

Abstract
Major efforts have been devoted to the development of constructs that enable sequence-specific recognition of double-stranded (ds) DNA, fueled by the promise for enabling tools for applications in molecular biology, diagnostics, and medicine. Towards this end, we have previously introduced Invader probes, i.e., short DNA duplexes with +1 interstrand zipper arrangements of intercalator-functionalized nucleotides. The individual strands of these labile probes display high affinity towards complementary DNA (cDNA), which drives sequence-unrestricted dsDNA-recognition. However, recognition of long targets is challenging due to the high stability of the corresponding probes. To address this, we recently introduced toehold Invader probes, i.e., Invader probes with 5’-single-stranded overhangs. The toehold architecture allows for shorter double-stranded segments to be used, which facilitates probe dissociation and dsDNA-recognition. As an extension thereof, we here report the biophysical and dsDNA-targeting properties of nicked Invader probes. In this probe architecture, the single-stranded overhangs of toehold Invader probes are hybridized to short intercalator-modified auxiliary strands, leading to formation of additional labile segments. The extra binding potential from the auxiliary strands imparts nicked Invader probes with greater dsDNA-affinity than the corresponding toehold or blunt-ended probes. Recognition of chromosomal DNA targets, refractory to recognition by conventional Invader probes, is demonstrated for nicked Invader probes in the context of non-denaturing FISH experiments, which highlights their utility as dsDNA-targeting tools.
Graphical Abstract
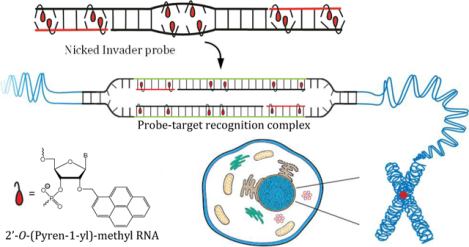
Nicked Invader probes, featuring three destabilized double-stranded segments, allow for recognition of long dsDNA targets (~25 bps) under non-denaturing conditions, including chromosomal targets previously found to be refractory to recognition by conventional Invader probes.
INTRODUCTION.
Considerable attention has been devoted to the development of molecules that recognize specific sequences of double-stranded DNA (dsDNA) due to the prospect for tools that enable regulation of gene expression at the transcriptional level, detection of diagnostically important sequences, and gene editing. Long before the advent of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-associated (Cas) endonucleases,1,2 oligomeric probes recognizing nucleobase-specific features accessible from the duplex grooves, such as triplex forming oligonucleotides (TFOs)3,4 and peptide nucleic acids (PNAs),5–7 or pyrrole-imidazole polyamides,8,9 were extensively studied. However, these dsDNA-targeting probes have a limited target scope. Triplex formation necessitates the presence of extended polypurine stretches, whereas minor-groove binding polyamides rely on binding- and shape-complementarity for efficient dsDNA-recognition, the latter of which is compromised when dsDNA regions longer than approximately eight base-pairs are targeted. These limitations have spurred development of strand-invading dsDNA-targeting strategies, i.e., probes capable of unzipping the Watson-Crick base-pairs of dsDNA targets and forming new, more stable Watson-Crick base-pairs between individual probe strands and complementary DNA (cDNA). Representative examples include modified PNA,10–12 Zorro LNAs (locked nucleic acids),13 and easily denaturing double-stranded probes such as pseudo-complementary (pc) DNA,14 pcPNA,15–17 heteroduplexes between intercalator-modified oligonucleotides and RNA, LNA or PNA strands,18–20 and related approaches.21–23
Our group has focused on the development of so-called Invader probes,24,25 which are short DNA duplexes modified with one or more +1 interstrand zipper arrangements26 of intercalator-functionalized nucleotides like 2′-O-(pyren-1-yl)methyl-RNA (Fig. 1). This monomer arrangement – termed an “energetic hotspot” for simplicity – forces a pair of intercalating moieties to vie for the same inter-base-pair region. As a result, the nearest-neighbor exclusion principle27 is violated and the duplex is unwound and destabilized.28,29 The principle asserts that a local intercalator density of more than one intercalator per two base-pairs in DNA duplexes is energetically unfavorable due to limitations in local helix expandability (duplex unwound by ~3.4 Å per intercalation event) and because stabilizing stacking interactions between neighboring base-pairs and the first intercalating moiety are perturbed.30–32 The two probe strands, in turn, display high affinity towards cDNA since probe dissociation and target binding reverses violation of the nearest-neighbor exclusion principle, leading to strongly stabilizing stacking interactions between intercalators and flanking base-pairs. The greater stability of the double-stranded probe-target regions formed vis-à-vis the double-stranded probe and the dsDNA target region, provides the driving force for dsDNA-recognition via double-duplex strand invasion (Fig. 1a).24,25,33 Conventional Invader probes have been used to detect i) DNA fragments from specific food pathogens (28-mer mixed-sequence dsDNA fragments detected at 20 pM using Invader capture/signaling probes in sandwich assays),34 ii) telomeric DNA of individual chromosomes in metaphasic spreads,35 and iii) gender-specific chromosomal-targets in interphase and metaphase nuclei under non-denaturing conditions.25
Figure 1.

Schematic of the dsDNA-recognition process using a) blunt-ended (conventional), b) toehold, or c) nicked Invader probes. d) Structure of 2’-O-(pyren-1-yl)methyl RNA monomer used to generate energetic hotspots.
Our optimization efforts have focused on the development of new intercalator-functionalized nucleotide monomers28,29,36 and different probe architectures37–39. Specifically concerning the latter, we have recently introduced so-called toehold Invader probes, in which Invader probes are asymmetrically extended to feature 5’-single-stranded overhangs (Fig. 1b).40 The advantage of toehold Invader probes vis-à-vis the corresponding blunt-ended Invader probes is that longer dsDNA regions can be targeted since the double-stranded region can be shortened, thus facilitating probe dissociation. Moreover, affinity-enhancing modifications such as LNA41 monomers can be incorporated in the single-stranded overhangs to further increase cDNA affinity. Toehold Invader probes have been shown to result in more efficient recognition of complementary dsDNA regions, allowing for recognition of chromosomal DNA targets that were refractory to recognition by conventional blunt-ended Invader probes.40
These encouraging results led us to contemplate the feasibility of hybridizing short auxiliary strands to the single-stranded overhangs, yielding a new probe architecture that we have dubbed nicked Invader probes (NIPs) since they can be viewed as long conventional Invader probes in which one nick per probe strand has been introduced (Fig. 1c). The expected benefit of these probes vis-à-vis toehold Invader probes is that a greater number of base-pairs are formed upon dsDNA-recognition. To ensure facile denaturation of the double-stranded probe, the auxiliary strands must be modified with intercalator-functionalized monomers in a manner that results in the formation of additional destabilizing energetic hotspots.
