report that the BACE1-dependent NRG1 activity's alteration is a substrate-specific event in schizophrenia shows: I) plasma BACE1-NRG1 levels are significantly higher in schizophrenia patients than healthy control individuals, II) a positive correlation between BACE1-NRG1 activity and both BACE1 and NRG1 protein expressions, III) BACE1-dependent NRG1 pathway activity is also associated with clinical severity and duration of schizophrenia [121]. The study indicates that an increased BACE1 gene expression leads to downstream increased cleavage of NRG1 substrate, as reflected from biomarker-based readouts [121]. Moreover, individuals with shorter-term courses and worse negative/positive symptoms display higher enzymatic activity levels than those with a longer course and milder forms of the disease [121]. Hence, plasma BACE1-NRG1 activity has been proposed as a candidate mechanistic biomarker for the diagnosis and prognosis of schizophrenia [121].
Circulating plasma concentrations of NRG1 subunit β1 (NRG1β1) are significantly decreased in young patients with early-onset schizophrenia compared with healthy control individuals [122]. Also, sexual dimorphism was reported with males having lower levels of NRG1β coupled with higher levels of BACE1 protein concentrations and a negative association between NRG1β1 and clinical measures [122]. Wang and colleagues found lower serum concentrations of NRG1β1 in drug-naïve individuals with first-episode and chronic schizophrenia, but not in patients with bipolar I/II disorder(s) or major depressive disorder (MDD) compared to healthy control individuals. The relevance of this finding, in line with other available data [123], lies in the necessity of developing a diagnostic biomarker to reduce diagnostic pitfalls and better discriminate schizophrenia from seer forms of bipolar disorders that often manifest with remarkable clinical overlap.
3.1.2.1. Animal and in-vitro Studies Confirm BACE1-mediated Processing of NRG1 in Schizophrenia
The link between BACE1 (and related pathways) and schizophrenic biological phenotypes has gained momentum due to schizophrenia-like endophenotypes observed in BACE1-/- mice and BACE1-dependent cleavage of NRG1 [113]. Gene KO NRG1 mouse models show schizophrenia-like clinical phenotypes with aberrant and neuroleptic-responsive behavior and cognitive dysfunction coupled with lower functionality rates of NMDA receptors and defective myelination [105, 108].
Another study reports that BACE1 is involved in calcyon-mediated pathways, including stimulation of NRG1 cleavage and shedding [124-126]. Calcyon is a neuronal protein regulating vesicle trafficking signaling associated with synaptic plasticity, neural development, and neurodegeneration. Mice overexpressing calcyon and mice overexpressing NRG1-type I share behavioral deficits relevant to schizophrenia [16, 127-129], thus suggesting the possibility of functional convergence between two seemingly distinct biological pathways [130].
HEK293 cells (expressing calcyon and NRG1) treated with both BACE1 inhibitor IV exhibit suppression of the enhancement of NRG1 cleavage by calcyon [130]; such an effect is not observed when the same cell lineage is treated with α-secretase inhibitors. Furthermore, BACE1-/- mice treated with a glutamatergic psychostimulant, MK-801, showed deficits in pre-pulse inhibition, a typical feature of several schizophrenic endophenotypes [131, 132], including cognitive impairments, deficits in social recognition, hypersensitivity to glutamatergic psychostimulants, and novelty-induced hyperactivity as schizophrenia-like behaviors [108]. BACE1-/- mice brains displayed a significant decrease in both ErbB4 binding to postsynaptic density protein 95 (PSD95) protein and spine density in hippocampal pyramidal neurons, indicating that altered BACE1-dependent NRG1/ErbB4 signaling pathways may contribute to the pathophysiology of schizophrenia [108].
Both in NRG1-/- and BACE1-/- mice, there is a decreased expression of Disrupted-in-Schizophrenia-1 (DISC1) protein, a susceptibility factor for various mental disorders, including schizophrenia [17,133], and this net effect might be due to impaired NRG1/ErbB/Akt signaling pathway, which is involved in neurodevelopment [17]. Specifically, DISC1 reduction may underlie the alterations in spine morphology and density, as well as LTP impairment, observed in BACE1-/- mice [134, 135].
Midbrain dopaminergic transmission plays a significant role in schizophreniform behaviors; amphetamine administration increases dopamine release in parallel with hyperlocomotion and hyper-reactivity; the latter sign is abolished in BACE1-/- mice and this may be related to increased amph-induced D2 autoreceptor-mediated inhibition [136]. Such experimental evidence is consistent with the link between BACE1-NRG1 and ErbB4 and has contributed to the understanding that NRG1 plays a fundamental role in regulating mGluR1 responses on dopaminergic cells and consequently the glutamatergic LTD [137, 138].
Finally, transcriptome profiling and gene pathway analysis in BACE1-/- mice hippocampus identified 91 differentially expressed genes mainly involved in inflammation, such as IL-9 and the nuclear factor κB (NF-κB) activation signaling pathways [139]. Immune system alterations have been consistently implicated in schizophrenia [140, 141], and network analysis has shown a close link between immune response via NF-κB and BACE1 signaling via the NRG1/Akt1 pathway.
4. BACE1 AND AUTISM SPECTRUM DISORDER: PRELIMINARY IN-HUMAN EVIDENCE
Autism spectrum disorder (ASD) is a multi-faceted psychiatric disorder whose biological and genetic underpinnings are still largely uncharacterized [2, 142]. Overlapping clinical and electrophysiological features as well as shared genetic architecture and related endophenotypes, mainly related to neurodevelopment pathways, exist between ASD and schizophrenia at the individual and experimental level [2, 142, 143].
Although the pathophysiological link between BACE1 and ASD pathogenesis has not been investigated systematically as of schizophrenia, mainly because standardized ASD animal models are still lacking, recent in-human data suggest a potential BACE-dependent involvement in ASD pathogenesis and pathophysiology [144].
Abnormal amyloidogenic and non-amyloidogenic pathways with distinct peripheral (blood) patterns of APP by-products levels, including sAPPα and sAPPβ (proteolytic cleavage products of APP by α- and β-secretase, respectively), were reported in children suffering from different clinical forms of ASD [145]. The high levels of sAPPα cleavage products in the bodily fluids of ASD children have led to the hypothesis of the anabolic pathogenesis of ASD whereby loss of proteostasis may hinder both neuronal homeostasis (by sAPPα-mediated cell overgrowth) and proper neurodevelopment [146]. According to this hypothesis, a downregulation of BACE and/or a functional shift toward α-secretase would characterize ASD in a stage-dependent fashion [19].
In contrast with this pathophysiological model of anabolic pathogenesis, a recent study reported that BACE1 gene expression levels (transcript concentrations assessed in the blood) are increased in ASD and are correlated with levels of BACE1 long non-coding RNAs ([lnRNA], a subset of non-sense RNA molecules greater than 200 nt in length) in ASD individuals. The observed significant correlations between BACE1 gene expression and its antisense comply with the proposed feed-forward loop between these two transcripts [21]. The authors reported an age-related variance of BACE1 expression levels only in the female group, suggesting a potential sexual dimorphism [21].
This BACE-oriented evidence is supported by a recent investigation of the BACE1-dependent NRG1 pathway as a potential biological readout of aberrant neuronal development underlying ASD. NRG1 mRNA blood levels are downregulated in patients affected by ASD, where NRG1 expression shows a sex-biased fashion and correlates with severity of behavioral symptoms and impairment in executive functions impairment [20].
Moreover, serum concentrations of NRG1 were reported significantly higher in non-medicated ASD patients than in healthy control individuals, thus paving the way for developing diagnostic mechanistic biomarkers [20]. Such findings are in line with results stemming from different single cohort-based studies and large-scale GWAS investigations that indicate SNPs in the NRG1 gene are associated with syndromic features of ASD. This suggests that NRG1 and related neurodevelopmental molecular pathways may play a role in ASD pathophysiology [1,2,144].
Experimental evidence of overexpression of NRG1 signaling and downstream GABAergic dysfunction has been reported in association with autistic syndromic features [147]. Whether a link between these NRG alterations and BACE1 homeostasis exists is to be investigated.
5. DISCUSSION: LESSONS, CHALLENGES, PITFALLS, AND PERSPECTIVES
5.1. Expanding on Molecular Orchestrators Biology is a Critical Step for Successful Drug R&D, Including BACE in the Neuropsychiatric Spectrum
Any attempt to achieve a holistic understanding of BACE physiology and pathophysiology, from molecular to brain macroscale structural and functional endophenotypes, is grounded on a granular description of the enzyme(s) biology. In the present article, we have reported extensive evidence from cellular and molecular studies of BACE1 and BACE2 KO mice and biomarker-based and genetic studies in humans.
However, a systematic research approach is needed to systematize such fragmented molecular knowledge on BACE biology and homeostasis to design substrate selective compounds that spare unwanted side effects. There is poorly explored but valuable in vitro evidence present that stems from studies conducted in human mutant neurons, showing that augmented retention of BACE1 in distal axons by modulation of autophagy is associated with increased β-cleavage of APP [148]. Recent data from both animal models and AD brain also suggest that the earliest intraneuronal Aβ accumulation causes DNA hypomethylation at 12 CpGs sites in the BACE-1 promoter, thus contributing to BACE-1 overexpression [149].
Moreover, the physiological functions of BACE1 and the homolog BACE2, alongside the cell biology of Ab and synaptic by-products in neurons and glia, deserve further investigation to I) provide a more comprehensive understanding of the biological functions of these critical molecular orchestrators, II) identify all their critical substrates, and III) track the association between their pathophysiological alterations and disease clinical-biological phenotypes.
Such insights will enable the expansion of the set of diseases BACE isoforms may be involved in, given the emerging evidence on other substrates such as SEZ6, CHL1 and their potential implication in pathological conditions other than those touched in the present article, such as seizures and epilepsy [45, 150, 151].
The catalyzing impact of new methodological approaches, such as systems pharmacology, will support individualized time-sensitive and effective pathway (a mechanism)-based and disease-modifying therapeutic strategies, agnostic for the clinical phenotypes, as the recent breakthrough in oncology indicates.
5.2. Lessons from Systems Biology Approaches in Oncology
Adopting this theoretical framework in neuroscience is supported by other medical areas, such as oncology, where tumor- and site-agnostic treatments have already been approved or are under late-stage development [152-154].
In 2018-2019, the U.S. Food and Drug Administration (FDA) authorized larotrectinib and pembrolizumab as immunotherapy treatments for different phenotypes of cancers grouped by a common pathophysiological alteration, regardless of the clinical manifestation such as tumor site and histology [3].
Larotrectinib targets a wide set of tumors displaying genetic alterations of neurotrophic tropomyosin or tyrosine receptor kinase (NTRK) genes that cause the formation of TRK fusion proteins. Such a mutation and downstream molecular aberrant pathways are associated with a broad spectrum of distinct cell/tissue cancerous lineages, which in turn manifest with clinically divergent adult and child forms of cancer [155]. Larotrectinib has shown remarkable efficacy in patients of different ages, exhibiting different TRK fusion-positive cancer phenotypes. Hence, it represents a strong candidate for a tumor-agnostic treatment of TRK fusion-positive cancers [155].
Pembrolizumab targets tumors expressing anti-programmed cell death protein 1 Pembrolizumab indications span a broad set of advanced solid tumors, with a therapeutic work-up guided by the investigation of microsatellite-high/DNA mismatch repair-deficient biomarkers rather than clinical phenotypes [4, 154].
These compounds represented the first examples of an innovative therapeutic paradigm, namely tissue-and tumor-agnostic, whereby the patient's tumor biomarker-based profiling, rather than “traditional” assessments such as histology and site, plays a critical role in the decision-making process. Other compounds are under development in line with this approach.
Such a therapeutic breakthrough has been achievable only by a stepwise, consistent, and systematic implementation of modern technologies in new-generation drug-biomarker co-development pipelines.
First, the development of reliable and replicable preclinical models, including animal and patient-derived organoids used for functional annotation of genetic and pharmacogenomic data, have significantly contributed to accelerating the translation of promising preclinical data into clinical trials without comprising rates of success [4, 156]. Such a paradigm lies in the key features of the experimental models that recapitulate the cancer microenvironment as well as the related “peripheral” systemic immune response, both of which underlie primary tumor genesis, locals spreading, and metastatic dynamics [156, 157].
Second, progress in clinical sequencing technologies, such as tumor DNA and RNA sequencing and plasma cell-free DNA profiling, led to the development of non-invasive, widely accessible, omics-based liquid biopsies serving for different context-of-use, including trial enrollment, target engagement, and outcome prediction [158]. Liquid biopsy is highly suitable for time series, which can boost the discovery and better understanding of pivotal molecular pathways relevant to the disease’s natural history, such as drivers of oncogenesis [158,159]. The processing of such vast datasets, containing multi-dimensional variables, and hundreds of observations, has been possible only through the integration of omics-based technology and traditional approaches such as assays, including fluorescence in-situ hybridization (FISH) or immunohistochemistry [160], embedded in machine learning algorithm-based blueprints for accurate and fast classification (i.e., outcome prediction) [161].
To follow, basket trial design paradigms have been executed to test oncogene-matched therapeutic efficacy in patients with different tumors harboring a distinctive genomic alteration while displaying different histology or manifesting in different organs [4, 162]. The biomarker-guided assessment of activity rates of the compound against a range of cancers harboring the qualifying genomic aberration, stratifying by which tumor and in which subpopulation there is responsiveness regardless of histological features, is another novel feature of oncology basket trials.
5.3. Challenges and Pitfalls in the Holistic Understanding of Neuropsychiatric Diseases: The Need for a Biomarker-driven, Systems-scaled Integrative Approach to BACE Pathways
It is conceivable that, in neuroscience, traditional study designs have been unable to capture complex interactions among distinct biological levels, from molecular pathways to synapsis and large-scale functional/structural networks, taking place in complex and multi-factorial neuropsychiatric diseases [163-165]. Therefore, the perspective of an integrative approach is suitable for the systems-level integration of the upstream and downstream factors impacting definite pathomechanistic dynamics, including BACE pathways, across different disorders, for developing effective screening, diagnostic and therapeutic strategies.
