

Abstract
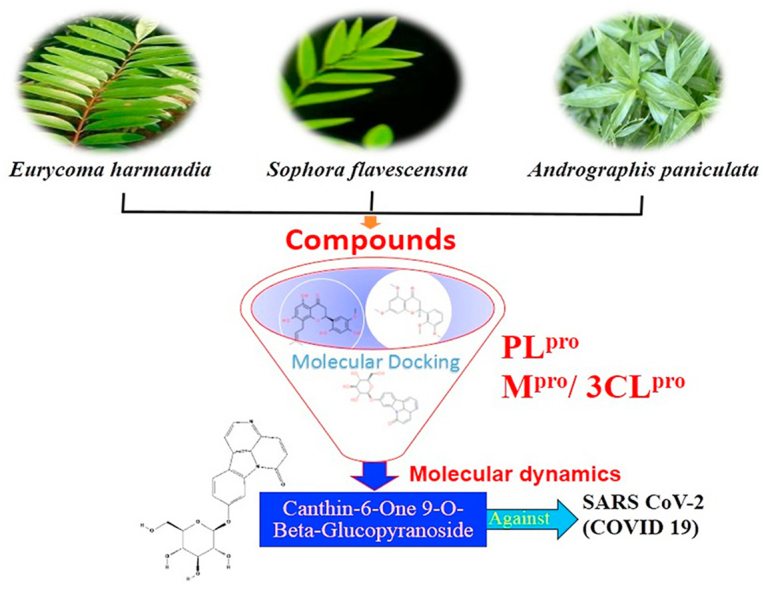
The rapid spread of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) - coronavirus disease 2019 (COVID-19) has raised a severe global public health issue and creates a pandemic situation. The present work aims to study the molecular -docking and dynamic of three pertinent medicinal plants i.e. Eurycoma harmandiana, Sophora flavescens and Andrographis paniculata phyto-compounds against SARS-COV-2 papain-like protease (PLpro) and main protease (Mpro)/3-chymotrypsin-like protease (3CLpro). The interaction of protein targets and ligands was performed through AutoDock-Vina visualized using PyMOL and BIOVIA-Discovery Studio 2020. Molecular docking with canthin-6-one 9-O-beta-glucopyranoside showed highest binding affinity and less binding energy with both PLpro and Mpro/3CLpro proteases and was subjected to molecular dynamic (MD) simulations for a period of 100ns. Stability of the protein-ligand complexes was evaluated by different analyses. The binding free energy calculated using MM-PBSA and the results showed that the molecule must have stable interactions with the protein binding site. ADMET analysis of the compounds suggested that it is having drug-like properties like high gastrointestinal (GI) absorption, no blood-brain barrier permeability and high lipophilicity. The outcome revealed that canthin-6-one 9-O-beta-glucopyranoside can be used as a potential natural drug against COVID-19 protease.
Keywords: SARS-CoV-2 (COVID-19), PLpro-Mpro/3CLpro, Canthin-6-one 9-O-Beta-glucopyranoside, Natural inhibitor, Molecular dynamics
Graphical abstract
1. Introduction
The illness caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is named coronavirus disease 2019 (COVID-19) (Zhang et al., 2020a; Mohamed and Fatima, 2020; Chaudhary and Janmeda, 2020). By 24th April 2021, beyond one hundred forty-four million sufferers were documented globally, with greater than three million sixty-six thousands of demises (223 countries, areas or territories with cases) which resume to rising, majorly in India, USA, Western Pacific, South-east Asia, Europe, Africa, and Eastern Mediterranean, as these are nations with a huge fragment of elderly inhabitants who have been infected with the SARS-CoV-2 (WHO, 2020; https://www.who.int/emergencies/diseases/novel-coronavirus-2019). SARS-CoV-2 is a non-segmented, enveloped, and positive-sense RNA virus. Structurally, SARS-CoV-2 comprises of four structural proteins including nucleocapsid protein (N), membrane glycol protein (M), small envelope glycoprotein (E), and spike protein (S). S protein generates homotrimers which are protruding on the surface of the virus and facilitate its binding to host cells by attracting angiotensin-converting enzyme 2 presented in lower respiratory tract cells. N protein is involved in the interaction with the viral genome and M protein and in virion assembly. M protein determines the shape of the viral envelope. Basically, it stabilizes the N protein and allows the completion of viral assembly by stabilizing the N protein-RNA complex. The last structural protein is E protein which plays an important role in the maturation and production of the virus (Astuti and Ysrafil, 2020). After successful fusion and entry, the viral genome is released and ORF1a and ORF1b of genomic RNA are translated in two kinds of big polyproteins (pp1a and pp1 ab) that are cleaved in several functional viral proteins by viral proteases, main protease or 3-chymotrypsin-like protease (Mpro/3CLpro) and papain-like proteases (PLpro) that are encoded in nsp5 and nsp 3. (Mitra et al., 2021; de Oliveira et al., 2020; Khan et al., 2020). pp1a and pp1ab are cleaved in into nonstructural proteins (1–11 and 1–10, and 12–16) which play an important role in various processes during the virus life cycle (V’Kovski et al., 2020). Mpro is recognized for its significant role in enzymatic activity which leads to post-translational processing of replicase protein. It consists of 306 amino acids and shows a great resemblance with SARS-CoV-2 Mpro. It also comprises of three domains (C-terminal domain-III, N-terminal domain-II, and N-terminal domain-I) and two catalytic dyads (H41 and C145) (Amin et al., 2021). PLpro is one of the main components of the SARS-CoV-2 replicase-transcriptase complex and its topological structure is divided into ubiquitin-like fingers, palm, and thumb. Basically, it carries out the processing of viral protein by identifying the Leu-X-Gly-Gly tertrapeptide motif and functions as de-ISGylating and de-ubiquitinating enzyme that inhibits the inflammatory signaling in the host cell (Li et al., 2021). Because of the pivotal roles and crucial function, the proteases perform in the life-cycle of the virus, they are a crucial target for the discovery of antiviral agents for SARS-CoV-2 (Arya et al., 2020; Kumar and Singh, 2020; Zhang et al., 2020b). At present, no drug has been invented, and hence because of the risk factors related to this illness, there is a critical requirement for a treatment (Enayatkhani et al., 2020). Currently, the idea of drug reuse is popularly utilized for the cure of variable disorders as it lessens the time, cost, and uncertainty of the drug generation operation by using a rapid computational perspective (Boopathi et al., 2019; Muralidharan et al., 2020).
Eurycoma harmandiana Pierre is a small plant, belongs to the Simaroubaceae family, allocated in the dividing line between Laos and Thailand (Kanchanapoom et al., 2001). Two alkaloids, 7-hydroxy-β-carboline 1-propionic acid and canthin-6-one 9-O-beta-glucopyranoside from the root part of E. harmandiana used as anti-viral agents (Kanchanapoom et al., 2001). Docking study of canthin-6-one 9-O-beta-glucopyranoside with NS1 protease to find out its effectiveness against the dengue virus, showed hydrogen bonds with the backbone and a side chain of SER80 and to THR87 and ASN130 side chains (ul Qamar et al., 2019). Sophora flavescens is a small Fabaceous plant, related to the Sophora genus. The roots of S. flavescens are termed as Kushen. It is broadly distributed in the regions of the Pacific Islands, Oceanica and the Asia, and has been greatly utilized as conventional herbal medication for the therapy of pain, scabies, myocarditis, cancer, dysentery, fever, and viral hepatitis (Kim et al., 2018). Many of the compounds were isolated from the roots of S. flavescens but out of them, kushenol W and kushenol K are utilized for carried the study of molecular docking in case of dengue virus. Andrographis paniculata (Burm. F) Nees, is an herbaceous plant, belongs to the Acanthaceae family. It has conventionally been utilized in China, Sri Lanka, India, and some other Southeast Asian countries. This plant is broadly acknowledged for its anti-inflammatory action against the infection of an upper respiratory tract (Mussard et al., 2019). Several compounds such as 3α, 14, 15, 18-Tetrahydroxy-5β, 9βH, 10α-labda-8, 12-dien-16-oic acid γ-lactone, deoxyandrographolide, neoandrographolide, 14-deoxy-11, 12-didehydroandrographolide, deoxyandrographolide19β-d-glucoside, 5,7,2′,3′-tetramethoxyflavanone, 5-Hydroxy-7,2′,3′-trimethoxyflavone, 14-Deoxy-11-oxoandrographolide, 5-Hydroxy-7,8,2′,3′-Tetramethoxyflavone, 5-Hydroxy-7,8,2′-Trimethoxyflavone, andrographine, panicoline, paniculide-A, paniculide-B, and paniculide-C have been reported from A. paniculata. According to JayaKumar et al. (2013) many of the physicians have manufactured several derivatives of andrographolide, which manifests remarkable remedial activities such as antiviral, antifeedant, anti-HIV, antidiabetic, antitumor, antibacterial, and anti-inflammatory. The concentration and composition of chemical constituents in plants are greatly affected by variable factors like harvesting condition, growth condition, genetic factors, types of extraction solvent, and concentration of solvents (Rafi et al., 2020). So, the present investigation is concentrated on the identification of natural composite with an exclusive objective to speed up the recognition of particular medication for the treatment of SARS-CoV-2.
2. Materials and methods
2.1. Medicinal plant and their natural components
A. paniculata, E. harmandiana and S. flavescens are highly valuable medicinal plants as reported by many researchers (ul Qamar et al., 2019; Mussard et al., 2019; JayaKumar et al., 2013; Rafi et al., 2020). They are mainly used as an herbal remedy for the treatments of lung, sore throat, flu and upper respiratory tract infections, and possess inhibitors activity of NS1, NS3/NS2B and NS5 proteins of dengue virus and anti-inflammatory, anti-tumor, anti-HIV, antibacterial, antifeedant, antidiabetic, antiviral & other pharmacological activities. The molecular properties of the natural components of three pertinent medicinal plants were retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) as listed in Supplementary Table 1 (Kim et al., 2016).