Here, we report the biophysical and dsDNA-targeting properties of different nicked Invader probe architectures and compare them to the corresponding conventional and toehold Invader probes. The nicked Invader probes display higher dsDNA-affinity and excellent binding specificity, which allows for recognition of chromosomal DNA targets that have proven refractory to recognition by conventional Invader probes.
RESULTS AND DISCUSSION
Design and synthesis of probes.
The 2’-O-(pyren-1-yl)methyl-RNA-modified oligodeoxyribonucleotides (ONs) used herein were available from prior studies or synthesized using established protocols42,43 (Table S1† and Figs. S1–S21†). Access to these ONs allowed for the assembly of four nicked Invader probes (NIP1-NIP4), designed to recognize a 25-base-pair (bp) mixed-sequence DNA region embedded within longer targets (Fig. 2). Each nicked Invader probe is comprised of four strands and features six identically positioned energetic hotspots. The position of the nicks, however, varies among the probes. Nicked Invader probes have three double-stranded segments, i.e., a central segment that is flanked on either side by an auxiliary segment. Hence, nicked Invader probes can be described according to the number of base-pairs in each segment. Thus, NIP1-NIP4 are referred to as 6-13-6, 8–9-8, 10-5-10, and 12-1-12 constructs, respectively (Fig. 2). Alternatively, nicked Invader probes can be viewed as toehold Invader probes that are additionally hybridized to two auxiliary strands. Thus, NIP1 is a toehold Invader probe (TIP) comprised of two 19-mer main strands and two auxiliary 6-mer strands designed to hybridize to the 5’-single-stranded overhangs of the toehold probe. Similarly, the auxiliary strands of NIP2-NIP4 are designed to hybridize the 8-, 10- and 12-mer single-stranded overhangs of the corresponding toehold probes, respectively.
Figure 2.
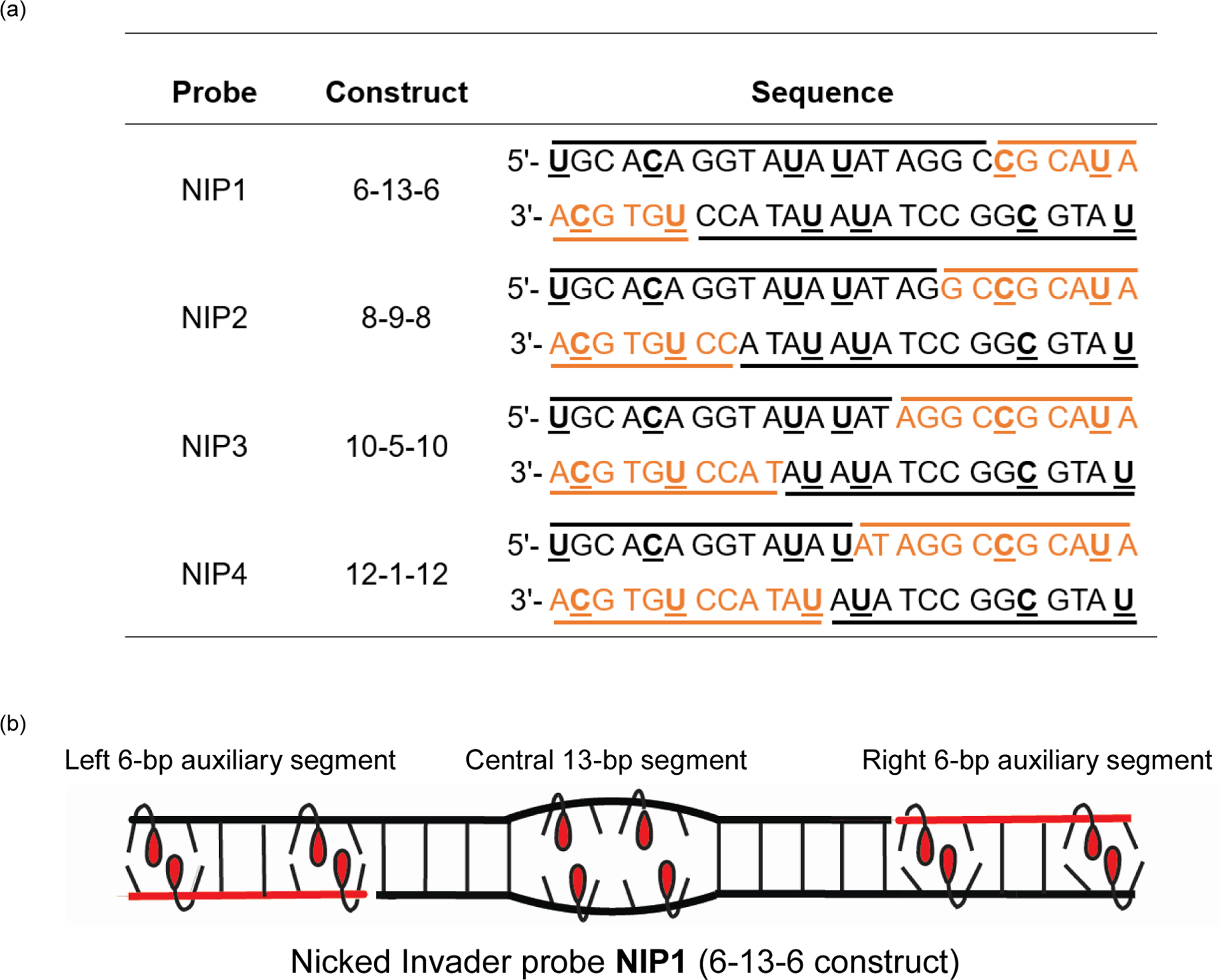
a) Sequences of nicked Invader probes used herein. Main and auxiliary strands are shown in black and red, respectively. The corresponding toehold Invader probes are comprised of main strands only. U = 2′-O-(pyren-1-yl)methyluridine. C = 2′-O-(pyren-1-yl)methylcytidine monomers. b) Illustration of a representative nicked Invader probe and its three duplex segments.
Thermal denaturation properties.
Given the multistranded nature of the nicked Invader probes and their three distinct double-stranded segments, it is not feasible to determine thermal denaturation temperatures (Tms) for fully assembled probes as overlapping transitions are likely. Instead, the stability of the left, central and right segments was approximated by determining Tms for duplexes formed by the i) 5’-main probe and 3’-auxiliary probe, ii) 5’-main probe and 3’-main probe, and iii) 5’-auxiliary probe and 3’-main probe, respectively (1st-3rd Tm columns, Table 1). As expected for a 13-bp duplex with two energetic hotspots, the central segment of NIP1 is relatively labile as indicated by the moderate Tm value for ON1:ON4 (Tm = 49.0 °C). The value decreases as the number of base-pairs in the central segment is reduced, in steps of four, from thirteen to one (note the Tm trend for ON1:ON4 → ON5:ON8 → ON9:ON12 → ON13:ON16, 2nd Tm column, Table 1). Similarly, the stability of the “left” and “right” auxiliary segments increases as the number of base-pairs increases, in steps of two, from six to twelve (e.g., note the Tm trend on going from ON1:ON3 → ON5:ON7 → ON9:ON11 → ON13:ON15, 1st Tm column, Table 1). Only the central segments of NIP1 and NIP2 and the auxiliary segments of NIP2-NIP4 are likely to be stable at the temperature of the subsequent dsDNA-recognition experiments (37 °C, vide infra). Thus, only NIP2 is expected to be substantially assembled at 37 °C.