In the AD landscape, BACE plasma concentrations, supposed to reflect levels of gene expression [166, 167], are associated with multi-modal biomarkers tracking brain Aβ accumulation [166, 168], grey matter loss in the cholinergic circuitry [167], synaptic dysfunction [169], neurodegeneration [170, 171], and axonal damage [167], in AD dementia, mild cognitive impairment, and cognitively healthy individuals at risk for AD. In the latter clinical population, a negative association between brain Aβ levels and miRNA-125b, a molecular regulator of BACE1 gene expression [172], has recently been reported.
In the schizophrenia and ASD clinical research space, magnetic resonance imaging-based studies show that specific patterns of macroscale network functional connectivity and/or grey matter changes underlie different clinical phenotypes that can be grouped in different clusters based on presence/absence of psychosis, alterations in the affective/emotional dimensions, and mono-multidomain cognitive impairment [152, 173-176]. Enriching brain endophenotypes with a biological molecular layer, such as the BACE1-NRG1 axis, and its association with clinical outcomes may clarify whether the genetic evidence pointing out this pathway in schizophrenia and ASD has a correlate in brain structural and functional organization.
The assessment of BACE1 parameters (protein concentration, enzymatic activity rates, miRNA, exosomes) in peripheral matrixes, such as plasma, serum, platelets, allows the integration of BACE biomarkers in a non-invasive blood-based liquid biopsy for CNS diseases [177, 178].
Moreover, brain-penetrant PET radiotracers for in-vivo BACE1 imaging are under development or clinical validation and could be used to investigate spatial and temporal co-localization of BACE1-related pathways and brain structural/functional damage [148].
In summary, the reshaping of AD definition as a clinical-biological construct, reflected by the symptoms-agnostic ATN classification system coupled with progress in the development of drugs with putative biological effects, paves the way for enriching clinical research of all neuropsychiatric diseases with consistent biomarker-based protocols, using exploratory outcomes and endpoints while leveraging modern computational approaches.
5.4. Systems Pharmacology and Precision Pharmacology for Pathway-based Therapies
In order to overcome the CNS anatomical barrier and bridge liquid biopsy readouts with brain functional and structural endophenotypes, a systems-scaled approach is needed. Hence a multi-modal, biomarkers-guided paradigm, informed by machine and deep learning ranking, classification, and clustering algorithms, will enable identifying sub-population of individuals sharing pathophysiological commonalities (similar biological signatures or biotypes) (Fig. 4).
Fig. (4).
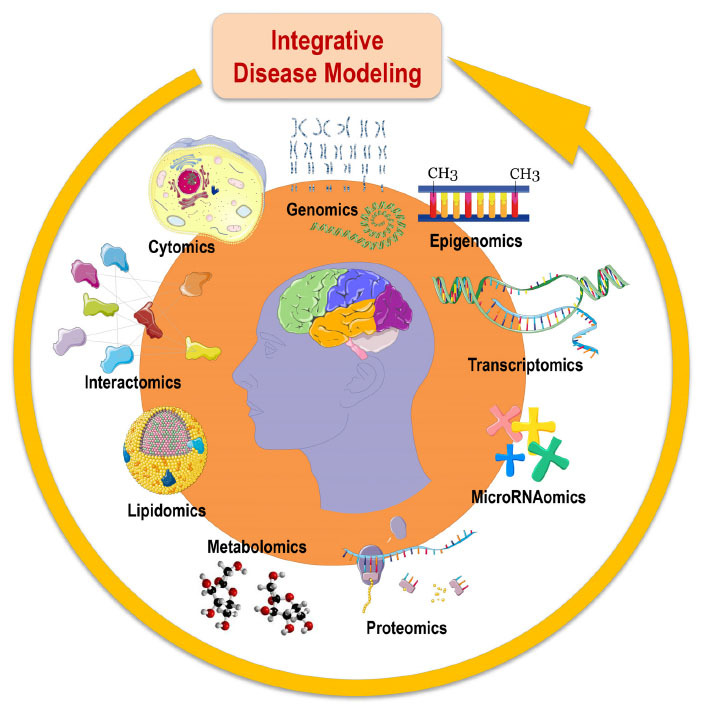
Cohorts stratified according to different multimodal-throughput technological platforms (“omic” sciences) are integrated into the disease modeling for classification and prediction of subsets of Alzheimer’s disease and patients with other brain proteinopathies and neurodegenerative diseases. Systems biology is an evolving hypothesis-free, exploratory, holistic (non-reductionistic), global, integrative, and interdisciplinary paradigm using advances in multimodal high-throughput technological platforms that enable the examination of networks of biological pathways, where elevated amounts of structurally and functionally different molecules are simultaneously explored overtime at a system-level (i.e., at the level of molecules and subcellular compartments, cells, group of cells, tissues, organs, apparatuses, or even whole organisms). According to systems biology, organisms are made of systems which are entities consisting in hierarchically self-organized levels with increasing structural complexity resulting in different emerging properties. Adopted with permission from: The Alzheimer Precision Medicine Initiative. J Alzheimers Dis. 2019;68(1):1-24. doi: 10.3233/JAD-181121.
Such integrative approaches in clinical research will then inform drug discovery and development pipelines through the simulation of traditional pharmacodynamic and pharmacokinetic indexes with modern in-silico computer-assisted drug investigations [179-181]. Systems pharmacology enables modeling snapshots of human drug-induced interactomes and intracellular signals, drug-related (epi)genetic regulation, transcription regulatory networks, drug-induced immune responses, drug-drug interactions, and drugs-multi targets interaction [179, 182, 183].
Thus, albeit still under development, quantitative systems pharmacology represents the most viable integrative paradigm to model a holistic drug effect capitalizing on structure- and ligand-based techniques coupled with non-a-priori feature selection and output prediction from extensive datasets that can capture the biological heterogeneity of large-scale population without losing accuracy and clinical insights [182, 184].
The clinical and pharmacological perspective of quantitative systems pharmacology lies in the notion of capitalizing on the successful oncology new-generation drug R&D, i.e., multi-targets and stage-specific treatments that can be tailored to the individuals genetic/biological profile, overcoming the limitation inherent in the traditional “one-drug-one-target” model [179, 180].
A tangible systems pharmacology paradigm in AD tauopathy is represented by MAPTA (Modeling Alliance for Systems Pharmacologies in Tauopathies), a program based on public-domain knowledge about tau biology and pathophysiology [180, 185]. MAPTA platform leverages the value of observations from preclinical, experimental models or human-derived organoids that may generate knowledge relevant to human tau pathology [180, 185]. A significant example of MAPTA-based disease modeling is represented by the simulation of glycogen synthase kinase-3β (GSK-3β)-mediated tau phosphorylation at different tau sites to explore the subsequent changes in ATP, tau structure, phosphatase, or GSK-3β levels on the phosphorylation state [180, 185]. Such computer-assisted pharmacodynamic study can estimate the indirect effect of specific tau kinase inhibitors on the overall tau-related biological signature with possible implications for biomarkers related to target engagement in novel therapeutics.
Moreover, GSK-3β inhibition through BACE1 is associated with anti-AD and neuroprotective effects [186]. Several mechanisms underlie the neuroprotection, spanning control over protein life-cycle, immune/inflammatory response and oxidative-redox balance restoring [7, 9, 36, 142, 187]. With regard to oxidative stress, age-related, genetic-driven, and stochastic factors have been reported to alter mitochondrial functions and raise oxidative stress levels, a critical trigger and driver of neuronal degeneration in a broad spectrum of clinical phenotypes [1, 7, 9, 142, 188].
Systems biology and pharmacology could also help refine the understanding of the therapeutic value of modulating BACE genes expression/activity for inducing hormesis, which is based on the principle that low and controlled exposure to a given stressor can set off and sustain bio-positive effects in the form of evolutionarily conserved cellular anti-stress responses (adaptation and vitagene pathways) [7, 9, 12, 189]. Proteostasis machinery involved in the full life-cycle of protein, from synthesis to folding up to degradation and clearance, as well as intracellular transduction pathways, e.g., GSK-3β and the mammalian target of rapamycin (mTOR), both aberrantly regulated in experimental models of neurodegenerative diseases [1, 7, 142, 186], are established anti-stress responses. We argue that it would be worthwhile to investigate whether the upstream pharmacological manipulation of BACE1/2 may influence proteostasis components, including heat shock proteins (prominent members of vitagene and neuroprotective networks) and GSK-3β and mTOR molecular axes [7, 9, 12, 142, 186, 189]. Such a scientific endeavor would help capture hidden layers of BACE biology, thus addressing caveats of previous BACE-modulators drugs and opening up viable therapeutic solutions upon previously hypothesized novel paradigms, including hormesis [7, 9, 12, 186-189].
In summary, systems-scaled approaches in biology, neuroscience, and pharmacology hold the potential to untangle the spatial and temporal architecture of BACE-related molecular-cellular pathways and biological dynamics in neurodevelopment, adulthood, and aging. Integration of explainable biological readouts with large-scale brain network functional and structural data could help capture the role of BACE in the cognitive/behavioral/functional decline occurring in neuropsychiatric diseases.
Systems pharmacology and quantitative approaches may help understand pitfalls in AD BACE1 trials whose dramatic failure indicates that a deeper understanding of BACE1 biology, aimed at developing compounds with a high substrate selectivity, is needed. Setting aside a molecular target because of the failure of clinical trials may represent a tremendous mistake if a critical revision of study designs is not put in place and in case biomarker-based outcome/endpoints were not part of the protocol. Hence, a few strategies may inform future clinical trials, such as I) the utilization of BACE1 biomarkers for target engagement and treatment monitoring studies (drug-biomarkers co-development pipelines), II) investigating different dose regimens, III) using dose-titration, IV) enrolling early-stages patient population.
There are potential limitations of this approach that may hinder its implementation in specific settings. First of all, despite the tremendous evolution of computing power, the implementation of multimodal analyses faces technical limitations. In addition, the larger the scale of big-data sets, the higher is the possibility of encountering false-positive findings due to multiple comparisons and several predictors that may cause model overfitting and effect size inflation [8, 179, 190]. Strategies must be put in place to extract relevant but subtle associations while limiting these risks. The curse of dimensionality stipulates that, in a high dimensional space, the data becomes sparse [191]. Thus, traditional algorithmic techniques fail, and increasing the number of subjects is generally not a sufficient or viable solution. Finally, pre-defined outcome labels (e.g., supervised learning) may constrain the set of hypotheses and/or influence the predictors’ selection, with all issues that this approach carries with itself, e.g., multicollinearity issue, violation of orthogonality, and homoscedasticity assumptions, to name a few [8, 9, 190]. Models which do not respect these conditions of validity may provide an erroneous interpretation of the assumptions. Moreover, investigations of features interaction and nonlinear relationships are often missing, thus not considering the complexity of the system [192]. In this sense, we value the blooming of machine and deep learning-based study design in clinical research. Automatic features selections, linear and nonlinear dimensionality reduction, and cross-validation methods, as well as more harmonized sensitivity analysis criteria and unsupervised learning algorithms, hold the promise to overcome traditional operator-dependent statistic caveats and minimize the risk of misleading clinical stances [8, 9, 179, 190]. However, since these approaches act as black boxes, particular attention must be given to the interpretability of the models [193, 194], which are not only predictive but also explanatory tools. This is of key importance for adoption in clinical trials [195].
CONCLUSION
We have provided a state-of-the-art review of translational research demonstrating the involvement of BACE isoforms in a comprehensive set of clinically heterogeneous neuropsychiatric diseases. Pivoting on this body of evidence, we propose how future studies can inform experimental models and in-human biomarker-based clinical research for next-generation pharmacological trials, in line with the precision medicine paradigm.
If the pathophysiological role of BACE enzymes and downstream pathways in different neuropsychiatric diseases were fully elucidated, biomarker-drug co-development pipelines would be suitable for investigating compounds with a quantifiable biological effect. This approach will circumvent the limitations of current late-stage symptoms-based diagnostic/therapeutic decision-making that do not capture the neurobiological complexity of multi-factorial, chronic neuropsychiatric diseases.
Table 2.
