

Abstract
Idiopathic inflammatory myopathies (IIM), also known as myositis, are a heterogeneous group of autoimmune disorders with varying clinical manifestations, treatment responses and prognoses. Muscle weakness is usually the classical clinical manifestation but other organs can be affected, including the skin, joints, lungs, heart and gastrointestinal tract, and they can even result in the predominant manifestations, supporting that these are systemic inflammatory disorders. Different myositis-specific autoantibodies have been identified and, on the basis of clinical, histopathological and serological features, IIMs can be classified into several subgroups — dermatomyositis (including amyopathic dermatomyositis), antisynthetase syndrome, immune-mediated necrotizing myopathy, inclusion body myositis, polymyositis and overlap myositis. The prognoses, treatment responses and organ manifestations vary among these groups, implicating different pathophysiological mechanisms in each subtype. A deeper understanding of the molecular pathways underlying the pathogenesis and identifying the autoantigens of the immune reactions in these subgroups is crucial to improve outcomes. New, more homogeneous subgroups defined by autoantibodies may help define disease mechanisms, and will also be important in future clinical trials to develop targeted therapies and in identifying biomarkers to guide treatment decisions for the individual patient.
Introduction
Idiopathic inflammatory myopathies (IIM), also known as myositis, are a heterogenous group of autoimmune disorders usually characterized by chronic inflammation of the muscle with varying clinical manifestations, treatment responses and prognoses. Muscle weakness, low muscle endurance and myalgia are frequent and shared symptoms, but extramuscular manifestations, such as skin rash, arthritis, interstitial lung disease (ILD), and heart involvement, are common, emphasizing the systemic inflammatory nature of these disorders (Figure 1). In some patients, extramuscular manifestations dominate the clinical picture and muscle weakness may be absent; diagnosing the disease in these patients can be particularly challenging. A major advancement in the field of myositis was the discovery of autoantibodies that are specific for myositis, called myositis-specific autoantibodies (MSAs; present in up to 60% of patients with IIM); which are helpful in establishing a diagnosis of IIM (Table 1)1. Furthermore, MSAs are strongly associated with distinct clinical phenotypes and, therefore, are important to predict organ manifestations and may predict prognosis. In addition, patients with IIM may also have autoantibodies which are present in other autoimmune disorders, such as systemic lupus erythematosus (SLE), systemic sclerosis or Sjögren syndrome. These autoantibodies are often named myositis-associated autoantibodies (MAA) and the most frequent are anti-Ro52, anti-PMScl, anti-Ku and anti-U1RNP. Around 20–30% of patients with IIM have no known autoantibodies, which can be classified as seronegative IIM2.
Figure 1. Manifestations of idiopathic inflammatory myopathies (IIM).

IIM exhibits not only myositis but also various extra-muscular complications. Some manifestations are strongly associated with the presence of specific circulating autoantibodies. MDA5, melanoma differentiation-associated gene 5; NXP2, nuclear matrix protein 2; SAE, small ubiquitin-like modifier activating enzyme; SRP, signal recognition particle; TIF1, transcriptional intermediary factor 1.
Table 1.
Main clinical associations of autoantibodies in adult IIM252
| Autoantibody | Target antigen | Frequency in myositis | Main clinical associations | Indirect immunofluorescence pattern on HEp-2 cells |
|---|---|---|---|---|
| Myositis-specific antibodies | ||||
| Anti-Jo1 | Histidyl-tRNA synthetase | 15–30% | ASyS | Cytoplasmatic fine speckled |
| Anti-Ha/YRS | Tyrosyl-tRNA synthetase | < 1% | ASyS | Cytoplasmic fine speckled |
| Anti-Zo | Phenylalanyl-tRNA synthetase | < 1% | ASyS | Cytoplasmic fine speckled |
| Anti-EJ | Glycyl-tRNA synthetase | < 2% | ASyS | Cytoplasmic fine speckled |
| Anti-PL-7 | Threonyl-tRNA synthetase | 5–10% | ASyS | Inconsistent cytoplasmic dense fine speckled |
| Anti-OJ | Isoleucyl-tRNA synthetase | < 2% | ASyS | Cytoplasmic fine speckled |
| Anti-KS | Asparaginyl-tRNA synthetase | < 2% | ASyS, often only ILD, arthritis | Cytoplasmic fine speckled |
| Anti-PL-12 | Alanyl-tRNA synthetase | < 5% | ASS, often only ILD | Cytoplasmic dense fine speckled |
| Anti-Mi-2 | Nucleosome remodelling deacetylase complex | 4–20% | Classical DM | Nuclear fine speckled |
| Anti-TIF1 | Transcription intermediary factor 1 | 10–20% | Only DM, often CDM in adults (40–75%), more severe disease | Nuclear fine speckled |
| Anti-MDA5 | Melanoma differentiation associated protein 5 | 13–30% | C-ADM (ILD), severe skin manifestation | Negative or cytoplasmic |
| Anti-SAE | Small ubiquitin-like modifier activating enzyme | < 10% | Initially classic skin disease, myositis often develops later | Nuclear fine speckled |
| Anti-NXP2 | Nuclear matrix protein 2 | 3–24% | DM, calcinosis, increased risk of CDM in adults | Nuclear fine speckled, nuclear multiple dots |
| Anti-SRP | Signal recognition particle | 5–15% | IMNM | Cytoplasmic fine speckled |
| Anti-HMGCR | 3-hydroxy-3-methylglutaryl CoA reductase | 6–10% | IMNM, often statin associated | Negative |
| Anti-FHL1 a | Four and a half LIM domains 1 | 5–10% | IIM often with severe muscle involvement | Not known |
| Myositis-associated antibodies | ||||
| Anti-PM-Scl | Human exosome protein complex | 8–10% | PM, DM, overlap with scleroderma | Nucleolar homogeneous |
| Anti-U1RNP | U1 small nuclear RNP | 10% | MCTD | Nuclear coarse speckled |
| Anti-Ku | Regulatory subunit of DNA-dependent protein kinase | <2% | Overlap with scleroderma, ILD | Nuclear fine speckled |
| Anti-RuvBL1 and anti-RuvBL2 a | Nuclear proteins RuvBL1/2 | 3% | Overlap with scleroderma | Nuclear speckled |
| Anti-Ro (52, 60) | TRIM21, ring-shaped protein binding Y RNAs | 10–40% | Often second autoantibody present, more severe disease including ILD | Negative or cytoplasmic |
| Anti-cN-1A | cytosolic 5’-nucleotidase 1A | 4–21% | IBM, pSS, SLE | Negative |
Myositis-specific autoantibodies are found only in patients with IIM and not in other diseases. Myositis-associated antibodies are not specific for IIM and can be detected in other connective tissue diseases, such as SLE, pSS or scleroderma.
Detection not routinely available. ARS – aminoacyl-tRNA synthetase, ASS – antisynthetase syndrome, PM – polymyositis, DM – dermatomyositis, IBM – inclusion body myositis, IIM – idiopathic inflammatory myopathy, ILD – interstitial lung disease, CDM – cancer associated DM, IMNM – immune mediated necrotizing myopathy, C-ADM – clinically amyopathic DM, MCTD – mixed connective tissue disease, pSS- primary Sjögren’s syndrome, SLE systemic lupus erythematosus
PM-Scl – polymyositis-scleroderma, RNP – ribonucleoprotein.
Historically, polymyositis was the overarching name for these disorders. In 1863, a disease subgroup with skin rash was identified and named dermatomyositis. A clinical definition of polymyositis and dermatomyositis was not proposed until the 1950s3. Later, based on clinical and histopathological manifestations of muscle tissue, IIMs were categorized into the subgroups dermatomyositis (DM; juvenile and adult onset), polymyositis (PM), inclusion body myositis (IBM), and amyopathic dermatomyositis (ADM). The definition of PM has become controversial and varied from the initially clinically defined to a definition based on specific histopathological features, including presence of T lymphocytes in the infiltrates requiring staining of muscle tissue sections by immunohistochemistry4. On the basis of MSAs, new subgroups of IIMs with distinct clinical manifestations and muscle histopathological features have been identified, which include antisynthetase syndrome (ASyS) in the 1990s and immune-mediated necrotizing myopathy (IMNM) in 2006. Many patients previously classified as having PM fall into these new subgroups, resulting in a decreasing number of patients included in the PM subgroup5. Additional suggested subgroups of IIM include cancer-associated myositis and overlap myositis (myositis that coexists with another autoimmune disorder).
Owing to the new observations that autoantibodies are frequently present in patients with IIM and that some patients may not have lymphocytic infiltrates in muscle biopsy samples, a change of the nomenclature from IIM to systemic autoimmune myopathies (SAM) has been proposed to better reflect the pathogenesis6. However, as this name change has not yet been widely recognized in the myositis community, we have retained the IIM nomenclature in this Primer.
The diagnosis of IIM and its subgroups is based on a combination of clinical symptoms and signs, including muscle biopsy features, MRI pattern, serological assessment and serum levels of muscle enzymes. Treatment is largely based on high doses of glucocorticoids in combination with other immunosuppressive drugs, but treatment responses vary and there is a high unmet need for new therapeutic approaches. Research to date has mainly focused on patients categorized as having DM, PM and IBM. A refined subgrouping of myositis that enables more homogenous classification defined by clinical, histopathological and serological data is important to improve understanding of the disease mechanisms that lead to myositis and the extramuscular manifestations to improve treatment and patient prognosis. Furthermore, data from clinical trials are mainly based on the traditional subgroups PM, DM and IBM, emphasizing the need for more clinical trials that include MSA-defined subgroups1,7–10.
In this Primer, we discuss the traditional myositis subgroups recognized in adults: DM, ADM, IBM and PM, as well as the newly classified subgroups ASyS and IMNM, focusing on their epidemiology, pathophysiology and diagnosis if data are available. Additionally, we outline the clinical management of IIM and highlight emerging therapeutic options. Finally, we review the pressing questions in the field, suggesting directions for future research.
Epidemiology
Studying the distribution and determinants of inflammatory myopathies is challenging, partly owing to the difficulty in identifying relevant disease entities representative of inflammatory myopathies in population-based data. Studies based on clinical cohorts from university hospitals, where severe cases of IIM are usually monitored, or studies based on biopsy-verified IIM could be underestimating the true incidence and prevalence. By contrast, register-based studies including all types of health care could be overestimating the true incidence and prevalence, as the potential for misclassification is increased among physicians who are not specialized in IIM. A limitation of register-based epidemiological studies to date is the lack of specific International Classification of Diseases (ICD) codes and changing classification systems for the new subgroups of IIM. For example, patients with ASyS and IMNM are often coded with ICD codes for PM.
Incidence and prevalence
A wide range of estimates of incidence and prevalence, as well as risk factors for disease, have been published. Globally, but with a majority of studies from Asia, Europe and North America, the incidence estimates range from 11 to 660 patients with newly diagnosed IIM per 1,000,000 person-years11–14 and between 2.9 and 34 individuals per 100,000 population are suggested to have the disease15–17. There are no clear signals suggesting differences in incidence and prevalence across different regions or ethnicities, but a north to south gradient has been suggested in both Europe and North America, with a higher prevalence of the subtype DM closer to the equator18,19. All available data suggest that PM, DM and IMNM are more common in women than men16,17,20,21 and that IBM is more common in men22–24. The incidence increases with age, and the peak age of incidence is ~ 50 years of age in both Europe20 and North America11,20.
Environmental risk factors
A few environmental factors have been associated with IIMs, of which several are infectious agents, suggested on the basis of data from animal models and clinical observations. These observations include the co-occurrence of hepatitis B virus infection in PM25, as well as infection with HIV or human T-cell leukemia virus type 1 (HTLV-1) as triggers for PM26, DM and IBM27,28. In small observational studies that have not been reproduced, hepatitis C virus infection has been associated with IBM29, gastrointestinal and lower respiratory infections have been suggested as risk factors for IIM (primarily PM or DM), whereas upper respiratory tract infections have been suggested to lower the risk of disease30,31. Of note, seasonality has been observed in the birthdates32 and onset33,34 of some clinical and autoantibody groups. Also, an increase was observed in autoimmune diseases, including myositis, in World Trade Center prolonged rescue and recovery workers35. Extensive physical exertion has been suggested as a risk factor for PM and DM in a small case-control study30. Other important findings include no association with vaccines and PM and DM30, whereas exposure to UV radiation is associated with development of DM36 and smoking is associated with anti-Jo1 autoantibody-positive ASyS37.
Genetic risk factors
The major genetic risk factors are involved in immune regulation and the strongest associations have been identified in the HLA region. In a study by the Myositis Genetics Consortium, in which >2,500 patients mainly of European ancestry with inflammatory myopathies were included, a strong association was noted among the alleles of the 8.1 ancestral haplotype and PM and DM38. In addition, a few non-HLA loci have been associated with inflammatory myopathy in European populations: STAT4, TRAF6 and UBE2L3 with IIM overall39; PTPN22 with PM40; PLCL1 and BLK with DM41; and CCR5 with IBM42.
Mortality
The mortality estimates also show large variations. 10-year survival rates vary between 20% and 90%43–51 in studies from Europe, North America and Japan, depending on study design and patient selection. In a large population-based cohort study that compared death rates in 326 patients with IIM diagnosed between 2003 and 2012 in southern parts of Norway with death rates in the general population48, the mortality ratio was 2.4, 2.6 and 1.7 for PM, DM and IBM, respectively, compared with the general population. A subsequent study suggested that the overall mortality in IIM is highest during the first year after diagnosis and then decreases, whereas mortality in the general population was increasing during the >10-year follow-up period52. The reason for this development is yet unknown. The main causes of death in patients with IIM were malignancies, cardiovascular diseases and lung diseases52, although it could be speculated that deaths occurring shortly after diagnosis are due to factors related to IIM, for example rapidly progressive ILD, and that later deaths are due to factors such as malignancy.
Mechanisms/pathophysiology
As with other types of autoimmunity, IIM phenotypes are thought to develop as a result of interactions between the necessary and sufficient genetic and environmental risk factors in the relative absence of protective factors53. Although understanding of these processes is limited, it seems that both adaptive and innate immune mechanisms, as well as non-immune mechanisms, are involved to a varying extent in the different types of IIM. Of note, early pathological observations were made in patients diagnosed as having PM; however, many of these patients would now be classified as having IMNM or ASyS instead. Emerging data now suggest that various risk factors result in each of the major clinical and myositis autoantibody-defined phenotypes, which also differ in many clinical features, responses to therapy and outcomes; hence, it seems that the pathogenic pathways differ between the various phenotypes. Thus, it is appropriate to focus the discussion of pathogenesis on the major IIM phenotypes DM, ASyS, IMNM, IBM and PM, for which most information is currently available. Due to the heterogeneity of overlap myositis, the pathogenesis of this subgroup will not be discussed in this Primer. Importantly, each of the subtypes has a different spectrum of target tissues: IMNM and IBM predominantly affect skeletal muscle, whereas DM and ASyS are multiorgan diseases, which also often affect the skin, lungs and/or joints. In addition, IMNM and IBM muscle biopsy samples have characteristic histopathological features that enable distinction from DM or ASyS samples. Furthermore, although DM and ASyS muscle samples share histological similarities, such as prominent perifascicular involvement, transcriptomic analyses have revealed that each has a unique gene expression profile. For example, the expression of type 1 interferon-inducible genes is markedly higher in DM than in ASyS54,55. Indeed, transcriptomic data alone can be used to identify a muscle biopsy sample from a patient with DM, ASyS, IMNM or IBM with >90% accuracy56.