Table 1.
Sequences of probes used in this study, as well as Tms of probe duplexes and duplexes with DNA targets, and thermal advantages (TAs) of probes.a
| Tm (°C) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ONs | Sequence | 5’-main probe vs 3’-aux probe | 5’-main probe vs 3’-main probe |
5’-aux probe vs 3’-main probe | 5’-main probe vs DNA2 | 5’-aux probe vs DNA2 | 3’-aux probe vs DNA1 | 3’-main probe vs DNA1 | TA (°C) |
|
1/2
3/4 |
5’-UGCACAGGTAUAUATAGGCCGCAUA 3’-ACGTGUCCATAUAUATCCGGCGTAU |
ntb | 49.0c,d | nt | 75.5 | nt | 25.5e | 75.5 | - |
|
5/6
7/8 |
5’-UGCACAGGTAUAUATAGGCCGCAUA 3’-ACGTGUCCATAUAUATCCGGCGTAU |
36.0 | 37.0 | 36.0f | 73.0 | 57.0f | 46.0g | 74.5 | 69.5 |
|
9/10
11/12 |
5’-UGCACAGGTAUAUATAGGCCGCAUA 3’-ACGTGUCCATAUAUATCCGGCGTAU |
45.5f | nt | 52.0 | 72.5 | 66.0 | 57.5g | 74.0 | >80.5h |
|
13/14
15/16 |
5’-UGCACAGGTAUAUATAGGCCGCAUA 3’-ACGTGUCCATAUAUATCCGGCGTAU |
54.0 | nt | 62.5 | 72.0 | 66.5 | 65.5 | 70.0 | >65.5h |
|
1
4 |
5’-UGCACAGGTAUAUATAGGC 3’-CCATAUAUATCCGGCGTAU |
- | 49.0c | - | 75.5c | - | - | 75.5c | 30.0c,i |
|
5
8 |
5’-UGCACAGGTAUAUATAG 3’-ATAUAUATCCGGCGTAU |
- | 37.0 | - | 73.0 | - | - | 74.5 | 38.5i |
|
9
12 |
5’-UGCACAGGTAUAUAT 3’-AUAUATCCGGCGTAU |
- | nt | - | 72.5 | - | - | 74.0 | >54.5i |
|
13
16 |
5’-UGCACAGGTAUAU 3’-AUATCCGGCGTAU |
- | nt | - | 72.0 | - | - | 70.0 | >50.0i |
|
1
18 |
5’-UGCACAGGTAUAUATAGGC 3’-ACGTGUCCATAUAUATCCG |
- | 66.0c | - | 75.5c | - | - | 76.0c | 13.5c |
|
17
4 |
5’-GGTAUAUATAGGCCGCAUA 3’-CCATAUAUATCCGGCGTAU |
- | 66.0c | - | 76.5c | - | - | 75.5c | 14.0c |
|
19
20 |
5’-GGTAUAUATAGGC 3’-CCATAUAUATCCG |
- | 42.5c | - | 54.5c | - | - | 54.0c | −6.0c |
NIP1 = ON1/2:ON3/4, NIP2 = ON5/6:ON7/8, NIP3 = ON9/10:ON11/12 and NIP4 = ON13/14:ON15/16. TIP1 = ON1:ON4, TIP2 = ON5:ON8, TIP3 = ON9:ON12, and TIP4 = ON13:ON16. DNA1 = 5’-AAGCTGCACAGGTATATATAGGCCGCATATGCA and DNA2 = 3’-TTCGACGTGTCCATATATATCCGGCGTATACGT. Tm (DNA1:DNA2) = 72.0 °C. Denaturation curves were recorded in medium salt phosphate buffer ([Na+] = 110 mM, [Cl−] = 100 mM, pH 7.0 (NaH2PO4/Na2HPO4), [EDTA] = 0.2 mM), with each strand at 0.5 μM concentration. Representative thermal denaturation curves shown in Figs. S22† and S23†. A/C/G/T = adenin-9-yl, cytosin-1-yl, guanin-9-yl, and thymin-1-yl DNA monomers, respectively. Structures of U and C are shown in Fig. 1. “nt” = no transition. “-“ = not applicable. Definition of TA given in main text.
A transition at ~45 °C is attributed to denaturation of a secondary structure entailing ON1 only (Fig. S26†).
Data previously reported in reference 40.
See discussion of Figs. S26† and S27† in ESI.
Differential denaturation experiments confirm this transition (Fig. S28†).
Weak transition.
A transition below 30 °C is attributed to denaturation of a secondary structure entailing DNA1 only (Fig. S28†).
nt assumed to be < 20 °C in TA calculation.
TA calculation simplifies to TA = Tm (5’-probe vs. DNA2) + Tm (3’-probe vs. DNA1) - Tm (toehold Invader probe) - Tm (dsDNA target).
To estimate the stability of the four double-stranded probe-target segments that would form upon successful dsDNA-recognition by nicked Invader probes (Fig. 1), we determined the Tm values for duplexes between individual probe strands and a 33-mer single-stranded (ss) DNA harboring the complementary target regions (4th-7th Tm columns, Table 1). Unsurprisingly, the Tm values decrease as the length of the probe strand – and, thus, the number of base-pairs in that segment – decreases (e.g., note decreasing Tm trend for duplexes between DNA2 and ON1, ON5, ON9 or ON13, 4th Tm column, or – equivalently – the increasing Tm trend for duplexes between DNA2 and ON2, ON6, ON10 or ON14, 5th Tm column). Recognition of complementary dsDNA regions is expected to result in the formation of four stable double-stranded probe-target segments when using NIP2-NIP4 as the segments display Tms > 45 °C. In contrast, probe-target segments entailing the 6-mer auxiliary strands of NIP1 are labile (no transition or Tm = 25.5 °C, for ON2:DNA2 and ON3:DNA1, respectively).
