

Abstract
Recent findings have improved our understanding of the multifactorial nature of AD. While in early asymptomatic stages of AD, increased amyloid-β synthesis and tau hyperphosphorylation play a key role, while in the latter stages of the disease, numerous dysfunctions of homeostatic mechanisms in neurons, glial cells, and cerebrovascular endothelium determine the rate of progression of clinical symptoms. The main driving forces of advanced neurodegeneration include increased inflammatory reactions in neurons and glial cells, oxidative stress, deficiencies in neurotrophic growth and regenerative capacity of neurons, brain insulin resistance with disturbed metabolism in neurons, or reduction of the activity of the Wnt-β catenin pathway, which should integrate the homeostatic mechanisms of brain tissue. In order to more effectively inhibit the progress of neurodegeneration, combination therapies consisting of drugs that rectify several above-mentioned dysfunctions should be used. It should be noted that many widely-used drugs from various pharmacological groups, “in addition” to the main therapeutic indications, have a beneficial effect on neurodegeneration and may be introduced into clinical practice in combination therapy of AD. There is hope that complex treatment will effectively inhibit the progression of AD and turn it into a slowly progressing chronic disease. Moreover, as the mechanisms of bidirectional communication between the brain and microbiota are better understood, it is expected that these pathways will be harnessed to provide novel methods to enhance health and treat AD.
Keywords: Alzheimer’s disease, inflammation, oxidative stress, Wnt-β catenin pathway, combination therapy, brain insulin resistance
1. INTRODUCTION
One of the biggest problems of modern western societies is the increasing number of senile dementia, particularly Alzheimer’s disease (AD). An estimated 60-70% of all cases of dementia are AD, which, according to the Alzheimer’s Association and World Health Organization (WHO), affects more than 55 million people worldwide. Unfortunately, it is expected to increase twice by 2050 [1]. Globally each year, over 10 million new cases of dementia are noticed. Currently, AD and related dementias have elevated to one of the world’s most pressing public health issues [2, 3]. AD is a more complicated disease than we previously assumed; we cannot prevent or stop it or even successfully cure it. Researchers are exploring a wide range of processes that contribute to the disease, including neuroinflammation, immune response, cell signaling, and communication, and also the participation of infectious agents.
The leading view on the causes of AD is that the main driving force behind this neurodegeneration is the overproduction of amyloid-β (Aβ) protein and hyperphosphorylated tau protein, which are prone to aggregation, form neurotoxic oligomers and, finally, build protein aggregates: extracellular - senile plaques (Aβ) or intracellular - tangles neurofibrillary (P-tau) [3]. The lack of effective AD treatment and prevention results mainly from our incomplete understanding of AD mechanisms which are heterogeneous [4]. According to the latest, widely accepted findings, it is believed that the therapy that could inhibit neurodegeneration may be effective only in the case of early diagnosis (before the onset of symptomatic symptoms. i.e., changes in behavior noticeable by the physician) [5, 6]. The main research efforts, in recent years, have been focused on attempts to reduce the generation of Aβ and tau, increase their degradation, and inhibit the aggregation of these proteins in the brain tissue. However, drugs that reduce brain amyloid levels, as well as protease inhibitors that cleave the gamma amyloid precursor protein and beta-secretase, have shown very little effect on disease progression and cognitive impairment in patients in many clinical studies [7]. They do not modify the course of the disease and are not universally beneficial because they provide mainly symptomatic relief [8]. Moreover, in family diseases caused by amyloid precursor protein mutations with increased generation of Aβ, the reduction of Aβ level turned out to be ineffective. Besides all, many clinical trials, despite promising pre-clinical studies, have failed to access full therapeutic achievement in AD [9]. The aforementioned therapeutic hypotheses are being investigated in clinical trials, and so far, none of them have proven to have expected clinical effectiveness, including immunotherapy against Aβ, which has not shown any benefits in AD patients [10, 11]. The results of these studies show that Aβ still cannot be considered the main driving force behind the pathomechanism of AD; rather, it is one of the many contributors to disease progression. It seems that the amyloid hypothesis appears to be necessary, but not sufficient, to cause AD, because Aβ acts primarily as a trigger of other downstream processes, in particular tau aggregation, which mediate neurodegeneration [4, 7]. It is currently assumed that AD is a multifactorial disease in which the role of the Aβ and P-tau proteins is as important as many other factors contributing to the pathogenesis of the disease, such as neuroinflammation, oxidative stress, and faulty growth factors/trophic pathways. The interaction of all of these factors determines the progression of this neurodegeneration. The main factors involved in the pathogenesis of AD are listed in Fig. (1) (explanatory box) [12-23].
Fig. (1).
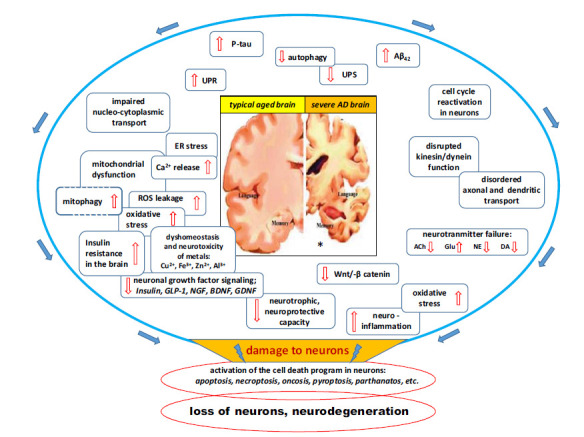
Major factors and intracellular pathways involved in the progression of Alzheimer's disease. At the present stage of knowledge about the pathomechanism of AD, it can be assumed that an important role in driving the pathomechanism of this disease is played by: 1 / high level of inflammation in the brain tissue along with oxidative stress, 2 / deficits in growth signaling pathways and neuroprotective factors, and 3 / reduced Wnt pathway / catenin, which integrates multiple pathways of neuron-glia interaction. The above three dysfunctions can be seen as a junction between many elements of the molecular pathomechanism of AD and are mainly responsible for disease progression. Therefore, any future therapeutic strategies for AD must also effectively reduce the level of inflammatory responses in brain tissue, improve neuronal signaling by growth factors, and enhance Wnt-catenin activity to near-physiological levels.
It is worth mentioning that among the risk factors of dementia, there are agents common for cardiovascular disease and dementia associated with metabolic syndrome (MetS). Their coexistence accelerates the development of diabetes, hypertension, vascular disease, and obesity. The pivotal role in the relationship between particular components, or connective with MetS diseases and central nervous system (CNS) dysfunction, plays vascular disintegrate with increasing blood-brain barrier (BBB) permeability for pro-inflammatory factors [24]. These factors, activated by MetS with insulin resistance, lead to neuronal and glial damage. Some of the common activated molecular pathways included altered brain insulin signaling, which induces neuroinflammatory cascade, leading to structural and cognitive impairment of CNS [24, 25]. Thus, systemic inflammation (peripheral, low grade), characteristic for MetS, in the case of BBB dysfunction, causes local inflammation resulting in CNS damage (neurodegeneration) due to neuroinflammation [25]. The final effect of these pathogenic events is neurodegenerative diseases, leading to dementia [26]. Many findings suggest that the managing components of the metabolic syndrome, lifestyle modification, regular physical activity, and dietary interventions, as well as social interventions, are logical targets for contemporary and future trials [27]. Pharmacological therapy should be implemented for persons at high risk for cardiovascular and cerebrovascular diseases and type 2 diabetes. Current management of metabolic syndrome includes reduction of obesity, insulin resistance, normalization of dyslipidemia, and hypertension control [28]. Among the most often suggested pharmacological agents are those which decrease insulin resistance (e.g., metformin), glucagon-like peptide-1 receptor agonist, dipeptidyl peptidase-4-inhibitors, alleviate dyslipidemia (fibrates, statins), and normalize blood pressure, such as angiotensin-converting enzyme inhibitors (ACEI) and angiotensin receptor blockers (ATRB) [27, 29].
2. CURRENT AD TREATMENT
Although considerable efforts have been made to develop effective therapeutic agents for AD therapy, contemporary pharmacotherapy of AD is substantially limited to cholinesterase inhibitors and the N-methylo-D-aspartate antagonist (NMDA)-memantine [30-32]. 1 shows the approved therapy in the treatment of AD. Cholinesterase inhibitors interfere with the neurotransmitters system (cholinergic, glutamatergic), which has been associated with the course of AD, especially slowing down the development of memory impairment, cognitive dysfunctions, and deficits in behavioral domains [32]. This led to the development and approval for use in mild to moderate stage of AD four cholinesterase inhibitors (tacrine, donepezil, rivastigmine, galantamine), which are sought to work by reducing the breakdown of the neurotransmitter acetylcholine [33, 34]. Tacrine, the first drug for the treatment of severe AD, is not recommended nowadays because of its hepatotoxicity [33]. Galantamine is characterised by a dual mechanism of action, which acts as a competitive, reversible cholinesterase inhibitor and allosterically potentiates nicotinic acetylcholine receptors, having a therapeutic benefit in AD [33, 35]. Cholinesterase inhibitors increase the level of synaptic acetylcholine, enable this important neurotransmitter, associated with memory, to bind to cholinergic receptors on the postsynaptic cells, and this central inhibition mechanism may temporarily improve cognition [32, 33]. Benefits of treatment were also seen on measures of activities of daily living and behavioral symptoms, like apathy; however, only one-third of the patients showed a clinically essential benefit, meanwhile another third demonstrated clinical worsening during the first 6 months of therapy; moreover, drop-out rates of 29% due to adverse symptoms were noted [32]. Memantine (NMDA) inhibits the effects of pathologically elevated tonic levels of glutamate, which allows physiological transmission and improvement of cognitive function, including language, memory, and praxis, as well as behavior and ability to perform everyday activities [33]. This drug is approved for moderate to severe AD, while it is ineffective in mild AD and does not modify disease progression according to recent clinical trial evidence [34, 35]. In 2014, the US Food and Drug Administration (FDA) approved another therapeutical option, which is a combination of NMDAR antagonist - memantine and ChEI - donepezil in fixed-dose combination product for moderate and severe AD treatment to get the benefits of targeting cholinergic as well as glutaminergic mechanisms of AD [35].