Substrates of BACE2, their physiological functions, and the effects related to enzyme-substrate interaction.
|
BACE2
Substrates |
Substrate Physiological Function(s) | BACE2-substrate Interaction | References* |
|---|---|---|---|
| VCAM-1 | ▪ Adhesion of white blood cells to the vascular endothelium; ▪ Triggering of endothelial signaling through NADPH oxidase-generated ROS |
▪ Specific processing (sheddase activity) of VCAM1; ▪ The physiological cleavage of VCAM1 by BACE2 is strongly up-regulated (additional shedding of VCAM1) under pro-inflammatory conditions |
Voytyuk et al. 2018 [26]; Kong et al. 2018 [71]; Cook-Mills et al. 2011 [72]; |
| DNER | ▪ Central nervous system development, neuron migration, and synapse assembly; ▪ Differentiation and proliferation during cancer and stemness; ▪ Regulation of IFN-γ expression in activated macrophages |
▪ Specific processing (sheddase activity) of DNER | Voytyuk et al. 2018 [26]; Ballester-López et al. 2019 [73]; Wang et al. 2017 [74]; Sun et al. 2009 [75]; Eiraku et al. 2002 [76]. |
| PLXDC2 | ▪ Protein receptor for pigment epithelium-derived factor; ▪ Involved in cell proliferation, senescence, and cell death |
▪ Cleavage of PLXDC2 | Voytyuk et al. 2018 [26]; Nanda et al. 2004 [77]; Miller-Delaney et al. 2011 [78]; Schwarze et al. 2005 [79]; McMurray et al. 2008 [80]; Hallstrom et al. 2008 [81]. |
| FGFR1 | ▪ Protein receptor for fibroblast growth factor; ▪ Midbrain development and neuron migration. ▪ Epithelial to mesenchymal transition |
▪ Cleavage of FGFR1 | Voytyuk et al. 2018 [26]; Gaudet et al. 2011 [82]; Ornitz et al. 1996 [83]; Eswarakumar et al. 2005 [84]; Wang et al. 2015 [85]. |
| SEZ6 & SEZ6L | ▪ Cerebellar Purkinje cell layer development and adult locomotory behavior; ▪ Dendrite development and synapse maturation; ▪ Involvement in the activity-dependent plasticity of learning and memory |
▪ Cleavage of both SEZ6 and SEZ6L | Pigoni et al. 2016 [43]; Gunnersen et al. 2007 [86]; Håvik et al. 2007 [87]; Causevic et al. 2018 [88]. |
| Tmem27 | ▪ Positive regulation of L-proline import across the plasma membrane, SNARE complex assembly, amino acid transport, calcium ion-dependent exocytosis, and insulin secretion involved in the cellular response to a glucose stimulus | ▪ Regulation of pancreatic β-cell function and mass | Gaudet et al. 2011 [82]; Esterházy et al. 2011 [89]; Zhang et al. 2001 [90]; Danilczyk et al. 2006 [91]; Fukui et al. 2005 [92]; Vuille-dit-Bille et al. 2015 [93]. |
| Amylin | ▪ Amyloid-beta binding and regulation of amyloid fibril formation; ▪ Positive regulation of MAPK cascade, protein kinases A and B signaling, and neurotrophic factors release; ▪ Negative regulation of bone resorption, cell differentiation, and mitochondrion organization |
▪ BACE2-amylin interaction lowers human amylin aggregation rate in a dose-dependent manner and reduces its intracellular concentration in β-cells | Fu et al. 2012 [94]; Caruso et al. 2018 [95]; Lorenzo et al. 1994 [96]; Nishi et al. 1989 [97]; Rulifson et al. 2016 [98]. |
| PMEL | ▪ Melanin biosynthetic process; ▪ Melanosome organization |
▪ Proteolytic processing of PMEL during melanogenesis; ▪ Modulation of pigmentation process |
Gaudet et al. 2011 [82]; Hoashi et al. 2005 [99]; Rochin et al. 2013 [100]. |
Abbreviations: DNER = delta and notch-like epidermal growth factor-related receptor; FGFR1 = fibroblast growth factor receptor 1; IFN-γ = interferon-γ; MAPK = mitogen-activated protein kinase; NRG2 = neuregulin 2; PLXDC2 = plexin domain-containing protein 2; PMEL = pigment cell-specific melanocyte protein; ROS = reactive oxygen species; SEZ6 = seizure protein 6; SEZ6L = seizure-related 6 homolog-like; SNARE = soluble NSF attachment protein receptor; Tmem27 = transmembrane protein 27; VCAM-1 = vascular cell adhesion molecule 1.
* Additional source: https://www.ncbi.nlm.nih.gov
ACKNOWLEDGEMENTS
HH is an employee of Eisai Inc. He declares no competing or financial interests related to the present article. This work was performed during his previous position at Sorbonne University, Paris, France. At Sorbonne University, he was supported by the AXA Research Fund, the “Fondation partenariale Sorbonne Université” and the “Fondation pour la Recherche sur Alzheimer”, Paris, France.
The Neurodegeneration Precision Medicine Initiative (NPMI) is a group of researchers sharing an intellectual interest in precision medicine for neurodegenerative diseases and inclined to engage in academic scientific collaborations for precision medicine-oriented clinical and medical projects. More information, including the list of contributors, available at https://npmiweb.net/.
LIST OF ABBREVIATIONS
- Aβ
Amyloid-β
- Aβ40
40-amino Acid-long Amyloid-β Peptide
- Aβ42
42-amino Acid-long Amyloid-β Peptide
- AβDP
Aβ-degrading Protease
- AD
Alzheimer’s Disease
- APOE ε4
ε4 Allele of the Gene Encoding for the Apolipoprotein E
- APP
Amyloid Precursor Protein
- ASD
Autism Spectrum Disorder
- BA6
Brodmann Area 6
- BACE1
β-site Amyloid Precursor Protein Cleaving Enzyme 1
- CHL1
Neural Cell Adhesion Molecule L1
- CNS
Central Nervous System
- CTFβ
β-C-terminal Fragment
- DISC1
Isrupted-in-Schizophrenia-1 Protein
- DNER
Delta and Notch-like Epidermal Growth Factor-related Receptor
- DS
Down Syndrome
- EGF
Epidermal Growth Factor
- EGFR
Epidermal Growth Factor Receptor
- FDA
U.S. Food and Drug Administration
- FGFR1
Fibroblast Growth Factor Receptor 1
- GABABR
γ-aminobutyric Acid Type B Receptor
- GSK-3β
Glycogen Synthase Kinase-3β
- IDE
Insulin-degrading Enzyme
- IFN-γ
Interferon-γ
- Jag1
Jagged-1
- KO
Knockout
- L1CAM
L1 Cell Adhesion Molecule
- lnRNA
Long non-coding RNA
- LTP
Long-term Potentiation
- MAPK
Mitogen-activated Protein Kinase
- MAPTA
Modeling Alliance for Systems Pharmacologies in Tauopathies
- MDD
Major Depressive Disorder
- mGluRI
Group 1 Metabotropic Glutamatergic Receptors
- mTOR
Mammalian Target Of Rapamycin
- NaV
Voltage-gated Sodium Channels
- NaVβ2
Voltage-gated Sodium Channels Beta 2
- nf-κb
Nuclear Factor Kappa-light-chain-enhancer of Activated B Cells
- NMDA
N-methyl-D-aspartate
- NRG1
Neuregulin-1
- NRG1β1
NRG1 Subunit β1
- NRG1-CTF
NRG1 C-terminal Fragment
- NRG1-ICD
NRG1 Intracellular Domain
- NRG1-NGF
NRG1 N-terminal Fragment
- NRG2
Neuregulin 2
- NTRK
Neurotrophic Tropomyosin or Tyrosine Receptor Kinase
- PLXDC2
Plexin Domain-containing Protein 2
- PMEL
Pigment Cell-specific Melanocyte Protein
- PNS
Peripheral Nervous System
- PSD95
Postsynaptic Density Protein 95
- ROS
Reactive Oxygen Species
- sAPP
Soluble Amyloid Precursor Protein
- Sema3A
Semaphorin 3A
- SEZ6
Seizure-related Protein 6
- SEZ6L
Seizure-related 6 Homolog-like
- SNARE
Soluble NSF Attachment Protein Receptor
- SNPs
Single Nucleotide Polymorphisms
- T2DM
Type 2 Diabetes Mellitus
- Tmem27
Transmembrane Protein 27
- TNF-α
Tumor Necrosis Factor Alfa
- VCAM1
Vascular Cell Adhesion Molecule 1
- VEGFR1
Vascular Endothelial Growth Factor Receptor 1
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
HH is an employee of Eisai Inc. The present article has been initiated and prepared as part of an academic position at Sorbonne University, Paris, France and it reflects entirely and exclusively his own opinion. HH serves as Senior Associate Editor for the Journal Alzheimer’s & Dementia and does not receive any fees or honoraria since May 2019; before May 2019, he had received lecture fees from Servier, Biogen, and Roche, research grants from Pfizer, Avid, and MSD Avenir (paid to the institution), travel funding from Eisai, Functional Neuromodulation, Axovant, Eli Lilly and Company, Takeda and Zinfandel, GE-Healthcare and Oryzon Genomics, consultancy fees from Qynapse, Jung Diagnostics, Cytox Ltd., Axovant, Anavex, Takeda and Zinfandel, GE Healthcare, Oryzon Genomics, and Functional Neuromodulation, and participated in scientific advisory boards of Functional Neuromodulation, Axovant, Eisai, Eli Lilly and Company, Cytox Ltd., GE Healthcare, Takeda and Zinfandel, Oryzon Genomics, and Roche Diagnostics. He is the inventor of 11 patents and has received no royalties:
•In Vitro Multiparameter Determination Method for The Diagnosis and Early Diagnosis of Neurodegenerative Disorders Patent Number: 8916388.
•In Vitro Procedure for Diagnosis and Early Diagnosis of Neurodegenerative Diseases Patent Number: 8298784.
•Neurodegenerative Markers for Psychiatric Conditions Publication Number: 20120196300.
•In Vitro Multiparameter Determination Method for The Diagnosis and Early Diagnosis of Neurodegenerative Disorders Publication Number: 20100062463.
•In Vitro Method for The Diagnosis and Early Diagnosis of Neurodegenerative Disorders Publication Number: 20100035286.
•In Vitro Procedure for Diagnosis and Early Diagnosis of Neurodegenerative Diseases Publication Number: 20090263822.
•In Vitro Method for The Diagnosis of Neurodegenerative Diseases Patent Number: 7547553.
•CSF Diagnostic in Vitro Method for Diagnosis of Dementias and Neuroinflammatory Diseases Publication Number: 20080206797.
•In Vitro Method for The Diagnosis of Neurodegenerative Diseases Publication Number: 20080199966.
•Neurodegenerative Markers for Psychiatric Conditions Publication Number: 20080131921.
•Method for diagnosis of dementias and neuroinflammatory diseases based on an increased level of procalcitonin in cerebrospinal fluid: Publication number: United States Patent 10921330.
SL received lecture honoraria from Roche and Servier.
AV declares no competing financial interests related to the present article, and his contribution to this article reflects only and exclusively his own academic expertise on the matter. This work was conceptualized and initiated during his previous academic position at Sorbonne University, Paris, France. AV was an employee of Eisai Inc. [Nov 2019 - June 2021]. AV does not receive any fees or honoraria since November 2019. Before November 2019 he had he received lecture honoraria from Roche, MagQu LLC, and Servier.
GC, RN, GP, NBM, FSG, FF, PL, and FC declare no conflict of interest.
REFERENCES
- 1.Anttila V., Bulik-Sullivan B., Finucane H.K., Walters R.K., Bras J., Duncan L., Escott-Price V., Falcone G.J., Gormley P., Malik R., Patsopoulos N.A., Ripke S., Wei Z., Yu D., Lee P.H., Turley P., Grenier-Boley B., Chouraki V., Kamatani Y., Berr C., Letenneur L., Hannequin D., Amouyel P., Boland A., Deleuze J.F., Duron E., Vardarajan B.N., Reitz C., Goate A.M., Huentelman M.J., Kamboh M.I., Larson E.B., Rogaeva E., St George-Hyslop P., Hakonarson H., Kukull W.A., Farrer L.A., Barnes L.L., Beach T.G., Demirci F.Y., Head E., Hulette C.M., Jicha G.A., Kauwe J.S.K., Kaye J.A., Leverenz J.B., Levey A.I., Lieberman A.P., Pankratz V.S., Poon W.W., Quinn J.F., Saykin A.J., Schneider L.S., Smith A.G., Sonnen J.A., Stern R.A., Van Deerlin V.M., Van Eldik L.J., Harold D., Russo G., Rubinsztein D.C., Bayer A., Tsolaki M., Proitsi P., Fox N.C., Hampel H., Owen M.J., Mead S., Passmore P., Morgan K., Nöthen M.M., Rossor M., Lupton M.K., Hoffmann P., Kornhuber J., Lawlor B., McQuillin A., Al-Chalabi A., Bis J.C., Ruiz A., Boada M., Seshadri S., Beiser A., Rice K., van der Lee S.J., De Jager P.L., Geschwind D.H., Riemenschneider M., Riedel-Heller S., Rotter J.I., Ransmayr G., Hyman B.T., Cruchaga C., Alegret M., Winsvold B., Palta P., Farh K.H., Cuenca-Leon E., Furlotte N., Kurth T., Ligthart L., Terwindt G.M., Freilinger T., Ran C., Gordon S.D., Borck G., Adams H.H.H., Lehtimäki T., Wedenoja J., Buring J.E., Schürks M., Hrafnsdottir M., Hottenga J.J., Penninx B., Artto V., Kaunisto M., Vepsäläinen S., Martin N.G., Montgomery G.W., Kurki M.I., Hämäläinen E., Huang H., Huang J., Sandor C., Webber C., Muller-Myhsok B., Schreiber S., Salomaa V., Loehrer E., Göbel H., Macaya A., Pozo-Rosich P., Hansen T., Werge T., Kaprio J., Metspalu A., Kubisch C., Ferrari M.D., Belin A.C., van den Maagdenberg A.M.J.M., Zwart J.A., Boomsma D., Eriksson N., Olesen J., Chasman D.I., Nyholt D.R., Avbersek A., Baum L., Berkovic S., Bradfield J., Buono R.J., Catarino C.B., Cossette P., De Jonghe P., Depondt C., Dlugos D., Ferraro T.N., French J., Hjalgrim H., Jamnadas-Khoda J., Kälviäinen R., Kunz W.S., Lerche H., Leu C., Lindhout D., Lo W., Lowenstein D., McCormack M., Møller R.S., Molloy A., Ng P.W., Oliver K., Privitera M., Radtke R., Ruppert A.K., Sander T., Schachter S., Schankin C., Scheffer I., Schoch S., Sisodiya S.M., Smith P., Sperling M., Striano P., Surges R., Thomas G.N., Visscher F., Whelan C.D., Zara F., Heinzen E.L., Marson A., Becker F., Stroink H., Zimprich F., Gasser T., Gibbs R., Heutink P., Martinez M., Morris H.R., Sharma M., Ryten M., Mok K.Y., Pulit S., Bevan S., Holliday E., Attia J., Battey T., Boncoraglio G., Thijs V., Chen W.M., Mitchell