As previously reported for conventional and toehold Invader probes,25,40 duplexes between individual probe strands and ssDNA harboring complementary target regions are much more stable than the corresponding double-stranded probe segments (e.g., compare Tm = 73.0 °C, 74.5 °C, and 37.0 °C, for ON5:DNA2, ON8:DNA1 and ON5:ON8, respectively). This is due to the difference in local intercalator density. The nearest-neighbor exclusion principle is violated in double-stranded probes (high local intercalator density) but not in probe-target duplexes (low local intercalator density). Accordingly, dsDNA-recognition is expected to be energetically favorable. The driving force for recognition of DNA1:DNA2, i.e., a 33-mer model dsDNA harboring the complementary 25-bp target region (sequence shown in footnote of Table 1), can be assessed by the term thermal advantage (TA) defined as TA = Tm (5’-main probe vs. DNA2) + Tm (5’-aux probe vs. DNA2) + Tm (3’-main probe vs. DNA1) + Tm (3’-aux probe vs. DNA1) − Tm (5’-main probe vs. 3’-aux probe) – Tm (5’-main probe vs. 3’-main probe) – Tm (5’-aux probe vs. 3’-main probe) − Tm (dsDNA target), with large positive values indicating a strongly activated probe. NIP2-NIP4 display TA values > 65.5 °C, which suggests that the driving force for recognition of DNA1:DNA2 is far greater than with conventional 13-mer Invader probe ON19:ON20 featuring two energetic hotspots (TA = −6 °C, Table 1), conventional 19-mer Invader probes ON1:ON18 and ON17:ON4 featuring four energetic hotspots (TA ~ 14 °C, Table 1), or the corresponding toehold analogues of NIP1-NIP4, i.e., TA ~ 30 °C, ~ 38.5 °C, TA > 54.5 °C, and TA > 50 °C, for TIP1-TIP4, respectively (Table 1).
We have previously shown that ONs modified with 2’-O-(pyren-1-yl)methyl RNA monomers (i.e., individual Invader strands) display much lower affinity towards complementary RNA than towards cDNA.29,42 Hence, binding to potential dsRNA targets is not a concern and Tm values for duplexes with cRNA segments were, therefore, not determined.
Characterization of dsDNA-targeting properties of nicked Invader probes.
Initial screen.
The dsDNA-targeting properties of NIP1-NIP4 were evaluated using an electrophoretic mobility shift assay in which a 3’-digoxigenin (DIG)-labeled DNA hairpin (DH) DH1 was used as a model dsDNA target (Fig. 3a). DH1 features a 33-mer double-stranded stem, which has the same sequence as DNA1:DNA2 used in the denaturation experiments, and a decameric thymidine loop (T10) that links the two stem strands at one end (Table S2†). The 25-bp region that is complementary to the nicked Invader probes is embedded within the stem of DH1. The unimolecular nature of DH1 renders it a high-melting target (Tm = 81 °C).40
Figure 3.
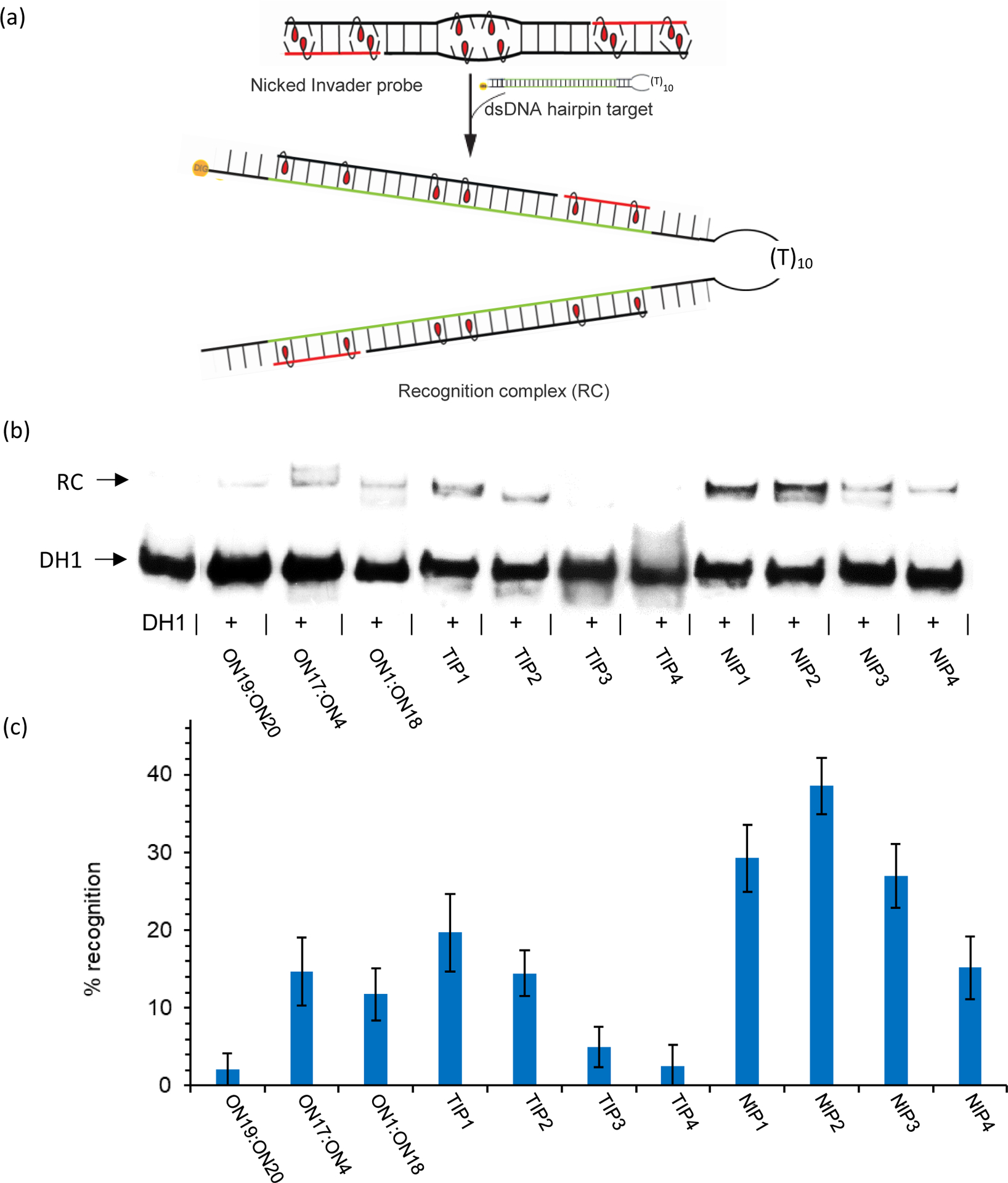
a) Illustration of the electrophoretic mobility shift assay used to evaluate dsDNA-recognition of Invader probes. b) Representative gel electrophoretograms from recognition experiments in which DH1 was incubated with a 50-fold molar excess of different probes. RC = recognition complex. c) Histogram depicting averaged results from at least three independent experiments with error bars representing standard deviation. Conditions: DIG-labeled DH1 (50 nM) incubated with a 50-fold molar excess of the specified probe in HEPES buffer (50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, pH 7.2, 10% sucrose, 1.44 mM spermine tetrahydrochloride) for 17 h at 37 °C. The sequence of DH1 is shown in Table S2†. The electrophoretogram is a composite image from different runs.