2.2. Protein preparation
The crystal structure of papain-like protease (PLpro), and 3-chymotrypsin-like protease (3CL pro/M pro) with PDB IDs 6W9C and 6M2N, respectively were retrieved from the Research Collaboratory for Structural Bioinformatics Protein Databank (RCSB PDB), https://www.rcsb.org/(Berman et al., 2000). All the crystal structures were prepared by removing existing ligands and water molecules by PyMOLv2.3.3, which is comprehensive molecular visualization software that enables users to observe the three-dimensional (3D) structures of the compounds (PyMOL, 2020; https://pymol.org/2/).
2.3. Ligand preparation
The 3D structures of phytochemicals and drugs were retrieved from the PubChem database, www.pubchem.ncbi.nlm.nih.gov (Kim et al., 2016). The ligands were then further converted into the.pdbqt format using an open babel tool for molecular docking (Dallakyan and Olson, 2015).
2.4. Protein-molecular docking and building complexes
Molecular docking approaches are the best techniques for forecasting the reactions between drugs and macromolecules. The blind docking process includes a quest for binding sites on the entire surface of the macromolecule. The blind molecular docking of some recognized medications was screened with SARS-CoV-2 proteases along with some bioactive natural compounds. Docking of the protein targets and ligands was carried out by using Auto-Dock Vina (Trott and Olson, 2010) and the interaction of the protein-ligand complex was visualized using PyMOL and BIOVIA Discovery Studio 2020 (Dassault Systèmes BIOVIA, 2020).
2.5. Absorption, distribution, metabolism, excretion and toxicity (ADMET)
The prediction of the ADMET properties plays an important role in drug discovery and development. Properties such as ADMET of compounds were determined using SWISS ADME (http://www.swissadme.ch/) and are represented in supplementary table 2. The 2D representation of the compounds was downloaded from the PubChem database (Berman et al., 2000).
3. Ligand parameterization
Before performing the molecular dynamics simulation of protein-ligand complex, parameterization of ligand molecule is necessary to generate its topology and parameter files which would be used during the simulation of the complex system. For the parameterization of ligand, PRODRG2 server was used (Schüttelkopf and van Aalten, 2004).
4. Molecular dynamics (MD) simulation
Two complex structures containing canthin-6-one 9-O-beta-glucopyranoside (ligand) bound with 3-chymotrypsin-like protease (Mpro/3CLpro) and papain-like protease (PLpro) of SARS-COV-2 were taken to perform the MD simulation. All MD simulations were conducted using the GROMACS 5.12 MD (Páll et al., 2015) and GROMOS96 43a1 force field (van Gunsteren et al., 1996) implemented on Intel Xeon Quad Core W3530 2.8 8 M 1366 Processor with LINUX environment. The Mpro/3CLpro –ligand complex was solvated in a cubic box with a dimension of 9.96146 X 9.96146 × 9.96146 nm3 while the PLpro-ligand complex was solvated in a box with a dimension of 10.85766 X 10.85766 × 10.85766 nm3. The simulation box of two complexes, Mpro/3CLpro –ligand and PLpro-ligand showed to contain 31154 and 40531 SPC216 water molecules respectively (Miyamoto and Kollman, 1992). Four water molecules were replaced by Na + to neutralize the net charge of the complex system Mpro/3CLpro –ligand while for the neutralization of PLpro-ligand complex 3 Cl- were needed to replace solvent molecules. All protein atoms were maintained at a distance equal to 1.0 nm from the solvate box edges. The solvated systems were subjected to energy minimization for a maximum of 50000 steps by the steepest descent minimization method. After performing the energy minimization, both the minimized systems were equilibrated for 100 ps at 300K temperature by position restrained MD simulation in order to maintain the pressure and temperature of systems. Following equilibration, both the systems were subjected to a final production run of 100ns MD simulations at 300K temperatures. Periodic boundary conditions were applied under isothermal and isobaric conditions using Berendsen Coupling algorithm with a relaxation time of 0.1 and 0.2 ps, respectively (Weber et al., 2000). In both the systems, bond length was constrained using the LINCS algorithm at a time step of 2 fs (Hess et al., 1997). Particle Mesh Ewald method was used for the analyzing the electrostatic interactions. van der Waals and coulombic interactions were calculated with a cutoff at 1.0 nm (Darden et al., 1999). The tools provided by GROMACS program package such as RMSD, RMSF were utilized to analyse the MD trajectories. XMGrace (Vaught et al., 1996bib_Vaught_1996bib_Vaught_1996bib_Vaught_1996) program implemented in GROMACS was used to prepare the graphs.
5. Calculation of binding free energy
Binding affinity of the ligand canthin-6-one 9-O-beta-glucopyranoside in the docked complexes with the enzymes Mpro/3CLpro and PLpro has been individually computed using a parallelized metadocking method implemented in the DINC server (Antunes, 2017). Coordinates of protein-ligand complexes as obtained from the MD simulation trajectories at an interval of 10 ns time scale were used for the calculation of binding energy. The working principle of the DINC server is based on the algorithm used in AutoDock-Vina. The grid center and dimension determined for each protein-ligand complex from the previous Autodock grid parameterization have been used during the calculation of binding energy.
6. MM-PBSA calculation
Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) calculation was performed for the canthin-6-one 9-O-beta-glucopyranoside bound Mpro/3CLpro and PLpro system using the mmpbsa.py package implemented in GROMACS version 2018.1 (Kumari et al., 2014).
7. Result and Discussion
The novel coronavirus has a viral genome that encodes for the several proteins, which includes two proteases PLpro and Mpro/3CLpro that play a crucial role in the viral life cycle and gained tremendous attention as a drug target. In the current study, these two proteases have been taken as a potential target. Eighteen phyto-compounds from the three medicinal plants viz. E. harmandiana, S. flavescens and A. paniculate were considered for the molecular docking analysis (Supplementary table 1). Chloroquine, oseltamivir, remdesivir and ribavirin has been used as a control. These drugs have been recently used in the treatment of COVID-19 patients and have been evaluated in clinical trials. There is no potential drug molecule approved by the FDA and this is the only reason that these repurposed drugs have been taken as a control in the current study. Oseltamivir in combination with hydroxychloroquine is also found to inhibit the coronavirus to some extent (Mitjà and Clotet, 2020; Wu et al., 2020). Remdesivir and ribavirin are the FDA-approved anti-RdRp drugs that were found through molecular docking approach (Pirzada et al., 2021; Hasan et al., 2021). Binding energies of various above-mentioned naturally occurring phytochemicals were obtained through Auto-Dock Vina. The interaction of inhibitor and protein receptor was predicted in 2D and 3D, with the help of BIOVIA Discovery study visualizer. The docked poses clearly showed that drug molecules and phyto-compounds bound to the active sites of SARS-COV-2 macromolecule structures.
7.1. Interaction with PLpro
Panicoline bound firmly with the residue GLY160 through a conventional hydrogen bond, GLN269 through carbon-hydrogen bond and GLY160 via. van der Waals interactions with the surrounding residues of the PLpro protein of SARS-CoV-2 (Fig. 1a). The 14-Deoxy-11,12-didehydroandrographolide stabilized the active site through conventional hydrogen bond (GLN269), carbon-hydrogen bond (GLY160) and van der Waals interactions with the rest of the residues (Fig. 1b). Canthin-6-one 9-O-beta-glucopyranoside appended within the active site of SARS-CoV-2 through pi-anion interaction with residues GLU161, conventional hydrogen bond with residue LEU162, GLY160, ASN109, Pi donor hydrogen bond with LEU162 and van der Waals interactions with the rest of the residues (Fig. 1c). Andrographine secured the active sites with amide-pi stacked interaction with residues GLY161, conventional hydrogen bond with ASN109, carbon hydrogen bond with ASP108, pi-alkyl, alkyl interaction with the residues LEU162, HIS89, VAL159 and van der Waals interactions with the surrounding residues (Fig. 1d). Andrographolide interacted with the active sites through conventional hydrogen bonds (VAL159 and GLN269) and with van der Waals interactions with the rest of the residues as shown in Fig. 1e. 5-Hydroxy-7,8,2′,3′-Tetramethoxyflavone firmly stabilized the active sites through amide pi-stacked interaction with residues GLY160, conventional hydrogen bond with ASN109, carbon hydrogen bond with ASP108, GLN269, further it underwent alkyl, pi-alkyl interactions with residues LEU162, VAL159, HIS89 and through van der Waals interactions with the rest of the residues (Fig. 1f). Neoandrographolide bound with the active sites through conventional hydrogen bonds (ASN109, GLN269, VA159) and carbon hydrogen bond (GLY160, THR158) (Fig. 1g). Paniculide-A stabilized the active site of the PLpro through alkyl interactions with residues VAL159 and conventional hydrogen bond with residues ASN109 (Fig. 1h). The 5-Hydroxy-7,2′,3′-Trimethoxyflavone bound with the active sites with residues LEU162, VAL159, HIS89 through alkyl, pi-alkyl interactions, pi-anion interactions with residue GLY160, conventional hydrogen bonds with residues THR158, ASN109 and carbon-hydrogen bond with residues ASN109 and with rest of the residues through van der Waals interactions as shown in Fig. 1i. 5,7,2′,3′-tetramethoxyflavanone formed several noncovalent interactions at the active site, it formed a conventional hydrogen bond with THR158, ASN109, carbon-hydrogen bond with residue GLN269, alkyl and pi-alkyl bond with LEU162, HIS89, VAL159 and pi-anion interaction with residue GLU161 (Fig. 1j). Deoxyandrographolide stabilized the active site only through a single conventional hydrogen bond (GLN269) and surrounding residues with van der Waals interaction (Fig. 1k). Kushenol W formed pi-sigma interactions with GLU161, pi alkyl bond with LEU162, conventional hydrogen bond with GLY160, carbon-hydrogen bonds with residues ASP108 and GLY160 (Figure 1l). The kushenol K formed conventional hydrogen bonds with LEU162, and ASN109, carbon-hydrogen bond with VAL159 and van der Waals interactions with the surrounding residues (Figure 1m). 5-Hydroxy-7,8,2′-trimethoxyflavone 5-glucoside secured the active site through amide pi stacked interactions with residues GLY160, pi-anion interaction with GLU161 and conventional hydrogen bond with GLN269, and ASN109 (Figure 1n).