B., Rothwell P., Sharma P., Sudlow C., Vicente A., Markus H., Kourkoulis C., Pera J., Raffeld M., Silliman S., Boraska Perica V., Thornton L.M., Huckins L.M., William Rayner N., Lewis C.M., Gratacos M., Rybakowski F., Keski-Rahkonen A., Raevuori A., Hudson J.I., Reichborn-Kjennerud T., Monteleone P., Karwautz A., Mannik K., Baker J.H., O’Toole J.K., Trace S.E., Davis O.S.P., Helder S.G., Ehrlich S., Herpertz-Dahlmann B., Danner U.N., van Elburg A.A., Clementi M., Forzan M., Docampo E., Lissowska J., Hauser J., Tortorella A., Maj M., Gonidakis F., Tziouvas K., Papezova H., Yilmaz Z., Wagner G., Cohen-Woods S., Herms S., Julià A., Rabionet R., Dick D.M., Ripatti S., Andreassen O.A., Espeseth T., Lundervold A.J., Steen V.M., Pinto D., Scherer S.W., Aschauer H., Schosser A., Alfredsson L., Padyukov L., Halmi K.A., Mitchell J., Strober M., Bergen A.W., Kaye W., Szatkiewicz J.P., Cormand B., Ramos-Quiroga J.A., Sánchez-Mora C., Ribasés M., Casas M., Hervas A., Arranz M.J., Haavik J., Zayats T., Johansson S., Williams N., Dempfle A., Rothenberger A., Kuntsi J., Oades R.D., Banaschewski T., Franke B., Buitelaar J.K., Arias Vasquez A., Doyle A.E., Reif A., Lesch K.P., Freitag C., Rivero O., Palmason H., Romanos M., Langley K., Rietschel M., Witt S.H., Dalsgaard S., Børglum A.D., Waldman I., Wilmot B., Molly N., Bau C.H.D., Crosbie J., Schachar R., Loo S.K., McGough J.J., Grevet E.H., Medland S.E., Robinson E., Weiss L.A., Bacchelli E., Bailey A., Bal V., Battaglia A., Betancur C., Bolton P., Cantor R., Celestino-Soper P., Dawson G., De Rubeis S., Duque F., Green A., Klauck S.M., Leboyer M., Levitt P., Maestrini E., Mane S., De-Luca D.M., Parr J., Regan R., Reichenberg A., Sandin S., Vorstman J., Wassink T., Wijsman E., Cook E., Santangelo S., Delorme R., Rogé B., Magalhaes T., Arking D., Schulze T.G., Thompson R.C., Strohmaier J., Matthews K., Melle I., Morris D., Blackwood D., McIntosh A., Bergen S.E., Schalling M., Jamain S., Maaser A., Fischer S.B., Reinbold C.S., Fullerton J.M., Guzman-Parra J., Mayoral F., Schofield P.R., Cichon S., Mühleisen T.W., Degenhardt F., Schumacher J., Bauer M., Mitchell P.B., Gershon E.S., Rice J., Potash J.B., Zandi P.P., Craddock N., Ferrier I.N., Alda M., Rouleau G.A., Turecki G., Ophoff R., Pato C., Anjorin A., Stahl E., Leber M., Czerski P.M., Cruceanu C., Jones I.R., Posthuma D., Andlauer T.F.M., Forstner A.J., Streit F., Baune B.T., Air T., Sinnamon G., Wray N.R., MacIntyre D.J., Porteous D., Homuth G., Rivera M., Grove J., Middeldorp C.M., Hickie I., Pergadia M., Mehta D., Smit J.H., Jansen R., de Geus E., Dunn E., Li Q.S., Nauck M., Schoevers R.A., Beekman A.T., Knowles J.A., Viktorin A., Arnold P., Barr C.L., Bedoya-Berrio G., Bienvenu O.J., Brentani H., Burton C., Camarena B., Cappi C., Cath D., Cavallini M., Cusi D., Darrow S., Denys D., Derks E.M., Dietrich A., Fernandez T., Figee M., Freimer N., Gerber G., Grados M., Greenberg E., Hanna G.L., Hartmann A., Hirschtritt M.E., Hoekstra P.J., Huang A., Huyser C., Illmann C., Jenike M., Kuperman S., Leventhal B., Lochner C., Lyon G.J., Macciardi F., Madruga-Garrido M., Malaty I.A., Maras A., McGrath L., Miguel E.C., Mir P., Nestadt G., Nicolini H., Okun M.S., Pakstis A., Paschou P., Piacentini J., Pittenger C., Plessen K., Ramensky V., Ramos E.M., Reus V., Richter M.A., Riddle M.A., Robertson M.M., Roessner V., Rosário M., Samuels J.F., Sandor P., Stein D.J., Tsetsos F., Van Nieuwerburgh F., Weatherall S., Wendland J.R., Wolanczyk T., Worbe Y., Zai G., Goes F.S., McLaughlin N., Nestadt P.S., Grabe H.J., Depienne C., Konkashbaev A., Lanzagorta N., Valencia-Duarte A., Bramon E., Buccola N., Cahn W., Cairns M., Chong S.A., Cohen D., Crespo-Facorro B., Crowley J., Davidson M., DeLisi L., Dinan T., Donohoe G., Drapeau E., Duan J., Haan L., Hougaard D., Karachanak-Yankova S., Khrunin A., Klovins J., Kučinskas V., Lee Chee Keong J., Limborska S., Loughland C., Lönnqvist J., Maher B., Mattheisen M., McDonald C., Murphy K.C., Nenadic I., van Os J., Pantelis C., Pato M., Petryshen T., Quested D., Roussos P., Sanders A.R., Schall U., Schwab S.G., Sim K., So H.C., Stögmann E., Subramaniam M., Toncheva D., Waddington J., Walters J., Weiser M., Cheng W., Cloninger R., Curtis D., Gejman P.V., Henskens F., Mattingsdal M., Oh S.Y., Scott R., Webb B., Breen G., Churchhouse C., Bulik C.M., Daly M., Dichgans M., Faraone S.V., Guerreiro R., Holmans P., Kendler K.S., Koeleman B., Mathews C.A., Price A., Scharf J., Sklar P., Williams J., Wood N.W., Cotsapas C., Palotie A., Smoller J.W., Sullivan P., Rosand J., Corvin A., Neale B.M., Schott J.M., Anney R., Elia J., Grigoroiu-Serbanescu M., Edenberg H.J., Murray R. Analysis of shared heritability in common disorders of the brain. Science. 2018;360(6395):eaap8757. doi: 10.1126/science.aap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hernandez L.M., Kim M., Hoftman G.D., Haney J.R., de la Torre-Ubieta L., Pasaniuc B., Gandal M.J. Transcriptomic insight into the polygenic mechanisms underlying psychiatric disorders. biol. Psychiatry. 2021;89(1):54–64. doi: 10.1016/j.biopsych.2020.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan L., Zhang W. Precision medicine becomes reality-tumor type-agnostic therapy. Cancer Commun. (Lond.) 2018;38(1):6. doi: 10.1186/s40880-018-0274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldberg K.B., Blumenthal G.M., McKee A.E., Pazdur R. The FDA Oncology Center of Excellence and precision medicine. Exp. Biol. Med. (Maywood) 2018;243(3):308–312. doi: 10.1177/1535370217740861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blumenthal G.M., Goldberg K.B., Pazdur R. Drug development, trial design, and endpoints in oncology: Adapting to rapidly changing science. Clin. Pharmacol. Ther. 2017;101(5):572–574. doi: 10.1002/cpt.623. [DOI] [PubMed] [Google Scholar]
- 6.Arenaza-Urquijo E.M., Vemuri P. Resistance vs resilience to Alzheimer disease: Clarifying terminology for preclinical studies. Neurology. 2018;90(15):695–703. doi: 10.1212/WNL.0000000000005303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hampel H., Lista S., Neri C., Vergallo A. Time for the systems-level integration of aging: Resilience enhancing strategies to prevent Alzheimer’s disease. Prog. Neurobiol. 2019;181:101662. doi: 10.1016/j.pneurobio.2019.101662. [DOI] [PubMed] [Google Scholar]
- 8.Ressler K.J., Williams L.M. Big data in psychiatry: multiomics, neuroimaging, computational modeling, and digital phenotyping. Neuropsychopharmacology. 2021;46(1):1–2. doi: 10.1038/s41386-020-00862-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hampel H., Nisticò R., Seyfried N.T., Levey A.I., Modeste E., Lemercier P., Baldacci F., Toschi N., Garaci F., Perry G., Emanuele E., Valenzuela P.L., Lucia A., Urbani A., Sancesario G.M., Mapstone M., Corbo M., Vergallo A., Lista S. Omics sciences for systems biology in Alzheimer’s disease: State-of-the-art of the evidence. Ageing Res. Rev. 2021;69:101346. doi: 10.1016/j.arr.2021.101346. [DOI] [PubMed] [Google Scholar]
- 10.Yan R., Vassar R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014;13(3):319–329. doi: 10.1016/S1474-4422(13)70276-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vassar R. BACE1: the beta-secretase enzyme in Alzheimer’s disease. J. Mol. Neurosci. 2004;23(1-2):105–114. doi: 10.1385/JMN:23:1-2:105. [DOI] [PubMed] [Google Scholar]
- 12.Hampel H., Vassar R., De Strooper B., Hardy J., Willem M., Singh N. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry. 2021;89(8):745–756. doi: 10.1016/j.biopsych.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Head E., Powell D., Gold B.T., Schmitt F.A. Alzheimer’s Disease in Down Syndrome. Eur. J. Neurodegener. Dis. 2012;1(3):353–364. [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang Y., Rigoglioso A., Peterhoff C.M., Pawlik M., Sato Y., Bleiwas C., Stavrides P., Smiley J.F., Ginsberg S.D., Mathews P.M., Levy E., Nixon R.A. Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: role of APP-CTF. Neurobiol. Aging. 2016;39:90–98. doi: 10.1016/j.neurobiolaging.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mei L., Xiong W-C. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 2008;9(6):437–452. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo X., He W., Hu X., Yan R. Reversible overexpression of bace1-cleaved neuregulin-1 N-terminal fragment induces schizophrenia-like phenotypes in mice. Biol. Psychiatry. 2014;76(2):120–127. doi: 10.1016/j.biopsych.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seshadri S., Kamiya A., Yokota Y., Prikulis I., Kano S., Hayashi-Takagi A., Stanco A., Eom T.Y., Rao S., Ishizuka K., Wong P., Korth C., Anton E.S., Sawa A. Disrupted-in-Schizophrenia-1 expression is regulated by beta-site amyloid precursor protein cleaving enzyme-1-neuregulin cascade. Proc. Natl. Acad. Sci. USA. 2010;107(12):5622–5627. doi: 10.1073/pnas.0909284107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z., Huang J., Shen Y., Li R. BACE1-Dependent Neuregulin-1 Signaling: An Implication for Schizophrenia. Front. Mol. Neurosci. 2017;10:302. doi: 10.3389/fnmol.2017.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray B., Long J.M., Sokol D.K., Lahiri D.K. Increased secreted amyloid precursor protein-α (sAPPα) in severe autism: proposal of a specific, anabolic pathway and putative biomarker. PLoS One. 2011;6(6):e20405. doi: 10.1371/journal.pone.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esnafoglu E. Levels of peripheral Neuregulin 1 are increased in non-medicated autism spectrum disorder patients. J. Clin. Neurosci. 2018;57:43–45. doi: 10.1016/j.jocn.2018.08.043. [DOI] [PubMed] [Google Scholar]
- 21.Ghafouri-Fard S., Namvar A., Arsang-Jang S., Komaki A., Taheri M. Expression analysis of BDNF, BACE1, and their natural occurring antisenses in autistic patients. J. Mol. Neurosci. 2020;70(2):194–200. doi: 10.1007/s12031-019-01432-7. [DOI] [PubMed] [Google Scholar]
- 22.Hu X., Hicks C.W., He W., Wong P., Macklin W.B., Trapp B.D., Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat. Neurosci. 2006;9(12):1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- 23.Ou-Yang M-H., Kurz J.E., Nomura T., Popovic J., Rajapaksha T.W., Dong H., Contractor A., Chetkovich D.M., Tourtellotte W.G., Vassar R. Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci. Transl. Med. 2018;10(459):eaao5620. doi: 10.1126/scitranslmed.aao5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vassar R. Editorial: Implications for BACE1 inhibitor clinical trials: Adult conditional BACE1 knockout mice exhibit axonal organization defects in the hippocampus. J. Prev. Alzheimers Dis. 2019;6(2):78–84. doi: 10.14283/jpad.2019.3. [DOI] [PubMed] [Google Scholar]
- 25.Vassar R., Bennett B.D., Babu-Khan S., Kahn S., Mendiaz E.A., Denis P., Teplow D.B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M.A., Biere A.L., Curran E., Burgess T., Louis J.C., Collins F., Treanor J., Rogers G., Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 26.Voytyuk I., Mueller S.A., Herber J., Snellinx A., Moechars D., van Loo G., Lichtenthaler S.F., De Strooper B. BACE2 distribution in major brain cell types and identification of novel substrates. Life Sci. Alliance. 2018;1(1):e201800026. doi: 10.26508/lsa.201800026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huentelman M., De Both M., Jepsen W., Piras I.S., Talboom J.S., Willeman M., Reiman E.M., Hardy J., Myers A.J. Common BACE2 Polymorphisms are Associated with Altered Risk for Alzheimer’s Disease and CSF Amyloid Biomarkers in APOE ε4 Non-Carriers. Sci. Rep. 2019;9(1):9640. doi: 10.1038/s41598-019-45896-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett B.D., Babu-Khan S., Loeloff R., Louis J.C., Curran E., Citron M., Vassar R. Expression analysis of BACE2 in brain and peripheral tissues. J. Biol. Chem. 2000;275(27):20647–20651. doi: 10.1074/jbc.M002688200. [DOI] [PubMed] [Google Scholar]
- 29.Wilhelm B.G., Mandad S., Truckenbrodt S., Kröhnert K., Schäfer C., Rammner B., Koo S.J., Claßen G.A., Krauss M., Haucke V., Urlaub H., Rizzoli S.O. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 2014;344(6187):1023–1028. doi: 10.1126/science.1252884. [DOI] [PubMed] [Google Scholar]
- 30.Seabrook G.R., Smith D.W., Bowery B.J., Easter A., Reynolds T., Fitzjohn S.M., Morton R.A., Zheng H., Dawson G.R., Sirinathsinghji D.J., Davies C.H., Collingridge G.L., Hill R.G. Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology. 1999;38(3):349–359. doi: 10.1016/S0028-3908(98)00204-4. [DOI] [PubMed] [Google Scholar]