Another approach in AD treatment is immunotherapy. It involves two forms: active injection of Aβ42 antigenic peptides (active immunotherapy, vaccines) or administering preformed anti-Aβ antibodies, such as monoclonal antibodies (passive immunotherapy). Passive immunotherapy appears to be more successful than active vaccination [36]. On June 7, 2021, the FDA approved Aluhelm (aducanumab) for the treatment of AD, the first new Alzheimer drug in 18 years [37]. The decision was based on two clinical trial studies carried out on Alzheimer patients, one showing that the drug slows down cognitive decline and the other one showing that it does not. Aducanumab, developed 20 years ago, is one of the many monoclonal anti-Aβ antibodies that have been recently explored. Monoclonal antibodies (mAbs) are derived from humanized murine monoclonal antibodies and bind to a single epitope given their monovalent affinity [36]. The mechanism of action of anti-Aβ antibodies is not fully understood. It is assumed, however, that anti-Aβ antibodies pass through the BBB to the brain parenchyma and either inhibit toxic oligomerization of Aβ or promote microglial phagocytosis and clear Aβ plaques. It was also evidenced that they reduce the production of pro-inflammatory cytokines [38]. mAbs are amenable to dose and specificity modulation, which is one of its advantages. Insufficient therapeutic effects, low improvement in cognition, and some adverse effects of antibodies against Aβ need to be addressed [39]. This strategy might be the future direction for researchers in developing hybrid molecules that would affect the disease course by simultaneously targeting more than one biological cell pathway.
Explanatory box

|
3. ALTERNATIVE POTENTIAL THERAPIES OF AD
Oxidative stress and inflammation, as very important issues of AD, are the aim of alternative therapies, including both antioxidants and anti-inflammatory agents as potentially modifiable protective factors [11, 32, 40, 41]. Several markers of oxidative stress have been shown to be increased in AD together with impaired antioxidant potentials of the system observed in the brain, cerebrospinal fluid (CSF), and blood [41]. The therapeutic possibilities of universally recognized antioxidants like vitamin C and E, coenzyme Q 10, alpha-lipoic acid, omega-3 fatty acids, and selenium have been evaluated in clinical trials, which revealed the failure of clinical efficacy in AD [41, 42].
Nutritional supplements combining vitamins and lipids, which have been shown to be lower in AD patients and which, simultaneously, exhibit persuasive antioxidants and anti-inflammatory properties, have been the subject of a sequence investigation for the treatment of AD [41, 42]. Adequate nutrition plays an important role in the maintenance of cognitive function, particularly during aging and malnutrition, being amongst the risk factors for the development of mild cognitive impairment (MCI) and AD [43]. Special attention has been given to Ginkgo biloba, which has been the subject of intense investigations, both preclinical and clinical, as a drug capable of slowing down cognitive decline in AD patients, but the overall data remain inconclusive [44].
3.1. Multifunctional Drugs for Alzheimer's Disease
Nowadays, we have many new potential therapeutic agents to use in the treatment of AD (2). It is well known that late-onset AD, the so-called sporadic form, is a multifactorial neurodegenerative disease in which many genetic and epigenetic risk factors are involved [45, 46]. Brain tissue cells, neurons, astrocytes, and microglia, as well as brain microvascular cells and BBB, are involved in the pathomechanism of the disease, and leukocytes, including lymphocytes, which penetrate brain tissue through BBB barriers, showing reduced tightness in AD, are also often present. At the subcellular level, the pathogenesis of AD involves the dysregulation of multiple cellular pathways that influence each other to form a very complex orchestra of factors, ultimately leading to neuronal death and neurodegeneration [47]. In addition to the relatively well-known pathways of Aβ and P-tau formation, the following factors contribute to the pathomechanism of AD: oligomerization and fibrillization, also abnormalities of chaperones in unfolded protein response failure of the ubiquitin-proteasomal protein degradation (UPR) system, disturbances in Wnt-β/ catenin signaling pathways, structural and functional damage to the mitochondria along with disturbances in the cellular balance of free radicals, abnormalities in the regulation of autophagy and apoptosis pathways, abnormal reactivation of the cell cycle in neurons, activation of a low degree of neuroinflammatory reactions, as well as abnormalities in the structure and function of synapses [48].
Table 2.
New potential drugs in AD.
| Drug | Mechanism of Action | Target Type and Therapeutic Purpose |
|---|---|---|
| Rifampicin (semisynthetic antibiotic derived from Amycolatopsis rifamycinica) | Multifunctional | Inhibition oligomerization of amyloid-β, tau, and α-synuclein |
| Rasagiline (selective, irreversible monoamine oxidase B (MAO B) inhibitor | Multifunctional (neuroprotective, neurotrophic and anti-apoptotic) | Improvement of frontostriatal glucose metabolism |
| Mastinib (selective phenylaminothiazole-type tyrosine kinase inhibitor) | Anti-neuroinflammatory | Activation of the immune system via mast cells and microglia |
| Metformin (biguanide) | Multifunctional (activation of the AMP-activated protein kinase (AMPK), reduced inflammation,excitotixity and mitochondrial dysfunction) | Increases generation of both intracellular and extracellular Aβ species |
| Neurotrophic growth factors (nerve growth factor, brain-derived neurotrophic factor, glial cell-derived neurotrophic factor, insulin and glucagon-like peptide-1) |
Multifunctional (anti-inflammatory activity) | Improvement of neuronal homeostasis |
| Activation Wnt/catenin pathway (lithium chloride/sodium selenate/rosiglitazone/pioglitazone) |
Multifunctional (anti-neuroinflammatory, neuroprotective; symptomatic cognitive enhancers) |
Inhibition of the production/aggregation of amyloid-β, tau phosphorylation |
Rifampicin is an example of a multi-functional drug that affects many different factors/mechanisms of AD pathogenesis. This antibiotic has been used for over 40 years to treat tuberculosis and Mycobacterium avium complex (MAC), legionellosis, and leprosy in humans, and its pharmacokinetics, side effects, toxicity, and drug interactions are well understood. Orally administered rifampicin to Tg2576 mice (AD model) inhibited the formation of Aβ as well as the tau oligomer, decreased tau hyperphosphorylation, improved autophage-lysosomal function in neurons, reduced microglial activation in a dose-dependent manner, prevented synapse loss, and significantly improved memory in mice treated with 1 mg/day [49, 50]. In the study of Iizuka et al., rifampicin therapy in a dose of 450 mg/day for >12 months, before the onset of dementia, appeared to be able to stabilize or improve hipometabolism in typical for the AD region and was connected with a milder decline in the long-term follow-up after completion of therapy [51]. It can be concluded that rifampicin is a ready-to-use drug with great promise in the prevention and treatment of mild stages of AD, in the latter case as an adjuvant in combination with other anti-dementia drugs.
Another example of combination treatment in a single multifunctional agent is rasagiline, a selective, irreversible monoamine oxidase B (MAO B) inhibitor, which has been developed as an anti-Parkinson drug [52, 53]. In controlled monotherapy and as an adjunct to L-dopa, it has shown that anti-Parkinson activity [53] has a neuroprotective function in neuronal cell cultures in response to various neurotoxins, global ischemia, neurotrauma, head injury, etc. [54]. Rasagiline has positive effects on neuronal activity in frontalstriatal pathways with clinical benefit [55]. Its neuroprotective effect can be explained by the preservation of mitochondrial viability and the prevention of the opening of the mitochondrial permeability transition pores (MPTP), simultaneously with the activation of the anti-apoptotic protein Bcl2 and the downregulation of pro-apoptotic Bax proteins [54]. Rasagiline also exerts neurotrophic activity by stimulating alpha-secretase to produce soluble neurotrophic APP-alpha (sAPPalpha), which is supported by PKC and MAP kinases. Rasagiline's neuroprotective, neurotrophic, and anti-apoptotic effects are independent of MAO inhibition [54]. Rasagiline is currently in the second phase of clinical trials in patients with mild to moderate AD [52].
3.2. Neuroinflammation in Alzheimer’s Disease Progression
A common feature of AD pathology is a complex inflammatory component that could be a target for treatment. Inflammation is associated directly with deficits in cognitive function and with diseases that are risk factors for cognitive decline. Interventions that reduce inflammation may improve cognition [56, 57]. However, the results of numerous clinical trials to date using first-line anti-inflammatory drugs to inhibit the progression of AD symptoms have failed. In the Cochrane database, the results of numerous studies on this topic have been clearly summarized as unambiguously indicating that aspirin, steroids, and non-steroidal anti-inflammatory drugs (NSAIDs) (also the selective cyclooxygenase-2 inhibitors) showed no significant benefit in the treatment of AD [58]. Clinical trials on the effects of classical anti-inflammatory drugs belonging to the NSAIDs group (e.g., diclofenac, naproxen, refecoxib), which could prevent neuroinflammation, failed to produce any therapeutic benefit dependent on treatment timing, dose-dependent, and agent choice [59]. Miguel-Alvares et al. underscored the role of NSAIDs in the prevention of AD [59]. Therefore, the use of these drugs cannot be recommended for the treatment of AD [58, 60].