Dermatomyositis
Although certain genetic and environmental factors have been associated with adult onset DM, the trigger of autoimmunity in patients with DM remains unclear. Similarly, the mechanisms of underlying autoimmunity remain obscure. Indeed, within muscle tissue, whether muscle cells, blood vessels and/or connective tissue are directly targeted by the immune system remains to be shown. Nevertheless, perifascicular myofibre abnormalities, including atrophy and necrosis, are typical histological features of muscle specimens from patients with DM (and may also be seen in some patients with ASyS). By contrast, myofibre necrosis and inflammation are distributed evenly throughout muscle fascicles in IMNM and IBM, respectively. One detailed histological study of DM muscle specimens revealed that endomysial capillaries in regions of perifascicular atrophy are reduced in number and size and that the remaining endothelial cells stain positive for components of the activated membrane attack complex57. However, whether membrane attack complex deposition is a cause or effect of endothelial cell damage in DM is not clear. Of note, the abnormal perifascicular regions are preferentially located near areas of perimysium with infiltrating inflammatory cells and remnants of blood vessels, but no intact intermediate-size vessels. Taken together, these observations raise the possibility that immune-mediated vascular damage may cause myofibre and capillary damage in regions of the endomysium that are most distant from an intact vascular supply.
It remains unclear how intermediate-size blood vessels in the perimysium are damaged and destroyed in patients with DM. Nevertheless, plasmacytoid dendritic cells, which are potent sources of interferon, may be found surrounding these vascular structures58. Furthermore, type 1 interferon-inducible genes are among the most upregulated of genes in the muscle58, skin59 and peripheral blood60,61 of patients with DM, further supporting a role for type 1 interferon in DM pathophysiology. This finding may help explain why JAK–STAT inhibitors, which target this pathway, are effective in treating DM-related damage in the skin62, muscles62 and lungs63.
Of note, most of the DM studies referenced above did not perform sub-analyses controlling for DM-specific autoantibody status. As it is now appreciated that each DM-specific autoantibody is associated with unique clinical manifestations, histologic features64, and transcriptomic profiles56, grouping these entities together may be a limitation of these studies.
Antisynthetase syndrome
ASyS is characterized by autoantibodies against one of many aminoacyl transfer RNA (tRNA) synthetases. As with most other forms of myositis, some genetic and environmental associations have been made with ASyS, but what specifically triggers this immune response remains unclear. One of the first examples of adaptive immune mechanisms involving antigen-driven B cell responses in myositis was the finding that anti-Jo1 autoantibodies directed against histidyl-tRNA synthetase arose months before clinical disease and, in addition to spectrotype broadening and class switching, underwent affinity maturation to that antigen65. Subsequent findings suggested that anti-Jo1 autoantibodies bound common autoepitopes, changed in titre with disease activity and were, therefore, modulated by an immune response closely linked to that responsible for myositis in these patients66.
Muscle biopsy samples from patients with ASyS more often include areas of perifascicular necrosis than those from patients with DM (79% versus 35%)67. In addition, ASyS samples include endomysial infiltration by clonally expanded T cells68, as observed in IBM. Interestingly, CD4+ T cells with reactivity against histidyl-tRNA synthetase are present in both blood and lungs of patients with ASyS69. Taken together, these observations suggest that the immune response in ASyS may be directed against endothelial cells, muscle cells and lung tissue. Of note, the finding that mice immunized with histidyl-tRNA synthetase develop antisynthetase autoantibodies, along with muscle and lung infiltration70, corroborates the possibility that an immune response against aminoacyl-tRNA synthetase proteins is associated with muscle and lung damage in patients with ASyS.
Most studies referenced above did not perform sub-analyses controlling for DM-specific rashes and, therefore, included patients traditionally defined as having PM or DM, who seem to have different risk factors and outcomes; hence, grouping these entities together may be a limitation of these studies.
Immune-mediated necrotizing myopathy
Patients with IMNM most often have autoantibodies recognizing either 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase (HMGCR) or the signal recognition particle (SRP). The HLA class II DRB1*11:01 allele is present in >70% of patients with IMNM and anti-HMGCR autoantibodies, but only in <20% of the general population71. Thus, DRB1*11:01 is one of the strongest known immunogenetic risk factors for developing an autoimmune disease. In addition, the use of statin medications is known to predispose individuals to developing anti-HMGCR-positive myositis72. As HMGCR is upregulated in muscle and other tissues in response to statin exposure, it has been hypothesized that increased expression and/or conformational changes of this protein caused by statins may contribute to breaking tolerance in genetically susceptible individuals, who then develop a necrotizing myopathy.
Muscle specimens from patients positive for autoantibodies against HMGCR and SRP are histologically indistinguishable. Furthermore, gene expression profiles from these two types of IMNM show only minor differences56. Thus, the mechanisms underlying muscle damage in each of these IMNM subtypes may be similar, if not identical.
The observation that both anti-SRP and anti-HMGCR autoantibody titres are highly correlated with muscle weakness and elevated levels of creatinine kinase, which is released into the blood stream from damaged muscle fibers, is consistent with their pathological role in the disease process73,74. Additional evidence has been provided by studies showing that SRP and HMGCR proteins are localized to the muscle cell membrane surface75, that anti-SRP and anti-HMGCR autoantibodies bind to the surface of cultured muscle cells75, and that membrane attack complex (which can be activated by antibodies) is observed on the surface of non-necrotic muscle fibres in IMNM muscle specimens76. Most importantly, one study showed that the passive transfer of anti-SRP or anti-HMGCR autoantibodies into immunodeficient mice caused myofibre necrosis and weakness that is dependent on an intact complement system77. Given the strength of evidence that autoantibody-mediated complement activation may cause muscle damage in IMNM, a phase II multicentre placebo-controlled trial of a C5 complement inhibitor was conducted in anti-SRP-positive and anti-HMGCR-positive patients78. Surprisingly, the complement inhibitor did not improve creatinine kinase levels, muscle strength or other relevant outcome measures in IMNM patients79. Consequently, the pathophysiological mechanisms underlying muscle damage in IMNM still remain to be elucidated.
Inclusion body myositis
Evidence that IBM is an autoimmune disease includes the presence of predisposing immunogenetic risk factors80, a large number of antibody-secreting plasma cells within IBM muscle tissue81, and the frequent occurrence of autoantibodies recognizing the NT5C1A protein in the blood of patients with IBM82,83. Furthermore, the observation that cytotoxic CD8+ T cells surround and invade muscle fibres in IBM muscle specimens provided early evidence that muscle damage could be mediated by T cells84–86. Indeed, subsequent studies revealed that CD8+ T cells are clonally expanded in muscle tissue87,88 and that the same clones are found both in blood and in multiple muscles from the same patient, where they persist89–91. Although the T cell targets remain unknown, these findings suggest that T cell stimulation by the relevant autoantigen persists for years in these patients. Interestingly, some of the T cell clone identities are shared between different patients with IBM, suggesting a common as yet undefined target autoantigen among those with IBM68. Importantly, studies showed that both CD4+ and CD8+ T cells in patients with IBM have unusual properties, including aberrant loss of CD28 or CD5 expression with the gain of CD16, CD94 and CD57 expression that is associated with terminally differentiated T cells92,93. Phenotypically similar to the abnormal lymphocytes seen in patients with T-cell large granulocytic leukaemia, the infiltrating T cells in IBM would also be expected to have increased cytotoxic potential and resistance to apoptosis. These features may help explain why IBM is refractory to glucocorticoids and other immunomodulatory therapies, but this population of T cells could also be a promising target for therapeutic intervention.
In addition to the invasion of myofibres by CD8+ CD57+ T cells, IBM muscle specimens are notable for the presence of rimmed vacuoles and protein inclusions within muscle fibres. For example, in one study, aggregates of p62 and TDP43 proteins were found in 12% of IBM myofibres, but only rarely in those of other IIM subtypes94. Although other reports suggest that p62 accumulation may be a non-specific feature of IIM,95–97 TDP43 positivity is recognized as highly specific for IBM98. Hence, IBM might have a considerable degenerative component, but it has not been shown whether accumulation of these proteins would lead to muscle cell degeneration. Furthermore, it remains unclear whether these changes occur in response to intensive immune-mediated damage or reflect some other underlying non-immune pathological process.
Polymyositis
PM could be defined as a myositis phenotype with chronic muscle weakness without skin involvement and involving predominant cytotoxic T cell mechanisms. During the past two decades, it has become clear that PM has been over-diagnosed99 and that most patients previously diagnosed with PM can now be classified as having IBM, IMNM, ASyS or genetic muscle disease; however, some patients with IIM do not have DM, ASyS, IBM, IMNM or myositis autoantibodies and have the classic clinical and pathologic findings of PM5. These patients, although now a small subgroup of those with IIM, are therefore appropriately classified as PM by currently accepted criteria8,100. However, most historical studies of PM included samples from patients now classified as IMNM and ASyS without a rash, diseases now recognized to be pathologically distinct from each other. Thus, future studies will be required to define the risk factors and mechanisms underlying muscle inflammation and damage more completely in PM.
Diagnosis, screening and prevention
Clinical subgroups
Dermatomyositis
DM is defined by the presence of characteristic cutaneous manifestations and myositis. While muscle and skin involvement coexist in the prototype of DM (classic DM), DM can exist without muscle disease (ADM) or overt muscle symptoms despite evidence of myositis on laboratory testing (hypomyopathic DM, HDM)101. ADM and HDM are defined when the conditions last for ≥6 months, and are collectively termed as clinically ADM (CADM)101. Skin manifestations in CADM are identical to those in classic DM. Patients with CADM can still have systemic involvement, such as ILD and dysphagia. Conversely, patients who have no rash but have muscle disease with classic histopathologic features of DM on muscle biopsy have been described as having dermatomyositis sine dermatitis102. Malignancy is occasionally associated with DM (cancer-associated DM), but can also occur in other forms of IIM103–105.
Most patients with DM have one of the myositis-specific autoantibodies, anti-Mi-2106, anti-melanoma differentiation-associated gene 5 (MDA5)107,108, anti-transcriptional intermediary factor 1 (TIF1)109,110, anti-nuclear matrix protein 2 (NXP2)111, or anti-small ubiquitin-like modifier activating enzyme (SAE)112 antibodies (Table 1). These autoantibodies are closely associated with distinct clinical phenotypes, leading to the proposal to subclassify DM into six different subtypes —anti-Mi-2 DM, anti-MDA5 DM, anti-TIF1-γ DM, anti-NXP2 DM, anti-SAE DM, and autoantibody-negative DM (Figure 2, 3)7,8,113,114. Patients with anti-Mi-2 DM and anti-NXP2 DM tend to have prominent myositis, whereas it is milder, often asymptomatic, in those with anti-MDA5, anti-TIF1, and anti-SAE DM. These different subtypes of DM also have associations with characteristic rashes in addition to the severity115,116.
Figure 2. Clinical manifestations in patients with dermatomyositis with anti-TIF1gamma, anti.NXP2 and anti.Mi-2 autoantibodies.

Dermatomyositis (DM), including amyopathic DM, is one of the classical subgroups of IIMs. DM can be further subclassified based on specific autoantibodies, which demonstrate various characteristic manifestations such as distribution of muscle weakness and extramuscular manifestations: a. anti-transcriptional intermediary 1 (TIF1) DM; b. anti-nuclear matrix protein 2 (NXP2) DM; c. anti-Mi-2 DM.
Figure 3. Clinical manifestations in patients with dermatomyositis with anti-MDA5 and anti-SAE autoantibodies.
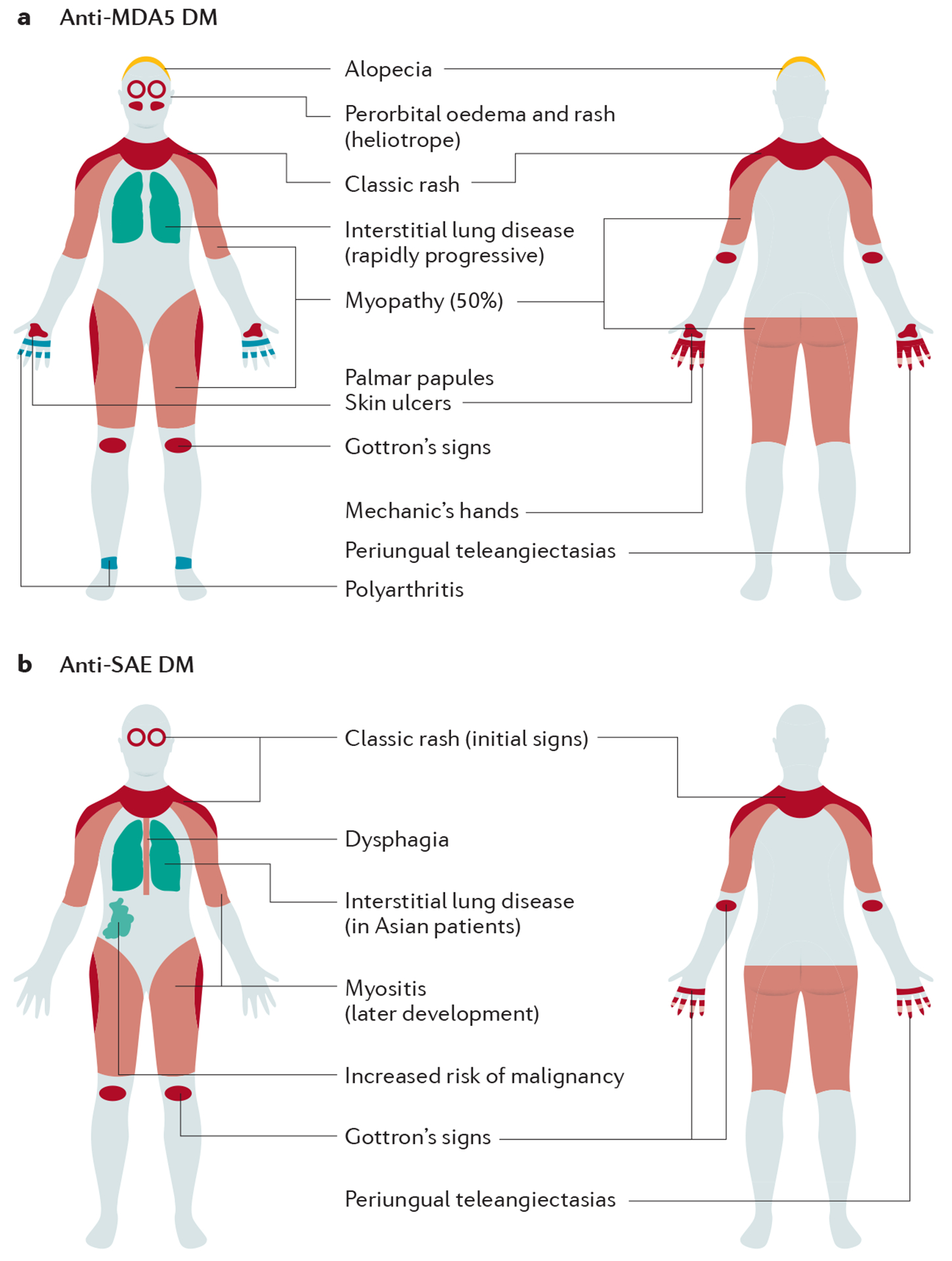
Dermatomyositis (DM), including amyopathic DM, is one of the classical subgroups of IIMs. DM can be further subclassified based on specific autoantibodies, which demonstrate various characteristic manifestations such as distribution of muscle weakness and extramuscular manifestations: a. anti-melanoma differentiation-associated gene 5 (MDA5) DM; and b. anti-small ubiquitin-like modifier activating enzyme (SAE) DM.