Successful recognition of DH1 is expected to result in the formation of a five-stranded recognition complex with lower mobility on non-denaturing polyacrylamide gels than DH1. Indeed, low-mobility bands are observed when a 50-fold molar excess of pre-annealed NIP1-NIP4 is incubated with DH1 at 37 °C (Fig. 3b). Varying levels of DH1 recognition are observed, ranging from minor (~15% with NIP4) to moderate recognition (~40% with NIP2) (Table S3†). Interestingly, the results suggest that nicked Invader probes with labile double-stranded segments recognize DH1 less efficiently than probes in which all segments are stable and of comparable stability (compare 6-13-6, 10-5-10 and 12-1-12 constructs relative to the 8-9-8 construct).
Nicked Invader probes result in more efficient recognition of DH1 than the corresponding toehold probes. The relative differences are more pronounced in nicked Invader probes with longer auxiliary strands (e.g., compare NIP1 vs TIP1 vis-à-vis NIP3 vs TIP3, Fig. 3b and Table S3†). This, in turn, is a consequence of toehold Invader trends. Thus, toehold probes with extensive overlaps result in more efficient recognition of DH1 than toehold Invader probes with small overlaps (note recognition efficiency trend for TIP1 → TIP2 → TIP3/TIP4, Fig. 3b and Table S3†). Presumably, this is linked to the greater number of base-pairs formed upon dsDNA-recognition. For example, the use of TIP1 (= ON1:ON4) results in the formation of two 19-bp probe-target duplex segments, while TIP4 (= ON13:ON16) would be expected to form two 13-bp probe-target duplex segments (~19% and <5% recognition, respectively, Fig. 3b and Table S3†). However, given the minor stability differences among the probe-target duplexes (e.g., compare Tms for duplexes between DNA2 and ON1, ON5, ON9 or ON13, 4th Tm column, Table 1), other factors likely impact dsDNA-recognition efficiency. Indeed, recognition of DH1 appears to require toehold probes to form stable double-stranded overlaps (correlate data in 2nd Tm column of Table 1 with efficiency of DH1-recognition shown in Fig. 3b and Table S3†). Thus, the results suggest that toehold and nicked Invader probes should be fully assembled to ensure efficient dsDNA-recognition.
Conventional 19-mer Invader probes ON1:ON18 and ON17:ON4 only result in low levels of DH1-recognition (<15%) even though two stable 19-bp probe-target duplex segments would be formed (Fig. 3b and Table S3†). We attribute the marginal dsDNA-recognition to the high probe stability (Tm ~66 °C, Table 1), which is expected to impact recognition kinetics. Conventional 13-mer Invader probe ON19:ON20, which can be viewed as a truncated version of NIP1, does not result in appreciable levels of DH1-recognition (Fig. 3b and Table S3†). This is as expected given the unfavorable driving force observed for ON19:ON20 (TA = −6°C, Table 1).
To sum up, the preliminary screen suggests that appropriately designed nicked Invader probes offer advantages with respect to dsDNA-recognition relative to toehold and short/long conventional Invader probes.
Dose-response profiles.
Next, dose-response profiles were determined for nicked Invader probes NIP1-NIP4 and the corresponding toehold probes (Figs. 4 and S29–S30†). In agreement with the results from the initial screen, NIP2 displays the lowest C30 value (i.e., probe concentration resulting in 30% recognition of DH1), whereas NIP3 and NIP4 display C30 values that are nearly seven- and fifteen-fold higher, respectively (~0.5 μM vs ~3.4 μM and 7.4 μM). Recognition of DH1 reaches a plateau below 30% when using NIP1, ~35% when using NIP4, and 40–45% when using NIP2 or NIP3. Less than 20% recognition is observed with the corresponding toehold probes (Fig. S31†). It is not clear why the plateaus cannot be overcome with a greater probe excess of probe. We speculate that the probes experience a similar form of self-inhibition as has been reported for double-stranded pcPNA probes.44
Figure 4.
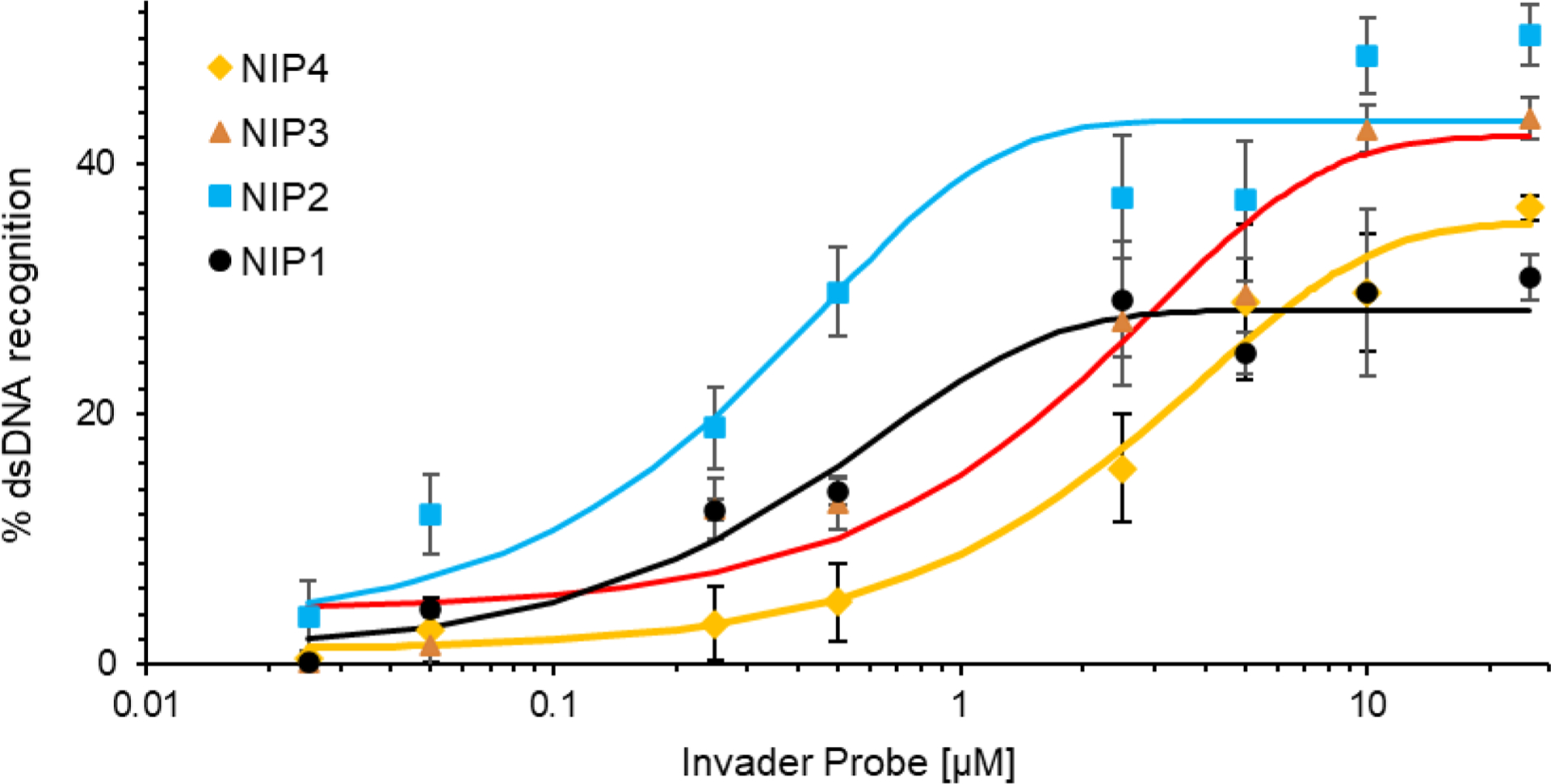
Dose-response profiles for recognition of DH1 by nicked Invader probes NIP1-NIP4 at 37 °C. Profiles are constructed based on the electrophoretograms shown in Figs. S29 and S30†. Experimental conditions are as described in Fig. 3.