It is widely accepted that the pathogenesis of neurodegenerative diseases is related to an underlying inflammatory process in the affected areas of the CNS, known as “inflammation of the nervous system”. Primary damage and neurodegenerative processes lead to the release of endogenous neurons, release of trophic factors and mediators of inflammatory reactions, e.g., TGF-β, IFN-γ as well as Fas/FasL and in response to neuronal cytokine signals, glial cells, including microglia and astrocytes, are activated and migrate to damaged neurons, to the inflammatory microfocus [61]. Inflammatory reactions/pathways are common even in classic non-inflammatory brain diseases such as neurodegeneration or trauma, including AD, Parkinson's disease, or stroke. In these cases, inflammatory responses show more subtle patterns than in the case of severe microglia and astrocyte stimulation in classic states of neuroinflammation. However, these subtle neuronal signals are sufficient to stimulate the migration of activated glial cells to local neuroinflammatory foci [62]. Concepts of direct protection of neurons in neuroinflammatory diseases also require a more subtle, preferably indirect modulation of inflammatory responses in neurons, rather than focusing on simple, potent first-line anti-inflammatory drugs that have proven ineffective in inhibiting AD progression. Epidemiological evidence and animal experimental studies, as well as first clinical trials, show the benefit of immunomodulating therapy in classical non-inflammatory brain diseases [61].
An example of a drug that indirectly modulates inflammatory responses in neurons is masatinib, a selective inhibitor of tyrosine kinases, in Phase III clinical trials for the treatment of malignant melanoma, multiple myeloma, gastrointestinal and pancreatic cancer, and also in Phase II/III clinical trials for the treatment of a number of neuroinflammatory diseases, among them also in AD. It has been well established that the drug strongly inhibits inflammatory responses in mast cells [52]. Mast cells located on both sides of the BBB play a significant role in sustaining the inflammatory network of the CNS [63, 64]. Acting both as sensors and effectors in communication among nervous, vascular, and immune systems [63], Masitinib mesilate is a selective tyrosine kinase inhibitor that targets c-Kit, platelet-derived growth factor receptors (PDGFR), and also Lyn, Fyn, and the FAK pathway [52, 65]. By combined targeting of c-Kit and Lyn, masitinib is particularly efficient in controlling the survival, differentiation, and degranulation of mast cells, and thus indirectly controlling the array of inflammatory and vasoactive mediators the cells can release [65]. Masitinib administered in combination with standard choliesterase inhibitors/memantine for 24 weeks led to significantly slower cognitive decline in AD with an accepable tolerance profile and showed significant improvement in the ADAS-Cog and MMSE cognitive tests [52, 65, 66]. Masitinib may be a promising innovative treatment strategy for AD [65].
Metformin, at doses that lead to the activation of the AMP-activated protein kinase (AMPK), significantly increases the generation of both intracellular and extracellular Aβ species. Furthermore, the effect of metformin on Aβ generation is mediated by transcriptional up-regulation of β-secretase (BACE1), which results in an elevated protein level and increased enzymatic activity [67]. Although insulin and metformin have opposite effects on Aβ production, when combined, metformin sensitizes/enhances the effect of insulin in lowering Aβ levels [67]. Literature data describe significantly fewer neuritic plaques in the brains of diabetics taking a combination of insulin and oral medications compared to those taking a single medication (insulin or oral medication) [68]. Combining metformin therapy with thiazolidinediones (glitazones), e.g., pioglitazone or rosiglitazone, may also be beneficial. The complementary mechanisms of action of metformin and thiazolidinediones significantly increase cell sensitivity to insulin, inhibit inflammatory responses, and reduce the risk of cardiovascular disease in patients with type 2 diabetes [67]. Therefore, the Food and Drug Administration (FDA) and European Medicines Agency (EMA) have approved the use of combined drugs containing metformin and rosiglitazone (Avandamet) and metformin plus pioglitazone (Competact) for the treatment of diabetes and prevention of vascular complications of the disease. Importantly, literature data indicate a beneficial effect of rosiglitazone and pioglitazone on the improvement of memory and learning abilities in murine AD models [69]. Additionally, it was found that in cultures of neuronal cells, the studied glitazones inhibited the key pathways of AD pathomechanism; rosiglitazone significantly decreased BACE1 transcription, thereby decreasing amyloidogenesis [69], and pioglitazone inhibited tau hyperphosphorylation and its oligomerization [70]. Glitazones are agonists of peroxisome proliferator-activated receptors (PPARγ) and activation of PPARγ reduces BACE1 transcription and activity [67, 70]. It remains to be determined if they will also be effective in suppressing the stimulating effect of metformin on BACE1 transcription and activity in human brains. In this case, the indications for the use of combination drugs containing metformin and glitazones may be extended to the prevention and supportive treatment of AD in patients without diabetes.
3.3. Insulin Resistance of the Brain in Alzheimer’s Disease
Brain insulin resistance is frequently found in AD patients [71], and numerous clinical studies have provided evidence for the beneficial effect of insulin therapy on cognition, memory functions, daily activity, as well as on Aβ and P-tau metabolism [71, 72]. In recent years, special attention has been paid to the administration of insulin by the intranasal route, as this method of drug administration increases the concentration of insulin in the brain and cerebrospinal fluid (CSF) and simultaneously does not significantly affect the plasma insulin level, and therefore does not cause hypoglycemia, the main side effect with systemic insulin administration. Intranasal insulin administration has also been found to positively affect regional vascular reactivity, causing vasodilation in the anterior regions of the brain, especially in the insular cortex, which regulates the performance of attention tasks. Such an effect was found in healthy people and in patients with type 2 diabetes [73]. Importantly, it has been shown that brain insulin resistance leads to a number of changes in neuronal metabolism, which result in an increase in the formation of Aβ42 and an increase of tau phosphorylation [72, 74]. Conversely, hyperphosphorylated tau protein leads to chronic insulin resistance, especially in those regions of the brain where neurons initially express a lower expression of insulin signaling proteins (insulin receptor substrate 1; IRS-1, Akt); therefore, these brain regions are particularly prone to developing insulin resistance and induced tau hyperphosphorylation [74]. This may explain the phenomenon of the selective increase in P-tau accumulation and the formation of neurofibrillary tangles in some regions of the brain, occupied earlier by neurodegeneration processes. Aβ42 and P-tau, when produced in excess, inhibit the activity of insulin signal transduction proteins and lead to the development of insulin resistance [69, 70, 75, 76]. In summary, brain insulin resistance increases the production of Aβ and P-tau, and in the case of normal insulin sensitivity, the primary increase in Aβ and P-tau formation disrupts the proteins of the insulin signaling pathways, which induces insulin resistance in neurons. In both cases, restoration of insulin signaling can be effective. However, it should be emphasized that the use of insulin in the treatment of AD requires regular and long-term use, and there may be side effects of such prolonged therapy, including an increase in blood pressure and hypoglycaemia [77]. An important challenge is to better optimize insulin dosing, especially in the case of intranasal administration, where poorly controlled absorption of the drug from the nasal cavity can lead to significant differences, increasing the local concentration of insulin in the brain, which could damage neurons and increase neurodegeneration [77]. Additionally, one should be aware that insulin has a growth-stimulating effect, and its long-term use may promote neoplastic proliferation in the brain tissue [77, 78].
3.4. Neurotrophic Growth Factor Substitution
A regular feature of neurodegenerative diseases, including AD, is the dysfunction of the major trophic growth factors signaling in the brain tissue, i.e., nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), glial cell-derived neurotrophic factor (GDNF), as well as insulin and glucagon-like peptide-1 (GLP-1) [79, 80]. In the course of neurodegeneration, a gradual decrease in the transcription of mRNA proteins and growth factors, as well as a decrease in the density of their receptors on brain neurons, has been described [81]. Improvement in signaling by growth factors has a pronounced neuroprotective effect in various animal models of AD [80]. It has been suggested that physiological, basal differences in the activity of growth factor signaling pathways in different regions of the brain determine their varying degrees of susceptibility to neurotoxic agents (such as Aβ oligomers, oxidative stress, some metal ions, etc.). This may explain regional differences in susceptibility to neurodegenerative changes reported in autopsied brain tissue in AD. For example, it has been documented that cholinergic neurons in certain regions of the brain, such as the basal nucleus, medial septum, and striatum, contain high levels of both known NGF receptors, i.e., the NGF-specific tyrosine kinase receptor, tropomyosin receptor kinase A (TrkA) and the non-specific panneurotropin-p75 receptor [81]. This indicates that the homeostasis of neurons in these brain regions is largely dependent on NGF signaling. The decrease in TrkA receptor expression found in AD [82] will lead to trophic dysfunction in these areas of the brain, resulting in a significant reduction in cholinergic signaling. The therapeutic administration of growth factors causes significant inhibition of the disease progression, and the therapeutic effect is long lasting. This may indicate that the dysfunction of growth factor signaling pathways in the brain is one of the important mechanisms of neurodegeneration and that improved growth factor signaling could enhance regenerative processes and restore their functions [80]. The administration of growth factors to patients with AD is a very promising therapeutic strategy; however, thus far, the main obstacle to wide clinical application of this method of treatment is the poor passage of NGF, BDNF, and GDNF through the brain's barriers [79]. Therefore, technologies for the delivery of growth factor proteins in nanotechnological platforms (lipid/liposome and non-lipid nanostructures), as well as viral transfer (adenoviruses, lentiviruses), are being intensively developed [79]. Among the routes for the administration of growth factors to the brain, the intranasal method is highly anticipated; drugs applied to the posterior upper surface of the nasal mucosa travel to the brain, bypassing the BBB. Although this method requires some optimization work, it is undoubtedly a good route of drug administration directly to the brain. Unlike the aforementioned growth factors, insulins, IGF-1, GLP-1, and GIP can cross the BBB, so they already offer a promising treatment strategy for AD [78]. The main drawback and the biggest challenge, however, is the way to effectively deliver the therapeutic agents from the bloodstream across the BBB without inducing systemic effects. The advantage of such an approach is that it can improve neuronal homeostasis, enhance neurodegenerative abilities, and thus improve the functions of a range of pathological processes observed in AD, not just one parameter [78]. Administration of a growth factor significantly reduces chronic inflammatory reactions in the brain tissue, reduces the release of inflammatory cytokines from glial cells, restores energy utilization by neurons, normalizes mitochondrial functions and increases the level of autophagy, reduces apoptosis, and improves synaptic plasticity, memory and cognitive functions [78]. With regard to the aforementioned considerations, it can be assumed that improving the safety of the neurotrophic factors administration in the near future may have a critical effect on successful AD therapy.