Anti-Mi-2 is associated with the classic DM phenotype with high creatinine kinase levels, good response to treatment and good prognosis, although relapses often occur. ILD is rare in anti-Mi-2 DM.
Patients with anti-MDA5 DM typically have mild muscle disease or no muscle disease (ADM). Anti-MDA5 DM is strongly associated with ILD in most regions and ethnicities108,117. Rapidly progressive types of ILD with poor prognosis tend to occur especially in Asia. Patients often have characteristic cutaneous manifestations, including palmar papules and deep ulcerations over joints. Arthritis is also common.
Anti-TIF1 DM is characterized by extensive rash and relatively mild myositis, whereas dysphagia is frequent and sometimes severe. Anti-TIF1 DM is associated with a high risk of malignancy109,118,119. Patients with anti-NXP2 DM tend to have prominent muscle disease. By contrast, cutaneous manifestations are relatively modest, and pathognomonic rash may be absent in some patients (DM sine dermatitis). Patients with anti-NXP2 DM have an increased risk of calcinosis. Subcutaneous edema is also characteristic. Anti-NXP2 DM has also been reported to have a high risk of malignancy109,118,119, although this remains controversial.
In patients with anti-SAE DM, skin manifestations usually precede muscle manifestations120. Rash may become extensive and dysphagia is common120. Patients may have mild ILD121,122. An association with malignancies has also been reported122.
Antisynthetase syndrome
Antisynthetase autoantibodies bind to and inhibit the function of aminoacyl-tRNA synthetases1. To date, eight autoantibodies have been identified in patients with ASyS (Table 1). ASyS is a relatively homogeneous but multisystem disease (Figure 4)66 and is usually classified as a IIM, although myositis is not always present123. Instead, ILD, usually chronic but often progressive, is the most frequent manifestation. Patients with ASyS and anti-PL-7 or anti-PL-12 autoantibodies have a higher rate of ILD and higher mortality than those with anti-Jo1 autoantibodies124,125.
Figure 4. Clinical manifestations of anti-synthetase syndrome.

Antisynthetase syndrome is characterized by the presence of one of the eight known autoantibodies against aminoacyl transfer RNA (tRNA) synthetases. The frequency and combination of symptoms may vary according to the type of aminoacyl tRNA synthetase autoantibody. Some patients can have only lung disease with no muscle involvement.
Immune-mediated necrotising myopathy
IMNM is characterised by proximal muscle weakness, with symmetrical distribution, with extremely high muscle enzyme serum levels, a myopathic electromyography pattern, and muscle specimens showing necrosis or regeneration with minimal lymphocytic infiltrates (Figure 5)9. Patients only rarely have prominent systemic manifestations, such as rash, arthritis or ILD. IMNM is not a homogeneous entity and three subgroups are recognized: anti-HMGCR51, anti-SRP126 and autoantibody-negative IMNM, in which myopathy associated with malignancy or induced by drugs or toxins needs to be excluded127.
Figure 5. Clinical manifestations in immune-mediated necrotizing myopathy.

A majority of patients with immune mediated necrotizing myopathy (IMNM) have autoantibodies recognizing either 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) or the signal recognition particle (SRP). Severe muscle disease is the predominant manifestation of IMNM. Anti-SRP positive patients usually have more serious disease often accompanied with dysphagia and sometimes with cardiac involvement. In patients positive for anti-HMGCR necrotizing myopathy prevails.
Patients with anti-SRP myopathy tend to have more severe muscle disease and extramuscular manifestations, including cardiac involvement and dysphagia, than those with anti-HMGCR myopathy128.
Patients with anti-HMGCR myopathy have predominantly skeletal muscle disease with variable, but mostly severe muscle weakness, without other organ manifestations. HMGCR is the pharmacological target of statin medications and anti-HMGCR myopathy is associated with statin use, but can also be found in individuals without previous statin use128.
Autoantibody-negative IMNM remains poorly described but has been reported to be characterized by frequent occurrences of associated connective tissue disorders, especially systemic sclerosis, and substantially higher rates of extramuscular manifestations than seen in patients with seropositive IMNM129.
Inclusion body myositis
IBM is clinically characterised by asymmetrical weakness of both proximal and distal muscles which often includes the quadriceps and long finger flexors (Figure 6)130. IBM occurs mainly in individuals >50 years of age. Dysphagia occurs in >50% of patients, whereas other extramuscular manifestations are rare. Hallmarks of muscle histopathological findings include endomysial T cell infiltrates and vacuoles rimmed by membranous cytoplasmic material131. IBM can be associated with Sjogren’s syndrome and other connective tissue diseases24. The co-occurrence of IBM with sarcoid myopathy has also been reported132. IBM progresses slowly over decades and usually does not respond to immunosuppressive therapy.
Figure 6. Clinical manifestations of inclusion body myositis.

Inclusion body myositis (IBM) is characterized by slowly progressive muscle weakness affecting mainly quadriceps muscle and forearm muscles. Muscle involvement can be asymmetrical. The disease leads to significant muscle atrophy and severe disability. Dysphagia is present in more than 50% of patients.
Polymyositis
PM is defined by inflammatory disease in the proximal muscles, endomysial infiltrates of CD4+ and CD8+ T-cell on muscle specimens and absence of skin involvement. However, PM is now considered a rare form of IIM8,10,133. Most patients previously diagnosed with PM are currently classified as having ASyS, IMNM, IBM or overlap myositis and, on the basis of this reclassification, no specific autoantibody is associated with PM.
Overlap myositis
Myositis can occur together with other connective tissue diseases, such as SLE, systemic sclerosis, Sjogren’s syndrome, or rheumatoid arthritis8,134,135. Overlap myositis is a heterogeneous subgroup, as clinical and histological findings vary. Autoantibodies detected in overlap myositis include anti-U1RNP, anti-Ku, anti-PM/Scl, anti-RuvBL1 and anti-RuvBL2136, anti-Ro/SS-A and anti-La/SS-B autoantibodies. Patients who have features of scleroderma and myositis are referred to as having scleromyositis. Head drop and distal weakness are common and an increased rate of extramuscular manifestations are found in those without scleroderma-associated antibodies. These patients have variable histopathological findings including necrotizing myopathy137.
Clinical presentation
The most typical symptom to suspect IIM is proximal muscle weakness with a symmetrical distribution, sometimes accompanied by pain. Muscles in the upper and lower extremities can be involved with hip and thigh muscles being affected most often. Neck muscles, most typically flexors but sometimes also extensors, can be involved. The onset can be acute, subacute or chronic. Patients complain of difficulty getting up from a seated position, climbing steps, or raising their arms or head. Serum levels of creatine kinase and aldolase, as well as lactate dehydrogenase and aspartate transaminase, are often elevated. Abnormal electrical activities of muscle fibres, signs of muscle edema, or immune-mediated changes in histopathological specimens can be detected by electromyography, imaging and muscle biopsy. In DM and ASyS, muscle involvement can be lacking. Many patients can have extramuscular symptoms as part of the presenting symptoms, particularly skin rash, (poly)arthritis and ILD.
Extramuscular manifestations
Skin manifestations
In DM, pathognomonic or highly characteristic skin lesions include periorbital violaceous erythema often associated with oedema (heliotrope rash), erythematous to violaceous papules (Gottron’s papules) and macules (Gottron’s sign) overlying the extensor surfaces of joints116. Moreover, periungual nailfold erythema with cuticular punctate haemorrhage is frequently observed. Erythema over lower anterior neck and upper chest (V sign) and over posterior shoulders, neck, and upper back (shawl sign) are also characteristic. These skin changes can result in poikiloderma, which involves a concurrence of hyperpigmentation, hypopigmentation, telangiectasia and superficial atrophy. Pruritus is also an important symptom. Patients with anti-MDA5 DM frequently develop ulcerations over the joints and palmar papules138,139. Anti-TIF1 DM and anti-SAE DM are associated with extensive skin involvement122,140. Nonpruritic, hyperkeratotic eruptions on the lateral surfaces of the digits called Mechanic’s hands are the hallmark skin feature in patients with ASyS but are also seen in those with DM and in patients with anti-PM/Scl antibodies. In patients with DM, and more frequently in juvenile DM, abnormal depositions of insoluble calcium salts can be found in skin and subcutaneous tissue141. Calcinosis occurs more frequently in sites subjected to micro trauma and is most frequently present in anti-NXP2 DM.
Histologic findings in DM skin specimens include interface dermatitis with dyskeratosis, dermal mucin deposition, perivascular inflammatory infiltrates, and vascular dilatation and/or damage116, although these findings are indistinguishable from those in SLE. A skin biopsy is recommended in patients without muscle involvement (CADM)100,116.
Pulmonary involvement
ILD is found in up to 78% of patients with IIM, typically in patients with ASyS and anti-MDA5 DM142,143. The most clinically alarming form is acute rapidly progressive ILD (RP-ILD) associated with anti-MDA5 autoantibodies, which can develop into adult respiratory distress syndrome and respiratory failure and has a poor prognosis. High-resolution CT shows perilobular opacities that progress rapidly to ground glass consolidations and, in later stages, traction bronchiectasis. Consolidation in the lower lung zones of high-resolution CT findings is particularly related to RP-ILD144. Some patients with ASyS who have the high-resolution CT pattern combining organizing pneumonia plus nonspecific interstitial pneumonia can develop RP-ILD but, usually, ILD is less severe and RP-ILD considerably less frequent in patients with ASyS than in those positive for anti-MDA5 (DM). In general, a nonspecific interstitial pneumonia pattern on CT is a favourable prognostic factor for patients with IIM. Most patients with ILD in IIM have subacute or chronic courses. ILD may even be asymptomatic or occult and can have a reasonable response to treatment, which also includes ILD in patients with anti-PM/Scl or anti-SAE autoantibodies.
Cardiac involvement
Heart involvement comprises myocarditis, inflammatory infiltration in the cardiac conduction system and replacement fibrosis145. Only ~10% of patients with IIM have clinically overt cardiac disease; subclinical involvement is more frequent (up to 75% of patients). Initial description that cardiac involvement was more common in patients positive for anti-SRP autoantibodies is now controversial, but those with overlap myositis and systemic sclerosis are more likely to be affected21. Cardiac MRI is a sensitive technique that can detect inflammation in the myocardium or irreparable changes very early, even when no symptoms are present146. Serum cardiac troponin I is a sensitive and specific measure of cardiac involvement, whereas troponin T levels may be elevated also due to inflammation in non-cardiac striated muscles147.
Oesophageal involvement
It can be argued whether dysphagia should be regarded as a muscle manifestation or an extramuscular manifestation. Dysphagia occurs in up to 60% of patients with myositis and is especially prevalent in IBM21,148. It may be caused by muscle weakness in the pharyngeal muscles or upper portion of oesophagus and may occasionally be very severe preventing the ability to swallow liquids. Dry mouth as in patients with secondary Sjögren syndrome may also contribute to dysphagia. Dysphagia is a risk factor for aspiration pneumonia.
Joint involvement.
Arthritis is commonly found in patients with IIM, particularly in those with ASyS21,149. In some patients it may be a presenting symptom and, because it is usually symmetric and affects small joints of the hands, it can initially be misdiagnosed as rheumatoid arthritis. In some cases, arthritis may be a dominant symptom in ASyS.
Diagnosis
Diagnosis of myositis is made when typical clinical and laboratory parameters are present and other possible causes are excluded. However, formal diagnostic criteria do not exist and classification criteria are used as a guidance instead. The most recent classification criteria developed by the European League Against Rheumatism (EULAR) and the American College of Rheumatology (ACR)100 published in 2017 can aid in identifying the major myositis subgroups: DM, PM, ADM, juvenile myositis and IBM. The probability of the disease can be evaluated using an internet-based calculator [http://www.imm.ki.se/biostatistics/calculators/iim/]. Muscle performance is usually assessed via muscle weakness using manual muscle testing or via muscle endurance using a functional index150.
As IIM are heterogeneous conditions, they are diagnosed into subgroups on the combined basis of clinical phenotypes, autoantibody profile and histopathological findings (Table 2, Figure 7)1,7,8. For the new subgroup IMNM, classification criteria have been proposed127. ASyS is usually diagnosed when one of the eight antisynthetase autoantibodies is detected together with a single or a combination of clinical features: myositis, arthritis, ILD, mechanic’s hands or Raynaud’s phenomenon151. No established diagnostic or classification criteria for ASyS are available yet. The European Neuromuscular Centre (ENMC) IBM research diagnostic criteria were proposed in 2011 and recognize clinicopathologically-defined IBM, as well as clinically defined IBM and probable IBM152. Diagnosis is based on the variable presence of combined or separate knee extension weakness more than hip flexion weakness, and finger flexion weakness more than shoulder abduction weakness, together with age and duration limits and histopathological features in muscle tissue. The newest IBM diagnostic criteria use evaluation of finger flexor or quadriceps, weakness, endomysial inflammation, and either invasion of nonnecrotic muscle fibres or presence of rimmed vacuoles153.
Table 2.
Histopathological findings in different IIM subtypes
| Myositis subtype | Muscle fibres and tissue | Inflammatory cell infiltrates | MHC-I expression | MAC depositions | Other specific findings |
|---|---|---|---|---|---|
| Dermatomyositis | Perifascicular atrophy. Reduced number of capillaries | Perivascular, perimysial, T cells, B cells, macrophages, plasmacytoid dendritic cells | Perifascicular fibres | Small blood vessels | Sarcoplasmic MxA expression |
| Polymyositis | Degeneration, necrosis, regeneration | Endomysial inflammatory infiltrate with T cells often surrounding and/or invading non-necrotic muscle fibres | Diffuse distribution | No specific findings | Absence of rimmed vacuoles |
| Immune-mediated necrotizing myopathy | Necrotic fibres with scattered distribution, different stages of necrosis and myophagocytosis and regeneration, endomysial fibrosis and proliferation | Macrophage predominant, paucilymphocytic infiltrates | Diffuse distribution, sometimes only faint | Sarcolemmal and/or on small blood vessels | No specific findings |
| Antisynthetase syndrome | Oedematous and/or fragmented perimysium that stains with alkaline phosphatase, sometimes perifascicular myofiber necrosis | Scattered perimysial CD68+, CD4+, CD8+ cells | Perifascicular predominance | Fibres adjacent to the perimysium, sarcolemmal on non-necrotic fibres | Myonuclear actin filament inclusions in electron microscopy, absence of MxA expression |
| Inclusion body myositis | Rimmed vacuoles, ragged red fibres, cytochrome oxidase-negative fibres, groups of atrophic fibres | Endomysial inflammatory infiltrate with mainly CD8+ cells surrounding and/or invading non-necrotic muscle fibres | Diffuse distribution | No specific findings | TDP-43, p62 aggregates, 15–18 nm filaments in electron microscopy |
TDP-43 - TAR DNA binding protein-43, MxA - Myxovirus-resistance protein A, p62 – ubiquitin binding protein p62, IIM - idiopathic inflammatory myopathy.
Figure 7. Typical histopathological changes in IIM subgroups.

Dermatomyositis: a) perifascicular atrophy and perivascular inflammatory infiltrate, b) inflammatory infiltrate with CD4+ cells, c) inflammatory infiltrate with B cells, d) MHC I expression on muscle fibres, particularly in perifascicular distribution, e) membrane attack complex depositions in capillaries.
Antisynthetase syndrome: f) perifascicular myofiber necrosis, perifascicular atrophy, perimysial fragmentation.