Binding specificity.
Next, the binding specificities of the different nicked Invader probes were evaluated by incubating a 50-fold molar probe excess with:
MM1, which differs in sequence at one position relative to DH1 (corresponding to the position of the third energetic hotspot of the probes as drawn); binding would result in the formation of a recognition complex, in which two of the four formed probe-target duplex segments harbor one mismatched base-pair each (Fig. 5),
MM2, which differs in sequence at two positions relative to DH1 (corresponding to the positions between the 1st and 2nd, and 5th and 6th energetic hotspot of the probes as drawn); binding would result in the formation of a recognition complex, in which each of the four formed probe-target duplex segments harbor one mismatched base-pair (Fig. 5),
MM3, which differs in sequence at three positions relative to DH1 (corresponding to the position of the third energetic hotpot, and between the 1st and 2nd, and 5th and 6th energetic hotspot of the probes as drawn); binding would result in the formation of a recognition complex, in which two of the four formed probe-target duplex segments harbor two mismatched base-pairs each, while the remaining two probe-target duplex segments harbor one mismatched base-pair each (Fig. 5).
Figure 5.
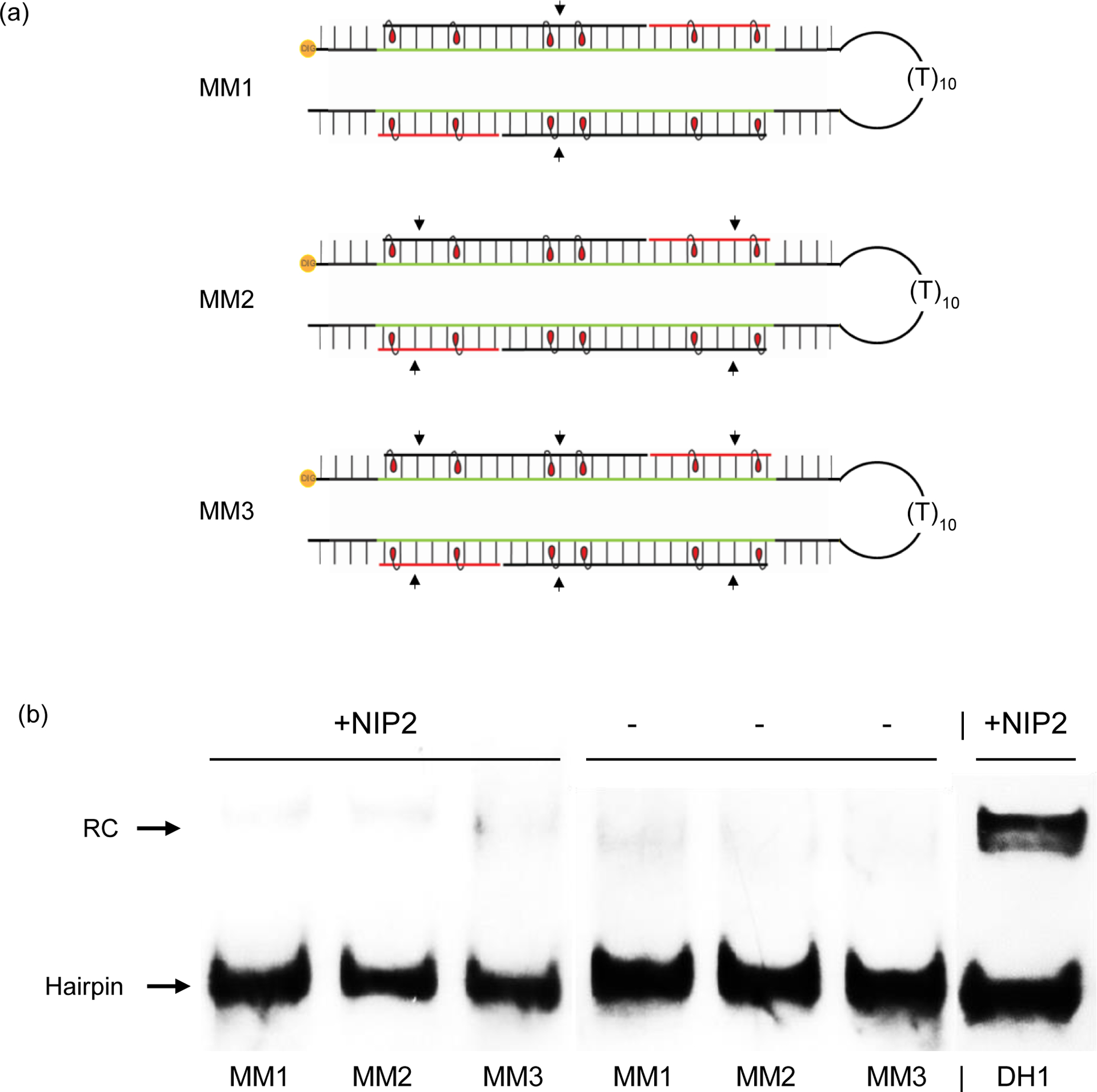
Binding specificity of nicked Invader probe NIP2. (a) Illustration of the mismatched recognition complexes that would ensue upon recognition of MM1-MM3 by NIP2; arrows indicate the position of mismatched base-pairs. For sequences of MM1-MM3, see Table S2†. Equivalent illustrations for recognition of MM1-MM3 with NIP1, NIP3 and NIP4 are shown in Fig. S33†. (b) Representative electrophoretograms from experiments in which NIP2 was incubated with non-complementary targets MM1-MM3 or complementary target DH1. For equivalent electrophoretograms entailing NIP1, NIP3 and NIP4, see Fig. S33†. Pre-annealed 3’-DIG-labelled hairpins (50 nM) were incubated with a 50-fold molar excess of pre-annealed probe at 37 °C for 17 h in HEPES buffer as outlined in Fig. 3. The electrophoretogram is a composite image from different runs.