3.5. Reactivation of the Wnt-β Catenin Pathway
The Wnt signaling pathways are a group of signal transduction pathways that begin with Wnt proteins binding to one of several frizzled family receptors and, in some cases, a co-receptor, such as a lipoprotein receptor-related protein (LRP-5/6) [83]. Wnt signals have several unique properties, including a short range of action. That is why Wnts predominantly mediate signaling locally between neighboring cells. In addition, Wnt signals give shape to tissues as cells are proliferating. This is a consequence of the ability of Wnt signaling to confer polarity and asymmetry to cells. The Wnt signaling pathway is a highly evolutionarily conserved pathway that regulates crucial aspects of cell fate determination, cell migration, cell polarity, neural patterning, neurogenesis and synaptogenesis, organogenesis during embryonic development [84]. Wnt ligands are a large family of secreted glycoproteins that are cysteine-rich and highly hydrophobic. In vertebrates, there are 19 different Wnt proteins whose expression is spatially and temporally regulated during development. Three Wnt signaling pathways have been characterized: the canonical Wnt- β catenin pathway, the noncanonical planar cell polarity (PCP) pathway, and the noncanonical Wnt/Calcium pathway [85]. The canonical Wnt pathway leads to the regulation of gene transcription of more than 50 Wnt-targeted genes. The noncanonical planar cell polarity pathway regulates actin fibril assembly of the cytoskeleton that is responsible for the shape of the cell. The noncanonical Wnt/calcium pathway regulates calcium inside the cell.
Wnt signaling has been implicated in axonal pathfinding, dendritogenesis, synapse formation, synaptic plasticity, and maintenance [86, 87]. In the canonical pathway, the Wnt ligands bind the transmembrane receptor (Frizzled) and the co-receptor LRP5/LRP6. This binding causes inactivation of the β-catenin destruction complex, formed by the proteins Axin, tumor suppressor adenomatous polyposis coli gene product (APC), Casein Kinase 1 (CK1), and GSK-3β. This inactivation prevents β-catenin phosphorylation by the destruction complex, and its proteasomal degradation and β-catenin migrate into the nucleus where it activates specific transcription factors and stimulates the transcription of > 50 Wnt target genes involved in cell proliferation, survival, differentiation, neurogenesis, and inflammation [87]. Instead, in the absence of the Wnt ligands, the β-catenin is constantly phosphorylated by CK-1 and glycogen synthase kinase 3 beta (GSK-3β), which promotes its ubiquitination and degradation through the proteasomal machinery, preventing in this way β-catenin regulation of transcription. GSK-3β is a crucial regulator of the canonical Wnt signaling involved in the regulation of neuronal polarity apoptosis and inflammation [87]. GSK-3β dysregulation has been associated with several pathological conditions, including obesity, diabetes mellitus, carcinogenesis, and neurodegenerative disorders [87]. The non-canonical, so-called planar cell polarity (PCP) Wnt pathway does not engage, which leads to transcriptional changes caused by c-Jun N-terminal kinase (JNK) activation and subsequent Jun phosphorylation and also to actin filaments remodeling by activated Rho-associated protein kinase (ROCK) [86]. A fine balance of canonical and non-canonical Wnt signaling is required for maintaining mature synapses, especially axonal pathfinding, dendritogenesis, synapse formation, synaptic plasticity, and dendritic spine maintenance [86]. Decreased levels of canonical Wnt signaling with the concomitant activation of the non-canonical PCP pathway leads to actin filament disorganization and synapse disassembly. The other non-canonical - the Wnt/Ca2+ pathway - enhances Ca2+ release from intracellular stores and activation of calcium/calmodulin-dependent protein kinase type II (CaMKII) and PKC, leading to transcriptional changes and actin remodeling. Wnt activation pathways participate in early LTP phases promoting AMPA receptor (AMPAR) recruitment to the synapses and also in the late phase of LTP maintaining basal N-methyl-D-aspartate receptor (NMDAR) currents. Altogether, these studies demonstrate that canonical and non-canonical Wnt signaling are important pathways maintaining mature synapse function [86].
Wnt canonical signaling dysfunction plays a key role in the development of neurodegenerative diseases [88]. Studies have shown that dysregulated Wnt-β catenin signaling plays an important role in the pathogenesis of AD. In AD, the Wnt canonical pathway is significantly reduced, mainly by the potent Wnt antagonist, extracellular DKK1 protein (Dickkopf1), which interacts with the LRP5/6 co-receptor preventing it from binding (together with the Frizzled receptor) to the Wnt ligand. This results in reduced transcription of Wnt target genes as well as an increase in GSK-3β activity, which further promotes inflammation and apoptosis, contributing to disease progression. In neuronal cell cultures, the addition of Aß rapidly elevated Dkk1 expression in hippocampal neurons, resulting in reduced canonical Wnt signaling concomitant elevation of Wnt/PCP pathway and synapse loss [89]. Most of the collected data indicate that Aβ-induced inhibition of Wnt signaling by DKK1 contributes to synaptic and cognitive deficits in AD [86]. From previous studies, it can be concluded that Aβ enhances the activity of GSK-3β, and, conversely, GSK-3β increased the inactivation of the Wnt canonical pathway and the formation of neurofibrillary tangles by phosphorylation of β-catenin and tau [90]. Instead, the activation of the canonical Wnt pathway suppresses GSK-3β and reduces AD pathology by preserving cells from Aβ and tau toxicity. Hence, it would be suggested that restoring the Wnt canonical pathway to normal levels has a neuroprotective effect. The Wnt canonical pathway was shown to play a role in modulating both innate and adaptive immune responses during inflammation [87, 91-94]. This evidence indicates that the Wnt-β-catenin pathway can exert its anti-inflammatory activity through the inhibition of the nuclear factor kappa B (NF-κB) pathway, by promoting the expression of anti-inflammatory genes such as peroxisome proliferator-activated receptor γ (PPARγ), and through the negative regulation of immune cells differentiation and maturation (Fig. 2). In the context of neuroinflammation, GSK3-β represents the main player, since it can regulate the interaction between these signaling networks involving NF-κB, PPARγ, and β-catenin [95]. This suggests that activation of the Wnt-β catenin pathway to inhibit GSK3β activity may be a promising therapeutic strategy to reduce the inflammatory response in neurodegenerative diseases.
Fig. (2).
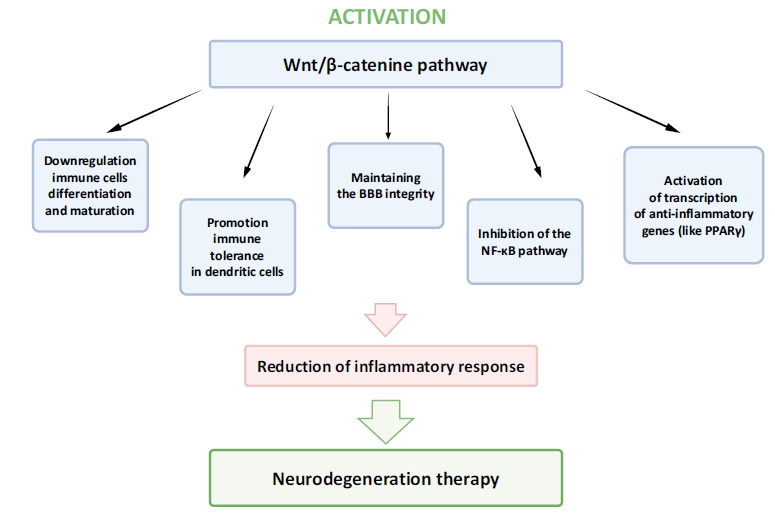
Activation of the Wnt-β catenin pathway. Drugs that activate the Wnt-β catenin pathway will meet the criteria of multitargeted drugs for treatment of neurodegeneration.