Immune mediated necrotizing myopathy: g) necrotic muscle fibres, h) MHC I hyperexpression on sarcolemma of muscle fibres, i) membrane attack complex deposition on capillaries and sarcolemma of muscle fibres.
Inclusion body myositis: j) lymphocytic and macrophage aggregates in endomysium and invasion into muscle fibres, k) CD8+ positive inflammatory infiltrate, l) rimmed vacuoles (arrows), m) p62 staining (arrows).
Polymyositis: n4) endomysial infiltrate surrounding and invading non-necrotic muscle fibres, o) CD8+ cells surrounding and invading non-necrotic muscle fibres.
Original magnification 40x (1, 6) and 200x (2–5, 7–15).
Laboratory testing
Muscle-derived enzymes in serum are elevated in most patients with active muscle disease. Creatinine kinase is the most sensitive marker and has diagnostic and monitoring utility, although its levels can be normal in some patients with active DM, ASyS or IBM. Rarely, serum aldolase levels are selectively increased without creatinine kinase level elevation154. Indirect immunofluorescence using HEp-2 cells as the substrate, the standard screening method for antinuclear antibodies, has limited value in the screening of MSAs and MAAs, as many MSAs and MAAs target cytoplasmic antigens yield weak or negative nuclear staining with indistinct pattern1,155. Immunoprecipitation is the gold standard technique to identify most MSAs and MAAs but is time-consuming and unsuitable for routine testing. Enzyme-linked immunosorbent assays and line blot assays for several MSAs and MAAs are becoming available, although their reliability needs further validation (except for anti-Jo1 antibody).
Histology
Muscle biopsy is an important tool to diagnose IIM, to confirm signs of inflammation, to identify signs of the different subtypes of IIM and, importantly, to exclude other myopathies. The main elements in the histopathological investigations of major subtypes are summarized in Table 2 and Figure 7156–159. Some findings are more common for each subtype and may enable diagnosis even in patients with uncharacteristic clinical manifestations. Some features may be nonspecific and found in various frequencies in different myositis subtypes as well as in other myopathies, for example necrotic or regenerating muscle fibres. Interpretation of pathological findings is sometimes difficult and there is interrater variability in diagnosing individual muscle specimen abnormalities160.
Differential diagnosis
Several different conditions may mimic myositis. Muscle weakness, the main clinical presentation of myositis, is a non-specific symptom and misdiagnosis is possible especially when typical skin manifestations and/or myositis-specific antibodies are absent. Most commonly, the difficulties are encountered in patients with muscular dystrophies, metabolic myopathies, mitochondrial myopathies, endocrine myopathies or toxic myopathies161,162. In particular, differentiating slowly progressive onset of myositis from muscle dystrophies is important. Careful medical history, physical examination, electromyography, laboratory and muscle biopsy analyses, as well as genetic testing are necessary to avoid incorrect diagnosis. Several warning signs exist that point to other conditions and their presence should increase the attention and prompt diagnosis verification, such as family history of muscle weakness, slowly progressive evolution of weakness over years, and asymmetric and considerable distal weakness161–164.
Cancer association and screening
All subgroups of adult-onset myositis except ASyS and IBM have a 2-fold to 7-fold increased risk of malignancy compared with the general population103–105. The risk of malignancy is particularly high in patients with DM, especially those with anti-TIF1118,165 or anti-NXP2119,166 autoantibodies, as well as in those with autoantibody-negative IMNM167. The risk is highest within 1 year before and after myositis diagnosis but remains elevated for an extended time period, up until 10 years in some subgroups.
Various types of malignancy can occur in patients with IIM. The most common are the same as seen in the general population: cancer of the lung, breast and ovary, as well as lymphoma168. Cancer screening includes physical examination, investigation of the breast, pelvis, testes, and prostate, CT of the chest, abdomen, and pelvis, ultrasonography of the abdomen, gastroscopy and colonoscopy, and, in women, pelvic ultrasonography and mammography. In some patients, PET imaging is useful, as it shows comparable performance to broad conventional screening, which requires multiple tests169.
Prevention
There are no scientific studies that define a clear approach to prevent the development of IIM. Some data suggest that smoking is associated with IIM in patients who are either anti-Jo1 antibody and/or HLA-DRB1*03 positive37. However, when disease is present, some steps can be taken to prevent flares of disease manifestations. In patients with anti-HMGCR IMNM, statin medications and statin-containing foods, such as oyster mushrooms, red yeast rice or Pu-Erh tea, should be avoided170. Few data are available to guide treatment with statins and other lipid-lowering agents in other myositis subtypes; however, most experts favour their use171. Additionally, immunomodulating drugs, such as immune checkpoint inhibitors, tumor necrosis factor alpha antagonists, and type I interferons, can cause myositis, but there is no recommendation not to use them when indicated, for example due to a malignancy172. Patients with rashes need to protect themselves from ultraviolet radiation173.
Management
Management of myositis is challenging owing to the rarity and heterogeneous nature of this disease. Complexity also arises from variable presentation and disease courses, as well as its multi-organ and systemic characteristics. No comprehensive consensus-driven guidelines or FDA-approved proven therapies exist, mostly due to lack of robust clinical evidence in form of clinical trials in myositis. Fortunately, an increasing number of novel therapeutics are currently undergoing phase 2 or 3 clinical trials, using validated disease classification and outcome measures. The goal of the therapy is to improve patient symptoms, so that patientś functional levels return to near baseline as well as non-muscular symptoms are manageable and do not interfere with activities of daily living. Evidence is from retrospective cohort studies for all the treatments except for intravenous immunoglobulin (IVIg), rituximab and exercise for which randomized controlled studies exist.
Glucocorticoids
Despite the lack of controlled clinical trials with proven efficacy, oral glucocorticoids are the first-line or initial treatment in most patients with PM, DM, ASyS or IMNM, especially for those with substantial muscle weakness and ILD (Figure 8)174. In patients with severe forms of myositis or extra-muscular manifestation, such as ILD, pulse glucocorticoid therapy with intravenous methyl prednisone is given. The chronic use of glucocorticoids should be limited, primarily because of the high rate of adverse effects and long-term complications. Clinical studies have shown normalization of muscle enzyme levels and improvement in muscle strength, but glucocorticoids alone are associated with high flare rates, low remission rates and lack of full recovery of muscle strength or function175.
Figure 8. Common pharmacological and other therapies for idiopathic inflammatory myopathies (IIM) except inclusion body myositis (IBM).

Note: IVIg: Intravenous Immunoglobulins; ILD: Interstitial Lung Disease; ASyS: Anti-synthetase syndrome.
Adrenocorticotropic hormone
Repository corticotropin injection (RCI) is a formulation containing adrenocorticotropic hormone and other pro-opiomelanocortin peptides that stimulates melanocortin receptors, leading to both glucocorticoid-dependent as well as glucocorticoid-independent mechanisms of anti-inflammatory action176–178. In an open-label trial of RCI biweekly for 24 weeks in 10 patients with refractory PM or DM, 70% of the patients met the primary efficacy end point of definition of improvement proposed by the International Myositis Assessment and Clinical Studies Group (IMACS)179,180. RCI is an FDA-approved therapy for PM and DM; however, it is only used as a third-line or later therapy owing to the lack of data from randomized controlled trials and high cost in the USA. Furthermore, the medication is not currently approved or available outside of the USA.
Traditional immunosuppressants
Traditional immunosuppressive drugs are commonly used together with glucocorticoids; however, the evidence for their efficacy in IIM is weak181. Most clinicians would start methotrexate or azathioprine in combination with glucocorticoids as an initial therapy in myositis unless contraindications exist (Figure 8). Several retrospective studies and one prospective open-label controlled study support their use in PM, DM and juvenile dermatomyositis for muscle and skin disease175,182–186. Azathioprine is preferred in patients with alcohol use, liver disease or concomitant ILD, and is relatively safe in pregnancy. The use of mycophenolate in myositis has been increasing over the years with more retrospective and prospective studies supporting its efficacy in this disease as well as in associated conditions, such as ILD and refractory DM rashes187–191. It is typically used as a second-line agent except in patients with moderate to severe myositis associated with ILD, in whom it can be the first-line agent192–195. Cyclosporine and tacrolimus are both calcineurin inhibitors that ultimately lead to inhibition of T cell activation. Both are used as second-line agents in patients with refractory myositis with either muscle weakness or associated ILD (Figure 9).196–200. Toxic effects of calcineurin inhibitors require an aggressive monitoring plan including checking trough blood levels periodically. Use of cyclophosphamide is limited to severe refractory muscle weakness, rapidly progressive ILD or systemic vasculitis associated with PM or DM201. Cyclophosphamide is sometimes used in combination with rituximab as reported in a cohort of patients with antisynthetase autoantibody positivity and severe ILD, such as in patients positive for anti-MDA5 autoantibodies; however, it may be associated with a high risk of infections202.
Figure 9. Treatment considerations in patients with refractory myositis based on the clinico-serologic presentation.

Note: IVIg: Intravenous Immunoglobulins; ILD: Interstitial Lung Disease; DM: Dermatomyositis; JDM: Juvenile Dermatomyositis; Anti-SRP: Anti-Signal Recognition Particle; Anti-HMGCR: 3-hydroxy-3-methylglutaryl CoA reductase
Immunoglobulins
IVIg is a medication with anti-inflammatory and immuno-modulating mechanisms without direct immunosuppressive actions. Currently, IVIg is used in IIM as a second-line or third-line treatment either as combination with or following failure of glucocorticoids and/or other immunosuppressive drugs. It is also increasingly used as first-line treatment in myositis, especially in IMNM203. The efficacy and safety of IVIg was first reported in a double-blind, crossover, controlled trial in 15 patients with refractory DM204. An important large randomized, placebo-controlled phase 3 study, recently published in abstract form205, confirmed the efficacy and safety of IVIg in refractory DM with muscle weakness and rash206. The trial has led to FDA and EMA approval for use of IVIg in adult dermatomyositis. A subcutaneous form of gamma immunoglobulin administered through a pump is increasingly being used in myositis207. IVIg has various advantages, as it can be given concomitantly with other immunosuppressive drugs or in the settings of pregnancy, infection or malignancy. However, the treatment is expensive, can be logistically challenging to administer and has limited availability in some countries.
Biologics and emerging drugs
Rituximab
Rituximab depletes CD20-positive B cells that are likely to be involved in the pathogenesis of some myositis subgroups. Several open-label studies have reported safety and efficacy in patients with severe and refractory myositis, including the subset of IMNM patients with anti-SRP autoantibodies, a poor prognostic marker208–210. A large randomized, double-blind, placebo-controlled clinical trial (Rituximab in Myositis (RIM) Trial) in DM, PM and juvenile DM failed to meet its primary or secondary end points211. However, 83% of patients who were refractory to multiple immunosuppressive agents showed clinical improvement in muscle and skin disease, as well as glucocorticoid reduction within 1 year. Furthermore, rituximab was considered relatively safe and well-tolerated in this patient population. The presence of anti-synthetase autoantibodies, anti-Mi2 autoantibodies, having juvenile DM, and low disease damage at trial entry were strong predictors of a beneficial response to rituximab212. Rituximab is increasingly being used in myositis-associated ILD, especially ASyS, with positive outcomes in retrospective and prospective studies; however, no randomized controlled trials have been performed to date.
Anti-TNF agents
Although tumour necrosis factor (TNF) has been implicated in the pathogenesis of myositis, the efficacy of anti-TNF agents (such as etanercept and infliximab) is somewhat disappointing213–217. A case series of five patients with DM using etanercept, an open label clinical trial of 13 patients with refractory myositis using infliximab and a randomized double-blind placebo-controlled trial in 12 patients with PM or DM using infliximab showed discouraging results215–217. However, a randomized double-blind placebo-controlled trial in patients with DM showed positive results with etanercept214. Currently, anti-TNF treatment is not typically recommended or considered in patients with adult myositis, although it may have a role in the treatment of calcinosis in juvenile dermatomyositis218.
Special therapeutic considerations
Most studies on myositis were performed in PM or DM; therefore, most data relate to these two clinical subsets in which muscle and/or skin is often the primary organ involved. However, slightly different treatment approaches may be beneficial in the new clinical and serological subtypes that have been identified in the past two decades. Furthermore, given the heterogeneous presentation of the disease, treatment often has to be specific to organ manifestations or associated conditions, such as ILD or dysphagia. Other treatments are being explored that may eventually be helpful. Three major randomized clinical trials have been completed with only top line or abstract results currently available. The trial of IVIg in adult dermatomyositis has a positive result leading to FDA and EMA approval205,206. Unfortunately, two other trials, abatacept in adult IIM, which included patients with DM, PM or IMNM, as well as lenabasum in adult DM, failed to reach their primary end point or key secondary end points.
Immune-mediated necrotizing myopathy
In patients with IMNM, high doses of glucocorticoids have traditionally been used as induction therapy together with methotrexate or azathioprine. However, several case series have suggested a more aggressive approach with the use of rituximab and/or IVIg in these patients, especially owing to the often refractory nature of disease, as well as risk of early muscle atrophy in patients who are anti-SRP positive (Figure 9). In addition, the risk of high doses of prolonged glucocorticoid treatment in elderly patients with anti-HMGCR autoantibodies needs to be considered127,208,219,220. Some reports suggest that the use of high doses of IVIg as monotherapy without glucocorticoids may be efficacious in patients with anti-HMGCR autoantibodies221.
ASyS-associated interstitial lung disease
Often the predominant and refractory condition in ASyS is ILD (Figure 9). Moreover, ILD is the major cause of mortality and morbidity in myositis in general and in ASyS specifically. Mycophenolate is preferentially used in patients with moderate to severe ILD as first-line therapy, including patients with ASyS192. Anti-T cell therapies, such as tacrolimus or cyclosporine, have been used with good success in patients with ASyS and, specifically, with ILD. In a small study including 13 patients with ILD who were positive for anti-synthetase autoantibodies, tacrolimus resulted in improvements in all pulmonary function testing parameters200. Furthermore, several studies in Asia have suggested survival benefits with the use of a combination of glucocorticoids with either tacrolimus or cyclosporine198,222. Rituximab has increasingly been used for myositis-associated ILD, especially in patients with ASyS223–225. A retrospective study of 24 patients with ASyS and severe ILD showed clinically significant improvement in pulmonary function after rituximab202. Similarly, 16 of 17 patients with anti-Jo1 autoantibodies treated with rituximab showed more rapid and marked responses compared with conventional immunosuppressive agents225.
CADM
CADM is a unique clinical phenotype highly associated with presence of anti-MDA5 autoantibodies and characterized by severe cutaneous rashes and frequently with RP-ILD but without objective muscle weakness. These patients often have high mortality and need aggressive combination immunosuppression as first-line therapy, which includes glucocorticoids in combination with calcineurin inhibitors as well as cyclophosphamide (Figure 9)226. Rituximab with or without cyclophosphamide is often used early in patients with worsening respiratory status227. A prospective, open-label study from China demonstrated efficacy and a survival benefit of glucocorticoids with tofacitinib compared with historical controls63.