Remarkably, each nicked Invader probe fully discriminates the singly, doubly, and triply mismatched DNA hairpin targets (Figs 5, S32† and S33†). In fact, binding is even more specific than what we previously observed with toehold Invader probes.40 Presumably, the high binding specificity is the consequence of multiple effects including i) stringency clamping effects, i.e., greater stability differences between matched and mismatched recognition complexes due to the metastable nature of the structured probes,44 ii) avoidance of forming multiple, energetically unfavorable, double-stranded segments containing mismatched base-pairs,45 and iii) high binding specificity of short oligonucleotides. Thus, these results strongly suggest that it is possible to improve both the binding affinity and specificity of Invader probes by using a nicked probe design.
Detection of chromosomal DNA using nicked Invader probes.
Motivated by these findings, we set out to demonstrate the use of nicked Invader probes for recognition of mixed-sequence chromosomal DNA regions. We have previously demonstrated the feasibility of conventional and toehold Invader probes for recognition of a complementary sequence in a highly repeated region of the DYZ-1 satellite gene (~6 × 104 tandem repeats of a ~1175 bp region) on the bovine (Bos taurus) Y-chromosome (NCBI code: M26067)46 in the context of non-denaturing fluorescence in situ hybridization (nd-FISH) experiments.25,40 One probe, however, i.e., conventional 15-mer Invader probe DYZ-REF (ON21:ON22, Table 2), has proven refractory to recognition of the DYZ-1 region.40 To overcome this limitation, we constructed a corresponding Cy3-labeled nicked Invader probe DYZ-NIP (ON23/24:ON25/26, Table 2), i.e., an 8-9-8 construct with six energetic hotspots that targets positions 862–886 of the DYZ-1 gene.
Table 2.
Sequences of probes used in FISH experiments and Tms of probe duplexes and duplexes between individual probe strands and DNA targets.a
| Tm (°C) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ONs | Sequence | 5’-main probe vs 3’-aux probe | 5’-main probe vs 3’-main probe | 5’-aux probe vs 3’-main probe | 5’-main probe vs DNA4 | 5’-aux probe vs DNA4 | 3’-aux probe vs DNA3 | 3’-main probe vs DNA3 | TA (°C) |
|
23/24 25/26 |
5’-Cy3-TGUGTUATAUGCUGTTCTCAGCCCT 3’-ACACAAUATACGACAAGAGUCGGGA-Cy3 |
<20.0 | nt | nt | 63.0 | 35.5 | 34.5 | 68.0 | nd |
|
21
22 |
5’-Cy3-TUA UAT GCT GUT CTC 3’-AAU AUA CGA CAA GAG-Cy3 |
- | 56.0 | - | 61.0 | - | - | 68.0 | +8.0 |
DNA3:DNA4 duplex is a model dsDNA target (Tm = 65.0 °C), where DNA3 = 5’- ACTGTGTGTTATATGCTGTTCTCAGCCCTACTG and DNA4 = 3’-TGACACACAATATACGACAAGAGTCGGGATGAC.
Experimental conditions are as stated in Table 1. ‘nt’= no clear sigmoidal transition. DYZ-NIP = ON23/24:ON25/26, DYZ-TIP = ON23:26and DYZ-REF = ON21:ON22. For representative denaturation profiles, see Figs. S24† and S25†.
Thermal denaturation experiments suggest that none of the double-stranded segments of DYZ-NIP display transitions above 20 °C (1st-3rd Tm columns, Table 2). Presumably, this is due to the low GC-content of the left and central sections (estimated by ON23:ON25 and ON23:ON26), whereas the presence of a G-triplet may impact the stability of the right segment (estimated by ON24:ON26). Transitions were observed at 36–37 °C in high salt buffer for the left and central sections, but no transitions were observed for the right segment (1st-3rd Tm columns, Table S4†). These observations suggest that DYZ-NIP is a labile probe.
All four DYZ-NIP probe strands form stable duplexes with 33-mer single-stranded DNA harboring complementary targets regions (4th-7th Tm columns, Table 2). The high affinity of the two main strands (ON23 and ON26) towards their cDNA target regions is noteworthy (Tm = 63 °C and 68 °C, respectively, Table 2). Recognition of the dsDNA target region is expected to be energetically feasible, given the greater stability of the probe-target duplexes vis-à-vis the double-stranded probe segments.
While the stability of the probe-target duplexes formed by the conventional Invader probe DYZ-REF are of similar stability relative to the dsDNA target (Tm = 61–68 °C vs 65 °C, Table 2), the probe duplex itself is also quite stable (Tm = 56 °C, Table 2). As a result, DYZ-REF only displays a minimally favorable driving force for recognition of dsDNA (TA = 8 °C, Table 2).
The dsDNA-targeting properties of DYZ-NIP were evaluated using the aforementioned DNA hairpin assay. Incubation of a 50-fold molar excess of DYZ-NIP with model dsDNA target DH2 - designed in a similar fashion as DH1 - results in observable recognition (~15%), whereas the corresponding conventional and toehold probes (i.e., DYZ-REF and DYZ-TIP (= ON23:ON26), respectively) do not lead to observable levels of recognition (Fig. 6). Incubation of DYZ-NIP with DH2-MM – a DNA hairpin designed akin to MM3 and differing in sequence at three positions vis-à-vis DYZ-NIP – does not result in recognition (Fig. 6). nd-FISH experiments were carried out next in which fixed interphase nuclei from a male bovine kidney cell line were incubated with DYZ-NIP. Gratifyingly, DYZ-NIP recognizes the Y-chromosome-specific DNA targets as evidenced by the formation of single, punctate Cy3-signals in ~40% of the nuclei, whereas conventional Invader probe DYZ-REF did not result in signal formation, even at high concentration (Fig. 7). Surprisingly, considering the results from the DNA hairpin experiments (Fig. 6), the corresponding toehold version of DYZ-NIP, i.e., DYZ-TIP, resulted in similar levels of recognition (Fig. S35†). The divergent results seen for DYZ-TIP are not fully understood.
Figure 6.