Therefore, activating Wnt signaling by administering exogenous Wnt agonists may be a promising therapeutic strategy against the development of neurodegenerative diseases [96]. Currently, drugs activating the Wnt-β catenin pathway for potential use in the treatment of AD are the following: lithium chloride, sodium selenate, glitazones rosiglitazone, and pioglitazone. Lithium chloride (LiCl) is an established therapeutic option for the treatment of bipolar disorder and major depression. Lithium has also been considered a neuroprotective agent and a candidate drug in some neurodegenerative diseases, including the treatment of AD. It activates the Wnt-β catenin signaling pathways and thus corrects several homeostatic mechanisms involved in the neurotrophic response, autophagy, oxidative stress, inflammation, and mitochondrial function. Lithium salts also strongly inhibit GSK-3β [97, 98]. Rosiglitazone and pioglitazone are currently used in diabetology as drugs increasing the sensitivity of cells to insulin, and, as agonists of PPAR-γ receptors, they exert an anti-inflammatory effect. Also, as recently proven, they stimulate the activity of the Wnt-β catenin pathway [97, 98] and, probably, in this way, inhibit the activity of GSK-3 beta, hyperphosphorylation, and oligomerization of the tau protein [98]. Interestingly, it was also found that selenium salts used chronically in the diet inhibit AD development by stimulating the Wnt-β catenin pathway [99]. In triple transgenic AD mice models (3 × Tg-AD), animals were treated with sodium selenate in drinking water for 10 months before the detection of hippocampal pathology. Sodium selenate could be a future new drug for AD treatment [99].
It should be noted that strong activation of the Wnt-β catenin pathway could enhance cell proliferation, which alternatively may stimulate potential neoplastic growth. However, numerous experimental studies in animals show that to enhance tumor growth, much higher activation of the Wnt pathway is needed than that proposed for substituting reactivation of this pathway in neurodegeneration [86]. Another experimental study on animals has shown that in tumors that may be dependent on the excessive activity of the Wnt-β catenin pathway (colon cancer, breast cancer, T-lympho-blastic leukemia), additional important mutations are necessary to reveal the pro-proliferative effect (APC, PTEN) [86]. Therefore, it can be assumed that the Wnt signaling stimulation proposed for AD treatment could provide a viable approach for the treatment of neurodegenerative diseases without increasing the incidence of cancer [86]. In our view, it is very likely that drugs activating the Wnt-β catenin pathway will meet the criteria of multitargeted drugs for the treatment of neurodegeneration.
4. POSSIBLE FUTURE TARGETS IN AD THERAPY
AD has long been considered the disease of only CNS without the important influence of periphery. Early human studies suggested that altering microbiota with beneficial bacteria or probiotics can lead to changes in brain function, including cognitive functions [100]. Recently, the identification of gastrointestinal microorganisms with metagenomics and metabolomics methods increased our ability to examine more subtle interactions between host and microbiome [101]. The gut provides the largest physical interface between the environment (including the microbiome) and the body. This bidirectional information flow between the microbiota and the brain suggests that brain development, function, mood, and cognition may be influenced by our gastrointestinal contents. We should underline that, with age, microbiota, with bacteria being its major component, can induce autoimmune and widespread neuroinflammation disorders such as AD [100]. Our experiences and review of literature allow us to examine other, relatively less often, until recently, considered fields of possible pathology with serious and perhaps predominant influence on the development of pathophysiological changes in the brain of AD patients. Our research leads us to expect that, as the mechanisms of this bidirectional communication between the brain and microbiota are better understood, these pathways will be harnessed to provide a novel method to enhance health and treat AD.
An association between poor oral health status and overall health has been debated for over 30 years. Intensively-studied periodontal disease (periodontitis, PD) is an oral infectious and chronic inflammatory disease of the periodontium that leads to periodontal ligament loss, destruction of alveolar bone, and of other structures that support the teeth. Finally, periodontitis results in tooth loss [102]. Oral inflammation and dysbiosis of oral microbiota are thought to be linked to several systemic diseases and conditions, including atherosclerosis, diabetes mellitus, and respiratory conditions [50]. Currently, PD is suspected to be involved also in the pathogenesis of AD. Infection results in local inflammation, leading to systemic inflammation as well as neuroinflammation. This, in turn, causes neurodegeneration and the senescence of the immune cells preceding the clinical manifestations [103]. Possible explanation for the association between PD and AD is the stimulation of chronic pro-inflammatory status in older adults. The close anatomical relationship between the oral cavity and the brain enables the translocation of oral pathogens to the brain, leading to local immune response [104, 105]. Recently published data showed that Porphyromonas gingivalis, mainly involved in PD, and its toxins were identified in the brain of AD patients. Thereby, P. gingivalis from the oral cavity may directly infect the brain, increasing the production of Aβ1-42 and leading to neurodegeneration and AD [106]. Perhaps not only P. gingivalis but also other periopathogens involved in dysbiosis of the oral microbiome may be linked to the exacerbation of the innate immune response and increased pro-inflammatory markers. The unrelenting progression of AD requires complex treatment and management efforts. Identifying patients with MCI or early stages of AD who have PD and/or innate immune deficits would be promising. This may be the basis for new interventions in AD. Opportunities for facing the challenges of AD therapy appear to come from recent progress in knowledge and methodological advances in the design, synthesis, and targeting of brain mRNA and microRNA with synthesis antisense oligonucleotides (ASOs) [107]. Several types of ASOs allow for the utilization of different mechanisms of posttranscriptional regulation and offer better effects than alternative therapeutics [108].
CONCLUSION
Recent studies have shown that many drugs used in clinical practice to treat various diseases can also inhibit the main mechanisms driving the progression of neurodegeneration. In this review, we focused on four pathways as contributors to neurodegeneration in AD, in which those drugs could be applied. The majority of these drugs have been used in medicine for years, so their pharmacokinetics, toxicity, and side effects, as well as their therapeutic dose range, are well understood. As a result, they can be relatively quickly introduced in the treatment of AD. Proposed drug combinations should include the following: drugs that reduce inflammation and oxidative stress, drugs that increase the sensitivity of neurons to insulin, drugs that improve the regenerative capacity of neurons, and drugs that restore Wnt-β catenin signaling in coordinating the homeostatic responses of brain tissue cells. The examples in this article suggest that the choice of such drugs seems to be large enough to develop at least some possible therapeutic strategies for AD. The challenge for modern medicine is to turn AD into a very slowly progressive chronic disease, and the implementation of individually tailored combination therapies will certainly improve the possibility of treating AD patients. To sum up, it is fair to say that the absence of disease-modifying treatments for Alzheimer’s is due to the amyloid hypothesis, a misguided hypothesis of Alzheimer’s disease etiology, which has dominated Alzheimer’s research, drug development, and clinical trials for 30 years. Reducing amyloid is not the same thing as mitigating symptoms of dementia. Dealing with Alzheimer's disease in a more integrated way has come of age, and future research should take advantage of such an integrative view.
Table 3.
Available, accepted drugs for Alzheimer's disease.
| Drug | Mechanism of Action | Target | Stage of AD |
|---|---|---|---|
| Donepezil, Rivastigmine, Galantamine |
Acetylcholinesterase inhibitors | Acetylocholine (AchE) | Early to moderate |
| Memantine | N-methyl D-aspartate (NMDA) antagonist | N-methyl D-aspartate (NMDA) | Moderate to severe |
| Aducanumab | Monoclonal antibody | Extracellular amyloid-β plague | Early |
ACKNOWLEDGEMENTS
Declared none.
LIST OF ABBREVIATIONS
- ACEI
Angiotensin-converting Enzyme Inhibitors
- AD
Alzheimer's Disease
- AMPAR
AMPA Receptor
- APC
Adenomatous Polyposis Coli Gene Product
- ATRB
Angiotensin Receptor Blockers
- Aβ
Amyloid-β
- BACE1
β-secretase 1
- BBB
Blood-brain Barrier
- BDNF
Brain-derived Neurotrophic Factor
- CaMKII
Calcium/calmodulin-dependent Protein Kinase type II
- CK1
Casein Kinase 1
- CNS
Central Nervous System
- CSF
Cerebrospinal Fluid
- EMA
European Medicines Agency
- FDA
Food and Drug Administration
- GDNF
Glial Cell-derived Neurotrophic Factor
- GLP-1
Insulin and Glucagon-like Peptide-1
- GSK-3β
Glycogen Synthase Kinase 3 beta
- IFNγ
Interferon Gamma
- IRS-1
Insulin Receptor Substrate 1
- JNK
c-Jun N-terminal Kinase
- LiCl
Lithium Chloride
- LRP
Lipoprotein Receptor-related Protein
- MAC
Mycobacteriumavium Complex
- MAO B
Monoamine Oxidase B
- MCI
Mild Cognitive Impairment
- MetS
Metabolic Syndrome
- MPTP
Mitochondrial Permeability Transition Pores
- NF-κB
Nuclear Factor Kappa B
- NGF
Nerve Growth Factor
- NMDA
N-methylo-D-aspartate
- NMDAR
N-methyl-D-aspartate receptor
- NSAIDs
Non-steroidal Anti-inflammatory Drugs
- PCP
Planar Cell Polarity
- PD
Periodontal Disease
- PDGFR
Platelet-derived Growth Factor Receptor
- PPARγ
Peroxisome Proliferator-activated Receptor γ
- ROCK
Rho-associated Protein Kinase
- sAPPalpha
Soluble Neurotrophic APP-alpha
- TGF-β
Transforming Growth Factor β
- TrkA
Tropomyosin Receptor Kinase A
- UPR
Ubiquitin-proteasomal Protein
- WHO
World Health Organization
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.2017 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2017;13(4):325–373. doi: 10.1016/j.jalz.2017.02.001. [DOI] [Google Scholar]
- 2. World Health Organization. Dementia. Available from: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed April 13, 2021).