Fig. 1.







Interactions established after docking the secondary metabolite against SARS-CoV-2 PLpro Protein (6W9C). The interacting residues of the protein are labeled in purple and the docking scores are listed under each of the complex, respectively. The receptor-ligand interaction is represented on a 3D diagram (Right) and 2D diagram (Left). Drugs are in cyan and interacting atoms of protein are represented red in the diagram, while green dotted lines represent the conventional h-bond interactions, light green dotted line represents weak van der Waals interactions. Additionally, dotted lines in sky blue display the pi-donor hydrogen bond, pi-sigma interaction is shown as violet dashed lines, pink dotted lines show alkyl and pi-alkyl interactions, respectively.
Deoxyandrographolide19β-D-glucoside bound active sites through a conventional hydrogen bond with residue THR158, carbon hydrogen bonds with residues GLY160, ASP108 and alkyl bond with residue LEU162 and it is depicted in form of 2D representation in Fig. 1o. Paniculide-B formed alkyl bond with VAL159, conventional hydrogen bond with ASN109, GLY160, and GLN269, and carbon hydrogen bond with GLN269 and where paniculide-C secured the active sites with alkyl (LEU162), and conventional hydrogen bond (ASN109) (Figure 1p and q). The interaction analysis of 14-Deoxy-11-oxoandrographolide depicted that it bound the active sites through conventional bond with residues LEU162, ASN109 and GLN269 (Figure 1r).
Van der wall interactions and hydrogen bonds played a major role in the binding process. van der Waals interaction is the weakest intermolecular attraction between two molecules. Though it is the weakest bond between two atoms, a lot of van der Waals forces can make the interaction very strong (Barratt et al., 2005). In the protein-ligand interactions, the hydrogen bond helped the ligand to stabilize but there are also other interactions such as hydrophobic or van der Waals interactions that helped in stabilization of the nonpolar ligands too. An analysis of the structure of ligands and docked poses usually helped in understanding the interactions and the binding energy of the compounds. Based on these basics, the molecular docking program is the most widely used tool in the drug discovery process as the result suggested the potential drug compound based on the less affinity (Meng et al., 2011). The comparison of the estimated free energy of binding (ΔG) or binding affinity inferred that the canthin-6-one 9-O-beta-glucopyranoside (−9.4 kcal/mol) have the less affinity among the studied plant natural components and can be used as a potential drug candidate against the PLpro of SARS-CoV-2 (Table 1).
Table 1.
Tabulated total free energy (kcal/mol) of docked protein PLpro.
| Compound name | Binding affinity (kcal/mol) |
|---|---|
| Canthin-6-one 9-O-beta-glucopyranoside | −9.4 |
| 5-Hydroxy-7,8,2′-trimethoxyflavone 5-glucoside | −8.8 |
| 14-Deoxy-11-oxoandrographolide | −8.7 |
| Kushenol K | −8.6 |
| Neoandrographolide | −8.6 |
| Kushenol W | −8.4 |
| 14-Deoxy-11,12-didehydroandrographolide | −8.2 |
| Deoxyandrographolide | −8.1 |
| Paniculine | −8.0 |
| Andrographolide | −7.9 |
| Deoxyandrographolide19β-D-glucoside | −7.9 |
| 5-Hydroxy-7,8,2′,3′-Tetramethoxyflavone | −7.6 |
| Andrographine | −7.6 |
| 5,7,2′,3′-tetramethoxyflavanone | −7.5 |
| 5-Hydroxy-7,2′,3′-Trimethoxyflavone | −7.5 |
| Paniculide-A | −7.1 |
| Paniculide-B | −7.2 |
| Paniculide-C | −6.7 |
The analysis of binding energies suggested that all the binding energy falls between −6.7 and 9.4 kcal/mol and 2D interaction in Fig. 1a-r depicted that the studied phyto-compounds have a lot of hydrogen bonds and van der Waals interactions that suggested that the ligands are stabilized within the complex.
7.2. Interaction with Mpro/3CLpro
Paniculine bound the active site of the main proteases (Mpro or 3CLpro) through hydrogen bonds and carbon hydrogen bonds with residues ASN151, THR111, GLN110, SER158 and ILE152, respectively (Fig. 2a). The 14-deoxy-11,12-didehydroandrographolide formed a hydrogen bond with residues THR111 and pi-alkyl interaction with ILE249 and van der Waals interaction with the surrounding residues (Fig. 2b). Canthin-6-one 9-O-beta-glucopyranoside firmly bound with the active site through conventional hydrogen bonds with residues THR111, ASP295, and THR292 and a pi-alkyl bond with VAL202 (Fig. 2c). Andrographine bound within the active sites through conventional hydrogen bond with the residue THR292, pi-pi stacked interaction with the residue PHE294 and pi-alkyl bonds with PRO293 and ILE294 (Fig. 2d). Andrographolide formed conventional hydrogen bond with residue ASN151, pi-sigma bond with residue PHE294 and van der Waals interaction with the surrounding residues (Fig. 2e). Neoandrographolide formed conventional hydrogen bond with the residues ASP295, THR111 and pi alkyl interaction with residues VAL202, PRO293 and ILE249 (Fig. 2f). Paniculide-A bound the active sites of the main protease (MPRO or 3CLPRO) with conventional hydrogen bond and alkyl, pi-alkyl interactions with the residues GLN110, THR111, THR292 and ILE249, PHE294, PHE8, respectively (Fig. 2g). 5-Hydroxy-7,8,2′,3′-Tetramethoxyflavone adhered to the active sites through conventional hydrogen bonds (ASN151 and THR111), pi donor hydrogen bond (THR292), pi-pi-stacked (PHE294), pi-alkyl interactions with residues PRO293, ILE294, and PHE294, depicted in Fig. 2h. 5,7,2′,3′-tetramethoxyflavanone formed Pi alkyl interactions, conventional hydrogen bonds pi-pi stacked interactions with residues ILE249, THR111 and PHE294, respectively (Fig. 2i). 5-Hydroxy-7,2′,3′-Trimethoxyflavone stabilized the active sites through conventional hydrogen bonds (ASN151 and THE111), pi donor hydrogen bond (THR292), pi-pi stacked interaction (PHE294), alkyl, pi-alkyl interaction (PHE294, ILE249, HIS246 and PRO293) and van der Waals interactions with the surrounding residues and the interactions can be seen in Fig. 2j. With a good efficiency of binding, deoxyandrographolide formed conventional hydrogen bond with ASN151 and pi-alkyl with PHE249 (Fig. 2k). Kushenol W bound with active sites residues THR111 through conventional hydrogen bond, pi-pi stacked interaction (PHE294) and pi alkyl interactions (ILE249 and PRO293) (Figure 2l). 5-Hydroxy-7,8,2′-trimethoxyflavone 5-glucoside formed conventional hydrogen bond with residue GLN110, pi-sigma interaction (PHE294), pi-pi interaction with PHE294, alkyl, pi-alkyl interaction with VAL297, PRO293, ILE249 and carbon hydrogen bond with ILE249 (Figure 2m). Kushenol K stabilized the active site with residues PHE294 through pi-pi stacked interaction, pi-pi sigma interaction (ILE249) and pi-alkyl interactions (PRO293 and PHE294) (Figure 2n). The deoxyandrographolide19β-d-glucoside formed conventional hydrogen bond with residue THR21, THR26, GLN69, ILE249 and carbon hydrogen bond with ASP245 (Figure 2o). Paniculide-B bound with the active sites through conventional hydrogen bonds (THR292, THR111 and GLN110) and alkyl interaction (PHE8 and PHE294) whereas paniculide-C formed conventional hydrogen bonds with THR111 and GLN110; alkyl and pi alkyl interactions with PRO293, HIS246, ILE249, VAL202 and the interactions are depicted in Fig. 2(p and q). In the case of 14-Deoxy-11-oxoandrographolide, it stabilized the active sites with residue GLN269, ASN109 and LEU162 through conventional hydrogen bond, alkyl bond with ILE249 and through van der Waals interaction with the rest of the residues (Figure 2r). The lower affinity of canthin-6-One 9-O-Beta-glucopyranoside (−8.5 kcal/mol) obtained from molecular docking study suggested that the molecule have good binding affinity against the Mpro/3CLpro (Table 2). The 2D and 3D interaction are depicted in Fig. 2 (a-r).
Fig. 2.







Interactions established after docking the secondary metabolite against SARS-CoV-2 Mpro/3CLpro Protein (6M2N). The interacting residues of the protein are labeled in purple and the docking scores are listed under each of the complex, respectively. The receptor-ligand interaction is represented on a 3D diagram (Right) and 2D diagram (Left). Drugs are in cyan and interacting atoms of protein are represented red in the diagram, while green dotted lines represent the conventional h-bond interactions, light green dotted line represents weak van der Waals interactions. Additionally, dotted lines in sky blue display the pi-donor hydrogen bond, pi-sigma interaction is shown as violet dashed lines, pink dotted lines show alkyl and pi-alkyl interactions, respectively.