Inclusion body myositis
Unlike DM and PM, IBM is typically refractory to immunotherapy. Although glucocorticoids and other immunosuppressive therapies have not been tested in randomized controlled trials, the general consensus is that they are not efficacious, even though glucocorticoids may improve muscle enzyme levels in the short term and may improve dysphagia in some. IVIg might slow disease progression but its long-term effectiveness remains unclear. Methotrexate, which is commonly used in other forms of myositis, failed to slow the progression of muscle weakness in a small randomized double-blind placebo-controlled study in patients with IBM228. Intravenous bimagrumab, an anti-activin type II receptor antibody, was evaluated in the largest phase 3 clinical trial in IBM to date and failed to meet its primary end point of 6-min walk distance at 52 weeks229. Pilot studies of arimoclomol, which co-induces the heat shock response by prolonging activation of heat shock factor-1 and may promote normalization of protein handling within muscle, and rapamycin (sirolimus), which inhibits protein kinase that regulates several intracellular processes including survival, protein synthesis and autophagy, have shown encouraging results, but these have not been confirmed. Unfortunately, a phase 2/3 clinical trial of arimoclomol in IBM failed to meet its primary end point; however, a phase 3 clinical trial of rapamycin is ongoing230,231. Exercise is currently the only treatment, which has consistently shown a varied degree of benefit in IBM, although the optimal type of exercise programme is yet to be determined.
Dysphagia
Proximal dysphagia can be a severe and life-threatening manifestation of myositis, and patients with refractory dysphagia should undergo full investigation to determine the cause of dysphagia and appropriate management. Although most patients would respond to conventional doses of glucocorticoids and immunosuppressive agents, some studies have suggested the use of IVIg for refractory dysphagia232.
Calcinosis
Calcinosis is the most severe and challenging presentation of DM. Control of active disease with traditional immunosuppressive agents is necessary but is often not sufficient for the treatment of calcinosis. Bisphosphonates, diltiazem, rituximab, IVIg and sodium thiosulfate have been tried with some success in patients with calcinosis233,234.
Exercise and Physical Therapy
Emerging data support the safety and effectiveness of exercise in adults with IIM. Muscle function as well as quality of life improves after exercise regimens235. Exercise programmes to improve strength and function have been shown to activate molecular pathways regulating aerobic capacity, capillary growth and muscle remodelling while concomitantly mitigating the inflammatory response in patients’ muscles236,237. Thus, exercise, especially directed by physical therapists, should be recommended early with gradual progression based on the individual response. Most patients tolerate exercise without adverse effects. Physical exercise should be considered as standard treatment for adult and juvenile myositis.
Outcome measures
International networks of myositis researchers called IMACS developed and validated standardized measure to assess disease activity, known as core set measures, for use in clinical trials. These are also useful tools in clinical practice. The core set includes physician and patient global disease activity as well as extra-muscular disease activity on a 10 cm visual analogue scale as well as muscle enzyme analyses, manual muscle testing and patient-reported health assessment questionnaires238. Composite response criteria using weighted changes in these core set measures have been developed and validated for adult and juvenile myositis patients239. There are several additional measures of muscle strength (such as handheld dynamometry) and function (functional index, timed up and go or sit to stand), as well as assessment of other organ manifestations, such as ILD (lung function study) or DM rash (Cutaneous Disease Area and Severity Index (CDASI)), which are increasingly being used as secondary outcome measures. Furthermore, various patient-reported outcome measures, especially well validated Patient-Reported Outcomes Measurement Information System (PROMIS) instruments, are being evaluated for use in patients with myositis. MRI and ultrasonography of muscle as well as physical activity monitors are promising tools that can provide much needed objective outcome measures in myositis.
Quality of life
It has been increasingly highlighted that clinical trials and observational studies should include an assessment of the outcomes that matter most to patients240. Myositis unquestionably affects patients’ quality of life (QoL) and may lead to long-term disability for some241,242; however, assessing life quality in a systematic, validated and quantifiable fashion in IIM is challenging. As the disease may affect organs beyond the muscle, health-related quality of life (HRQoL) indices that encompass the systemic nature of the disease are desirable but remain largely unexplored and are not validated.
Most instruments evaluated in IIM have been either generic or adopted from use in other rheumatic or neuromuscular diseases. Only the Myositis Activities Profile (MAP) was created specifically for adult PM and DM, and the McMaster-Toronto Arthritis Patient Preference Disability Questionnaire (MACTAR) was adapted for these two diagnoses132,243. In addition, the IBM Functional Rating Scale (IBMFRS) was created and validated for patients with IBM244. However, the indices were assembled from the provider viewpoint with little patient involvement in their construction or improvement, and review of the survey instrument by myositis patients revealed that they deemed some questions vague or irrelevant245. Patient engagement via focus groups and subsequent Delphi processes has now demonstrated the need to include pain and fatigue in addition to physical function246.
In addition to physical examination and laboratory measures, the IMACS core set of measures includes patient-reported outcome measures (PROMS) such as the patient/parent report of disease activity. IMACS also recommends including a measure of HRQoL, such as the Short Form-36 survey (SF-36) in clinical studies247. Disease activity and damage core set measures have been validated, but no specific PROM has been developed under IMACS auspices. Similarly, the ACR–EULAR criteria for clinical response in DM and PM were formulated using data-driven methods in the absence of patient input and include only a generic patient visual analog scale248. The Outcome Measures in Rheumatology (OMERACT) framework provides a complementary, non-overlapping approach to bringing the major stakeholders together within a ‘patient research partner’ framework to derive PROMS249.
Until myositis-specific instruments will be developed, generic instruments to evaluate HRQoL may be used; however, ongoing future efforts must focus on standardizing use of PROMs for research and clinical practice to avoid ambiguity when interpreting data between different cohorts and across trials. International organizations will need to take a central role in the promotion and use of high-quality, validated PROMs in IIM research. This approach is imperative for the aggregation of collected data, for example for meta-analysis, which is necessary when studying a collection of rare diseases250.
PROMs, when used in conjunction with clinical measures, add an important and too often dismissed patient’s perspective of the disease. PROMs implementation can improve the patient–physician relationship, increasing physician satisfaction and improving patient care by furthering shared decision making251. Given that the beliefs and perspectives of the patient and physician may not be comparable, validation and adoption of myositis-specific HRQoL instruments derived through the patient–provider partnership are fundamental for both high-quality care and research.
Outlook
Since the early 2000s, major advances have been made in understanding the pathophysiology of IIM and its subgroups. This has been possible by identification of myositis-specific autoantibodies and due to advances in molecular biology. The common occurrence of autoantibodies in IIM emphasises the influence of the immune system in the pathophysiology of these diseases. The phenotypes of inflammatory cells and their products in the most often affected organs muscle and skin, but also the lungs in the newly identified subsets of IIM defined by MSAs, have been reported. However, several important gaps remain in our understanding of IIM that need to be addressed in future research to improve treatment and outcome. One important point for future research and clinical management is a nomenclature of IIM and its subgroups that is widely accepted. This could be a task for the international networks on myositis, such as the IMACS network.
A major challenge in the clinical management of myositis is that we do not know which patient will respond to which treatment. Currently used immunosuppressive treatment has broad effects on the immune system and adverse effects are common. Furthermore, in one cohort of patients with PM or DM, only 20% obtained drug-free remission45. The chronic inflammatory process often leads to irreversible tissue damage. Thus, the need for new therapies is high. For more specific targeted therapies, more detailed information on molecular pathways, including the specificities of the targets of the immune reaction, is required. Furthermore, biomarkers for treatment response and prognosis are lacking. Availability of data to predict treatment response, prognosis and outcome to customize treatment for personalized medicine approaches would be a major advancement. To avoid chronicity and tissue damage, patients probably need to be identified earlier and treated more aggressively. Earlier diagnosis may be accomplished through increased physician awareness of IIM and its clinical phenotypes and through referral to specialist clinics for diagnostic evaluation when IIM is suspected. When diagnosis is confirmed, intense and specific treatment should be initiated early.
Treatment can be improved through several approaches. A better understanding of the pathophysiology of IIM is an essential aspect. Elucidating the immune specificities of the IIM subtypes is critical, as the target of the immune system is likely to vary between the subtypes with different clinical features and different MSAs associated with different HLA DRB1 alleles. Knowing the site where the immune reaction leading to the autoimmune disease takes place is also important to understand the molecular pathways leading to chronic disease and to stop the disease process early. Here, both the skin and mucosal sites in the airways or gut are of interest as niches for possible initiating immune reactions. An interesting IIM subgroup in this context are patients with anti-synthetase autoantibodies with a high frequency of ILD and myositis, in particular patients with anti-Jo1 autoantibodies that have a strong association with the HLA 8.1 ancestral haplotype, where smoking is a risk factor for disease and lung disease is often an early manifestation37. In this subgroup, antigen-reactive, proinflammatory CD4+ T cells targeting fragments and peptides of histidyl-tRNA synthetase, the target of anti-Jo1 autoantibodies, have been identified in peripheral blood and with highly reactive CD4+T cells in the bronchoalveolar lavage69. Identification of antigen-specific peptides that activate T cells is an important step to develop immune tolerance therapy, which could possibly ‘re-educate’ the immune system to cure the patients. In patients with IIM, several specific autoantigens targeted by autoantibodies and associated with distinct clinical phenotypes and genotypes are now known. These autoantigens are potential candidates to identify peptides that induce T cell reactivity. Identification of autoantigen-specific T cells will be an important step to develop immune tolerance therapy and should have high priority in research in the coming 5–10 years. Identification of autoantigen-specific T cells by T cell tetramers would be an excellent tool to monitor future specific immune therapies.
Identification of biomarkers for treatment response and prognosis is another important approach to improve treatment. This could be achieved through observational studies using large cohorts of well-characterized patients with longitudinal data. As IIM are rare disorders with several clinical and serological subgroups, international multicenter collaborations are needed, which could be facilitated via international web-based registries using validated classification criteria and outcome measures. Some major steps have been taken via the IMACS network through which tools to score disease activity and damage, as well as Response criteria, have been developed239. The EULAR–ACR classification criteria for IIM100 is also an important step towards harmonization of patient cohorts for multicenter studies.
Muscle biopsies are still important for the diagnosis of IIM and its subgroups and to exclude mimicking conditions. Muscle biopsies, including biopsies from longitudinal studies, are also important to achieve an improved understanding of IIM pathophysiology by careful investigations of molecular expression in clinically well-defined patients. Whether muscle specimen features will have a role as biomarkers in adults with IIM is yet to be understood. However, both in the clinical and the research setting, it is essential to standardize muscle biopsy assessments as reflected by considerable variations in the evaluation of biopsy samples among experienced muscle pathologists160. An important future project would be to advance the area of standardization of muscle biopsy assessment, including an international panel of muscle pathologists to review muscle biopsies together as is now often a routine in cancer diagnosis.
The performance of commercially available autoantibody tests is another area in need of evaluation and standardization. Discovery of new MSAs has enabled identification of new IIM subgroups, such as IMNM associated with anti-HMGCR or anti-SRP autoantibodies, and that anti-TIF1 gamma autoantibodies are strongly associated with risk of cancer in adult patients with DM. These MSAs are examples of biomarkers that could predict prognosis. However, the sensitivity and specificity of the different autoantibody assays vary for anti-TIF1 gamma autoantibodies and several other MSAs. Thus, validation of commercially available autoantibody assays in large cohorts with longitudinal data is required, which should have a high priority for the international myositis community in the coming years.
Acknowledgements
I.E.L. was funded by the Swedish Research council, the Swedish Rheumatism Association, King Gustav V 80 Year Foundation and Stockholm County Council (ALF Project). J.V. acknowledges the support from the Czech Ministry of Health - Conceptual Development of Research Organization 00023728 (Institute of Rheumatology). M.H. was funded by the Swedish Research Council, The Marianne and Marcus Wallenberg Foundation, The Professor Nanna Svartz Foundation and the King Gustaf V:s 80-year Foundation. A.L.M. and F.W.M. were supported in part by the Intramural Research Program of the NIH, National Institute of Arthritis and Musculoskeletal and Skin Diseases and/or the National Institute of Environmental Health Sciences.
Competing interests
I.E.L. has received research funding from Astra Zeneca and Bristol Myers Squibb and has served on advisory boards for Corbus Pharmaceutical, EMD Serono Research & Development Institute, Argenx, Octapharma, Kezaar, Orphazyme, Janssen and Pfizer, and has stock shares in Roche and Novartis.
M.F. has received research funding and speakers fee from Abbvie, Amgen, Eli-Lilly, Japan Blood Product Organization, Kyowa-Kirin, Maruho, Novartis, Sanofi and Taiho.
J.V. has been on Speakers Bureaus of Abbvie, Biogen, MSD, Pfizer, Roche, Sanofi and UCB, and has consulted for Abbvie, Argenx, Boehringer, Eli Lilly and Octapharma.
R.A. receives research funding from BMS, Mallinckrodt, Genentech and Pfizer, and has served as consultant for BMS, Mallinckrodt, Pfizer, Orphazyme, Octapharma, Kyverna, Kezar, Janssen, Csl Behring, Corbus, Boehringer Ingelheim, AstraZeneca, Argenx, Alexion, Q32 and Abbvie.
L.C-S. has research support from Pfizer, Corbus and Kezar, and has served on advisory boards for UCB (not myositis related), Janssen, Boehringer-Ingelheim, Mallinckrodt, Serono, Argenx, Roivant and Dysimmune Disease Foundation (DDF), and received compensation for consultancy work from Guidepoint and Allogene. She is also a patent holder on an assay for anti-HMGCR autoantibodies for which she received royalty payments from Inova Diagnostics.
A.L.M is a patent holder on an assay for anti-HMGCR autoantibodies, but does not receive any royalties.
All other authors declare no competing interests.