- 3.Du X., Wang X., Geng M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018;7:2. doi: 10.1186/s40035-018-0107-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendiola-Precoma J., Berumen L.C., Padilla K., Garcia-Alcocer G. Therapies for prevention and treatment of Alzheimer’s disease. BioMed Res. Int. 2016;2016:2589276. doi: 10.1155/2016/2589276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stephenson D., Perry D., Bens C., Bain L.J., Berry D., Krams M., Sperling R., Dilts D., Luthman J., Hanna D., McKew J., Temple R., Fields F.O., Salloway S., Katz R. Charting a path toward combination therapy for Alzheimer’s disease. Expert Rev. Neurother. 2015;15(1):107–113. doi: 10.1586/14737175.2015.995168. [DOI] [PubMed] [Google Scholar]
- 6.Bredesen D.E., John V. Next generation therapeutics for Alzheimer’s disease. EMBO Mol. Med. 2013;5(6):795–798. doi: 10.1002/emmm.201202307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musiek E.S., Holtzman D.M. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 2015;18(6):800–806. doi: 10.1038/nn.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bredesen D.E. Reversal of cognitive decline: A novel therapeutic program. Aging (Albany NY) 2014;6(9):707–717. doi: 10.18632/aging.100690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kametani F., Hasegawa M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018;12:25. doi: 10.3389/fnins.2018.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Godyń J., Jończyk J., Panek D., Malawska B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 2016;68(1):127–138. doi: 10.1016/j.pharep.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 11.Pakavathkumar P., Sharma G., Kaushal V., Foveau B., LeBlanc A.C. Methylene blue inhibits caspases by oxidation of the catalytic cysteine. Sci. Rep. 2015;5:13730. doi: 10.1038/srep13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elmore S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007;35(4):495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Obulesu M., Lakshmi M.J. Apoptosis in Alzheimer’s disease: An understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 2014;39(12):2301–2312. doi: 10.1007/s11064-014-1454-4. [DOI] [PubMed] [Google Scholar]
- 14.Miller D.R., Cramer S.D., Thorburn A. International Review of Cell and Molecular Biology. The Interplay of Autophagy and Non-Apoptotic Cell Death Pathways. In: Spetz, J. K.E.; Galluzzi, L. Academic Press, 2020; 353, pp. 159-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhen X., Zhao G. Pyroptosis and neurological diseases. Neuroimmunol. Neuroinflamm. 2014;1:60–65. doi: 10.4103/2347-8659.139716. [DOI] [Google Scholar]
- 16.Han C., Yang Y., Guan Q., Zhang X., Shen H., Sheng Y., Wang J., Zhou X., Li W., Guo L., Jiao Q. New mechanism of nerve injury in Alzheimer’s disease: β-amyloid-induced neuronal pyroptosis. J. Cell. Mol. Med. 2020;24(14):8078–8090. doi: 10.1111/jcmm.15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.David K.K., Andrabi S.A., Dawson T.M., Dawson V.L. Parthanatos, a messenger of death. Front. Biosci. 2009;14:1116–1128. doi: 10.2741/3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X., Ge P. Parthanatos in the pathogenesis of nervous system diseases. Neuroscience. 2020;449:241–250. doi: 10.1016/j.neuroscience.2020.09.049. [DOI] [PubMed] [Google Scholar]
- 19.Weerasinghe P., Buja L.M. Oncosis: An important non-apoptotic mode of cell death. Exp. Mol. Pathol. 2012;93(3):302–308. doi: 10.1016/j.yexmp.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 20.Nirmala J.G., Lopus M. Cell death mechanisms in eukaryotes. Cell Biol. Toxicol. 2020;36(2):145–164. doi: 10.1007/s10565-019-09496-2. [DOI] [PubMed] [Google Scholar]
- 21.Hikari T., Homma H., Fujita K., Kondo K., Yamada S., Jin X., Waragai M., Ohtomo G., Iwata A., Tagawa K., Atsuta N., Katsuno M., Tomita N., Furukawa K., Saito Y., Saito T., Ichise A., Shibata S., Arai H., Saido T., Sudol M., Muramatsu S., Okano H., Mufson E.J., Sobue G., Murayama S., Okazawa H. YAP-dependent necrosis occurs in early stages of Alzheimer’s disease and regulates mouse model pathology. Nat. Commun. 2020;11(1):1–22. doi: 10.1038/s41467-020-14353-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Callus B.A., Vaux D.L. Caspase inhibitors: viral, cellular and chemical. Cell Death Differ. 2007;14(1):73–78. doi: 10.1038/sj.cdd.4402034. [DOI] [PubMed] [Google Scholar]
- 23.Caccamo A., Branca C., Piras I.S., Ferreira E., Huentelman M.J., Liang W.S., Readhead B., Dudley J.T., Spangenberg E.E., Green K.N., Belfiore R., Winslow W., Oddo S. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017;20(9):1236–1246. doi: 10.1038/nn.4608. [DOI] [PubMed] [Google Scholar]
- 24.Leszek J., Trypka E., Tarasov V.V., Ashraf G.M., Aliev G. Type 3 diabetes mellitus: A novel implication of Alzheimers disease. Curr. Top. Med. Chem. 2017;17(12):1331–1335. doi: 10.2174/1568026617666170103163403. [DOI] [PubMed] [Google Scholar]
- 25.Ferreira L.S.S., Fernandes C.S., Vieira M.N.N., De Felice F.G. Insulin resistance in Alzheimer’s disease. Front. Neurosci. 2018;12:830. doi: 10.3389/fnins.2018.00830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim B., Feldman E.L. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome. Exp. Mol. Med. 2015;47:e149. doi: 10.1038/emm.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Accardi G., Caruso C., Colonna-Romano G., Camarda C., Monastero R., Candore G. Can Alzheimer disease be a form of type 3 diabetes? Rejuvenation Res. 2012;15(2):217–221. doi: 10.1089/rej.2011.1289. [DOI] [PubMed] [Google Scholar]
- 28.Fernando W.M.D.B., Martins I.J., Goozee K.G., Brennan C.S., Jayasena V., Martins R.N. The role of dietary coconut for the prevention and treatment of Alzheimer’s disease: Potential mechanisms of action. Br. J. Nutr. 2015;114(1):1–14. doi: 10.1017/S0007114515001452. [DOI] [PubMed] [Google Scholar]
- 29.Kosik K.S. Personalized medicine for effective Alzheimer disease treatment. JAMA Neurol. 2015;72(5):497–498. doi: 10.1001/jamaneurol.2014.3445. [DOI] [PubMed] [Google Scholar]
- 30.Gao C., Ding Y., Zhong L., Jiang L., Geng C., Yao X., Cao J. Tacrine induces apoptosis through lysosome- and mitochondria-dependent pathway in HepG2 cells. Toxicol. Vitro Int. J. Publ. Assoc. BIBRA. 2014;28(4):667–674. doi: 10.1016/j.tiv.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 31.Minarini A., Milelli A., Simoni E., Rosini M., Bolognesi M.L., Marchetti C., Tumiatti V. Multifunctional tacrine derivatives in Alzheimer’s disease. Curr. Top. Med. Chem. 2013;13(15):1771–1786. doi: 10.2174/15680266113139990136. [DOI] [PubMed] [Google Scholar]
- 32.Campos C., Rocha N.B., Vieira R.T., Rocha S.A., Telles-Correia D., Paes F., Yuan T., Nardi A.E., Arias-Carrión O., Machado S., Caixeta L. Treatment of Cognitive Deficits in Alzheimer’s disease: A psychopharmacological review. Psychiatr. Danub. 2016;28(1):2–12. [PubMed] [Google Scholar]
- 33.Sharma K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019;20(2):1479–1487. doi: 10.3892/mmr.2019.10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molino I., Colucci L., Fasanaro A.M., Traini E., Amenta F. Efficacy of memantine, donepezil, or their association in moderate-severe Alzheimer’s disease: A review of clinical trials. Scientific World J. 2013;2013:925702. doi: 10.1155/2013/925702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farrimond L.E., Roberts E., McShane R. Memantine and cholinesterase inhibitor combination therapy for Alzheimer’s disease: A systematic review. BMJ Open. 2012;2(3):e000917. doi: 10.1136/bmjopen-2012-000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Usman M.B., Bhardwaj S., Roychoudhury S., Kumar D., Alexiou A., Kumar P., Ambasta R.K., Prasher P., Shukla S., Upadhye V., Khan F.A., Awasthi R., Shastri M.D., Singh S.K., Gupta G., Chellappan D.K., Dua K., Jha S.K., Ruokolainen J., Kesari K.K., Ojha S., Jha N.K. Immunotherapy for Alzheimer’s disease: current scenario and future perspectives. J. Prev. Alzheimers Dis. 2021;8(4):534–551. doi: 10.14283/jpad.2021.52. [DOI] [PubMed] [Google Scholar]
- 37.Mullard A. FDA approval for Biogen’s aducanumab sparks Alzheimer’s disease firestorm. Nat. Rev. Drug Discov. 2021 doi: 10.1038/d41573-021-00099-3. [DOI] [PubMed] [Google Scholar]
- 38.Jeremic D., Jiménez-Díaz L., Navarro-López J.D. Past, present and future of therapeutic strategies against amyloid-β peptides in Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2021;72:101496. doi: 10.1016/j.arr.2021.101496. [DOI] [PubMed] [Google Scholar]
- 39.Selkoe D.J. Alzheimer’s drugs: Does reducing amyloid work?-Response. Science. 2021;374(6567):545–546. doi: 10.1126/science.abm3288. [DOI] [PubMed] [Google Scholar]