Table 2.
Tabulated total free energy (kcal/mol) of docked protein Mpro/3CLpro.
| Compound name | Binding affinity (kcal/mol) |
|---|---|
| Canthin-6-One 9-O-Beta-Glucopyranoside | −8.5 |
| Neoandrographolide | −8.4 |
| Kushenol W | −7.3 |
| 14-deoxy-11,12-didehydroandrographolide | −7.1 |
| 14-deoxy-11-oxoandrographolide | −7.1 |
| 5-hydroxy-7,8,2′-trimethoxyflavone 5-glucoside | −6.9 |
| Andrographolide | −6.9 |
| 5-hydroxy-7,2′,3′-trimethoxyflavone | −6.8 |
| 5,7,2′,3′-tetramethoxyflavanone | −6.5 |
| 5-hydroxy-7,8,2′,3′-tetramethoxyflavone | −6.5 |
| Andrographine | −6.5 |
| Paniculine | −6.5 |
| Kushenol K | −6.4 |
| Deoxyandrographolide19β-d-glucoside | −6.2 |
| Diterpene glucoside | −6.2 |
| Paniculide-A | −6.1 |
| Paniculide-B | −5.9 |
| Paniculide-C | −6.0 |
7.3. Interaction with control drugs
The docked pose of minimum energies conformers of four control drugs viz. chloroquine, oseltamivir, remdesivir and ribavirin [Fig. 3 (a-d)] showed chloroquine binds firmly with PLpro active site and formed a conventional hydrogen bond with THR158 residue and carbon-hydrogen bonds with residue GLU161 and VAL159. Oseltamivir bound within the active site through conventional bonds with residue ASN109, carbon-hydrogen bond with residue GLY160 and alkyl interaction with residue LEU162. Remdesivir bound the active site through pi-anion (GLU161), amide-pi stacked interaction (VAL159 and GLY160), conventional a hydrogen bond (ASN109) and pi-alkyl interaction (LEU162). Ribavirin formed conventional hydrogen bond with residue GLY160, GLN269, LEU162, and ASN109, carbon-hydrogen bond with residue GLN269, and ASN109 and among all the interactions van der waals interaction plays a major role. The comparison of binding affinity or estimated free energy (ΔG value) of binding showed that the remedisivir (−8.4 kcal/mol) have lower affinity with active sites of the PLpro. The order of binding energy of other drugs with PLpro is as follows: ribavirin (−6.8 kcal/mol) > oseltamivir (−6.4 kcal/mol) > chloroquine (−6.3 kcal/mol).
Fig. 3.


Interactions established after docking the drugs against SARS-CoV-2 PLpro protein (6W9C). The receptor-ligand interaction is represented on a 3D diagram (Right) and 2D diagram (Left). The interacting residues of the protein are labeled in purple and the docking scores are listed under each of the complex, respectively. Drugs are in cyan and interacting atoms of protein are represented red in the diagram, while green dotted lines represent the conventional h-bond interactions, light green dotted lined represents weak van der Waals interactions. Additionally, dotted lines in sky blue display the pi-donor hydrogen bond, pi-sigma interaction is shown as violet dashed lines, pink dotted lines show alkyl and pi-alkyl interactions, respectively.
Interactions of the drugs with the Mpro/3CLpro showed that chloroquine firmly bound within the active sites through pi-anion interaction with the residues ASP295, pi-pi stacked interaction with residues PHE294 and pi-alkyl interaction with residue PHE294 and carbon-hydrogen bond with residue THR111. It did not form any hydrogen bonds within the active site. Ribavirin formed a conventional hydrogen bond with residues GLN110, PHE294, THR111, and THR292. Oseltamivir stabilized the pi-alkyl interaction with PHE294 and conventional hydrogen bonds with residues GLN110, THR111 and ASN151. Remdesivir bound the active site through conventional hydrogen bonds (GLN110, ASN151 and PHE294) and pi-alkyl interaction (ILE249, PRO252, VAL297 and PRO293) with the active sites of Mpro/3CLpro. The pi-interaction played a major role instability of a particular drug within the active site (Arthur and Uzairu, 2019). The binding affinity of the drugs with Mpro/3CLpro suggested that the remdesivir have high affinity to inhibit the protein with most negative ΔG value i.e. - 8.3 kcal/mol and all the other studied drugs showed similar ΔG value i.e. −5.8 kcal/mol, the 3D and 2D interactions of drugs with the active sites of Mpro/3CLpro are depicted in Fig. 4 (a-d).
Fig. 4.


Interactions established after docking the drugs against SARS-CoV-2 Mpro/3CLpro Protein (6M2N). The interacting residues of the protein are labeled in purple and the docking scores are listed under each of the complex, respectively. The receptor-ligand interaction is represented on a 3D diagram (right) and 2D diagram (left). Drugs are in cyan and interacting atoms of protein are represented red in the diagram, while green dotted lines represent the conventional h-bond interactions, light green dotted lined represents weak van der Waals interactions. Additionally, dotted lines in sky blue display the pi-donor hydrogen bond, pi-sigma interaction is shown as violet dashed lines, pink dotted lines show alkyl and pi-alkyl interactions, respectively.
7.4. Pharmacokinetics
The prediction of the absorption, distribution, metabolism, excretion and toxicity (ADMET) properties played an important role in drug discovery and development. The absorption of drugs depends on membrane permeability, intestinal absorption, skin permeability levels, P-glycoprotein substrate or inhibitor. The distribution of drugs depends on factors that include the blood–brain barrier (logBB), central nervous system (CNS) permeability, and the volume of distribution (VDss). Metabolism is predicted based on the CYP models for substrate or inhibitors (CYP2D6, CYP3A4, CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4). Excretion is predicted which is based on the total clearance model and renal OCT2 substrate. The toxicity of drugs is predicted based on Ames toxicity, human ether-a-go-go-related gene (hERG) inhibition, hepatotoxicity, and skin sensitization. These parameters were calculated and checked for compliance with their standard ranges (Han et al., 2019). Properties such as ADMET profiling of compounds were determined using SwissADME, http://www.swissadme.ch/(Daina et al., 2017). Intestinal epithelium barrier/gut–blood barrier is a barrier that is a crucial barrier for all the compounds to overcome. Intestinal epithelium barrier regulates nutrients absorption, water and ion fluxes, and denotes the first defensive barrier against toxins and enteric pathogens and the gut-blood barrier (GBB) controls the passage of drugs from intestinal lumen to the bloodstream. Except, kushenol K, GI absorption of all the phytochemicals was high which means that it was predicted to be absorbed easily in gut and intestine epithelium. Distribution of compounds through various compartments of the body was accessed using its blood-brain barrier (BBB) penetration (Daneman and Prat, 2015). Among all the phytocompounds, only panculide-A, andrographine, deoxyandrographolide, 5,7,2′,3′-tetramethoxyflavanone, 5-Hydroxy-7,2′,3′-Trimethoxyflavone were predicted to have blood brain barriers penetration. P-glycoprotein is a drug transporter having broad substrate specificity, drugs that are substrates of P-gp are subject to low intestinal absorption, low blood-brain barrier permeability, and face the risk of increased metabolism in intestinal cells. 14-deoxy-11,12-didehydroandrographolide, 5-hydroxy-7,8,2′-trimethoxyflavone 5-glucoside, andrographolide, paniculine A, neoandrographolide, kushenol K, 14-deoxy-11-oxoandrographolide, and deoxyandrographolide were predicted to have affinity for p-glycoprotein. The Result showed that except these phytochemicals, rest of the other phytochemicals may act as a non-substrate of p-glycoprotein (Pajouhesh and Lenz, 2005). Metabolism mainly depends on the CYP450 enzyme and its iso forms which are CYP 3A4, 2D6, 1A2, 2C9 and 2C19. These enzymes are responsible for the detoxification of drugs passing the liver. Therefore, any compound blocking P450 can cause toxicity. Out of all the 18 phyto-compounds, 5-hydroxy-7,8,2′-trimethoxyflavone 5-glucoside, neoandrographolide, kushenol W, kushenol K, andrographine, 5-Hydroxy-7,8,2′,3′-tetramethoxyflavone, 14-deoxy-11,12-didehydroandrographolide, 5,7,2′,3′-tetramethoxyflavanone, and 5-hydroxy-7,2′,3′-trimethoxyflavone compounds blocked the enzyme responsible for detoxification and hence, can be responsible for the toxicity. Skin permeation coefficient (log Kp) is the measure of skin to absorb a certain drug or chemical. In the given supplementary table 2, log kp of skin permeation of all the compounds lies between the standard value of log Kp −8.0 to −1.0 cm/s except 5-Hydroxy-7,8,2′-trimethoxyflavone 5-glucoside, deoxyandrographolide19β-D-glucoside, and paniculine, which showed that these possess an ability to absorb less in the membrane (Chen et al., 2018).
ADMET study of all these molecules suggested that the molecules canthin-6-one 9-O-beta-glucopyranoside, deoxyandrographolide 19β-d-glucoside or diterpene glucoside, paniculide-B, and paniculide-C have passed all the barriers of ADMET and indicated that these molecules can be a successful drug molecule. All these compounds have high gastrointestinal absorption which is an important and decision making standard of oral dosing, this implied that the phyto-compounds can be used as an oral drug. These drugs also passed the blood brain barrier (BBB) permeability test, therefore, these compounds qualify a fundamental index of drug distribution. High negative value of log Kp suggested that the phyto-compounds have less skin permeation. Binding of CYP450 enzyme may cause degradation of phyto-compounds inside the body and can lead to toxicity. Bioavailability of these compounds are high and it indicated high potential as drug molecule. The above mentioned parameters for all these molecules are in the range found for successful drug molecule (Supplementary table 2).