References
- 1.McHugh NJ & Tansley SL Autoantibodies in myositis. Nat Rev Rheumatol 14, 290–302 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Betteridge Z, et al. Frequency, mutual exclusivity and clinical associations of myositis autoantibodies in a combined European cohort of idiopathic inflammatory myopathy patients. J Autoimmun 101, 48–55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walton JN Some Diseases of Muscle. Lancet 1, 447–452 (1964). [DOI] [PubMed] [Google Scholar]
- 4.Hoogendijk JE, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 14, 337–345 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Loarce-Martos J, Lilleker JB, Parker M, McHugh N & Chinoy H Polymyositis: is there anything left? A retrospective diagnostic review from a tertiary myositis centre. Rheumatology (Oxford) 60, 3398–3403 (2021). [DOI] [PubMed] [Google Scholar]
- 6.de Souza FHC, et al. Guidelines of the Brazilian Society of Rheumatology for the treatment of systemic autoimmune myopathies. Adv Rheumatol 59, 6 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Fujimoto M, Watanabe R, Ishitsuka Y & Okiyama N Recent advances in dermatomyositis-specific autoantibodies. Curr Opin Rheumatol 28, 636–644 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Schmidt J Current Classification and Management of Inflammatory Myopathies. J Neuromuscul Dis 5, 109–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinal-Fernandez I, Casal-Dominguez M & Mammen AL Immune-Mediated Necrotizing Myopathy. Curr Rheumatol Rep 20, 21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selva-O’Callaghan A, et al. Classification and management of adult inflammatory myopathies. Lancet Neurol 17, 816–828 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furst DE, Amato AA, Iorga SR, Gajria K & Fernandes AW Epidemiology of adult idiopathic inflammatory myopathies in a U.S. managed care plan. Muscle Nerve 45, 676–683 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Oddis CV, Conte CG, Steen VD & Medsger TA Jr. Incidence of polymyositis-dermatomyositis: a 20-year study of hospital diagnosed cases in Allegheny County, PA 1963–1982. J Rheumatol 17, 1329–1334 (1990). [PubMed] [Google Scholar]
- 13.Pearson CM Polymyositis. Annu Rev Med 17, 63–82 (1966). [DOI] [PubMed] [Google Scholar]
- 14.Yu KH, See LC, Kuo CF, Chou IJ & Chou MJ Prevalence and incidence in patients with autoimmune rheumatic diseases: a nationwide population-based study in Taiwan. Arthritis Care Res (Hoboken) 65, 244–250 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Barnabe C, et al. Prevalence of autoimmune inflammatory myopathy in the first nations population of Alberta, Canada. Arthritis Care Res (Hoboken) 64, 1715–1719 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Dobloug C, et al. Prevalence and clinical characteristics of adult polymyositis and dermatomyositis; data from a large and unselected Norwegian cohort. Ann Rheum Dis 74, 1551–1556 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Bernatsky S, et al. Estimating the prevalence of polymyositis and dermatomyositis from administrative data: age, sex and regional differences. Ann Rheum Dis 68, 1192–1196 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Hengstman GJ, van Venrooij WJ, Vencovsky J, Moutsopoulos HM & van Engelen BG The relative prevalence of dermatomyositis and polymyositis in Europe exhibits a latitudinal gradient. Ann Rheum Dis 59, 141–142 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love LA, et al. Ultraviolet radiation intensity predicts the relative distribution of dermatomyositis and anti-Mi-2 autoantibodies in women. Arthritis Rheum 60, 2499–2504 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Svensson J, Arkema EV, Lundberg IE & Holmqvist M Incidence and prevalence of idiopathic inflammatory myopathies in Sweden: a nationwide population-based study. Rheumatology (Oxford) 56, 802–810 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Lilleker JB, et al. The EuroMyositis registry: an international collaborative tool to facilitate myositis research. Ann Rheum Dis 77, 30–39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molberg O & Dobloug C Epidemiology of sporadic inclusion body myositis. Curr Opin Rheumatol 28, 657–660 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Badrising UA, et al. Epidemiology of inclusion body myositis in the Netherlands: a nationwide study. Neurology 55, 1385–1387 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Dobloug GC, et al. High prevalence of inclusion body myositis in Norway; a population-based clinical epidemiology study. Eur J Neurol 22, 672–e641 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Nojima T, et al. A case of polymyositis associated with hepatitis B infection. Clin Exp Rheumatol 18, 86–88 (2000). [PubMed] [Google Scholar]
- 26.Johnson RW, Williams FM, Kazi S, Dimachkie MM & Reveille JD Human immunodeficiency virus-associated polymyositis: a longitudinal study of outcome. Arthritis Rheum 49, 172–178 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Dalakas MC, et al. Inclusion body myositis with human immunodeficiency virus infection: four cases with clonal expansion of viral-specific T cells. Ann Neurol 61, 466–475 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Matsuura E, et al. Inclusion body myositis associated with human T-lymphotropic virus-type I infection: eleven patients from an endemic area in Japan. J Neuropathol Exp Neurol 67, 41–49 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Uruha A, et al. Hepatitis C virus infection in inclusion body myositis: A case-control study. Neurology 86, 211–217 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Lyon MG, Bloch DA, Hollak B & Fries JF Predisposing factors in polymyositis-dermatomyositis: results of a nationwide survey. J Rheumatol 16, 1218–1224 (1989). [PubMed] [Google Scholar]
- 31.Svensson J, Holmqvist M, Lundberg IE & Arkema EV Infections and respiratory tract disease as risk factors for idiopathic inflammatory myopathies: a population-based case-control study. Ann Rheum Dis 76, 1803–1808 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Vegosen LJ, et al. Seasonal birth patterns in myositis subgroups suggest an etiologic role of early environmental exposures. Arthritis Rheum 56, 2719–2728 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szabo K, et al. Effect of Genetic and Laboratory Findings on Clinical Course of Antisynthetase Syndrome in a Hungarian Cohort. Biomed Res Int 2018, 6416378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toquet S, et al. The seasonality of Dermatomyositis associated with anti-MDA5 antibody: An argument for a respiratory viral trigger. Autoimmun Rev 20, 102788 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Webber MP, et al. Nested case-control study of selected systemic autoimmune diseases in World Trade Center rescue/recovery workers. Arthritis Rheumatol 67, 1369–1376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson C, Piguet V & Choy E The pathogenesis of dermatomyositis. Br J Dermatol 179, 1256–1262 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Chinoy H, et al. Interaction of HLA-DRB1*03 and smoking for the development of anti-Jo-1 antibodies in adult idiopathic inflammatory myopathies: a European-wide case study. Ann Rheum Dis 71, 961–965 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rothwell S, et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann Rheum Dis 75, 1558–1566 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugiura T, et al. Positive association between STAT4 polymorphisms and polymyositis/dermatomyositis in a Japanese population. Ann Rheum Dis 71, 1646–1650 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Chinoy H, et al. The protein tyrosine phosphatase N22 gene is associated with juvenile and adult idiopathic inflammatory myopathy independent of the HLA 8.1 haplotype in British Caucasian patients. Arthritis Rheum 58, 3247–3254 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Q, et al. Positive association of genetic variations in the phospholipase C-like 1 gene with dermatomyositis in Chinese Han. Immunol Res 64, 204–212 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Rothwell S, et al. Immune-Array Analysis in Sporadic Inclusion Body Myositis Reveals HLA-DRB1 Amino Acid Heterogeneity Across the Myositis Spectrum. Arthritis Rheumatol 69, 1090–1099 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marie I Morbidity and mortality in adult polymyositis and dermatomyositis. Curr Rheumatol Rep 14, 275–285 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Yamasaki Y, et al. Longterm survival and associated risk factors in patients with adult-onset idiopathic inflammatory myopathies and amyopathic dermatomyositis: experience in a single institute in Japan. J Rheumatol 38, 1636–1643 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Bronner IM, et al. Long-term outcome in polymyositis and dermatomyositis. Ann Rheum Dis 65, 1456–1461 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DeVere R & Bradley WG Polymyositis: its presentation, morbidity and mortality. Brain 98, 637–666 (1975). [DOI] [PubMed] [Google Scholar]
- 47.Danko K, Ponyi A, Constantin T, Borgulya G & Szegedi G Long-term survival of patients with idiopathic inflammatory myopathies according to clinical features: a longitudinal study of 162 cases. Medicine (Baltimore) 83, 35–42 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Dobloug GC, Garen T, Brunborg C, Gran JT & Molberg O Survival and cancer risk in an unselected and complete Norwegian idiopathic inflammatory myopathy cohort. Semin Arthritis Rheum 45, 301–308 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Sultan SM, Ioannou Y, Moss K & Isenberg DA Outcome in patients with idiopathic inflammatory myositis: morbidity and mortality. Rheumatology (Oxford) 41, 22–26 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Torres C, et al. Survival, mortality and causes of death in inflammatory myopathies. Autoimmunity 39, 205–215 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Johnson C, et al. Assessment of Mortality in Autoimmune Myositis With and Without Associated Interstitial Lung Disease. Lung 194, 733–737 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dobloug GC, Svensson J, Lundberg IE & Holmqvist M Mortality in idiopathic inflammatory myopathy: results from a Swedish nationwide population-based cohort study. Ann Rheum Dis 77, 40–47 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Miller FW, Lamb JA, Schmidt J & Nagaraju K Risk factors and disease mechanisms in myositis. Nat Rev Rheumatol 14, 255–268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pinal-Fernandez I, et al. Identification of distinctive interferon gene signatures in different types of myositis. Neurology 93, e1193–e1204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rigolet M, et al. Distinct interferon signatures stratify inflammatory and dysimmune myopathies. RMD Open 5, e000811 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pinal-Fernandez I, et al. Machine learning algorithms reveal unique gene expression profiles in muscle biopsies from patients with different types of myositis. Ann Rheum Dis 79, 1234–1242 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pestronk A, Schmidt RE & Choksi R Vascular pathology in dermatomyositis and anatomic relations to myopathology. Muscle Nerve 42, 53–61 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Greenberg SA, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol 57, 664–678 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Wong D, et al. Interferon and biologic signatures in dermatomyositis skin: specificity and heterogeneity across diseases. PLoS One 7, e29161 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walsh RJ, et al. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum 56, 3784–3792 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baechler EC, et al. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med 13, 59–68 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ladislau L, et al. JAK inhibitor improves type I interferon induced damage: proof of concept in dermatomyositis. Brain 141, 1609–1621 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Chen Z, Wang X & Ye S Tofacitinib in Amyopathic Dermatomyositis-Associated Interstitial Lung Disease. N Engl J Med 381, 291–293 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Pinal-Fernandez I, Casciola-Rosen LA, Christopher-Stine L, Corse AM & Mammen AL The Prevalence of Individual Histopathologic Features Varies according to Autoantibody Status in Muscle Biopsies from Patients with Dermatomyositis. J Rheumatol 42, 1448–1454 (2015). [PMC free article] [PubMed] [Google Scholar]
- 65.Miller FW, Waite KA, Biswas T & Plotz PH The role of an autoantigen, histidyl-tRNA synthetase, in the induction and maintenance of autoimmunity. Proc Natl Acad Sci U S A 87, 9933–9937 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mahler M, Miller FW & Fritzler MJ Idiopathic inflammatory myopathies and the anti-synthetase syndrome: a comprehensive review. Autoimmun Rev 13, 367–371 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mescam-Mancini L, et al. Anti-Jo-1 antibody-positive patients show a characteristic necrotizing perifascicular myositis. Brain 138, 2485–2492 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Montagne JMZ, et al. Ultra-Efficient Short Read Sequencing of T Cell Receptor Repertoires. bioRxiv, doi: 10.1101/494062 (2020). [DOI] [Google Scholar]
- 69.Galindo-Feria AS, et al. Proinflammatory Histidyl-Transfer RNA Synthetase-Specific CD4+ T Cells in the Blood and Lungs of Patients With Idiopathic Inflammatory Myopathies. Arthritis Rheumatol 72, 179–191 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Katsumata Y, et al. Species-specific immune responses generated by histidyl-tRNA synthetase immunization are associated with muscle and lung inflammation. J Autoimmun 29, 174–186 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mammen AL, et al. Increased frequency of DRB1*11:01 in anti-hydroxymethylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Care Res (Hoboken) 64, 1233–1237 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mammen AL, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 63, 713–721 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Benveniste O, et al. Correlation of anti-signal recognition particle autoantibody levels with creatine kinase activity in patients with necrotizing myopathy. Arthritis Rheum 63, 1961–1971 (2011). [DOI] [PubMed] [Google Scholar]
- 74.Werner JL, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum 64, 4087–4093 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Allenbach Y, et al. Necrosis in anti-SRP(+) and anti-HMGCR(+)myopathies: Role of autoantibodies and complement. Neurology 90, e507–e517 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Christopher-Stine L, et al. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 62, 2757–2766 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Arouche-Delaperche L, et al. Pathogenic role of anti-signal recognition protein and anti-3-Hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: Myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann Neurol 81, 538–548 (2017). [DOI] [PubMed] [Google Scholar]
- 78.US National Library of Medicine. Clinicaltrials.gov, https://clinicaltrials.gov/ct2/show/NCT04025632 (2021). [DOI] [PubMed]
- 79. https://www.ucb.com/stories-media/Press-Releases/article/UCB-s-zilucoplan-shows-no-relevant-effect-in-immune-mediated-necrotizing-myopathy-IMNM.