- 31.Rice H.C., de Malmazet D., Schreurs A., Frere S., Van Molle I., Volkov A.N., Creemers E., Vertkin I., Nys J., Ranaivoson F.M., Comoletti D., Savas J.N., Remaut H., Balschun D., Wierda K.D., Slutsky I., Farrow K., De Strooper B., de Wit J. Secreted amyloid-β precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science. 2019;363(6423):eaao4827. doi: 10.1126/science.aao4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giuffrida M.L., Tomasello M.F., Pandini G., Caraci F., Battaglia G., Busceti C., Di Pietro P., Pappalardo G., Attanasio F., Chiechio S., Bagnoli S., Nacmias B., Sorbi S., Vigneri R., Rizzarelli E., Nicoletti F., Copani A. Monomeric ß-amyloid interacts with type-1 insulin-like growth factor receptors to provide energy supply to neurons. Front. Cell. Neurosci. 2015;9:297. doi: 10.3389/fncel.2015.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zimbone S., Monaco I., Gianì F., Pandini G., Copani A.G., Giuffrida M.L., Rizzarelli E. Amyloid Beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells. Aging Cell. 2018;17(1):e12684. doi: 10.1111/acel.12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tyan S-H., Shih A.Y-J., Walsh J.J., Maruyama H., Sarsoza F., Ku L., Eggert S., Hof P.R., Koo E.H., Dickstein D.L. Amyloid precursor protein (APP) regulates synaptic structure and function. Mol. Cell. Neurosci. 2012;51(1-2):43–52. doi: 10.1016/j.mcn.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abramov E., Dolev I., Fogel H., Ciccotosto G.D., Ruff E., Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 2009;12(12):1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- 36.Basi G., Frigon N., Barbour R., Doan T., Gordon G., McConlogue L., Sinha S., Zeller M. Antagonistic effects of beta-site amyloid precursor protein-cleaving enzymes 1 and 2 on beta-amyloid peptide production in cells. J. Biol. Chem. 2003;278(34):31512–31520. doi: 10.1074/jbc.M300169200. [DOI] [PubMed] [Google Scholar]
- 37.Fluhrer R., Capell A., Westmeyer G., Willem M., Hartung B., Condron M.M., Teplow D.B., Haass C., Walter J. A non-amyloidogenic function of BACE-2 in the secretory pathway. J. Neurochem. 2002;81(5):1011–1020. doi: 10.1046/j.1471-4159.2002.00908.x. [DOI] [PubMed] [Google Scholar]
- 38.Shi X-P., Tugusheva K., Bruce J.E., Lucka A., Wu G-X., Chen-Dodson E., Price E., Li Y., Xu M., Huang Q., Sardana M.K., Hazuda D.J. Beta-secretase cleavage at amino acid residue 34 in the amyloid beta peptide is dependent upon gamma-secretase activity. J. Biol. Chem. 2003;278(23):21286–21294. doi: 10.1074/jbc.M209859200. [DOI] [PubMed] [Google Scholar]
- 39.Abdul-Hay S.O., Sahara T., McBride M., Kang D., Leissring M.A. Identification of BACE2 as an avid ß-amyloid-degrading protease. Mol. Neurodegener. 2012;7:46. doi: 10.1186/1750-1326-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dominguez D., Tournoy J., Hartmann D., Huth T., Cryns K., Deforce S., Serneels L., Camacho I.E., Marjaux E., Craessaerts K., Roebroek A.J., Schwake M., D’Hooge R., Bach P., Kalinke U., Moechars D., Alzheimer C., Reiss K., Saftig P., De Strooper B. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J. Biol. Chem. 2005;280(35):30797–30806. doi: 10.1074/jbc.M505249200. [DOI] [PubMed] [Google Scholar]
- 41.Yang J.Z., Si T.M., Ruan Y., Ling Y.S., Han Y.H., Wang X.L., Zhou M., Zhang H.Y., Kong Q.M., Liu C., Zhang D.R., Yu Y.Q., Liu S.Z., Ju G.Z., Shu L., Ma D.L., Zhang D. Association study of neuregulin 1 gene with schizophrenia. Mol. Psychiatry. 2003;8(7):706–709. doi: 10.1038/sj.mp.4001377. [DOI] [PubMed] [Google Scholar]
- 42.Yan R., Fan Q., Zhou J., Vassar R. Inhibiting BACE1 to reverse synaptic dysfunctions in Alzheimer’s disease. Neurosci. Biobehav. Rev. 2016;65:326–340. doi: 10.1016/j.neubiorev.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pigoni M., Wanngren J., Kuhn P-H., Munro K.M., Gunnersen J.M., Takeshima H., Feederle R., Voytyuk I., De Strooper B., Levasseur M.D., Hrupka B.J., Müller S.A., Lichtenthaler S.F. Seizure protein 6 and its homolog seizure 6-like protein are physiological substrates of BACE1 in neurons. Mol. Neurodegener. 2016;11(1):67. doi: 10.1186/s13024-016-0134-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu K., Xiang X., Filser S., Marinković P., Dorostkar M.M., Crux S., Neumann U., Shimshek D.R., Rammes G., Haass C., Lichtenthaler S.F., Gunnersen J.M., Herms J. Beta-site amyloid precursor protein cleaving enzyme 1 inhibition impairs synaptic plasticity via seizure protein 6. Biol. Psychiatry. 2018;83(5):428–437. doi: 10.1016/j.biopsych.2016.12.023. [DOI] [PubMed] [Google Scholar]
- 45.Barão S., Gärtner A., Leyva-Díaz E., Demyanenko G., Munck S., Vanhoutvin T., Zhou L., Schachner M., López-Bendito G., Maness P.F., De Strooper B. Antagonistic effects of BACE1 and APH1B-γ-secretase control axonal guidance by regulating growth cone collapse. Cell Rep. 2015;12(9):1367–1376. doi: 10.1016/j.celrep.2015.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hitt B., Riordan S.M., Kukreja L., Eimer W.A., Rajapaksha T.W., Vassar R. β-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J. Biol. Chem. 2012;287(46):38408–38425. doi: 10.1074/jbc.M112.415505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z., Xu Q., Cai F., Liu X., Wu Y., Song W. BACE2, a conditional β-secretase, contributes to Alzheimer’s disease pathogenesis. JCI Insight. 2019;4(1):123431. doi: 10.1172/jci.insight.123431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Czarnek M., Bereta J. Proteolytic processing of neuregulin 2. Mol. Neurobiol. 2020;57(4):1799–1813. doi: 10.1007/s12035-019-01846-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martiskainen H., Herukka S-K., Stančáková A., Paananen J., Soininen H., Kuusisto J., Laakso M., Hiltunen M. Decreased plasma β-amyloid in the Alzheimer’s disease APP A673T variant carriers. Ann. Neurol. 2017;82(1):128–132. doi: 10.1002/ana.24969. [DOI] [PubMed] [Google Scholar]
- 50.Zhou L., Brouwers N., Benilova I., Vandersteen A., Mercken M., Van Laere K., Van Damme P., Demedts D., Van Leuven F., Sleegers K., Broersen K., Van Broeckhoven C., Vandenberghe R., De Strooper B. Amyloid precursor protein mutation E682K at the alternative β-secretase cleavage β′-site increases Aβ generation. EMBO Mol. Med. 2011;3(5):291–302. doi: 10.1002/emmm.201100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mullan M., Crawford F., Axelman K., Houlden H., Lilius L., Winblad B., Lannfelt L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1992;1(5):345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 52.Fukumoto H., Cheung B.S., Hyman B.T., Irizarry M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002;59(9):1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- 53.Zhao J., Fu Y., Yasvoina M., Shao P., Hitt B., O’Connor T., Logan S., Maus E., Citron M., Berry R., Binder L., Vassar R. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J. Neurosci. 2007;27(14):3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sadleir K.R., Kandalepas P.C., Buggia-Prévot V., Nicholson D.A., Thinakaran G., Vassar R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer’s disease. Acta Neuropathol. 2016;132(2):235–256. doi: 10.1007/s00401-016-1558-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wisniewski K.E., Wisniewski H.M., Wen G.Y. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann. Neurol. 1985;17(3):278–282. doi: 10.1002/ana.410170310. [DOI] [PubMed] [Google Scholar]
- 56.Alić I., Goh P.A., Murray A., Portelius E., Gkanatsiou E., Gough G., Mok K.Y., Koschut D., Brunmeir R., Yeap Y.J., O’Brien N.L., Groet J., Shao X., Havlicek S., Dunn N.R., Kvartsberg H., Brinkmalm G., Hithersay R., Startin C., Hamburg S., Phillips M., Pervushin K., Turmaine M., Wallon D., Rovelet-Lecrux A., Soininen H., Volpi E., Martin J.E., Foo J.N., Becker D.L., Rostagno A., Ghiso J., Krsnik Ž., Šimić G., Kostović I., Mitrečić D., Francis P.T., Blennow K., Strydom A., Hardy J., Zetterberg H., Nižetić D. Patient-specific Alzheimer-like pathology in trisomy 21 cerebral organoids reveals BACE2 as a gene dose-sensitive AD suppressor in human brain. Mol. Psychiatry. 2021;26(10):5766–5788. doi: 10.1038/s41380-020-0806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caraci F., Iulita M.F., Pentz R., Flores Aguilar L., Orciani C., Barone C., Romano C., Drago F., Cuello A.C. Searching for new pharmacological targets for the treatment of Alzheimer’s disease in Down syndrome. Eur. J. Pharmacol. 2017;817:7–19. doi: 10.1016/j.ejphar.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 58.Myllykangas L., Wavrant-De Vrièze F., Polvikoski T., Notkola I-L., Sulkava R., Niinistö L., Edland S.D., Arepalli S., Adighibe O., Compton D., Hardy J., Haltia M., Tienari P.J. Chromosome 21 BACE2 haplotype associates with Alzheimer’s disease: a two-stage study. J. Neurol. Sci. 2005;236(1-2):17–24. doi: 10.1016/j.jns.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 59.Mok K.Y., Jones E.L., Hanney M., Harold D., Sims R., Williams J., Ballard C., Hardy J. Polymorphisms in BACE2 may affect the age of onset Alzheimer’s dementia in Down syndrome. Neurobiol. Aging. 2014;35(6):1513.e1–1513.e5. doi: 10.1016/j.neurobiolaging.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rovelet-Lecrux A., Charbonnier C., Wallon D., Nicolas G., Seaman M.N.J., Pottier C., Breusegem S.Y., Mathur P.P., Jenardhanan P., Le Guennec K., Mukadam A.S., Quenez O., Coutant S., Rousseau S., Richard A.C., Boland A., Deleuze J.F., Frebourg T., Hannequin D., Campion D. De novo deleterious genetic variations target a biological network centered on Aβ peptide in early-onset Alzheimer disease. Mol. Psychiatry. 2015;20(9):1046–1056. doi: 10.1038/mp.2015.100. [DOI] [PubMed] [Google Scholar]
- 61.Holler C.J., Webb R.L., Laux A.L., Beckett T.L., Niedowicz D.M., Ahmed R.R., Liu Y., Simmons C.R., Dowling A.L., Spinelli A., Khurgel M., Estus S., Head E., Hersh L.B., Murphy M.P. BACE2 expression increases in human neurodegenerative disease. Am. J. Pathol. 2012;180(1):337–350. doi: 10.1016/j.ajpath.2011.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qiu K., Liang W., Wang S., Kong T., Wang X., Li C., Wang Z., Wu Y. BACE2 degradation is mediated by both the proteasome and lysosome pathways. BMC Mol. Cell Biol. 2020;21(1):13. doi: 10.1186/s12860-020-00260-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thibaudeau T.A., Anderson R.T., Smith D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018;9(1):1097. doi: 10.1038/s41467-018-03509-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bettens K., Vermeulen S., Van Cauwenberghe C., Heeman B., Asselbergh B., Robberecht C., Engelborghs S., Vandenbulcke M., Vandenberghe R., De Deyn P.P., Cruts M., Van Broeckhoven C., Sleegers K. Reduced secreted clusterin as a mechanism for Alzheimer-associated CLU mutations. Mol. Neurodegener. 2015;10:30. doi: 10.1186/s13024-015-0024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ge S., Wang Y., Song M., Li X., Yu X., Wang H., Wang J., Zeng Q., Wang W. Type 2 diabetes mellitus: Integrative analysis of multiomics data for biomarker discovery. OMICS. 2018;22(7):514–523. doi: 10.1089/omi.2018.0053. [DOI] [PubMed] [Google Scholar]
- 66.Ghosh A.K., Brindisi M., Yen Y-C., Lendy E.K., Kovela S., Cárdenas E.L., Reddy B.S., Rao K.V., Downs D., Huang X., Tang J., Mesecar A.D. Highly selective and potent human β-secretase 2 (BACE2) inhibitors against type 2 diabetes: Design, synthesis, X-ray structure and structure-activity relationship studies. ChemMedChem. 2019;14(5):545–560. doi: 10.1002/cmdc.201900100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen Z., Zhong C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013;108:21–43. doi: 10.1016/j.pneurobio.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 68.Biosa A., Outeiro T.F., Bubacco L., Bisaglia M. Diabetes mellitus as a risk factor for Parkinson’s disease: A molecular point of view. Mol. Neurobiol. 2018;55(11):8754–8763. doi: 10.1007/s12035-018-1025-9. [DOI] [PubMed] [Google Scholar]
- 69.Barbiero L., Benussi L., Ghidoni R., Alberici A., Russo C., Schettini G., Pagano S.F., Parati E.A., Mazzoli F., Nicosia F., Signorini S., Feudatari E., Binetti G. BACE-2 is overexpressed in Down’s syndrome. Exp. Neurol. 2003;182(2):335–345. doi: 10.1016/S0014-4886(03)00049-9. [DOI] [PubMed] [Google Scholar]
- 70.Sun X., He G., Song W. BACE2, as a novel APP theta-secretase, is not responsible for the pathogenesis of Alzheimer’s disease in Down syndrome. FASEB J. 2006;20(9):1369–1376. doi: 10.1096/fj.05-5632com. [DOI] [PubMed] [Google Scholar]