7.5. Potential drug against COVID 19
The molecular docking studies have been carried out using 18 phyto-compounds that might act as potential drug against SARS-CoV-2 Mpro/3CLpro and PLpro. The analysis of molecular interaction between the active sites of the studied protein and the phyto-compounds suggested that these compounds can inhibit the protein and will block the viral replication. The active site of Mpro/3CLpro consists of residues THR21, THR26, GLN69, GLN110, THR111, ASN151, HIS246, ILE249, THR292 and ASP295 whereas the active site residues for PLpro are ASN109, THR158, VAL159, GLY160, LEU162 and GLN269 (Supplementary table 3). The canthin-6-one 9-O-beta-glucopyranoside in comparison with the control drugs, showed lower affinity with active sites of both the target proteins i.e. Mpro/3CLpro and PLpro. The distance between the ligand and catalytic residues of the protein showed that the canthin-6-one 9-O-beta-glucopyranoside interacted within the active site of the protease proteins and have high affinity of binding. The ADMET study also suggested that the canthin-6-one 9-O-beta-glucopyranoside have all the drug like properties and can be considered as a potential drug against the infection caused by SARS-CoV-2.
8. MD simulation
8.1. Root mean Square deviation (RMSD)
Structural changes specifically the deviation between two structures can be best interpreted by using the graph obtained after calculating RMSD from the MD simulation trajectories. This calculation can determine the spatial differences between the backbone atoms present in the protein throughout the simulation time. The differences are calculated in respect with the initial structure at the start of the simulation. The smaller the differences, the more spatially equivalents the compared structures are, whereas more distantly related structures are defined by comparatively greater RMSD values. Thus, a greater RMSD value indicates the instability of a protein system.
Comparative RMSD pattern analysis between canthin-6-One 9-O-beta-Glucopyranoside bound Mpro/3CLpro and PLpro protein complexes demonstrated that, both complexes showed a more or less similar RMSD values up to first 19 ns of MD trajectory. After 19 ns of MD simulation, there is a comparative more deviation observed in case of PLpro protein than Mpro/3CLpro up to 32 ns time scale of MD simulation. After 32 ns and up to next 3 ns time scale of MD trajectory, relatively less RMSD has been recorded for PLpro protein than from Mpro/3CLpro when interacting with canthin-6-One 9-O-beta-Glucopyranoside. Again a greater RMSD value of PLpro protein backbone has been shown for next 13 ns and up to 48 ns of MD simulation compared to Mpro/3CLpro protein. From 48 ns to 100 ns of MD simulation, the backbone of PLpro protein deviated less than in case of Mpro/3CLpro protein (Supplementary Figure 1). The Result indicated that Mpro/3CLpro and PLpro protein have the occasional backbone stability throughout the MD simulation while bound with the ligand Canthin-6-One 9-O-beta-Glucopyranoside.
8.2. Root mean Square Fluctuation (RMSF)
A more detailed view of differences in residue mobility in both the Mpro/3CLpro protein and PLpro bound with ligand canthin-6-One 9-O-beta-Glucopyranoside has been obtained from the RMSF graph calculated for the Cα atoms in comparison with the respective average structure of the two complexes throughout the simulation. RMSF values increases with the depletion of secondary structure of a protein that sometime may be caused due to the decline in binding affinity between the protein and ligand. It has been noted that the ligand binding residue in Mpro/3CLpro Asn203 has the minimum RMSF value of 0.0881 among the 5 interacting residues of canthin-6-One 9-O-beta-Glucopyranoside while in PLpro protein, the residue Asn109 has the minimum RMSF value of 0.1038 among the 3 interacting residues with the ligand (Supplementary Figure 2; Supplementary table 4).
This relative Result from RMSF analysis revealed that Asn203 residue of the protein Mpro/3CLpro might have greater impact for a tight binding with the ligand canthin-6-One 9-O-beta-Glucopyranoside in compare with the residue Asn109 in PLpro protein as the residue Asn203 has comparatively lower RMSF value than that of Asn109. Bharatiy et al. (2016) has studied some stabilizing and destabilizing saltbridge forming residues of two homologues mesophilic and thermophilic α-carbonic anhydrase (α-CA) from Neisseria gonorrhoeae and Sulfurihydrogenibium sp. on aspect of their respective RMSF value. Likewise, as the result from the present study, Bhartiy et al. (2016) also reported about some salt bridge residues having notably less RMSF values in maintaining higher conformational stability of Sulfurihydrogenibium sp. α-CA at higher temperature compared to its mesophilic counterpart.
8.3. Free energy calculations
Comparative binding calculation between Mpro/3CLpro and PLpro bound with ligand Canthin-6-One 9-O-beta-Glucopyranoside throughout the 100 ns MD simulation presented that, the protein Mpro/3CLpro has comparatively more affinity with the ligand in relation to PLpro at most of the time scale in MD simulation. Binding energy calculation at the start of the simulation between two complexes of canthin-6-One 9-O-beta-Glucopyranoside bound Mpro/3CLpro and PLpro revealed that the Mpro/3CLpro protein has an affinity value of −8.70 kcal/mol which is much higher than that of PLpro with a value of −4.80 kcal/mol. While at the next two 10 ns time scale intervals, PLpro showed higher binding affinity (−8.20 and −5.40 kcal/mol) with the ligand in comparison to Mpro/3CLpro (−5.80 and −4.20 kcal/mol). Interestingly, at 30 ns and 40 ns of time scale in MD simulation, again Mpro/3CLpro protein exhibited comparative greater binding affinity (−5.00 and −5.20 kcal/mol) compared to PLpro (−3.40 and −4.90 kcal/mol). At 50 ns, a relatively higher binding energy towards ligand has been recorded for PLpro with a value of −8.10 kcal/mol than in the case of Mpro/3CLpro with a value of −7.60 kcal/mol. Significant difference in binding energy between Mpro/3CLpro and PLpro has been reported while interacting with the ligand at the time scale of 60 ns, 80 ns and 90 ns in MD simulation. For the protein Mpro/3CLpro binding energy values of −9.80, −9.00 and −10.50 kcal/mol has been calculated in place of −3.40, −3.60 and −3.60 kcal/mol for PLpro respectively at those three time scales. After the completion of the 100 ns MD simulation, a greater binding energy of −10.00 kcal/mol has been documented for Mpro/3CLpro towards the ligand canthin-6-one 9-O-beta-glucopyranoside while for PLpro a value of −8.20 kcal/mol was recorded (Fig. 5).
Fig. 5.

Comparative binding energy of canthin-6-one 9-O-beta-glucopyranoside with Mpro/3CLpro and PLpro (∗energy values are the smaller in Mpro/3CLpro and PLpro in respective time scale).
Binding energy vs. RMSD plots have been graphically represented for Mpro/3CLpro and PLpro while interacting with the ligand canthin-6-one 9-O-beta-glucopyranoside at 10 ns time interval up to 100 ns MD simulation (Fig. 6). Comparative higher binding affinity of Mpro/3CLpro with the ligand than PLpro is also might be resulted from the relatively less flexible residue Asn203 present in the protein Mpro/3CLpro which has lower RMSF value as compare to Asn109 of PLpro that was determined previously. Thus, the overall Result from this binding energy analysis suggested that, Mpro/3CLpro has remarkably greater binding affinity and hence stronger interaction with the ligand canthin-6-one 9-O-beta-glucopyranoside than PLpro throughout most of the time scales in their dynamics state.
Fig. 6.

Comparative RMSD vs. binding energy plots of canthin-6-One 9-O-beta-Glucopyranoside bound with Mpro/3CLpro and PLpro calculated from MD simulation trajectory.
8.4. MM-PBSA interaction energy
From the MM-PBSA calculation, it can be observed that in case of canthin-6-One 9-O-beta-Glucopyranoside bound with Mpro/3CLpro, there is comparatively higher degree of energy contribution than in the case of PLpro bound complex of the same ligand (Fig. 7). This observation is in agreement with the Result found from the previously performed RMSD vs. binding energy plots for the two protein complexes where Mpro/3CLpro presented higher binding affinity towards the ligand canthin-6-One 9-O-beta-Glucopyranoside. Shahbaaz et al. (2017), in their study has performed the same kind of MM-PBSA analysis for the protein Rab21 GTPase during the interaction with both the GDP and GTP through 50ns MD simulation. In their study, comparatively greater energy contribution has been recorded for the ligand GTP than the GDP during their interaction with Rab21 GTPase.
Fig. 7.

Plot showing MM-PBSA calculation of canthin-6-One 9-O-beta-Glucopyranoside bound with Mpro/3CLpro (black) and PLpro (red) calculated from MD simulation trajectory.