- 80.Lampe JB, et al. Analysis of HLA class I and II alleles in sporadic inclusion-body myositis. J Neurol 250, 1313–1317 (2003). [DOI] [PubMed] [Google Scholar]
- 81.Greenberg SA, et al. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology 65, 1782–1787 (2005). [DOI] [PubMed] [Google Scholar]
- 82.Larman HB, et al. Cytosolic 5’-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol 73, 408–418 (2013). [DOI] [PubMed] [Google Scholar]
- 83.Pluk H, et al. Autoantibodies to cytosolic 5’-nucleotidase 1A in inclusion body myositis. Ann Neurol 73, 397–407 (2013). [DOI] [PubMed] [Google Scholar]
- 84.Engel AG & Arahata K Monoclonal antibody analysis of mononuclear cells in myopathies. II: Phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann Neurol 16, 209–215 (1984). [DOI] [PubMed] [Google Scholar]
- 85.Arahata K & Engel AG Monoclonal antibody analysis of mononuclear cells in myopathies. V: Identification and quantitation of T8+ cytotoxic and T8+ suppressor cells. Ann Neurol 23, 493–499 (1988). [DOI] [PubMed] [Google Scholar]
- 86.Arahata K & Engel AG Monoclonal antibody analysis of mononuclear cells in myopathies. IV: Cell-mediated cytotoxicity and muscle fiber necrosis. Ann Neurol 23, 168–173 (1988). [DOI] [PubMed] [Google Scholar]
- 87.Fyhr IM, Moslemi AR, Lindberg C & Oldfors A T cell receptor beta-chain repertoire in inclusion body myositis. J Neuroimmunol 91, 129–134 (1998). [DOI] [PubMed] [Google Scholar]
- 88.Bender A, Behrens L, Engel AG & Hohlfeld R T-cell heterogeneity in muscle lesions of inclusion body myositis. J Neuroimmunol 84, 86–91 (1998). [DOI] [PubMed] [Google Scholar]
- 89.Muntzing K, Lindberg C, Moslemi AR & Oldfors A Inclusion body myositis: clonal expansions of muscle-infiltrating T cells persist over time. Scand J Immunol 58, 195–200 (2003). [DOI] [PubMed] [Google Scholar]
- 90.Amemiya K, Granger RP & Dalakas MC Clonal restriction of T-cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time. Studies in repeated muscle biopsies. Brain 123 (Pt 10), 2030–2039 (2000). [DOI] [PubMed] [Google Scholar]
- 91.Dimitri D, et al. Shared blood and muscle CD8+ T-cell expansions in inclusion body myositis. Brain 129, 986–995 (2006). [DOI] [PubMed] [Google Scholar]
- 92.Greenberg SA, Pinkus JL, Amato AA, Kristensen T & Dorfman DM Association of inclusion body myositis with T cell large granular lymphocytic leukaemia. Brain 139, 1348–1360 (2016). [DOI] [PubMed] [Google Scholar]
- 93.Pandya JM, et al. Expanded T cell receptor Vbeta-restricted T cells from patients with sporadic inclusion body myositis are proinflammatory and cytotoxic CD28null T cells. Arthritis Rheum 62, 3457–3466 (2010). [DOI] [PubMed] [Google Scholar]
- 94.Dubourg O, et al. Diagnostic value of markers of muscle degeneration in sporadic inclusion body myositis. Acta Myol 30, 103–108 (2011). [PMC free article] [PubMed] [Google Scholar]
- 95.Milisenda JC, et al. Accumulation of autophagosome cargo protein p62 is common in idiopathic inflammatory myopathies. Clin Exp Rheumatol 39, 351–356 (2021). [PMC free article] [PubMed] [Google Scholar]
- 96.Girolamo F, et al. Autophagy markers LC3 and p62 accumulate in immune-mediated necrotizing myopathy. Muscle Nerve 60, 315–327 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Fischer N, et al. Sequestosome-1 (p62) expression reveals chaperone-assisted selective autophagy in immune-mediated necrotizing myopathies. Brain Pathol 30, 261–271 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hiniker A, Daniels BH, Lee HS & Margeta M Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol Commun 1, 29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van der Meulen MF, et al. Polymyositis: an overdiagnosed entity. Neurology 61, 316–321 (2003). [DOI] [PubMed] [Google Scholar]
- 100.Lundberg IE, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis 76, 1955–1964 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gerami P, Schope JM, McDonald L, Walling HW & Sontheimer RD A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol 54, 597–613 (2006). [DOI] [PubMed] [Google Scholar]
- 102.Inoue M, et al. Association of Dermatomyositis Sine Dermatitis and With Anti-Nuclear Matrix Protein 2 Autoantibodies. JAMA Neurol (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stockton D, Doherty VR & Brewster DH Risk of cancer in patients with dermatomyositis or polymyositis, and follow-up implications: a Scottish population-based cohort study. Br J Cancer 85, 41–45 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Madan V, Chinoy H, Griffiths CE & Cooper RG Defining cancer risk in dermatomyositis. Part I. Clin Exp Dermatol 34, 451–455 (2009). [DOI] [PubMed] [Google Scholar]
- 105.Madan V, Chinoy H, Griffiths CE & Cooper RG Defining cancer risk in dermatomyositis. Part II. Assessing diagnostic usefulness of myositis serology. Clin Exp Dermatol 34, 561–565 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Reichlin M & Mattioli M Description of a serological reaction characteristic of polymyositis. Clin Immunol Immunopathol 5, 12–20 (1976). [DOI] [PubMed] [Google Scholar]
- 107.Sato S, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum 60, 2193–2200 (2009). [DOI] [PubMed] [Google Scholar]
- 108.Sato S, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 52, 1571–1576 (2005). [DOI] [PubMed] [Google Scholar]
- 109.Fujimoto M, et al. Myositis-specific anti-155/140 autoantibodies target transcription intermediary factor 1 family proteins. Arthritis Rheum 64, 513–522 (2012). [DOI] [PubMed] [Google Scholar]
- 110.Targoff IN, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum 54, 3682–3689 (2006). [DOI] [PubMed] [Google Scholar]
- 111.Gunawardena H, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum 60, 1807–1814 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Betteridge Z, Gunawardena H, North J, Slinn J & McHugh N Identification of a novel autoantibody directed against small ubiquitin-like modifier activating enzyme in dermatomyositis. Arthritis Rheum 56, 3132–3137 (2007). [DOI] [PubMed] [Google Scholar]
- 113.Mammen AL, Allenbach Y, Stenzel W, Benveniste O & Group, E.t.W.S. 239th ENMC International Workshop: Classification of dermatomyositis, Amsterdam, the Netherlands, 14–16 December 2018. Neuromuscul Disord 30, 70–92 (2020). [DOI] [PubMed] [Google Scholar]
- 114.Rider LG, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore) 92, 223–243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hamaguchi Y, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol 147, 391–398 (2011). [DOI] [PubMed] [Google Scholar]
- 116.Lundberg IE, de Visser M & Werth VP Classification of myositis. Nat Rev Rheumatol 14, 269–278 (2018). [DOI] [PubMed] [Google Scholar]
- 117.Moghadam-Kia S, Oddis CV & Aggarwal R Anti-MDA5 Antibody Spectrum in Western World. Curr Rheumatol Rep 20, 78 (2018). [DOI] [PubMed] [Google Scholar]
- 118.Trallero-Araguas E, et al. Usefulness of anti-p155 autoantibody for diagnosing cancer-associated dermatomyositis: a systematic review and meta-analysis. Arthritis Rheum 64, 523–532 (2012). [DOI] [PubMed] [Google Scholar]
- 119.Fiorentino DF, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1gamma. Arthritis Rheum 65, 2954–2962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Betteridge ZE, et al. Clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis 68, 1621–1625 (2009). [DOI] [PubMed] [Google Scholar]
- 121.Fujimoto M, et al. Autoantibodies to small ubiquitin-like modifier activating enzymes in Japanese patients with dermatomyositis: comparison with a UK Caucasian cohort. Ann Rheum Dis 72, 151–153 (2013). [DOI] [PubMed] [Google Scholar]
- 122.Ge Y, Lu X, Shu X, Peng Q & Wang G Clinical characteristics of anti-SAE antibodies in Chinese patients with dermatomyositis in comparison with different patient cohorts. Sci Rep 7, 188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Friedman AW, Targoff IN & Arnett FC Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum 26, 459–467 (1996). [DOI] [PubMed] [Google Scholar]
- 124.Hervier B, et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev 12, 210–217 (2012). [DOI] [PubMed] [Google Scholar]
- 125.Aggarwal R, et al. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann Rheum Dis 73, 227–232 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Targoff IN, Johnson AE & Miller FW Antibody to signal recognition particle in polymyositis. Arthritis Rheum 33, 1361–1370 (1990). [DOI] [PubMed] [Google Scholar]
- 127.Allenbach Y, Mammen AL, Benveniste O, Stenzel W & Immune-Mediated Necrotizing Myopathies Working, G. 224th ENMC International Workshop:: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14–16 October 2016. Neuromuscul Disord 28, 87–99 (2018). [DOI] [PubMed] [Google Scholar]
- 128.Allenbach Y, Benveniste O, Stenzel W & Boyer O Immune-mediated necrotizing myopathy: clinical features and pathogenesis. Nat Rev Rheumatol 16, 689–701 (2020). [DOI] [PubMed] [Google Scholar]
- 129.Lim J, et al. Seronegative patients form a distinctive subgroup of immune-mediated necrotizing myopathy. Neurol Neuroimmunol Neuroinflamm 6, e513 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Naddaf E, Barohn RJ & Dimachkie MM Inclusion Body Myositis: Update on Pathogenesis and Treatment. Neurotherapeutics 15, 995–1005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hilton-Jones D & Brady S Diagnostic criteria for inclusion body myositis. J Intern Med 280, 52–62 (2016). [DOI] [PubMed] [Google Scholar]
- 132.Alemo Munters L, van Vollenhoven RF & Alexanderson H Patient preference assessment reveals disease aspects not covered by recommended outcomes in polymyositis and dermatomyositis. ISRN Rheumatol 2011, 463124 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Loarce-Martos J, Lilleker JB, Parker M, McHugh N & Chinoy H Polymyositis: is there anything left? A retrospective diagnostic review from a tertiary myositis centre. Rheumatology (Oxford) (2020). [DOI] [PubMed] [Google Scholar]
- 134.Troyanov Y, et al. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore) 84, 231–249 (2005). [DOI] [PubMed] [Google Scholar]
- 135.Aguila LA, et al. Clinical and laboratory features of overlap syndromes of idiopathic inflammatory myopathies associated with systemic lupus erythematosus, systemic sclerosis, or rheumatoid arthritis. Clin Rheumatol 33, 1093–1098 (2014). [DOI] [PubMed] [Google Scholar]
- 136.Kaji K, et al. Autoantibodies to RuvBL1 and RuvBL2: a novel systemic sclerosis-related antibody associated with diffuse cutaneous and skeletal muscle involvement. Arthritis Care Res (Hoboken) 66, 575–584 (2014). [DOI] [PubMed] [Google Scholar]
- 137.Leclair V, et al. Autoantibody profiles delineate distinct subsets of scleromyositis. Rheumatology (Oxford) (2021). [DOI] [PubMed] [Google Scholar]
- 138.Fiorentino D, Chung L, Zwerner J, Rosen A & Casciola-Rosen L The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol 65, 25–34 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kurtzman DJB & Vleugels RA Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: A concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol 78, 776–785 (2018). [DOI] [PubMed] [Google Scholar]
- 140.Fiorentino DF, et al. Distinctive cutaneous and systemic features associated with antitranscriptional intermediary factor-1gamma antibodies in adults with dermatomyositis. J Am Acad Dermatol 72, 449–455 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chung MP, et al. Calcinosis Biomarkers in Adult and Juvenile Dermatomyositis. Autoimmun Rev 19, 102533 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Long K & Danoff SK Interstitial Lung Disease in Polymyositis and Dermatomyositis. Clin Chest Med 40, 561–572 (2019). [DOI] [PubMed] [Google Scholar]
- 143.Shappley C, Paik JJ & Saketkoo LA Myositis-Related Interstitial Lung Diseases: Diagnostic Features, Treatment, and Complications. Curr Treatm Opt Rheumatol 5, 56–83 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zuo Y, et al. Clinical significance of radiological patterns of HRCT and their association with macrophage activation in dermatomyositis. Rheumatology (Oxford) (2020). [DOI] [PubMed] [Google Scholar]
- 145.Gupta R, Wayangankar SA, Targoff IN & Hennebry TA Clinical cardiac involvement in idiopathic inflammatory myopathies: a systematic review. Int J Cardiol 148, 261–270 (2011). [DOI] [PubMed] [Google Scholar]
- 146.Khoo T, et al. Cardiac involvement in idiopathic inflammatory myopathies detected by cardiac magnetic resonance imaging. Clin Rheumatol 38, 3471–3476 (2019). [DOI] [PubMed] [Google Scholar]
- 147.Hughes M, Lilleker JB, Herrick AL & Chinoy H Cardiac troponin testing in idiopathic inflammatory myopathies and systemic sclerosis-spectrum disorders: biomarkers to distinguish between primary cardiac involvement and low-grade skeletal muscle disease activity. Ann Rheum Dis 74, 795–798 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Casal-Dominguez M, et al. High-resolution manometry in patients with idiopathic inflammatory myopathy: Elevated prevalence of esophageal involvement and differences according to autoantibody status and clinical subset. Muscle Nerve 56, 386–392 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Klein M, et al. Arthritis in idiopathic inflammatory myopathy: clinical features and autoantibody associations. J Rheumatol 41, 1133–1139 (2014). [DOI] [PubMed] [Google Scholar]
- 150.Rider LG, et al. International consensus on preliminary definitions of improvement in adult and juvenile myositis. Arthritis Rheum 50, 2281–2290 (2004). [DOI] [PubMed] [Google Scholar]
- 151.Marco JL & Collins BF Clinical manifestations and treatment of antisynthetase syndrome. Best Pract Res Clin Rheumatol, 101503 (2020). [DOI] [PubMed] [Google Scholar]
- 152.Rose MR & Group EIW 188th ENMC International Workshop: Inclusion Body Myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord 23, 1044–1055 (2013). [DOI] [PubMed] [Google Scholar]
- 153.Lloyd TE, et al. Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology 83, 426–433 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nozaki K & Pestronk A High aldolase with normal creatine kinase in serum predicts a myopathy with perimysial pathology. J Neurol Neurosurg Psychiatry 80, 904–908 (2009). [DOI] [PubMed] [Google Scholar]
- 155.Benveniste O, Stenzel W & Allenbach Y Advances in serological diagnostics of inflammatory myopathies. Curr Opin Neurol 29, 662–673 (2016). [DOI] [PubMed] [Google Scholar]
- 156.Tanboon J & Nishino I Classification of idiopathic inflammatory myopathies: pathology perspectives. Curr Opin Neurol 32, 704–714 (2019). [DOI] [PubMed] [Google Scholar]
- 157.De Bleecker JL, Lundberg IE, de Visser M & Group, E.M.M.B.S. 193rd ENMC International workshop Pathology diagnosis of idiopathic inflammatory myopathies 30 November - 2 December 2012, Naarden, The Netherlands. Neuromuscul Disord 23, 945–951 (2013). [DOI] [PubMed] [Google Scholar]
- 158.De Bleecker JL, et al. 205th ENMC International Workshop: Pathology diagnosis of idiopathic inflammatory myopathies part II 28–30 March 2014, Naarden, The Netherlands. Neuromuscul Disord 25, 268–272 (2015). [DOI] [PubMed] [Google Scholar]
- 159.Uruha A, et al. Diagnostic potential of sarcoplasmic myxovirus resistance protein A expression in subsets of dermatomyositis. Neuropathol Appl Neurobiol 45, 513–522 (2019). [DOI] [PubMed] [Google Scholar]
- 160.Olivier PA, et al. Idiopathic inflammatory myopathy: Interrater variability in muscle biopsy reading. Neurology 93, e889–e894 (2019). [DOI] [PubMed] [Google Scholar]
- 161.Michelle EH & Mammen AL Myositis Mimics. Curr Rheumatol Rep 17, 63 (2015). [DOI] [PubMed] [Google Scholar]
- 162.Michelle H & Mammen AL Myositis Mimics: The Differential Diagnosis of Myositis. in Managing Myositis (eds. Aggarwal R & Oddis C) 209–223 (Springer, 2020). [Google Scholar]
- 163.Vencovsky J Clinical Features of Myositis: Muscular Manifestations. in Managing Myositis (eds. Aggarwal R & Oddis C) 37–46 (Springer, 2020). [Google Scholar]
- 164.Lilleker JB & Roberts ME Metabolic myopathies. in Myositis (eds. Chinoy H & Cooper RG) 41–52 (Oxford University Press, 2018). [Google Scholar]
- 165.Kaji K, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 46, 25–28 (2007). [DOI] [PubMed] [Google Scholar]
- 166.Ichimura Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy. Ann Rheum Dis 71, 710–713 (2012). [DOI] [PubMed] [Google Scholar]
- 167.Allenbach Y, et al. High risk of cancer in autoimmune necrotizing myopathies: usefulness of myositis specific antibody. Brain 139, 2131–2135 (2016). [DOI] [PubMed] [Google Scholar]
- 168.Tiniakou E & Mammen AL Idiopathic Inflammatory Myopathies and Malignancy: a Comprehensive Review. Clin Rev Allergy Immunol 52, 20–33 (2017). [DOI] [PubMed] [Google Scholar]
- 169.Selva-O’Callaghan A, et al. Conventional cancer screening versus PET/CT in dermatomyositis/polymyositis. Am J Med 123, 558–562 (2010). [DOI] [PubMed] [Google Scholar]