- 71.Kong D.H., Kim Y.K., Kim M.R., Jang J.H., Lee S. Emerging roles of vascular cell adhesion molecule-1 (VCAM-1) in immunological disorders and cancer. Int. J. Mol. Sci. 2018;19(4):E1057. doi: 10.3390/ijms19041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cook-Mills J.M., Marchese M.E., Abdala-Valencia H. Vascular cell adhesion molecule-1 expression and signaling during disease: regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 2011;15(6):1607–1638. doi: 10.1089/ars.2010.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ballester-López C., Conlon T.M., Ertüz Z., Greiffo F.R., Irmler M., Verleden S.E., Beckers J., Fernandez I.E., Eickelberg O., Yildirim A.Ö. The Notch ligand DNER regulates macrophage IFNγ release in chronic obstructive pulmonary disease. EBioMedicine. 2019;43:562–575. doi: 10.1016/j.ebiom.2019.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang L., Wu Q., Zhu S., Li Z., Yuan J., Yu D., Xu Z., Li J., Sun S., Wang C. Delta/notch-like epidermal growth factor-related receptor (DNER) orchestrates stemness and cancer progression in prostate cancer. Am. J. Transl. Res. 2017;9(11):5031–5039. [PMC free article] [PubMed] [Google Scholar]
- 75.Sun P., Xia S., Lal B., Eberhart C.G., Quinones-Hinojosa A., Maciaczyk J., Matsui W., Dimeco F., Piccirillo S.M., Vescovi A.L., Laterra J. DNER, an epigenetically modulated gene, regulates glioblastoma-derived neurosphere cell differentiation and tumor propagation. Stem Cells. 2009;27(7):1473–1486. doi: 10.1002/stem.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eiraku M., Hirata Y., Takeshima H., Hirano T., Kengaku M. Delta/notch-like epidermal growth factor (EGF)-related receptor, a novel EGF-like repeat-containing protein targeted to dendrites of developing and adult central nervous system neurons. J. Biol. Chem. 2002;277(28):25400–25407. doi: 10.1074/jbc.M110793200. [DOI] [PubMed] [Google Scholar]
- 77.Nanda A., Buckhaults P., Seaman S., Agrawal N., Boutin P., Shankara S., Nacht M., Teicher B., Stampfl J., Singh S., Vogelstein B., Kinzler K.W., St Croix B. Identification of a binding partner for the endothelial cell surface proteins TEM7 and TEM7R. Cancer Res. 2004;64(23):8507–8511. doi: 10.1158/0008-5472.CAN-04-2716. [DOI] [PubMed] [Google Scholar]
- 78.Miller-Delaney S.F.C., Lieberam I., Murphy P., Mitchell K.J. Plxdc2 is a mitogen for neural progenitors. PLoS One. 2011;6(1):e14565. doi: 10.1371/journal.pone.0014565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schwarze S.R., Fu V.X., Desotelle J.A., Kenowski M.L., Jarrard D.F. The identification of senescence-specific genes during the induction of senescence in prostate cancer cells. Neoplasia. 2005;7(9):816–823. doi: 10.1593/neo.05250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McMurray H.R., Sampson E.R., Compitello G., Kinsey C., Newman L., Smith B., Chen S.R., Klebanov L., Salzman P., Yakovlev A., Land H. Synergistic response to oncogenic mutations defines gene class critical to cancer phenotype. Nature. 2008;453(7198):1112–1116. doi: 10.1038/nature06973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hallstrom T.C., Mori S., Nevins J.R. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell. 2008;13(1):11–22. doi: 10.1016/j.ccr.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gaudet P., Livstone M.S., Lewis S.E., Thomas P.D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief. Bioinform. 2011;12(5):449–462. doi: 10.1093/bib/bbr042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ornitz D.M., Xu J., Colvin J.S., McEwen D.G., MacArthur C.A., Coulier F., Gao G., Goldfarb M. Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 1996;271(25):15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 84.Eswarakumar V.P., Lax I., Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16(2):139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 85.Wang L., Song G., Tan W., Qi M., Zhang L., Chan J., Yu J., Han J., Han B. MiR-573 inhibits prostate cancer metastasis by regulating epithelial-mesenchymal transition. Oncotarget. 2015;6(34):35978–35990. doi: 10.18632/oncotarget.5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gunnersen J.M., Kim M.H., Fuller S.J., De Silva M., Britto J.M., Hammond V.E., Davies P.J., Petrou S., Faber E.S., Sah P., Tan S.S. Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron. 2007;56(4):621–639. doi: 10.1016/j.neuron.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 87.Håvik B., Røkke H., Dagyte G., Stavrum A.K., Bramham C.R., Steen V.M. Synaptic activity-induced global gene expression patterns in the dentate gyrus of adult behaving rats: induction of immunity-linked genes. Neuroscience. 2007;148(4):925–936. doi: 10.1016/j.neuroscience.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 88.Causevic M., Dominko K., Malnar M., Vidatic L., Cermak S., Pigoni M., Kuhn P.H., Colombo A., Havas D., Flunkert S., McDonald J., Gunnersen J.M., Hutter-Paier B., Tahirovic S., Windisch M., Krainc D., Lichtenthaler S.F., Hecimovic S. BACE1-cleavage of Sez6 and Sez6L is elevated in Niemann-Pick type C disease mouse brains. PLoS One. 2018;13(7):e0200344. doi: 10.1371/journal.pone.0200344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Esterházy D., Stützer I., Wang H., Rechsteiner M.P., Beauchamp J., Döbeli H., Hilpert H., Matile H., Prummer M., Schmidt A., Lieske N., Boehm B., Marselli L., Bosco D., Kerr-Conte J., Aebersold R., Spinas G.A., Moch H., Migliorini C., Stoffel M. Bace2 is a β cell-enriched protease that regulates pancreatic β cell function and mass. Cell Metab. 2011;14(3):365–377. doi: 10.1016/j.cmet.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 90.Zhang H., Wada J., Hida K., Tsuchiyama Y., Hiragushi K., Shikata K., Wang H., Lin S., Kanwar Y.S., Makino H. Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J. Biol. Chem. 2001;276(20):17132–17139. doi: 10.1074/jbc.M006723200. [DOI] [PubMed] [Google Scholar]
- 91.Danilczyk U., Sarao R., Remy C., Benabbas C., Stange G., Richter A., Arya S., Pospisilik J.A., Singer D., Camargo S.M., Makrides V., Ramadan T., Verrey F., Wagner C.A., Penninger J.M. Essential role for collectrin in renal amino acid transport. Nature. 2006;444(7122):1088–1091. doi: 10.1038/nature05475. [DOI] [PubMed] [Google Scholar]
- 92.Fukui K., Yang Q., Cao Y., Takahashi N., Hatakeyama H., Wang H., Wada J., Zhang Y., Marselli L., Nammo T., Yoneda K., Onishi M., Higashiyama S., Matsuzawa Y., Gonzalez F.J., Weir G.C., Kasai H., Shimomura I., Miyagawa J., Wollheim C.B., Yamagata K. The HNF-1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab. 2005;2(6):373–384. doi: 10.1016/j.cmet.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 93.Vuille-dit-Bille R.N.; Camargo, S.M.; Emmenegger, L.; Sasse, T.; Kummer, E.; Jando, J.; Hamie, Q.M.; Meier, C.F.; Hunziker, S.; Forras-Kaufmann, Z.; Kuyumcu, S.; Fox, M.; Schwizer, W.; Fried, M.; Lindenmeyer, M.; Götze, O.; Verrey, F. Human intestine luminal ACE2 and amino acid transporter expression increased by ACE-inhibitors. Amino Acids. 2015;47(4):693–705. doi: 10.1007/s00726-014-1889-6. [DOI] [PubMed] [Google Scholar]
- 94.Fu W., Ruangkittisakul A., MacTavish D., Shi J.Y., Ballanyi K., Jhamandas J.H. Amyloid β (Aβ) peptide directly activates amylin-3 receptor subtype by triggering multiple intracellular signaling pathways. J. Biol. Chem. 2012;287(22):18820–18830. doi: 10.1074/jbc.M111.331181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Caruso G., Fresta C.G., Lazzarino G., Distefano D.A., Parlascino P., Lunte S.M., Lazzarino G., Caraci F. Sub-toxic human amylin fragment concentrations promote the survival and proliferation of SH-SY5Y cells via the release of VEGF and HspB5 from endothelial RBE4 cells. Int. J. Mol. Sci. 2018;19(11):E3659. doi: 10.3390/ijms19113659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lorenzo A., Razzaboni B., Weir G.C., Yankner B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368(6473):756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- 97.Nishi M., Sanke T., Seino S., Eddy R.L., Fan Y.S., Byers M.G., Shows T.B., Bell G.I., Steiner D.F. Human islet amyloid polypeptide gene: complete nucleotide sequence, chromosomal localization, and evolutionary history. Mol. Endocrinol. 1989;3(11):1775–1781. doi: 10.1210/mend-3-11-1775. [DOI] [PubMed] [Google Scholar]
- 98.Rulifson I.C., Cao P., Miao L., Kopecky D., Huang L., White R.D., Samayoa K., Gardner J., Wu X., Chen K., Tsuruda T., Homann O., Baribault H., Yamane H., Carlson T., Wiltzius J., Li Y. Identification of Human Islet Amyloid Polypeptide as a BACE2 Substrate. PLoS One. 2016;11(2):e0147254. doi: 10.1371/journal.pone.0147254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hoashi T., Watabe H., Muller J., Yamaguchi Y., Vieira W.D., Hearing V.J. MART-1 is required for the function of the melanosomal matrix protein PMEL17/GP100 and the maturation of melanosomes. J. Biol. Chem. 2005;280(14):14006–14016. doi: 10.1074/jbc.M413692200. [DOI] [PubMed] [Google Scholar]
- 100.Rochin L., Hurbain I., Serneels L., Fort C., Watt B., Leblanc P., Marks M.S., De Strooper B., Raposo G., van Niel G. BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proc. Natl. Acad. Sci. USA. 2013;110(26):10658–10663. doi: 10.1073/pnas.1220748110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mathalon D.H., Sullivan E.V., Lim K.O., Pfefferbaum A. Progressive brain volume changes and the clinical course of schizophrenia in men: a longitudinal magnetic resonance imaging study. Arch. Gen. Psychiatry. 2001;58(2):148–157. doi: 10.1001/archpsyc.58.2.148. [DOI] [PubMed] [Google Scholar]
- 102.Harrison P.J., Law A.J. Neuregulin 1 and schizophrenia: genetics, gene expression, and neurobiology. Biol. Psychiatry. 2006;60(2):132–140. doi: 10.1016/j.biopsych.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 103.Pickard B.S. Schizophrenia biomarkers: translating the descriptive into the diagnostic. J. Psychopharmacol. 2015;29(2):138–143. doi: 10.1177/0269881114566631. [DOI] [PubMed] [Google Scholar]
- 104.Hayashi-Takagi A., Vawter M.P., Iwamoto K. Peripheral biomarkers revisited: integrative profiling of peripheral samples for psychiatric research. Biol. Psychiatry. 2014;75(12):920–928. doi: 10.1016/j.biopsych.2013.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stefansson H., Sigurdsson E., Steinthorsdottir V., Bjornsdottir S., Sigmundsson T., Ghosh S., Brynjolfsson J., Gunnarsdottir S., Ivarsson O., Chou T.T., Hjaltason O., Birgisdottir B., Jonsson H., Gudnadottir V.G., Gudmundsdottir E., Bjornsson A., Ingvarsson B., Ingason A., Sigfusson S., Hardardottir H., Harvey R.P., Lai D., Zhou M., Brunner D., Mutel V., Gonzalo A., Lemke G., Sainz J., Johannesson G., Andresson T., Gudbjartsson D., Manolescu A., Frigge M.L., Gurney M.E., Kong A., Gulcher J.R., Petursson H., Stefansson K. Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 2002;71(4):877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Walss-Bass C., Raventos H., Montero A.P., Armas R., Dassori A., Contreras S., Liu W., Medina R., Levinson D.F., Pereira M., Leach R.J., Almasy L., Escamilla M.A. Association analyses of the neuregulin 1 gene with schizophrenia and manic psychosis in a Hispanic population. Acta Psychiatr. Scand. 2006;113(4):314–321. doi: 10.1111/j.1600-0447.2005.00631.x. [DOI] [PubMed] [Google Scholar]
- 107.Fleck D., Voss M., Brankatschk B., Giudici C., Hampel H., Schwenk B., Edbauer D., Fukumori A., Steiner H., Kremmer E., Haug-Kröper M., Rossner M.J., Fluhrer R., Willem M., Haass C. Proteolytic Processing of Neuregulin 1 Type III by Three Intramembrane-cleaving Proteases. J. Biol. Chem. 2016;291(1):318–333. doi: 10.1074/jbc.M115.697995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Savonenko A.V., Melnikova T., Laird F.M., Stewart K-A., Price D.L., Wong P.C. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc. Natl. Acad. Sci. USA. 2008;105(14):5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Buonanno A. The neuregulin signaling pathway and schizophrenia: from genes to synapses and neural circuits. Brain Res. Bull. 2010;83(3-4):122–131. doi: 10.1016/j.brainresbull.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mei L., Nave K-A. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron. 2014;83(1):27–49. doi: 10.1016/j.neuron.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang H., Li R., Shen Y. β-Secretase: its biology as a therapeutic target in diseases. Trends Pharmacol. Sci. 2013;34(4):215–225. doi: 10.1016/j.tips.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Allen N.C., Bagade S., McQueen M.B., Ioannidis J.P.A., Kavvoura F.K., Khoury M.J., Tanzi R.E., Bertram L. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat. Genet. 2008;40(7):827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- 113.Hu X., Fan Q., Hou H., Yan R. Neurological dysfunctions associated with altered BACE1-dependent Neuregulin-1 signaling. J. Neurochem. 2016;136(2):234–249. doi: 10.1111/jnc.13395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hahn C-G., Wang H-Y., Cho D-S., Talbot K., Gur R.E., Berrettini W.H., Bakshi K., Kamins J., Borgmann-Winter K.E., Siegel S.J., Gallop R.J., Arnold S.E. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat. Med. 2006;12(7):824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- 115.Krivosheya D., Tapia L., Levinson J.N., Huang K., Kang Y., Hines R., Ting A.K., Craig A.M., Mei L., Bamji S.X., El-Husseini A. ErbB4-neuregulin signaling modulates synapse development and dendritic arborization through distinct mechanisms. J. Biol. Chem. 2008;283(47):32944–32956. doi: 10.1074/jbc.M800073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lang U.E., Hellweg R., Gallinat J. Association of BDNF serum concentrations with central serotonergic activity: evidence from auditory signal processing. Neuropsychopharmacology. 2005;30(6):1148–1153. doi: 10.1038/sj.npp.1300666. [DOI] [PubMed] [Google Scholar]