Das et al. (2020) studied 33 molecules through the help of docking approach, and find out that these molecules could bind near the Cys145 and His41 (catalytic residue) of the main protease. Salim and Noureddine (2020) reported that α-hederin and nigellidine are the major phytochemical obtained from Nigella sativa which are capable of restricting SARS-CoV-2 with great energy score as compared to clinical drugs based on molecular docking. Muralidharan et al. (2020) performed MD simulations to evaluate the interconnection among the protein and three drugs. The RMSD of interconnection is found to be 3 Å and remain firm while the simulations. Therefore, the amalgam of ritonavir, lopinavir, and oseltamivir are greatly efficacious against the protease of SARS-CoV-2, and these agents can be surveyed further for drug reusing purposes for the victorious restriction of SARS-CoV-2. Joshi et al. (2020) identified some natural compounds such as phyllaemblicin B, hesperidin, biorobin, afzelin, nympholide A, lacticopicrin 15-oxalate, myricitrin, nympholide A, and d-viniferin from the screening of ~7100 molecules which proved as a strong binders not only for proteases but also for other targets (RNA dependent RNA polymerase, and human angiotensin-converting enzyme of SARS-CoV-2. Adeoye et al. (2020) determined the inhibitory potentiality of chloroquine, lopinavir, remdesivir, ribavirin, azithromycin, and oseltamir towards viral proteases, V-ATPase, SARS-CoV spike glycoprotein/ACE-2 complex, and protein kinase A. It was evaluated that, lopinavir has the greatest affinities for the 3-chymotrypsin like protease, cyclic AMP-dependent protein kinase A, and SARS-CoV spike glycoprotein/ACE-2 complex whereas remdesivir has shown affinities for papain-like proteins, and vacuolar proton-translocating (V-ATPase), and chloroquine has affinities for cyclic AMP-dependent protein kinase A, and 3-chymotrypsin-like protease in comparison to ribavirin and oseltamivir (McKee et al., 2020). Aanouz et al. (2020) used the docking process for examining the interaction type and affinity at the binding site among the 67 compounds and the SARS-CoV-2 proteases. The results showed that, only three molecules (b-eudesmol, crocin, and digitoxigenin) are found as inhibitors in the control of SARS-CoV-2. Enmozhi et al. (2020) evaluated andrographolide from A. paniculata as a main protease inhibitor in case of SARS-CoV-2 through in silico studies. Kumar et al. (2020a, b) carried out molecular docking study of FDA approved drugs that are used in the cure of different viral ailments in order to investigate their binding affinity for the active site of Mpro (Lobo-Galo et al., 2020). Docking studies determined that drugs raltegravir, tipranavir, and lopinavir-ritonavir among others particularly binds to the proteases with similar affinity as of the α-ketoamide inhibitors (Muralidharan et al., 2020). From the earlier time, herbal plant products are successfully employed for the cure of several viral disorders (Mukhtar et al., 2008; Lin et al., 2014). Many of the promising lead natural compounds of our investigation are anti-viral, therefore, there is a big probability that these bioactive constituents would be appropriate in the control of SARS-CoV-2 and could be helpful for the discovery of multi-targeted agent against the SARS-CoV-2 infection (Sharma et al., 2020; Jyotisha et al., 2020; Vimal et al., 2020; Ribaudo et al., 2020).
The hunt for plant-derived antiviral for the control of SARS-CoV-2 is hopeful, as many plants have been reported to be effective against beta coronaviruses (Mitra et al., 2021; Tallei et al., 2020; Chowdhury et al., 2020; Babar et al., 2020; Gyebi et al., 2020, Gyebi et al., 2020; Yepes-Pérez et al., 2020; Lakshmi et al., 2020; Razzaghi-Asl et al., 2020; Subhomoi and Banerjee, 2020). Many reports to recognize the promising inhibitors of SARS-CoV-2 from plants have been published by using a computational approach (Pant et al., 2020; Elmezayen et al., 2020; Ghosh et al., 2020; Das et al., 2020; Maroli et al., 2020; Shree et al., 2020; Saravanan et al., 2020; Toluwase Hezekiah Fatoki et al., 2020; Rolta et al., 2020; Aanouz et al., 2020; Vimal et al., 2020). In the present study, after evaluating the multiple medicinal properties of the natural products, a total 18 compounds (canthin-6-one 9-O-beta-glucopyranoside from E. harmandian; kushenol W, and kushenol K from S. flavescens; and 3α, 14, 15, 18-Tetrahydroxy-5β, 9βH, 10α-labda-8, 12-dien-16-oic acid γ-lactone, deoxyandrographolide, neoandrographolide, 14-deoxy-11, 12-didehydroandrographolide, deoxyandrographolide19β-d-glucoside, 5,7,2′,3′-tetramethoxyflavanone, 5-hydroxy-7,2′,3′-trimethoxyflavone, 14-deoxy-11-oxoandrographolide, 5-hydroxy-7,8,2′,3′-tetramethoxyflavone, 5-hydroxy-7,8,2′, trimethoxyflavone, andrographine, panicoline, paniculide-A, paniculide-B, and paniculide-C from A. paniculate) were screened out to determine their inhibitory potentiality against Mpro and PLpro by molecular docking analysis.
9. Conclusion
The current study is a screening-based study performed with the assistance of molecular docking and ADMET analysis tools/server and is now subject of matter to examine the activity of the compound potency of canthin-6-one 9-O-beta-glucopyranoside in inhibiting SARS-CoV-2 Mpro/3CLpro and PLpro. The results of the MD simulation and binding free energy calculations indicated that canthin-6-one 9-O-beta-glucopyranoside compound interacted with the Mpro and PLpro and can also be proposed as a potential natural drug molecule for the treatment of SARS CoV-2 (COVID 19).
10. Summary points
-
•
SARS-CoV-2 is a beta coronavirus that spreads coronavirus disease 2019 (COVID-19) in the world leading to 74.29 million persons infected and 1.66 million death worldwide (222 countries, areas or territories with cases). The rapid spread of SARS-CoV-2 (COVID-19) has raised a severe global public health issue and creates a pandemic situation.
-
•
The continuing SARS-CoV-2 pandemic makes us worryingly noticed that our present choices for nursing against deadly coronavirus disease are very restricted. Therefore, there is an instant need for the discovery of novel inhibitors attacking the vital viral proteins.
-
•
The compounds of the three plants viz. E. harmandiana, S. flavescens and A. paniculata were screened for antiviral activity against the proteases of the 2019-nCov or SARS CoV-2.
-
•
The molecular docking and ADMET analysis suggested that with less affinity and drug-likeness properties, canthin-6-one 9-O-beta-glucopyranoside is a potential molecule that binds to the Mpro/3CLpro and PLpro and can inhibit the activity of replication and transcription of the virus and finally stop the multiplication of the virus. MD Simulation used to find best interaction and binding energy between the canthin-6-one 9-O-beta-glucopyranoside and Mpro/3CLpro and PLpro proteases of COVID19.
Author contributions
DM, MP and DV contributed equally as first authors. DV, DM, MP, PC, AK, DJ was involved in the writing; PKDM, HT, PJ, PP, RS, KP was involved in manuscript refinement & important intellectual content discussion; DV, AK was involved in molecular docking and DM initiated the idea of the study. MP carried out the MD simulation and related analyses along with binding energy calculation.
CRediT authorship contribution statement
Devvret Verma: Conception and design of study, Funding acquisition, Data curation, Formal analysis, Writing – original draft, and/or interpretation of data, Drafting the manuscript, Revising the manuscript critically for important intellectual content, Approval of the version of the manuscript to be published. Debasis Mitra: Conception and design of study, Funding acquisition, Data curation, Formal analysis, Writing – original draft, Acquisition of data, Analysis and/or interpretation of data, Drafting the manuscript, Revising the manuscript critically for important intellectual content, Approval of the version of the manuscript to be published. Manish Paul: Formal analysis, Data curation, Writing – original draft, Analysis and/or interpretation of data, Drafting the manuscript, Approval of the version of the manuscript to be published. Priya Chaudhary: Funding acquisition, Formal analysis, Data curation, Analysis and/or interpretation of data, Drafting the manuscript, Approval of the version of the manuscript to be published. Anshul Kamboj: Funding acquisition, Formal analysis, Data curation, Analysis and/or interpretation of data, Drafting the manuscript, Approval of the version of the manuscript to be published. Hrudayanath Thatoi: Revising the manuscript critically for important intellectual content, Approval of the version of the manuscript to be published. Pracheta Janmeda: Revising the manuscript critically for important intellectual content, Approval of the version of the manuscript to be published. Divya Jain: Approval of the version of the manuscript to be published. Periyasamy Panneerselvam: Revising the manuscript critically for important intellectual content, Approval of the version of the manuscript to be published. Rakesh Shrivastav: Revising the manuscript critically for important intellectual content, Approval of the version of the manuscript to be published. Kumud Pant: Revising the manuscript critically for important intellectual content, Approval of the version of the manuscript to be published. Pradeep K. Das Mohapatra: Revising the manuscript critically for important intellectual content, Approval of the version of the manuscript to be published.
Declaration of competing interest
The authors declare that there is no conflict of interest.
Acknowledgments
The authors are thankful to Graphic Era (Deemed to be University), Dehradun; Raiganj University, Raiganj; Maharaja Sriram Chandra Bhanja Deo University, Odisha; Banasthali Vidyapith, Vanasthali; ICAR- National Rice Research Institute, Cuttack; NGF College of Engineering and Technology, Palwal for the encouragement and support needed in the present investigation. Authors are grateful to the Bioinformatics Facility, Department of Biotechnology, Maharaja Sriram Chandra Bhanja Deo University, Baripada, India for the provision of computational support. The work was partially conducted in the context of the Bioinformatics Resources and Applications Facility (BRAF), C-DAC, Pune, India. D. Mitra is grateful to Government of West Bengal, India for Swami Vivekananda Merit Cum Means Ph.D. Scholarship (WBP191584588825). The authors are grateful to Dr. Isabella Zanella and Dr. Simon Pitchford, Guest Editor, Special Issue: COVID-19 therapy, from lung disease to systemic disorder, CRPHAR and reviewers for their valuable suggestions to increase the scientific quality of the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.crphar.2021.100038.
Contributor Information
Devvret Verma, Email: devvret@gmail.com.
Pradeep K. Das Mohapatra, Email: pkdmvu@gmail.com.
Funding
n/a.
Intellectual property
We confirm that we have given due consideration to the protection of intellectual property associated with this work and that there are no impediments to publication, including the timing of publication, with respect to intellectual property. In so doing we confirm that we have followed the regulations of our institutions concerning intellectual property.