- 40.Sydow A., Hochgräfe K., Könen S., Cadinu D., Matenia D., Petrova O., Joseph M., Dennissen F.J., Mandelkow E-M. Age-dependent neuroinflammation and cognitive decline in a novel Ala152Thr-Tau transgenic mouse model of PSP and AD. Acta Neuropathol. Commun. 2016;4:17. doi: 10.1186/s40478-016-0281-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang W-J., Zhang X., Chen W-W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016;4(5):519–522. doi: 10.3892/br.2016.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otaegui-Arrazola A., Amiano P., Elbusto A., Urdaneta E., Martínez-Lage P. Diet, cognition, and Alzheimer’s disease: Food for thought. Eur. J. Nutr. 2014;53(1):1–23. doi: 10.1007/s00394-013-0561-3. [DOI] [PubMed] [Google Scholar]
- 43.Szczechowiak K., Diniz B.S., Leszek J. Diet and Alzheimer’s dementia - Nutritional approach to modulate inflammation. Pharmacol. Biochem. Behav. 2019;184:172743. doi: 10.1016/j.pbb.2019.172743. [DOI] [PubMed] [Google Scholar]
- 44.Yang G., Wang Y., Sun J., Zhang K., Liu J. Ginkgo biloba for mild cognitive impairment and Alzheimer’s disease: A systematic review and meta-analysis of randomized controlled trials. Curr. Top. Med. Chem. 2016;16(5):520–528. doi: 10.2174/1568026615666150813143520. [DOI] [PubMed] [Google Scholar]
- 45.Lardenoije R., Iatrou A., Kenis G., Kompotis K., Steinbusch H.W.M., Mastroeni D., Coleman P., Lemere C.A., Hof P.R., van den Hove D.L.A., Rutten B.P.F. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015;131:21–64. doi: 10.1016/j.pneurobio.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hardy J., Escott-Price V. Genes, pathways and risk prediction in Alzheimer’s disease. Hum. Mol. Genet. 2019;28(R2):R235–R240. doi: 10.1093/hmg/ddz163. [DOI] [PubMed] [Google Scholar]
- 47.Sochocka M., Diniz B.S., Leszek J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017;54(10):8071–8089. doi: 10.1007/s12035-016-0297-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gadhave K., Kumar D., Uversky V.N., Giri R. A Multitude of signaling pathways associated with Alzheimer’s disease and their roles in AD pathogenesis and therapy. Med. Res. Rev. 2021;41(5):2689–2745. doi: 10.1002/med.21719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Umeda T., Ono K., Sakai A., Yamashita M., Mizuguchi M., Klein W.L., Yamada M., Mori H., Tomiyama T. Rifampicin is a candidate preventive medicine against amyloid-β and tau oligomers. Brain. 2016;139(Pt 5):1568–1586. doi: 10.1093/brain/aww042. [DOI] [PubMed] [Google Scholar]
- 50.Nazir M.A. Prevalence of periodontal disease, its association with systemic diseases and prevention. Int. J. Health Sci. (Qassim) 2017;11(2):72–80. [PMC free article] [PubMed] [Google Scholar]
- 51.Iizuka T., Morimoto K., Sasaki Y., Kameyama M., Kurashima A., Hayasaka K., Ogata H., Goto H. Preventive effect of rifampicin on Alzheimer disease needs at least 450 mg daily for 1 year: An FDG-PET follow-up study. Dement. Geriatr. Cogn. Disord. Extra. 2017;7(2):204–214. doi: 10.1159/000477343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cummings J.L., Tong G., Ballard C. Treatment combinations for Alzheimer’s disease: Current and future pharmacotherapy options. J. Alzheimers Dis. 2019;67(3):779–794. doi: 10.3233/JAD-180766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Youdim M.B., Weinstock M. Molecular basis of neuroprotective activities of rasagiline and the anti-Alzheimer drug TV3326 (N-propargyl-(3R)aminoindan-5-YL)-ethyl methyl carbamate. Cell. Mol. Neurobiol. 2001;21(6):555–573. doi: 10.1023/A:1015131516649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Youdim M.B.H., Bar Am O., Yogev-Falach M., Weinreb O., Maruyama W., Naoi M., Amit T. Rasagiline: Neurodegeneration, neuroprotection, and mitochondrial permeability transition. J. Neurosci. Res. 2005;79(1-2):172–179. doi: 10.1002/jnr.20350. [DOI] [PubMed] [Google Scholar]
- 55.Matthews D.C., Ritter A., Thomas R.G., Andrews R.D., Lukic A.S., Revta C., Kinney J.W., Tousi B., Leverenz J.B., Fillit H., Zhong K., Feldman H.H., Cummings J. Rasagiline effects on glucose metabolism, cognition, and tau in Alzheimer’s dementia. Alzheimers Dement. (N. Y.) 2021;7(1):e12106. doi: 10.1002/trc2.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liberman A.C., Trias E., da Silva Chagas L., Trindade P., Dos Santos Pereira M., Refojo D., Hedin-Pereira C., Serfaty C.A. Neuroimmune and inflammatory signals in complex disorders of the central nervous system. Neuroimmunomodulation. 2018;25(5-6):246–270. doi: 10.1159/000494761. [DOI] [PubMed] [Google Scholar]
- 57.Ownby R.L. Neuroinflammation and cognitive aging. Curr. Psychiatry Rep. 2010;12(1):39–45. doi: 10.1007/s11920-009-0082-1. [DOI] [PubMed] [Google Scholar]
- 58.Jaturapatporn D., Isaac M.G.E.K.N., McCleery J., Tabet N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2012;(2):CD006378. doi: 10.1002/14651858.CD006378.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miguel-Álvarez M., Santos-Lozano A., Sanchis-Gomar F., Fiuza-Luces C., Pareja-Galeano H., Garatachea N., Lucia A. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: A systematic review and meta-analysis of treatment effect. Drugs Aging. 2015;32(2):139–147. doi: 10.1007/s40266-015-0239-z. [DOI] [PubMed] [Google Scholar]
- 60.Meyer P-F., Tremblay-Mercier J., Leoutsakos J., Madjar C., Lafaille-Maignan M-É., Savard M., Rosa-Neto P., Poirier J., Etienne P., Breitner J. INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology. 2019;92(18):e2070–e2080. doi: 10.1212/WNL.0000000000007232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aktas O., Ullrich O., Infante-Duarte C., Nitsch R., Zipp F. Neuronal damage in brain inflammation. Arch. Neurol. 2007;64(2):185–189. doi: 10.1001/archneur.64.2.185. [DOI] [PubMed] [Google Scholar]
- 62.Wyss-Coray T., Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012;2(1):a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dong H., Zhang X., Qian Y. Mast cells and neuroinflammation. Med. Sci. Monit. Basic Res. 2014;20:200–206. doi: 10.12659/MSMBR.893093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shaik-Dasthagirisaheb Y.B., Conti P. The role of mast cells in Alzheimer’s disease. Adv. Clin. Exp. Med. 2016;25(4):781–787. doi: 10.17219/acem/61914. [DOI] [PubMed] [Google Scholar]
- 65.Piette F., Belmin J., Vincent H., Schmidt N., Pariel S., Verny M., Marquis C., Mely J., Hugonot-Diener L., Kinet J-P., Dubreuil P., Moussy A., Hermine O. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimers Res. Ther. 2011;3(2):16. doi: 10.1186/alzrt75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Folch J., Petrov D., Ettcheto M., Pedrós I., Abad S., Beas-Zarate C., Lazarowski A., Marin M., Olloquequi J., Auladell C., Camins A. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev. Neurother. 2015;15(6):587–596. doi: 10.1586/14737175.2015.1045419. [DOI] [PubMed] [Google Scholar]
- 67.Chen Y., Zhou K., Wang R., Liu Y., Kwak Y-D., Ma T., Thompson R.C., Zhao Y., Smith L., Gasparini L., Luo Z., Xu H., Liao F.F. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc. Natl. Acad. Sci. USA. 2009;106(10):3907–3912. doi: 10.1073/pnas.0807991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beeri M.S., Schmeidler J., Silverman J.M., Gandy S., Wysocki M., Hannigan C.M., Purohit D.P., Lesser G., Grossman H.T., Haroutunian V. Insulin in combination with other diabetes medication is associated with less Alzheimer neuropathology. Neurology. 2008;71(10):750–757. doi: 10.1212/01.wnl.0000324925.95210.6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pedersen W.A., McMillan P.J., Kulstad J.J., Leverenz J.B., Craft S., Haynatzki G.R. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp. Neurol. 2006;199(2):265–273. doi: 10.1016/j.expneurol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 70.Wang R., Li J.J., Diao S., Kwak Y-D., Liu L., Zhi L., Büeler H., Bhat N.R., Williams R.W., Park E.A., Liao F.F. Metabolic stress modulates Alzheimer’s β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab. 2013;17(5):685–694. doi: 10.1016/j.cmet.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Freiherr J., Hallschmid M., Frey W.H., II, Brünner Y.F., Chapman C.D., Hölscher C., Craft S., De Felice F.G., Benedict C. Intranasal insulin as a treatment for Alzheimer’s disease: A review of basic research and clinical evidence. CNS Drugs. 2013;27(7):505–514. doi: 10.1007/s40263-013-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benedict C., Grillo C.A. Insulin resistance as a therapeutic target in the treatment of Alzheimer’s disease: A state-of-the-art review. Front. Neurosci. 2018;12:215. doi: 10.3389/fnins.2018.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Novak V., Milberg W., Hao Y., Munshi M., Novak P., Galica A., Manor B., Roberson P., Craft S., Abduljalil A. Enhancement of vasoreactivity and cognition by intranasal insulin in type 2 diabetes. Diabetes Care. 2014;37(3):751–759. doi: 10.2337/dc13-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mullins R.J., Diehl T.C., Chia C.W., Kapogiannis D. Insulin resistance as a link between amyloid-beta and tau pathologies in Alzheimer’s disease. Front. Aging Neurosci. 2017;9:118. doi: 10.3389/fnagi.2017.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Felice F.G., Vieira M.N.N., Bomfim T.R., Decker H., Velasco P.T., Lambert M.P., Viola K.L., Zhao W-Q., Ferreira S.T., Klein W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA. 2009;106(6):1971–1976. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yarchoan M., Toledo J.B., Lee E.B., Arvanitakis Z., Kazi H., Han L-Y., Louneva N., Lee V.M-Y., Kim S.F., Trojanowski J.Q., Arnold S.E. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol. 2014;128(5):679–689. doi: 10.1007/s00401-014-1328-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dubey S.K., Lakshmi K.K., Krishna K.V., Agrawal M., Singhvi G., Saha R.N., Saraf S., Saraf S., Shukla R., Alexander A. Insulin mediated novel therapies for the treatment of Alzheimer’s disease. Life Sci. 2020;249:117540. doi: 10.1016/j.lfs.2020.117540. [DOI] [PubMed] [Google Scholar]