- 117.Corfas G., Roy K., Buxbaum J.D. Neuregulin 1-erbB signaling and the molecular/cellular basis of schizophrenia. Nat. Neurosci. 2004;7(6):575–580. doi: 10.1038/nn1258. [DOI] [PubMed] [Google Scholar]
- 118.Barakat A., Dean B., Scarr E., Evin G. Decreased Neuregulin 1 C-terminal fragment in Brodmann’s area 6 of patients with schizophrenia. Schizophr. Res. 2010;124(1-3):200–207. doi: 10.1016/j.schres.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 119.Dean B., Soulby A., Evin G.M., Scarr E. Levels of [(3)H]pirenzepine binding in Brodmann’s area 6 from subjects with schizophrenia is not associated with changes in the transcription factor SP1 or BACE1. Schizophr. Res. 2008;106(2-3):229–236. doi: 10.1016/j.schres.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 120.Marballi K., Cruz D., Thompson P., Walss-Bass C. Differential neuregulin 1 cleavage in the prefrontal cortex and hippocampus in schizophrenia and bipolar disorder: preliminary findings. PLoS One. 2012;7(5):e36431. doi: 10.1371/journal.pone.0036431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang Z., Cui J., Gao F., Li Y., Zhang G., Liu M., Yan R., Shen Y., Li R. Elevated cleavage of neuregulin-1 by beta-secretase 1 in plasma of schizophrenia patients. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2019;90:161–168. doi: 10.1016/j.pnpbp.2018.11.018. [DOI] [PubMed] [Google Scholar]
- 122.Zhang Z., Li Y., He F., Cui Y., Zheng Y., Li R. Sex differences in circulating neuregulin1-β1 and β-secretase 1 expression in childhood-onset schizophrenia. Compr. Psychiatry. 2020;100:152176. doi: 10.1016/j.comppsych.2020.152176. [DOI] [PubMed] [Google Scholar]
- 123.Shibuya M., Komi E., Wang R., Kato T., Watanabe Y., Sakai M., Ozaki M., Someya T., Nawa H. Measurement and comparison of serum neuregulin 1 immunoreactivity in control subjects and patients with schizophrenia: an influence of its genetic polymorphism. J. Neural Transm. (Vienna) 2010;117(7):887–895. doi: 10.1007/s00702-010-0418-3. [DOI] [PubMed] [Google Scholar]
- 124.Muthusamy N., Faundez V., Bergson C. Calcyon, a mammalian specific NEEP21 family member, interacts with adaptor protein complex 3 (AP-3) and regulates targeting of AP-3 cargoes. J. Neurochem. 2012;123(1):60–72. doi: 10.1111/j.1471-4159.2012.07814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Alberi S., Boda B., Steiner P., Nikonenko I., Hirling H., Muller D. The endosomal protein NEEP21 regulates AMPA receptor-mediated synaptic transmission and plasticity in the hippocampus. Mol. Cell. Neurosci. 2005;29(2):313–319. doi: 10.1016/j.mcn.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 126.Norstrom E.M., Zhang C., Tanzi R., Sisodia S.S. Identification of NEEP21 as a ß-amyloid precursor protein-interacting protein in vivo that modulates amyloidogenic processing in vitro. J. Neurosci. 2010;30(46):15677–15685. doi: 10.1523/JNEUROSCI.4464-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Trantham-Davidson H., Vazdarjanova A., Dai R., Terry A., Bergson C. Up-regulation of calcyon results in locomotor hyperactivity and reduced anxiety in mice. Behav. Brain Res. 2008;189(2):244–249. doi: 10.1016/j.bbr.2007.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vazdarjanova A., Bunting K., Muthusamy N., Bergson C. Calcyon upregulation in adolescence impairs response inhibition and working memory in adulthood. Mol. Psychiatry. 2011;16(6):672–684. doi: 10.1038/mp.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kato T., Kasai A., Mizuno M., Fengyi L., Shintani N., Maeda S., Yokoyama M., Ozaki M., Nawa H. Phenotypic characterization of transgenic mice overexpressing neuregulin-1. PLoS One. 2010;5(12):e14185. doi: 10.1371/journal.pone.0014185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yin D-M., Chen Y-J., Liu S., Jiao H., Shen C., Sathyamurthy A., Lin T.W., Xiong W.C., Li B.M., Mei L., Bergson C. Calcyon stimulates neuregulin 1 maturation and signaling. Mol. Psychiatry. 2015;20(10):1251–1260. doi: 10.1038/mp.2014.131. [DOI] [PubMed] [Google Scholar]
- 131.Kumari V., Das M., Zachariah E., Ettinger U., Sharma T. Reduced prepulse inhibition in unaffected siblings of schizophrenia patients. Psychophysiology. 2005;42(5):588–594. doi: 10.1111/j.1469-8986.2005.00346.x. [DOI] [PubMed] [Google Scholar]
- 132.Cadenhead K.S., Swerdlow N.R., Shafer K.M., Diaz M., Braff D.L. Modulation of the startle response and startle laterality in relatives of schizophrenic patients and in subjects with schizotypal personality disorder: evidence of inhibitory deficits. Am. J. Psychiatry. 2000;157(10):1660–1668. doi: 10.1176/appi.ajp.157.10.1660. [DOI] [PubMed] [Google Scholar]
- 133.Hennah W., Thomson P., Peltonen L., Porteous D. Genes and schizophrenia: beyond schizophrenia: the role of DISC1 in major mental illness. Schizophr. Bull. 2006;32(3):409–416. doi: 10.1093/schbul/sbj079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Deng Q-S., Dong X-Y., Wu H., Wang W., Wang Z-T., Zhu J-W., Liu C.F., Jia W.Q., Zhang Y., Schachner M., Ma Q.H., Xu R.X. Disrupted-in-schizophrenia-1 attenuates amyloid-β generation and cognitive deficits in APP/PS1 transgenic mice by reduction of β-Site APP-cleaving enzyme 1 levels. Neuropsychopharmacology. 2016;41(2):440–453. doi: 10.1038/npp.2015.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang Z-T., Lu M-H., Zhang Y., Ji W-L., Lei L., Wang W., Fang L.P., Wang L.W., Yu F., Wang J., Li Z.Y., Wang J.R., Wang T.H., Dou F., Wang Q.W., Wang X.L., Li S., Ma Q.H., Xu R.X. Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell. 2019;18(1):e12860. doi: 10.1111/acel.12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Paredes R.M., Piccart E., Navaira E., Cruz D., Javors M.A., Koek W., Beckstead M.J., Walss-Bass C. Physiological and behavioral effects of amphetamine in BACE1(-/-) mice. Genes Brain Behav. 2015;14(5):411–418. doi: 10.1111/gbb.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ledonne A., Mercuri N.B. mGluR1-dependent long term depression in rodent midbrain dopamine neurons is regulated by neuregulin 1/ErbB signaling. Front. Mol. Neurosci. 2018;11:346. doi: 10.3389/fnmol.2018.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ledonne A., Nobili A., Latagliata E.C., Cavallucci V., Guatteo E., Puglisi-Allegra S., D’Amelio M., Mercuri N.B. Neuregulin 1 signalling modulates mGluR1 function in mesencephalic dopaminergic neurons. Mol. Psychiatry. 2015;20(8):959–973. doi: 10.1038/mp.2014.109. [DOI] [PubMed] [Google Scholar]
- 139.Stertz L., Contreras-Shannon V., Monroy-Jaramillo N., Sun J., Walss-Bass C. BACE1-deficient mice exhibit alterations in immune system pathways. Mol. Neurobiol. 2018;55(1):709–717. doi: 10.1007/s12035-016-0341-1. [DOI] [PubMed] [Google Scholar]
- 140.Xu J., Sun J., Chen J., Wang L., Li A., Helm M., Dubovsky S.L., Bacanu S.A., Zhao Z., Chen X. RNA-Seq analysis implicates dysregulation of the immune system in schizophrenia. BMC Genomics. 2012;13(Suppl. 8):S2. doi: 10.1186/1471-2164-13-S8-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dimitrov D.H., Lee S., Yantis J., Valdez C., Paredes R.M., Braida N., Velligan D., Walss-Bass C. Differential correlations between inflammatory cytokines and psychopathology in veterans with schizophrenia: potential role for IL-17 pathway. Schizophr. Res. 2013;151(1-3):29–35. doi: 10.1016/j.schres.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 142.Gandal M.J., Haney J.R., Parikshak N.N., Leppa V., Ramaswami G., Hartl C., Schork A.J., Appadurai V., Buil A., Werge T.M., Liu C., White K.P., Horvath S., Geschwind D.H. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science. 2018;359(6376):693–697. doi: 10.1126/science.aad6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cheung C., Yu K., Fung G., Leung M., Wong C., Li Q., Sham P., Chua S., McAlonan G. Autistic disorders and schizophrenia: related or remote? An anatomical likelihood estimation. PLoS One. 2010;5(8):e12233. doi: 10.1371/journal.pone.0012233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yoo H.J., Woo R-S., Cho S-C., Kim B-N., Kim J-W., Shin M-S., Park T.W., Son J.W., Chung U.S., Park S., Park M., Kim S.A. Genetic association analyses of neuregulin 1 gene polymorphism with endopheontype for sociality of Korean autism spectrum disorders family. Psychiatry Res. 2015;227(2-3):366–368. doi: 10.1016/j.psychres.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 145.Sokol D.K., Chen D., Farlow M.R., Dunn D.W., Maloney B., Zimmer J.A., Lahiri D.K. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J. Child Neurol. 2006;21(6):444–449. doi: 10.1177/08830738060210062201. [DOI] [PubMed] [Google Scholar]
- 146.Lahiri D.K., Sokol D.K., Erickson C., Ray B., Ho C.Y., Maloney B. Autism as early neurodevelopmental disorder: evidence for an sAPPα-mediated anabolic pathway. Front. Cell. Neurosci. 2013;7:94. doi: 10.3389/fncel.2013.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang Y-Y., Zhao B., Wu M-M., Zheng X-L., Lin L., Yin D-M. Overexpression of neuregulin 1 in GABAergic interneurons results in reversible cortical disinhibition. Nat. Commun. 2021;12(1):278. doi: 10.1038/s41467-020-20552-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Feng T., Tammineni P., Agrawal C., Jeong Y.Y., Cai Q. Autophagy-mediated Regulation of BACE1 Protein Trafficking and Degradation. J. Biol. Chem. 2017;292(5):1679–1690. doi: 10.1074/jbc.M116.766584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Do Carmo S., Hanzel C.E., Jacobs M.L., Machnes Z., Iulita M.F., Yang J., Yu L., Ducatenzeiler A., Danik M., Breuillaud L.S., Bennett D.A., Szyf M., Cuello A.C. Rescue of early bace-1 and global DNA demethylation by S-adenosylmethionine reduces amyloid pathology and improves cognition in an Alzheimer’s model. Sci. Rep. 2016;6:34051. doi: 10.1038/srep34051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tan G-H., Liu Y-Y., Hu X-L., Yin D-M., Mei L., Xiong Z-Q. Neuregulin 1 represses limbic epileptogenesis through ErbB4 in parvalbumin-expressing interneurons. Nat. Neurosci. 2011;15(2):258–266. doi: 10.1038/nn.3005. [DOI] [PubMed] [Google Scholar]
- 151.Li K-X., Lu Y-M., Xu Z-H., Zhang J., Zhu J-M., Zhang J-M., Cao S.X., Chen X.J., Chen Z., Luo J.H., Duan S., Li X.M. Neuregulin 1 regulates excitability of fast-spiking neurons through Kv1.1 and acts in epilepsy. Nat. Neurosci. 2011;15(2):267–273. doi: 10.1038/nn.3006. [DOI] [PubMed] [Google Scholar]
- 152.Gabrielsen T.P., Anderson J.S., Stephenson K.G., Beck J., King J.B., Kellems R., Top D.N., Jr, Russell N.C.C., Anderberg E., Lundwall R.A., Hansen B., South M. Functional MRI connectivity of children with autism and low verbal and cognitive performance. Mol. Autism. 2018;9:67. doi: 10.1186/s13229-018-0248-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Penault-Llorca F., Rudzinski E.R., Sepulveda A.R. Testing algorithm for identification of patients with TRK fusion cancer. J. Clin. Pathol. 2019;72(7):460–467. doi: 10.1136/jclinpath-2018-205679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Le D.T., Durham J.N., Smith K.N., Wang H., Bartlett B.R., Aulakh L.K., Lu S., Kemberling H., Wilt C., Luber B.S., Wong F., Azad N.S., Rucki A.A., Laheru D., Donehower R., Zaheer A., Fisher G.A., Crocenzi T.S., Lee J.J., Greten T.F., Duffy A.G., Ciombor K.K., Eyring A.D., Lam B.H., Joe A., Kang S.P., Holdhoff M., Danilova L., Cope L., Meyer C., Zhou S., Goldberg R.M., Armstrong D.K., Bever K.M., Fader A.N., Taube J., Housseau F., Spetzler D., Xiao N., Pardoll D.M., Papadopoulos N., Kinzler K.W., Eshleman J.R., Vogelstein B., Anders R.A., Diaz L.A. Jr Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Drilon A., Laetsch T.W., Kummar S., DuBois S.G., Lassen U.N., Demetri G.D., Nathenson M., Doebele R.C., Farago A.F., Pappo A.S., Turpin B., Dowlati A., Brose M.S., Mascarenhas L., Federman N., Berlin J., El-Deiry W.S., Baik C., Deeken J., Boni V., Nagasubramanian R., Taylor M., Rudzinski E.R., Meric-Bernstam F., Sohal D.P.S., Ma P.C., Raez L.E., Hechtman J.F., Benayed R., Ladanyi M., Tuch B.B., Ebata K., Cruickshank S., Ku N.C., Cox M.C., Hawkins D.S., Hong D.S., Hyman D.M. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med. 2018;378(8):731–739. doi: 10.1056/NEJMoa1714448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Drost J., Clevers H. Organoids in cancer research. Nat. Rev. Cancer. 2018;18(7):407–418. doi: 10.1038/s41568-018-0007-6. [DOI] [PubMed] [Google Scholar]