Research ethics
We further confirm that any aspect of the work covered in this manuscript that has involved human patients has been conducted with the ethical approval of all relevant bodies and that such approvals are acknowledged within the manuscript.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Aanouz I., Belhassan A., Ei-Khatabi K., Lakhlifi T., Ei-Idrissi M., Bouachrine M. Moroccan medicinal plants as inhibitors against SARS-CoV-2 main protease: computational investigation. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1758790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeoye A.O., Oso B.J., Olaoye I.F., Tijjani H., Adebayo A.I. Repurposing of chloroquine and some clinically approved antiviral drugs as effective therapeutics to prevent cellular entry and replication of coronavirus. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1765876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin S.A., Banerjee S., Ghosh K., Gayen S., Jha T. Protease targeted COVID-19 drug discovery and its challenges: insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg. Med. Chem. 2021;29:115860. doi: 10.1016/j.bmc.2020.115860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres F. Yepes-Pérez, Oscar Herrera-Calderon, Quintero-Saumeth Jorge. Uncaria tomentosa (cat's claw): a promising herbal medicine against SARS-CoV-2/ACE-2 junction and SARS-CoV-2 spike protein based on molecular modeling. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1837676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes D.A., Moll M., Devaurs D., Jackson K.R., Lizée G., Kavraki L.E. Dinc 2.0: a new protein–peptide docking webserver using an incremental approach. Canc. Res. 2017;77(21):e55–e57. doi: 10.1158/0008-5472.CAN-17-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur D.E., Uzairu A. Molecular docking studies on the interaction of NCI anticancer analogues with human Phosphatidylinositol 4, 5-bisphosphate 3-kinase catalytic subunit. J. King Saud Univ. Sci. 2019;31:1151–1166. [Google Scholar]
- Arya R., Das A., Prashar V., Kumar M. Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs. ChemRxiv. 2020 doi: 10.26434/chemrxiv.1186001.v2. [DOI] [Google Scholar]
- Astuti I., Ysrafil Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): an overview of viral structure and host response. Diabetes metab Syndr. 2020;14(4):407–412. doi: 10.1016/j.dsx.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt E., Bingham R.J., Warner D.J., Laughton C.A., Phillips S.E., Homans S.W. Van der Waals interactions dominate ligand− protein association in a protein binding site occluded from solvent water. J. Am. Chem. Soc. 2005;127:11827–11834. doi: 10.1021/ja0527525. [DOI] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharatiy S.K., Hazra M., Paul M., Mohapatra S., Samantaray D., Dubey R.C., Sanyal S., Datta S., Hazra S. In silico designing of an industrially sustainable carbonic anhydrase using molecular dynamics simulation. ACS Omega. 2016;1(6):1081–1103. doi: 10.1021/acsomega.6b00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathi S., Poma A.B., Kolandaivel P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dyn. 2020;1–14 doi: 10.1080/07391102.2020.1758788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary P., Janmeda P. Spreadable virus of the 21st century: covid-19. Environ. Conserv. J. 2020;21(1&2):13–18. doi: 10.36953/ECJ.2020.211202. [DOI] [Google Scholar]
- Chen C.P., Chen C.C., Huang C.W., Chang Y.C. Evaluating molecular properties involved in transport of small molecules in stratum corneum: a quantitative structure-activity relationship for skin permeability. Molecules. 2018;23:911. doi: 10.3390/molecules23040911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury Papia. In silico investigation of phytoconstituents from Indian medicinal herb ‘Tinospora cordifolia (giloy)’ against SARS-CoV-2 (COVID-19) by molecular dynamics approach. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1803968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S., Olson A.J. Small-molecule library screening by docking with pyrx. Chem. Biol.: Methods Protocols. 2015 doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- Daneman R., Prat A. The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 2015;7:a020412. doi: 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T., Perera L., Li L., Pedersen L. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure. 1999;7(3):R55–R60. doi: 10.1016/s0969-2126(99)80033-1. [DOI] [PubMed] [Google Scholar]
- Das S., Sarmah S., Lyndem S., Roy A.S. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1763201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassault Systèmes Biovia . Dassault Systèmes; San Diego: 2020. Discovery Studio, Release 2020. [Google Scholar]
- de Oliveira O.V., Rocha G.B., Paluch A.S., Costa L.T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 2020;1–14 doi: 10.1080/07391102.2020.1772885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen Ammar D., Al-Obaidi Anas, Şahin Alp Tegin, Yelekçi Kemal. Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1758791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enayatkhani M., Hasaniazad M., Faezi S., Guklani H., Davoodian P., Ahmadi N., Ali Einakian M., Karmostaji A., Ahmadi K. Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: an in silico study. J. Biomol. Struct. Dyn. 2020:1–19. doi: 10.1080/07391102.2020.1756411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enmozhi S.K., Raja K., Sebastine I., Joseph J. Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: an in-silico approach. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1760136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh Rajesh, Chakraborty Ayon, Biswas Ashis, Chowdhuri Snehasis. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors – an in silico docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1779818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyebi Gideon A., Ogunro Olalekan B., Adegunloye Adegbenro P., Ogunyemi Oludare M., Afolabi Saheed O. Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): an in silico screening of alkaloids and terpenoids from African medicinal plants. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1764868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyebi G.A., Ogunro O.B., Adegunloye A.P., Ogunyemi O.M., Afolabi S.O. Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): an in silico screening of alkaloids and terpenoids from African medicinal plants. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1764868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Zhang X., Zhang J., Hu C.Q. In silico ADME and toxicity prediction of ceftazidime and its impurities. Front. Pharmacol. 2019;10:434. doi: 10.3389/fphar.2019.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan M.K., Kamruzzaman M., Manjur O.H.B., Mahmud A., Hussain N., Mondal M.S.A., Hosen M.I., Bello M., Rahman A. Structural analogues of existing anti-viral drugs inhibit SARS-CoV-2 RNA dependent RNA polymerase: a computational hierarchical investigation. Heliyon. 2021;7(3) doi: 10.1016/j.heliyon.2021.e06435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B., Bekker H., Berendsen H.J., Fraaije J.G. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18(12):1463–1472. [Google Scholar]
- JayaKumar T., Hsieh C.Y., Lee J.J., Sheu J.R. Experimental and clinical pharmacology of Andrographis paniculate and its major bioactive phytoconstituent Andrographolide. Evid. Based Complementary Altern. Med. 2013:1–16. doi: 10.1155/2013/846740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi R.S., Jagdale S.S., Bansode S.B., Shankar S.S., Tellis M.B., Pandya V.K., Chugh A., Giri A.P., Kulkarni M.J. Discovery of potential multi-targeted-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1760137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyotisha Samayaditya Singh, Ahmed Qureshi Insaf. Multi-epitope vaccine against SARS-CoV-2 applying immunoinformatics and molecular dynamics simulation approaches. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1844060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanchanapoom T., Kasai R., Chumsri P., Hiraga Y., Yamasaki K. Canthin-6-one and β-carboline alkaloids from Eurycoma harmandiana. Phytochemistry. 2001;56:383–386. doi: 10.1016/s0031-9422(00)00363-0. [DOI] [PubMed] [Google Scholar]
- Khan R.J., Jha R.K., Amera G.M., Jain M., Singh E., Pathak A., Singh R.P., Muthukumaran J., Singh A.K. Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J. Biomol. Struct. Dyn. 2020;1–14 doi: 10.1080/07391102.2020.1753577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Thiessen P.A., Bolton E.E., Chen J., Fu G., Gindulyte A., Han L., He J., He S., Shoemaker B.A., Wang J., Yu B., Zhang J., Bryant S.H. PubChem substance and compound databases. Nucleic Acids Res. 2016;44:D1202–D1213. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.H., Cho I.S., So Y.K., Kim H.H., Kim Yh Y.H. Kushenol A and 8-prenylkaempferol, tyrosinase inhibitors, derived from Sophora flavescens. J. Enzym. Inhib. Med. Chem. 2018;33:1048–1054. doi: 10.1080/14756366.2018.1477776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konda Mani Saravanan, Zhang Haiping, Renganathan Senthil, Kumar Vijayakumar Kevin, Sounderrajan Vignesh, Wei Yanjie, Shakila Harshavardhan. Structural basis for the inhibition of SARS-CoV2 main protease by Indian medicinal plant-derived antiviral compounds. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1834457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y., Singh H. ChemRxiv; 2020. In Silico Identification and Docking-Based Drug Repurposing against the Main Protease of SARS-CoV-2, Causative Agent of COVID-19. [Google Scholar]