- 78.Hölscher C. Brain insulin resistance: Role in neurodegenerative disease and potential for targeting. Expert Opin. Investig. Drugs. 2020;29(4):333–348. doi: 10.1080/13543784.2020.1738383. [DOI] [PubMed] [Google Scholar]
- 79.Allen S.J., Watson J.J., Shoemark D.K., Barua N.U., Patel N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013;138(2):155–175. doi: 10.1016/j.pharmthera.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 80.Hölscher C. Insulin, incretins and other growth factors as potential novel treatments for Alzheimer’s and Parkinson’s diseases. Biochem. Soc. Trans. 2014;42(2):593–599. doi: 10.1042/BST20140016. [DOI] [PubMed] [Google Scholar]
- 81.Williams B.J., Eriksdotter-Jonhagen M., Granholm A-C. Nerve growth factor in treatment and pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2006;80(3):114–128. doi: 10.1016/j.pneurobio.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 82.Counts S.E., Nadeem M., Wuu J., Ginsberg S.D., Saragovi H.U., Mufson E.J. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer’s disease. Ann. Neurol. 2004;56(4):520–531. doi: 10.1002/ana.20233. [DOI] [PubMed] [Google Scholar]
- 83.MacDonald B.T., He X. Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb. Perspect. Biol. 2012;4(12):a007880. doi: 10.1101/cshperspect.a007880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nusse R., Clevers H. Wnt/β-Catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 85.Komiya Y., Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4(2):68–75. doi: 10.4161/org.4.2.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Palomer E., Buechler J., Salinas P.C. Wnt signaling deregulation in the aging and Alzheimer’s brain. Front. Cell. Neurosci. 2019;13:227. doi: 10.3389/fncel.2019.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Libro R., Bramanti P., Mazzon E. The role of the Wnt canonical signaling in neurodegenerative diseases. Life Sci. 2016;158:78–88. doi: 10.1016/j.lfs.2016.06.024. [DOI] [PubMed] [Google Scholar]
- 88.Wan W., Xia S., Kalionis B., Liu L., Li Y. The role of Wnt signaling in the development of Alzheimer’s disease: A potential therapeutic target? BioMed Res. Int. 2014;2014:301575. doi: 10.1155/2014/301575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sellers K.J., Elliott C., Jackson J., Ghosh A., Ribe E., Rojo A.I., Jarosz-Griffiths H.H., Watson I.A., Xia W., Semenov M., Morin P., Hooper N.M., Porter R., Preston J., Al-Shawi R., Baillie G., Lovestone S., Cuadrado A., Harte M., Simons P., Srivastava D.P., Killick R. Amyloid β synaptotoxicity is Wnt-PCP dependent and blocked by fasudil. Alzheimers Dement. 2018;14(3):306–317. doi: 10.1016/j.jalz.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hadi F., Akrami H., Shahpasand K., Fattahi M.R. Wnt signalling pathway and tau phosphorylation: A comprehensive study on known connections. Cell Biochem. Funct. 2020;38(6):686–694. doi: 10.1002/cbf.3530. [DOI] [PubMed] [Google Scholar]
- 91.Xue H-H., Zhao D-M. Regulation of mature T cell responses by the Wnt signaling pathway. Ann. N. Y. Acad. Sci. 2012;1247:16–33. doi: 10.1111/j.1749-6632.2011.06302.x. [DOI] [PubMed] [Google Scholar]
- 92.Borrell-Pages M., Romero J.C., Crespo J., Juan-Babot O., Badimon L. LRP5 associates with specific subsets of macrophages: Molecular and functional effects. J. Mol. Cell. Cardiol. 2016;90:146–156. doi: 10.1016/j.yjmcc.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 93.Manoharan I., Hong Y., Suryawanshi A., Angus-Hill M. L., Sun Z., Mellor A. L., Munn D. H., Manicassamy S. TLR2-dependent activation of β-catenin pathway in dendritic cells induces regulatory responses and attenuates autoimmune inflammation. . Immunol. Baltim. Md 1950. 2014;193(8):4203–4213. doi: 10.4049/jimmunol.1400614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Swafford D., Manicassamy S. Wnt signaling in dendritic cells: Its role in regulation of immunity and tolerance. Discov. Med. 2015;19(105):303–310. [PMC free article] [PubMed] [Google Scholar]
- 95.Caricasole A., Copani A., Caraci F., Aronica E., Rozemuller A.J., Caruso A., Storto M., Gaviraghi G., Terstappen G.C., Nicoletti F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer’s brain. J. Neurosci. 2004;24(26):6021–6027. doi: 10.1523/JNEUROSCI.1381-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blagodatski A., Poteryaev D., Katanaev V.L. Targeting the Wnt pathways for therapies. Mol. Cell. Ther. 2014;2:28. doi: 10.1186/2052-8426-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Forlenza O.V., De-Paula V.J.R., Diniz B.S.O. Neuroprotective effects of lithium: Implications for the treatment of Alzheimer’s disease and related neurodegenerative disorders. ACS Chem. Neurosci. 2014;5(6):443–450. doi: 10.1021/cn5000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hamano T., Shirafuji N., Makino C., Yen S-H., Kanaan N.M., Ueno A., Suzuki J., Ikawa M., Matsunaga A., Yamamura O., Kuriyama M., Nakamoto Y. Pioglitazone prevents tau oligomerization. Biochem. Biophys. Res. Commun. 2016;478(3):1035–1042. doi: 10.1016/j.bbrc.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 99.Jin N., Zhu H., Liang X., Huang W., Xie Q., Xiao P., Ni J., Liu Q. Sodium selenate activated Wnt/β-catenin signaling and repressed amyloid-β formation in a triple transgenic mouse model of Alzheimer’s disease. Exp. Neurol. 2017;297:36–49. doi: 10.1016/j.expneurol.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 100.Sochocka M., Donskow-Łysoniewska K., Diniz B.S., Kurpas D., Brzozowska E., Leszek J. The gut microbiome alterations and inflammation-driven pathogenesis of Alzheimer’s disease-a critical review. Mol. Neurobiol. 2019;56(3):1841–1851. doi: 10.1007/s12035-018-1188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mallick H., Franzosa E.A., Mclver L.J., Banerjee S., Sirota-Madi A., Kostic A.D., Clish C.B., Vlamakis H., Xavier R.J., Huttenhower C. Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat. Commun. 2019;10(1):3136. doi: 10.1038/s41467-019-10927-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Slots J. Periodontitis: Facts, fallacies and the future. Periodontol. 2000. 2017;75(1):7–23. doi: 10.1111/prd.12221. [DOI] [PubMed] [Google Scholar]
- 103.Tohidpour A., Morgun A.V., Boitsova E.B., Malinovskaya N.A., Martynova G.P., Khilazheva E.D., Kopylevich N.V., Gertsog G.E., Salmina A.B. Neuroinflammation and infection: Molecular mechanisms associated with dysfunction of neurovascular unit. Front. Cell. Infect. Microbiol. 2017;7:276. doi: 10.3389/fcimb.2017.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cardoso E.M., Reis C., Manzanares-Céspedes M.C. Chronic periodontitis, inflammatory cytokines, and interrelationship with other chronic diseases. Postgrad. Med. 2018;130(1):98–104. doi: 10.1080/00325481.2018.1396876. [DOI] [PubMed] [Google Scholar]
- 105.Scannapieco F.A., Cantos A. Oral inflammation and infection, and chronic medical diseases: Implications for the elderly. Periodontol. 2000. 2016;72(1):153–175. doi: 10.1111/prd.12129. [DOI] [PubMed] [Google Scholar]
- 106.Dominy S.S., Lynch C., Ermini F., Benedyk M., Marczyk A., Konradi A., Nguyen M., Haditsch U., Raha D., Griffin C., Holsinger L.J., Arastu-Kapur S., Kaba S., Lee A., Ryder M.I., Potempa B., Mydel P., Hellvard A., Adamowicz K., Hasturk H., Walker G.D., Reynolds E.C., Faull R.L.M., Curtis M.A., Dragunow M., Potempa J. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019;5(1):eaau3333. doi: 10.1126/sciadv.aau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bennett C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019;70:307–321. doi: 10.1146/annurev-med-041217-010829. [DOI] [PubMed] [Google Scholar]
- 108.Bishop K.M. Progress and promise of antisense oligonucleotide therapeutics for central nervous system diseases. Neuropharmacology. 2017;120:56–62. doi: 10.1016/j.neuropharm.2016.12.015. [DOI] [PubMed] [Google Scholar]