- 157.Ballard D.H., Boyer C.J., Alexander J.S. Organoids - preclinical models of human disease. N. Engl. J. Med. 2019;380(20):1981–1982. doi: 10.1056/NEJMc1903253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Siravegna G., Marsoni S., Siena S., Bardelli A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017;14(9):531–548. doi: 10.1038/nrclinonc.2017.14. [DOI] [PubMed] [Google Scholar]
- 159.Parikh A.R., Leshchiner I., Elagina L., Goyal L., Levovitz C., Siravegna G., Livitz D., Rhrissorrakrai K., Martin E.E., Van Seventer E.E., Hanna M., Slowik K., Utro F., Pinto C.J., Wong A., Danysh B.P., de la Cruz F.F., Fetter I.J., Nadres B., Shahzade H.A., Allen J.N., Blaszkowsky L.S., Clark J.W., Giantonio B., Murphy J.E., Nipp R.D., Roeland E., Ryan D.P., Weekes C.D., Kwak E.L., Faris J.E., Wo J.Y., Aguet F., Dey-Guha I., Hazar-Rethinam M., Dias-Santagata D., Ting D.T., Zhu A.X., Hong T.S., Golub T.R., Iafrate A.J., Adalsteinsson V.A., Bardelli A., Parida L., Juric D., Getz G., Corcoran R.B. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019;25(9):1415–1421. doi: 10.1038/s41591-019-0561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Marchiò C., Scaltriti M., Ladanyi M., Iafrate A.J., Bibeau F., Dietel M., Hechtman J.F., Troiani T., López-Rios F., Douillard J.Y., Andrè F., Reis-Filho J.S. ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann. Oncol. 2019;30(9):1417–1427. doi: 10.1093/annonc/mdz204. [DOI] [PubMed] [Google Scholar]
- 161.Shah P., Kendall F., Khozin S., Goosen R., Hu J., Laramie J., Ringel M., Schork N. Artificial intelligence and machine learning in clinical development: a translational perspective. NPJ Digit. Med. 2019;2:69. doi: 10.1038/s41746-019-0148-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hong D.S., DuBois S.G., Kummar S., Farago A.F., Albert C.M., Rohrberg K.S., van Tilburg C.M., Nagasubramanian R., Berlin J.D., Federman N., Mascarenhas L., Geoerger B., Dowlati A., Pappo A.S., Bielack S., Doz F., McDermott R., Patel J.D., Schilder R.J., Tahara M., Pfister S.M., Witt O., Ladanyi M., Rudzinski E.R., Nanda S., Childs B.H., Laetsch T.W., Hyman D.M., Drilon A. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020;21(4):531–540. doi: 10.1016/S1470-2045(19)30856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bassett D.S., Sporns O. Network neuroscience. Nat. Neurosci. 2017;20(3):353–364. doi: 10.1038/nn.4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shine J.M., Breakspear M., Bell P.T., Ehgoetz Martens K.A., Shine R., Koyejo O., Sporns O., Poldrack R.A. Human cognition involves the dynamic integration of neural activity and neuromodulatory systems. Nat. Neurosci. 2019;22(2):289–296. doi: 10.1038/s41593-018-0312-0. [DOI] [PubMed] [Google Scholar]
- 165.Chiesa P.A., Houot M., Vergallo A., Cavedo E., Lista S., Potier M.C., Zetterberg H., Blennow K., Vanmechelen E., De Vos A., Dubois B., Hampel H. Association of brain network dynamics with plasma biomarkers in subjective memory complainers. Neurobiol. Aging. 2020;88:83–90. doi: 10.1016/j.neurobiolaging.2019.12.017. [DOI] [PubMed] [Google Scholar]
- 166.Shen Y., Wang H., Sun Q., Yao H., Keegan A.P., Mullan M., Wilson J., Lista S., Leyhe T., Laske C., Rujescu D., Levey A., Wallin A., Blennow K., Li R., Hampel H. Increased plasma beta-secretase 1 may predict conversion to Alzheimer’s disease dementia in individuals with mild cognitive impairment. Biol. Psychiatry. 2018;83(5):447–455. doi: 10.1016/j.biopsych.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Vergallo A., Lemercier P., Cavedo E., Lista S., Vanmechelen E., De Vos A., Zetterberg H., Blennow K., Habert M.O., Potier M.C., Dubois B., Teipel S., Hampel H. Plasma β-secretase1 concentrations correlate with basal forebrain atrophy and neurodegeneration in cognitively healthy individuals at risk for AD. Alzheimers Dement. 2021;17(4):629–640. doi: 10.1002/alz.12228. [DOI] [PubMed] [Google Scholar]
- 168.Vergallo A., Houot M., Cavedo E., Lemercier P., Vanmechelen E., De Vos A., Habert M.O., Potier M.C., Dubois B., Lista S., Hampel H. Brain Aβ load association and sexual dimorphism of plasma BACE1 concentrations in cognitively normal individuals at risk for AD. Alzheimers Dement. 2019;15(10):1274–1285. doi: 10.1016/j.jalz.2019.07.001. [DOI] [PubMed] [Google Scholar]
- 169.Schipke C.G., De Vos A., Fuentes M., Jacobs D., Vanmechelen E., Peters O. Neurogranin and BACE1 in CSF as potential biomarkers differentiating depression with cognitive deficits from early Alzheimer’s disease: A pilot study. Dement. Geriatr. Cogn. Disord. Extra. 2018;8(2):277–289. doi: 10.1159/000489847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zetterberg H., Andreasson U., Hansson O., Wu G., Sankaranarayanan S., Andersson M.E., Buchhave P., Londos E., Umek R.M., Minthon L., Simon A.J., Blennow K. Elevated cerebrospinal fluid BACE1 activity in incipient Alzheimer disease. Arch. Neurol. 2008;65(8):1102–1107. doi: 10.1001/archneur.65.8.1102. [DOI] [PubMed] [Google Scholar]
- 171.Ewers M., Cheng X., Zhong Z., Nural H.F., Walsh C., Meindl T., Teipel S.J., Buerger K., He P., Shen Y., Hampel H. Increased CSF-BACE1 activity associated with decreased hippocampus volume in Alzheimer’s disease. J. Alzheimers Dis. 2011;25(2):373–381. doi: 10.3233/JAD-2011-091153. [DOI] [PubMed] [Google Scholar]
- 172.Vergallo A., Lista S., Zhao Y., Lemercier P., Teipel S.J., Potier M-C., Habert M.O., Dubois B., Lukiw W.J., Hampel H. MiRNA-15b and miRNA-125b are associated with regional Aβ-PET and FDG-PET uptake in cognitively normal individuals with subjective memory complaints. Transl. Psychiatry. 2021;11(1):78. doi: 10.1038/s41398-020-01184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Williamson P.C., Allman J.M. A framework for interpreting functional networks in schizophrenia. Front. Hum. Neurosci. 2012;6:184. doi: 10.3389/fnhum.2012.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Vercammen A., Knegtering H., den Boer J.A., Liemburg E.J., Aleman A. Auditory hallucinations in schizophrenia are associated with reduced functional connectivity of the temporo-parietal area. Biol. Psychiatry. 2010;67(10):912–918. doi: 10.1016/j.biopsych.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 175.Dekhil O., Hajjdiab H., Shalaby A., Ali M.T., Ayinde B., Switala A., Elshamekh A., Ghazal M., Keynton R., Barnes G., El-Baz A. Using resting state functional MRI to build a personalized autism diagnosis system. PLoS One. 2018;13(10):e0206351. doi: 10.1371/journal.pone.0206351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Reininghaus U., Böhnke J.R., Chavez-Baldini U., Gibbons R., Ivleva E., Clementz B.A., Pearlson G.D., Keshavan M.S., Sweeney J.A., Tamminga C.A. Transdiagnostic dimensions of psychosis in the bipolar-schizophrenia network on intermediate phenotypes (B-SNIP). World Psychiatry. 2019;18(1):67–76. doi: 10.1002/wps.20607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hampel H., Lista S., Vergallo A. Commentary: Development of the blood-based Alzheimer’s disease liquid biopsy. J. Prev. Alzheimers Dis. 2019;6(3):182–184. doi: 10.14283/jpad.2019.22. [DOI] [PubMed] [Google Scholar]
- 178.Hampel H., Lista S., Vanmechelen E., Zetterberg H., Giorgi F.S., Galgani A., Blennow K., Caraci F., Das B., Yan R., Vergallo A. β-Secretase1 biological markers for Alzheimer’s disease: state-of-art of validation and qualification. Alzheimers Res. Ther. 2020;12(1):130. doi: 10.1186/s13195-020-00686-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Musante C.J., Ramanujan S., Schmidt B.J., Ghobrial O.G., Lu J., Heatherington A.C. Quantitative systems pharmacology: A case for disease models. Clin. Pharmacol. Ther. 2017;101(1):24–27. doi: 10.1002/cpt.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Geerts H., Hofmann-Apitius M., Anastasio T.J. Knowledgedriven computational modeling in Alzheimer’s disease research: Current state and future trends. Alzheimers Dement. 2017;13(11):1292–1302. doi: 10.1016/j.jalz.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 181.Lyu J., Wang S., Balius T.E., Singh I., Levit A., Moroz Y.S., O’Meara M.J., Che T., Algaa E., Tolmachova K., Tolmachev A.A., Shoichet B.K., Roth B.L., Irwin J.J. Ultra-large library docking for discovering new chemotypes. Nature. 2019;566(7743):224–229. doi: 10.1038/s41586-019-0917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Harrold J.M., Ramanathan M., Mager D.E. Network-based approaches in drug discovery and early development. Clin. Pharmacol. Ther. 2013;94(6):651–658. doi: 10.1038/clpt.2013.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Geerts H., Spiros A., Roberts P., Carr R. Towards the virtual human patient. Quantitative Systems Pharmacology in Alzheimer’s disease. Eur. J. Pharmacol. 2017;817:38–45. doi: 10.1016/j.ejphar.2017.05.062. [DOI] [PubMed] [Google Scholar]
- 184.Leil T.A., Bertz R. Quantitative Systems Pharmacology can reduce attrition and improve productivity in pharmaceutical research and development. Front. Pharmacol. 2014;5:247. doi: 10.3389/fphar.2014.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Diaz K., Lancashire L., Jeromin A., Haas M., Spiros A., Geerts H. P2-102: MAPTA: Modeling alliance of systems pharmacology in tauopathies. Alzheimers Dement. 2018;14(75):707. doi: 10.1016/j.jalz.2018.06.788. [DOI] [Google Scholar]
- 186.Ly P.T., Wu Y., Zou H., Wang R., Zhou W., Kinoshita A., Zhang M., Yang Y., Cai F., Woodgett J., Song W. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest. 2013;123(1):224–235. doi: 10.1172/JCI64516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Calabrese V., Cornelius C., Dinkova-Kostova A.T., Calabrese E.J., Mattson M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010;13(11):1763–1811. doi: 10.1089/ars.2009.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Calabrese V., Copani A., Testa D., Ravagna A., Spadaro F., Tendi E., Nicoletti V.G., Giuffrida Stella A.M. Nitric oxide synthase induction in astroglial cell cultures: effect on heat shock protein 70 synthesis and oxidant/antioxidant balance. J. Neurosci. Res. 2000;60(5):613–622. doi: 10.1002/(SICI)1097-4547(20000601)60:5<613:AID-JNR6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 189.Dattilo S., Mancuso C., Koverech G., Di Mauro P., Ontario M.L., Petralia C.C., Petralia A., Maiolino L., Serra A., Calabrese E.J., Calabrese V. Heat shock proteins and hormesis in the diagnosis and treatment of neurodegenerative diseases. Immun. Ageing. 2015;12:20. doi: 10.1186/s12979-015-0046-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Richards B.A., Lillicrap T.P., Beaudoin P., Bengio Y., Bogacz R., Christensen A., Clopath C., Costa R.P., de Berker A., Ganguli S., Gillon C.J., Hafner D., Kepecs A., Kriegeskorte N., Latham P., Lindsay G.W., Miller K.D., Naud R., Pack C.C., Poirazi P., Roelfsema P., Sacramento J., Saxe A., Scellier B., Schapiro A.C., Senn W., Wayne G., Yamins D., Zenke F., Zylberberg J., Therien D., Kording K.P. A deep learning framework for neuroscience. Nat. Neurosci. 2019;22(11):1761–1770. doi: 10.1038/s41593-019-0520-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Aggarwal C.C., Hinneburg A., Keim D.A. On the Surprising Behavior of Distance Metrics in High Dimensional Space. In: International Conference on Database Theory. Database Theory — ICDT 2001. Lecture Notes in Computer Science book series (LNCS); Van den Bussche, J; Vianu , V Springer: Berlin, Heidelberg, 2001; 1973, pp. 420-434. [DOI] [Google Scholar]
- 192.Lydon-Staley D.M., Cornblath E.J., Blevins A.S., Bassett D.S. Modeling brain, symptom, and behavior in the winds of change. Neuropsychopharmacology. 2021;46(1):20–32. doi: 10.1038/s41386-020-00805-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Saxe A., Nelli S., Summerfield C. If deep learning is the answer, what is the question? Nat. Rev. Neurosci. 2021;22(1):55–67. doi: 10.1038/s41583-020-00395-8. [DOI] [PubMed] [Google Scholar]
- 194.Molnar C. Interpretable machine learning. A Guide for Making Black Box Models Explainable 2019. Available from: https://christophm.github.io/interpretable-ml-book/ (Accessed November 17, 2021).
- 195.Kundu S. AI in medicine must be explainable. Nat. Med. 2021;27(8):1328. doi: 10.1038/s41591-021-01461-z. [DOI] [PubMed] [Google Scholar]