- Kumar D., Kumari K., Jayaraj A., Kumar V., Kumar R.V., Dass S.K., Chandra R., Singh P. Understanding the binding affinity of noscapines with protease of SARS-CoV-2 for COVID-19 using MD simulations at different temperatures. J. Biomol. Struct. Dyn. 2020:1–14. doi: 10.1080/07391102.2020.1752310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V., Dhanjal J.K., Kaul S.C., Wadhwa R., Sundar D. Withanone and Caffeic acid Phenethyl ester are predicted to Interact with main protease (Mpro) of SARS-CoV-2 and inhibit its activity. J. Biomol. Struct. Dyn. 2020:1–17. doi: 10.1080/07391102.2020.1772108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R., Kumar R. Open source drug discovery Consortium, & Lynn A. g_mmpbsa- A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- Li D., Luan J., Zhang L. Molecular docking of potential SARS-CoV-2 papain-like protease inhibitors. Biochem. Biophys. Res. Commun. 2021;538:72–79. doi: 10.1016/j.bbrc.2020.11.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L.-T., Hsu W.-C., Lin C.-C. Antiviral natural products and herbal medicines. J Tradit Complement Med. 2014;4(1):24–35. doi: 10.4103/2225-4110.124335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo-Galo N., Terrazas-López M., Martínez-Martínez A., Díaz-Sánchez A.G. FDA-approved thiol-reacting drugs that potentially bind into the SARS-CoV-2 main protease, essential for viral replication. J. Biomol. Struct. Dyn. 2020;1–12 doi: 10.1080/07391102.2020.1764393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroli Nikhil, Bhasuran Balu, Natarajan Jeyakumar, Kolandaivel Ponmalai. The potential role of procyanidin as a therapeutic agent against SARS-CoV-2: a text mining, molecular docking and molecular dynamics simulation approach. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1823887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee D.L., Sternberg A., Stange U., Laufer S., Naujokat C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol. Res. 2020;157:104859. doi: 10.1016/j.phrs.2020.104859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X.Y., Zhang H.X., Mezei M., Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput-Aid Drug. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitjà O., Clotet B. Use of antiviral drugs to reduce COVID-19 transmission. Lancet Glob Health. 2020;8:e639–e640. doi: 10.1016/S2214-109X(20)30114-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra D., Verma D., Mahakur B., Kamboj A., Srivastava R., Gupta S., Pandey A., Arora B., Pant K., Panneerselvam P., Ghosh A., Barik D., Das Mohapatra P.K. Molecular docking and simulation studies of natural compounds of Vitex negundo L. against papain-like protease (PLpro) of SARS CoV-2 (coronavirus) to conquer the pandemic situation in the world. J. Biomol. Struct. Dyn. 2021;39:1–22. doi: 10.1080/07391102.2021.1873185. [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Kollman P.A. Molecular dynamics studies of calixspherand complexes with alkali metal cations: calculation of the absolute and relative free energies of binding of cations to a calixspherand. J. Am. Chem. Soc. 1992;114(10):3668–3674. [Google Scholar]
- Mohamed A.H., Fatima A.J. Covid-19 induced superimposed bacterial infection. J. Biomol. Struct. Dyn. 2020:1–18. doi: 10.1080/07391102.2020.1772110. [DOI] [PubMed] [Google Scholar]
- Mukhtar M., Arshad M., Ahmad M., Pomerantz R.J., Wigdahl B., Parveen Z. Antiviral potentials of medicinal plants. Virus Res. 2008;131(2):111–120. doi: 10.1016/j.virusres.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan N., Sakthivel R., Velmurugan D., Gromiha M.M. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 Protease against COVID-19. J. Biomol. Struct. Dyn. 2020:1–6. doi: 10.1080/07391102.2020.1752802. [DOI] [PubMed] [Google Scholar]
- Mussard E., Cesaro A., Lespessailles E., Legrain B., Berteina-Raboin S., Toumi H. Andrographolide, a natural antioxidant: an update. Antioxidants. 2019;8:571. doi: 10.3390/qntiox8120571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajouhesh H., Lenz G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Páll S., Abraham M.J., Kutzner C., Hess B., Lindahl E. International Conference on Exascale Applications and Software. Springer; Cham: 2015. Tackling exascale software challenges in molecular dynamics simulations with GROMACS; pp. 3–27. [Google Scholar]
- Pirzada R.H., Haseeb M., Batool M., Kim M., Choi S. Remdesivir and Ledipasvir among the FDA-approved antiviral drugs have potential to inhibit SARS-CoV-2 replication. Cells. 2021;10(5):1052. doi: 10.3390/cells10051052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya Shree, Mishra Priyanka, Selvaraj Chandrabose, Singh Sanjeev Kumar, Chaube Radha, Garg Neha, Tripathi Yamini Bhusan. Targeting COVID-19 (SARS-CoV-2) main protease through active phytochemicals of ayurvedic medicinal plants – Withania somnifera (Ashwagandha), Tinospora cordifolia (Giloy) and Ocimum sanctum (Tulsi) – a molecular docking study. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1810778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafi M., Devi A.F., Syafitri U.D., Heryanto R., Suparto I.H., Amran M.B., Rohman A., Prajogo B., Lim L.W. Classification of Andrographis paniculate extracts by solvent extraction using HPLC fingerprint and chemometric analysis. BMC Res. Notes. 2020;13:56. doi: 10.1186/s13104-020-4920-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaghi-Asl Nima, Ahmad Ebadi, Shahabipour Sara, Gholamin Danial. Identification of a potential SARS-CoV2 inhibitor via molecular dynamics simulations and amino acid decomposition analysis. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1797536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribaudo Giovanni, Alberto Ongaro, Oselladore Erika, Zagotto Giuseppe, Memo Maurizio, Gianoncelli Alessandra. A computational approach to drug repurposing against SARS-CoV-2 RNA dependent RNA polymerase (RdRp) J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1822209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolta Rajan, Yadav Rohitash, Salaria Deeksha, Trivedi Shubham, Imran Mohammad, Sourirajan Anuradha, Baumler David J., Dev Kamal. In silico screening of hundred phytocompounds of ten medicinal plants as potential inhibitors of nucleocapsid phosphoprotein of COVID-19: an approach to prevent virus assembly. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1804457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim B., Noureddine M. Identification of compounds from Nigella sativa as new potential inhibitors of 2019 novel coronaviruses (covid-19): molecular docking study. ChemRxiv. 2020 doi: 10.20944/preprints202004.0079.v1. [DOI] [Google Scholar]
- Schüttelkopf A.W., van Aalten D.M.F. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. 2004;D60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Selvaraj Alagu Lakshmi, Raja Mohamed Beema Shafreen, Arumugam Priya, Karutha Pandian Shunmugiah. Ethnomedicines of Indian origin for combating COVID-19 infection by hampering the viral replication: using structure-based drug discovery approach. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1778537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbaaz Mohd, Amir Mohd, Rahman Safikur, Mustafa Hasan Gulam, Dohare Ravins, Krishna Bisetty, Ahmad Faizan, Kim Jihoe, Hassan Md Imtaiyaz. Structural insights into Rab21 GTPase activation mechanism by molecular dynamics simulations. Mol. Simulat. 2018;44(3):179–189. [Google Scholar]
- Sharma A., Tiwari S., Deb M.K., Marty J.L. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): a global pandemic and treatment strategies. Int. J. Antimicrob. Agents. 2020;10:106054. doi: 10.1016/j.ijantimicagh.2020.106054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subhomoi B., Banerjee M. A computational prediction of SARS-CoV-2 structural protein inhibitors from Azadirachta indica (Neem) J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1774419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyash Pant, Singh Meenakshi, Ravichandiran V., Murty U.S.N., Srivastava Hemant Kumar. Peptide-like and small-molecule inhibitors against Covid-19. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1757510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallei T., Tumilaar S.G., Niode N., Fatimawali F., Kepel B., Idroes R., Effendi Y. 2020. Potential of Plant Bioactive Compounds as SARS-CoV-2 Main Protease (Mpro) and Spike (S) Glycoprotein Inhibitors: a Molecular Docking Study. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System . Schrödinger, LLC; 2020. Version 1.2r3pre.https://pymol.org/2/ [Google Scholar]
- Fatoki Toluwase Hezekiah, Ibraheem Omodele, Ogunyemi Ibukun Oladejo, Akinmoladun Afolabi Clement, Ugboko Harriet U., Adeseko Catherine Joke, Awofisayo Oladoja A., Olusegun Sunday Joseph, Enibukun Jesupemi Mercy. Network analysis, sequence and structure dynamics of key proteins of coronavirus and human host, and molecular docking of selected phytochemicals of nine medicinal plants. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1794971. [DOI] [PubMed] [Google Scholar]
- Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ul Qamar M.T., Maryam A., Muneer I., Xing F., Ashfaq U.A., Khan F.A., Anwar F., Geesi M.H., Khalid R.R., Rauf S.A., Siddiqi A.R. Computational screening of medicinal plants phytochemicals to discover potent pan-seotype inhibitors against dengue virus. Sci. Rep. 2019;9:1433. doi: 10.1038/s41598-018-38450-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- V'kovski P., Kratzel A., Steiner S., Stalder H., Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 2020;19:155–170. doi: 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gunsteren W.F., Billeter S.R., Eising A.A., Hünenberger P.H., Krüger P.K.H.C., Mark A.E., Tironi I.G. Vdf Hochschulverlag AG an der ETH Zürich; Zürich: 1996. Biomolecular Simulation: the GROMOS96 Manual and User Guide; p. 86. [Google Scholar]
- Vaught A. Graphing with Gnuplot and Xmgr: two graphing packages available under linux. Linux J. 1996;1996(28es):7. [Google Scholar]
- Vimal K. Maurya, Kumar Swatantra, Madan L., Bhatt B., Saxena Shailendra K. Antiviral activity of traditional medicinal plants from Ayurveda against SARS-CoV-2 infection. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1832577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber W., Hünenberger P.H., McCammon J.A. Molecular dynamics simulations of a polyalanine octapeptide under Ewald boundary conditions: influence of artificial periodicity on peptide conformation. J. Phys. Chem. B. 2000;104(15):3668–3675. [Google Scholar]
- World Health Organization Coronavirus disease (COVID-19): situation report-107. 2020. https://www.who.int
- Wu R., Wang L., Dina Kuo H.C., Shannar A., Peter R., Chou P.J., Li S., Hudlikar R., Liu X., Liu Z., Poiani G.J., Amorosa L., Brunetti L., Kong A.N. An update on current therapeutic drugs treating COVID-19. Curr. Pharmacol. Rep. 2020;11:1–15. doi: 10.1007/s40495-020-00216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zainib Babar, Khan Mazhar, Zahra Mubeen, Anwar Munazza, Noor Kashif, Hashmi Huma Farooque, Suleman Muhammad, Waseem Muhammad, Shah Abdullah, Ali Shahid, Ali Syed Shujait. Drug similarity and structure-based screening of medicinal compounds to target macrodomain-I from SARS-CoV-2 to rescue the host immune system: a molecular dynamics study. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1815583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Shen F.M., Chen F., Lin Z. Origin and evolution of the 2019 novel coronavirus. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Penninger J., Li Y., Zhong N., Slutsky A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46:586–590. doi: 10.1007/s00134-020-05985-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.