- 170.Gerards MC, Terlou RJ, Yu H, Koks CH & Gerdes VE Traditional Chinese lipid-lowering agent red yeast rice results in significant LDL reduction but safety is uncertain - a systematic review and meta-analysis. Atherosclerosis 240, 415–423 (2015). [DOI] [PubMed] [Google Scholar]
- 171.Bae SS, Oganesian B, Golub I & Charles-Schoeman C Statin use in patients with non-HMGCR idiopathic inflammatory myopathies: A retrospective study. Clin Cardiol 43, 732–742 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Supakornnumporn S & Katirji B Autoimmune Neuromuscular Diseases Induced by Immunomodulating Drugs. J Clin Neuromuscul Dis 20, 28–34 (2018). [DOI] [PubMed] [Google Scholar]
- 173.Mamyrova G, et al. Environmental factors associated with disease flare in juvenile and adult dermatomyositis. Rheumatology (Oxford) 56, 1342–1347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chandra T & Aggarwal R Clinical trials and novel therapeutics in dermatomyositis. Expert Opin Emerg Drugs 25, 213–228 (2020). [DOI] [PubMed] [Google Scholar]
- 175.Joffe MM, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 94, 379–387 (1993). [DOI] [PubMed] [Google Scholar]
- 176.Catania A, et al. The melanocortin system in control of inflammation. ScientificWorldJournal 10, 1840–1853 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Getting SJ, Christian HC, Flower RJ & Perretti M Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis. Arthritis and rheumatism 46, 2765–2775 (2002). [DOI] [PubMed] [Google Scholar]
- 178.Catania A, Gatti S, Colombo G & Lipton JM Targeting melanocortin receptors as a novel strategy to control inflammation. Pharmacol Rev 56, 1–29 (2004). [DOI] [PubMed] [Google Scholar]
- 179.Aggarwal R, et al. Efficacy and safety of adrenocorticotropic hormone gel in refractory dermatomyositis and polymyositis. Annals of the rheumatic diseases 77, 720–727 (2018). [DOI] [PubMed] [Google Scholar]
- 180.Saygin D, et al. Follow-up results of myositis patients treated with H. P. Acthar gel. Rheumatology (Oxford, England) (2020). [DOI] [PubMed] [Google Scholar]
- 181.Gordon PA, Winer JB, Hoogendijk JE & Choy EH Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev, CD003643 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Newman ED & Scott DW The Use of Low-dose Oral Methotrexate in the Treatment of Polymyositis and Dermatomyositis. J Clin Rheumatol 1, 99–102 (1995). [DOI] [PubMed] [Google Scholar]
- 183.Ruperto N, et al. Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. Lancet 387, 671–678 (2016). [DOI] [PubMed] [Google Scholar]
- 184.Bunch TW Prednisone and azathioprine for polymyositis: long-term followup. Arthritis and rheumatism 24, 45–48 (1981). [DOI] [PubMed] [Google Scholar]
- 185.Bunch TW, Worthington JW, Combs JJ, Ilstrup DM & Engel AG Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med 92, 365–369 (1980). [DOI] [PubMed] [Google Scholar]
- 186.Villalba L, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis and rheumatism 41, 392–399 (1998). [DOI] [PubMed] [Google Scholar]
- 187.Majithia V & Harisdangkul V Mycophenolate mofetil (CellCept): an alternative therapy for autoimmune inflammatory myopathy. Rheumatology (Oxford, England) 44, 386–389 (2005). [DOI] [PubMed] [Google Scholar]
- 188.Pisoni CN, Cuadrado MJ, Khamashta MA, Hughes GR & D’Cruz DP Mycophenolate mofetil treatment in resistant myositis. Rheumatology (Oxford, England) 46, 516–518 (2007). [DOI] [PubMed] [Google Scholar]
- 189.Schneider C, Gold R, Schafers M & Toyka KV Mycophenolate mofetil in the therapy of polymyositis associated with a polyautoimmune syndrome. Muscle & nerve 25, 286–288 (2002). [DOI] [PubMed] [Google Scholar]
- 190.Danieli MG, et al. Intravenous immunoglobulin as add on treatment with mycophenolate mofetil in severe myositis. Autoimmun Rev 9, 124–127 (2009). [DOI] [PubMed] [Google Scholar]
- 191.Rowin J, Amato AA, Deisher N, Cursio J & Meriggioli MN Mycophenolate mofetil in dermatomyositis: is it safe? Neurology 66, 1245–1247 (2006). [DOI] [PubMed] [Google Scholar]
- 192.Fischer A, et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. The Journal of rheumatology 40, 640–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Morganroth PA, Kreider ME & Werth VP Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis Care Res (Hoboken) 62, 1496–1501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Saketkoo LA & Espinoza LR Experience of mycophenolate mofetil in 10 patients with autoimmune-related interstitial lung disease demonstrates promising effects. Am J Med Sci 337, 329–335 (2009). [DOI] [PubMed] [Google Scholar]
- 195.Swigris JJ, et al. Mycophenolate mofetil is safe, well tolerated, and preserves lung function in patients with connective tissue disease-related interstitial lung disease. Chest 130, 30–36 (2006). [DOI] [PubMed] [Google Scholar]
- 196.Oddis CV, Sciurba FC, Elmagd KA & Starzl TE Tacrolimus in refractory polymyositis with interstitial lung disease. Lancet 353, 1762–1763 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mitsui T, Kuroda Y, Ueno S & Kaji R The effects of FK506 on refractory inflammatory myopathies. Acta Neurol Belg 111, 188–194 (2011). [PubMed] [Google Scholar]
- 198.Kotani T, et al. Combination with corticosteroids and cyclosporin-A improves pulmonary function test results and chest HRCT findings in dermatomyositis patients with acute/subacute interstitial pneumonia. Clin Rheumatol 30, 1021–1028 (2011). [DOI] [PubMed] [Google Scholar]
- 199.Labirua-Iturburu A, et al. Calcineurin inhibitors in a cohort of patients with antisynthetase-associated interstitial lung disease. Clinical and experimental rheumatology 31, 436–439 (2013). [PubMed] [Google Scholar]
- 200.Wilkes MR, Sereika SM, Fertig N, Lucas MR & Oddis CV Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis and rheumatism 52, 2439–2446 (2005). [DOI] [PubMed] [Google Scholar]
- 201.Yamasaki Y, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford, England) 46, 124–130 (2007). [DOI] [PubMed] [Google Scholar]
- 202.Andersson H, et al. Long-term experience with rituximab in anti-synthetase syndrome-related interstitial lung disease. Rheumatology (Oxford, England) 54, 1420–1428 (2015). [DOI] [PubMed] [Google Scholar]
- 203.Lim J, et al. Intravenous immunoglobulins as first-line treatment in idiopathic inflammatory myopathies: a pilot study. Rheumatology (Oxford) 60, 1784–1792 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Dalakas MC, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 329, 1993–2000 (1993). [DOI] [PubMed] [Google Scholar]
- 205.Aggarwal R, et al. Safety and Tolerability of IVIg (Octagam 10%) in Patients with Active Dermatomyositis. Results of a Randomized, Double-Blind, Placebo-Controlled Phase III Trial [abstract 0695]. Arthritis Rheumatol 73, Suppl. S9 (2021). [Google Scholar]
- 206.US National Library of Medicine. Clinicaltrials.gov, https://clinicaltrials.gov/ct2/show/NCT02728752 (2021). [DOI] [PubMed]
- 207.Danieli MG, Pettinari L, Moretti R, Logullo F & Gabrielli A Subcutaneous immunoglobulin in polymyositis and dermatomyositis: a novel application. Autoimmun Rev 10, 144–149 (2011). [DOI] [PubMed] [Google Scholar]
- 208.Valiyil R, Casciola-Rosen L, Hong G, Mammen A & Christopher-Stine L Rituximab therapy for myopathy associated with anti-signal recognition particle antibodies: a case series. Arthritis Care Res (Hoboken) 62, 1328–1334 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mok CC, Ho LY & To CH Rituximab for refractory polymyositis: an open-label prospective study. The Journal of rheumatology 34, 1864–1868 (2007). [PubMed] [Google Scholar]
- 210.Chung L, Genovese MC & Fiorentino DF A pilot trial of rituximab in the treatment of patients with dermatomyositis. Arch Dermatol 143, 763–767 (2007). [DOI] [PubMed] [Google Scholar]
- 211.Oddis CV, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis and rheumatism 65, 314–324 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Aggarwal R, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis & rheumatology (Hoboken, N.J.) 66, 740–749 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Efthimiou P Tumor necrosis factor-alpha in inflammatory myopathies: pathophysiology and therapeutic implications. Seminars in arthritis and rheumatism 36, 168–172 (2006). [DOI] [PubMed] [Google Scholar]
- 214.Muscle Study G A randomized, pilot trial of etanercept in dermatomyositis. Ann Neurol 70, 427–436 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Iannone F, Scioscia C, Falappone PC, Covelli M & Lapadula G Use of etanercept in the treatment of dermatomyositis: a case series. J Rheumatol 33, 1802–1804 (2006). [PubMed] [Google Scholar]
- 216.Dastmalchi M, et al. A high incidence of disease flares in an open pilot study of infliximab in patients with refractory inflammatory myopathies. Ann Rheum Dis 67, 1670–1677 (2008). [DOI] [PubMed] [Google Scholar]
- 217.Schiffenbauer A, et al. A randomized, double-blind, placebo-controlled trial of infliximab in refractory polymyositis and dermatomyositis. Semin Arthritis Rheum 47, 858–864 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Riley P, et al. Effectiveness of infliximab in the treatment of refractory juvenile dermatomyositis with calcinosis. Rheumatology (Oxford) 47, 877–880 (2008). [DOI] [PubMed] [Google Scholar]
- 219.de Souza JM, Hoff LS & Shinjo SK Intravenous human immunoglobulin and/or methylprednisolone pulse therapies as a possible treat-to-target strategy in immune-mediated necrotizing myopathies. Rheumatol Int 39, 1201–1212 (2019). [DOI] [PubMed] [Google Scholar]
- 220.De Souza FHC, Miossi R & Shinjo SK Necrotising myopathy associated with anti-signal recognition particle (anti-SRP) antibody. Clin Exp Rheumatol 35, 766–771 (2017). [PubMed] [Google Scholar]
- 221.Mammen AL & Tiniakou E Intravenous Immune Globulin for Statin-Triggered Autoimmune Myopathy. N Engl J Med 373, 1680–1682 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Go DJ, et al. Survival benefit associated with early cyclosporine treatment for dermatomyositis-associated interstitial lung disease. Rheumatol Int 36, 125–131 (2016). [DOI] [PubMed] [Google Scholar]
- 223.Keir GJ, et al. Rituximab in severe, treatment-refractory interstitial lung disease. Respirology 19, 353–359 (2014). [DOI] [PubMed] [Google Scholar]
- 224.Allenbach Y, et al. Efficacy of Rituximab in Refractory Inflammatory Myopathies Associated with Anti- Synthetase Auto-Antibodies: An Open-Label, Phase II Trial. PLoS One 10, e0133702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Bauhammer J, et al. Rituximab in the Treatment of Jo1 Antibody-associated Antisynthetase Syndrome: Anti-Ro52 Positivity as a Marker for Severity and Treatment Response. The Journal of rheumatology 43, 1566–1574 (2016). [DOI] [PubMed] [Google Scholar]
- 226.Romero-Bueno F, et al. Recommendations for the treatment of anti-melanoma differentiation-associated gene 5-positive dermatomyositis-associated rapidly progressive interstitial lung disease. Seminars in arthritis and rheumatism 50, 776–790 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ogawa Y, Kishida D, Shimojima Y, Hayashi K & Sekijima Y Effective Administration of Rituximab in Anti-MDA5 Antibody-Positive Dermatomyositis with Rapidly Progressive Interstitial Lung Disease and Refractory Cutaneous Involvement: A Case Report and Literature Review. Case Rep Rheumatol 2017, 5386797 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Badrising UA, et al. Comparison of weakness progression in inclusion body myositis during treatment with methotrexate or placebo. Ann Neurol 51, 369–372 (2002). [DOI] [PubMed] [Google Scholar]
- 229.Hanna MG, et al. Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): a randomised, double-blind, placebo-controlled phase 2b trial. Lancet Neurol 18, 834–844 (2019). [DOI] [PubMed] [Google Scholar]
- 230.Ahmed M, et al. Targeting protein homeostasis in sporadic inclusion body myositis. Sci Transl Med 8, 331ra341 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Benveniste O, et al. Rapamycin Vs. Placebo for the Treatment of Inclusion Body Myositis: Improvement of the 6 Min Walking Distance, a Functional Scale, the FVC and Muscle Quantitative MRI [abstract 5L]. Arthritis Rheumatol. 69 Suppl.10 (2017). [Google Scholar]
- 232.Giannini M, et al. Long-term efficacy of adding intravenous immunoglobulins as treatment of refractory dysphagia related to myositis: a retrospective analysis. Rheumatology (Oxford) 60, 1234–1242 (2021). [DOI] [PubMed] [Google Scholar]
- 233.Traineau H, et al. Treatment of calcinosis cutis in systemic sclerosis and dermatomyositis: A review of the literature. J Am Acad Dermatol 82, 317–325 (2020). [DOI] [PubMed] [Google Scholar]
- 234.Reiter N, El-Shabrawi L, Leinweber B, Berghold A & Aberer E Calcinosis cutis: part II. Treatment options. J Am Acad Dermatol 65, 15–22; quiz 23–14 (2011). [DOI] [PubMed] [Google Scholar]
- 235.Alexanderson H & Bostrom C Exercise therapy in patients with idiopathic inflammatory myopathies and systemic lupus erythematosus - A systematic literature review. Best Pract Res Clin Rheumatol 34, 101547 (2020). [DOI] [PubMed] [Google Scholar]
- 236.Munters LA, et al. Endurance Exercise Improves Molecular Pathways of Aerobic Metabolism in Patients With Myositis. Arthritis & rheumatology (Hoboken, N.J.) 68, 1738–1750 (2016). [DOI] [PubMed] [Google Scholar]
- 237.Alemo Munters L, et al. Improvement in health and possible reduction in disease activity using endurance exercise in patients with established polymyositis and dermatomyositis: a multicenter randomized controlled trial with a 1-year open extension followup. Arthritis Care Res (Hoboken) 65, 1959–1968 (2013). [DOI] [PubMed] [Google Scholar]
- 238.Rider LG, et al. Update on outcome assessment in myositis. Nat Rev Rheumatol 14, 303–318 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Aggarwal R, et al. 2016 American College of Rheumatology/European League Against Rheumatism criteria for minimal, moderate, and major clinical response in adult dermatomyositis and polymyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis 76, 792–801 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Snyder CF, Jensen RE, Segal JB & Wu AW Patient-reported outcomes (PROs): putting the patient perspective in patient-centered outcomes research. Med Care 51, S73–79 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Feldon M, et al. Predictors of Reduced Health-Related Quality of Life in Adult Patients With Idiopathic Inflammatory Myopathies. Arthritis Care Res (Hoboken) 69, 1743–1750 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Opinc AH, Brzezinska OE & Makowska JS Disability in idiopathic inflammatory myopathies: questionnaire-based study. Rheumatol Int 39, 1213–1220 (2019). [DOI] [PubMed] [Google Scholar]
- 243.Alexanderson H, Lundberg IE & Stenstrom CH Development of the myositis activities profile--validity and reliability of a self-administered questionnaire to assess activity limitations in patients with polymyositis/dermatomyositis. J Rheumatol 29, 2386–2392 (2002). [PubMed] [Google Scholar]
- 244.Jackson CE, et al. Inclusion body myositis functional rating scale: a reliable and valid measure of disease severity. Muscle Nerve 37, 473–476 (2008). [DOI] [PubMed] [Google Scholar]
- 245.Regardt M, et al. Patients’ Experience of Myositis and Further Validation of a Myositis-specific Patient Reported Outcome Measure - Establishing Core Domains and Expanding Patient Input on Clinical Assessment in Myositis. Report from OMERACT 12. J Rheumatol 42, 2492–2495 (2015). [DOI] [PubMed] [Google Scholar]
- 246.Regardt M, et al. OMERACT 2018 Modified Patient-reported Outcome Domain Core Set in the Life Impact Area for Adult Idiopathic Inflammatory Myopathies. J Rheumatol 46, 1351–1354 (2019). [DOI] [PubMed] [Google Scholar]
- 247.Miller FW, et al. Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford) 40, 1262–1273 (2001). [DOI] [PubMed] [Google Scholar]
- 248.Rider LG, et al. 2016 ACR-EULAR adult dermatomyositis and polymyositis and juvenile dermatomyositis response criteria-methodological aspects. Rheumatology (Oxford) 56, 1884–1893 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.de Wit M, et al. Successful Stepwise Development of Patient Research Partnership: 14 Years’ Experience of Actions and Consequences in Outcome Measures in Rheumatology (OMERACT). Patient 10, 141–152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.DiRenzo D, Bingham CO 3rd & Mecoli CA Patient-Reported Outcomes in Adult Idiopathic Inflammatory Myopathies. Curr Rheumatol Rep 21, 62 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Rotenstein LS, Huckman RS & Wagle NW Making Patients and Doctors Happier - The Potential of Patient-Reported Outcomes. N Engl J Med 377, 1309–1312 (2017). [DOI] [PubMed] [Google Scholar]
- 252.Gono T & Kuwana M Current understanding and recent advances in myositis-specific and -associated autoantibodies detected in patients with dermatomyositis. Expert Rev Clin Immunol 16, 79–89 (2020). [DOI] [PubMed] [Google Scholar]