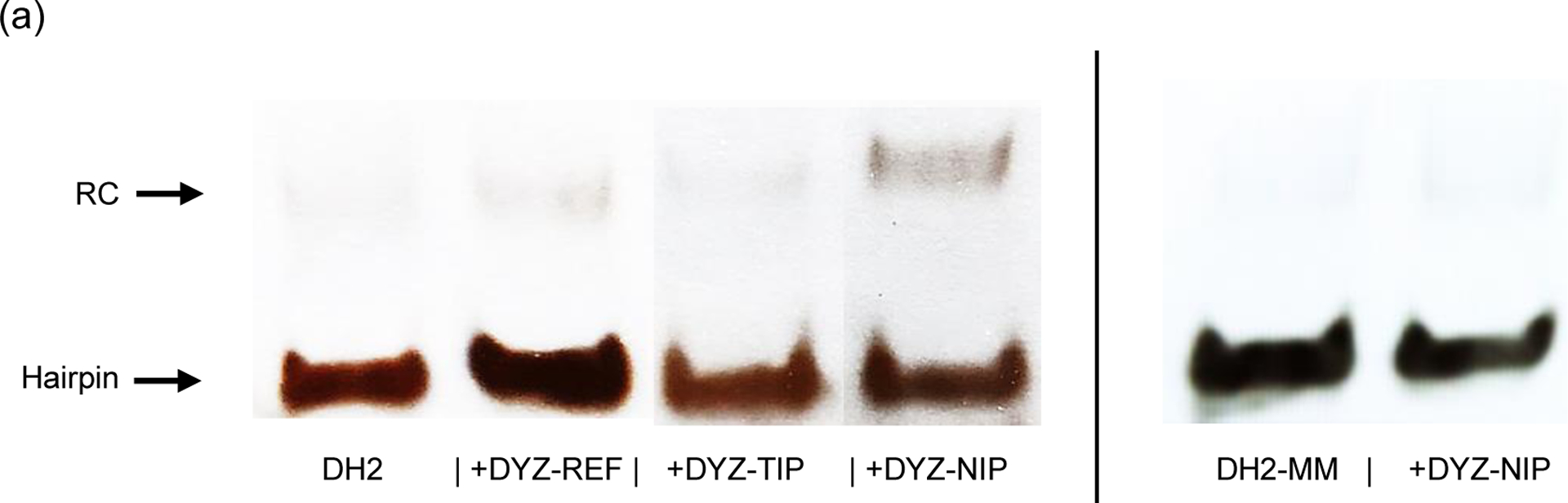
Representative gel electrophoretograms from recognition experiments in which a 50-fold molar excess of different DYZ-1-targeting probes was incubated with complementary target DH2 or non-complementary target DH2-MM at 37 °C for 17 h. For sequences of DH2 and DH2-MM and structures of recognition complexes formed, see Table S2† and Fig. S34†, respectively. Experimental conditions are as described in Fig. 2. The electrophoretogram is a composite image from different runs.
Figure 7.
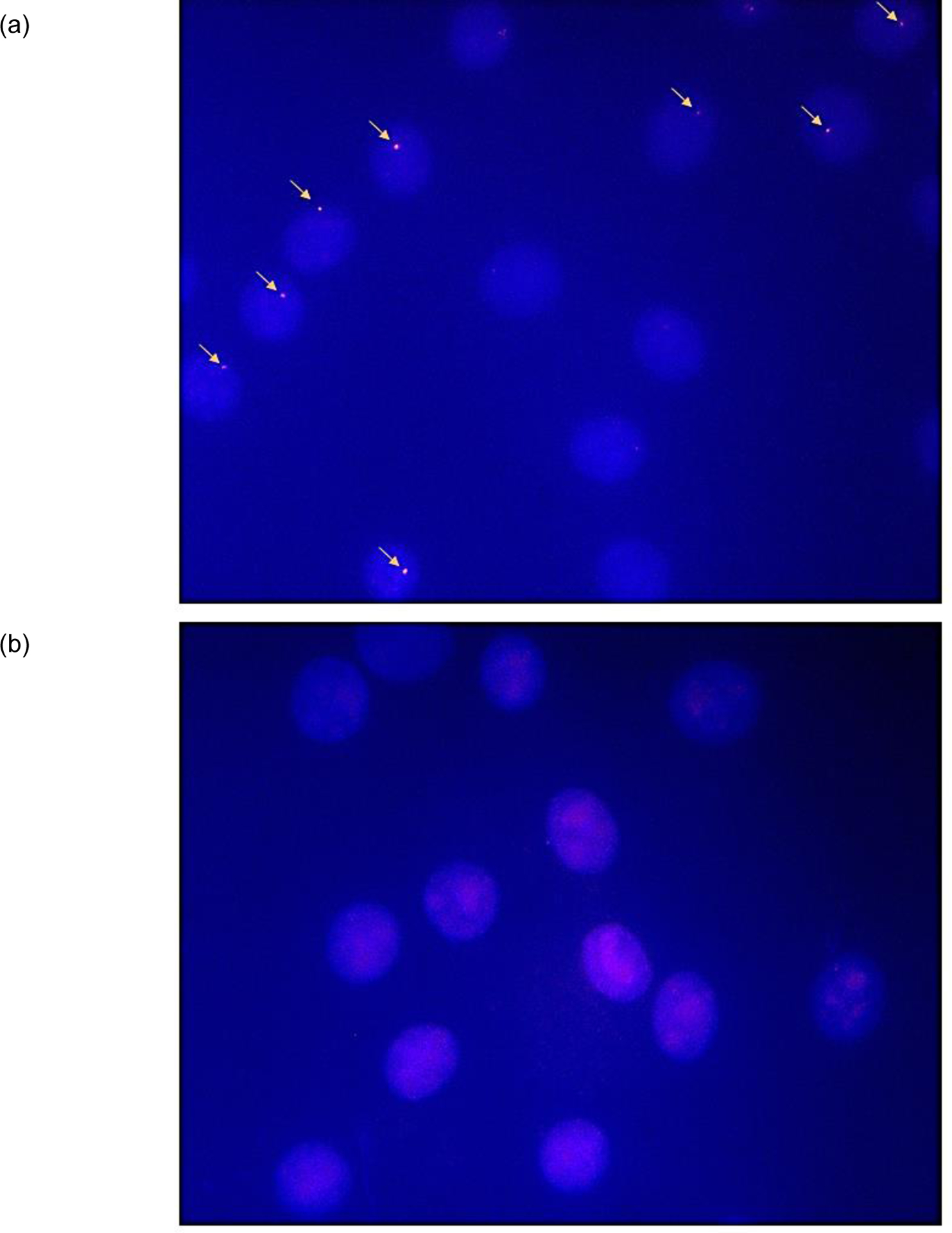
Images from nd-FISH experiments using (a) nicked Invader probe DYZ-NIP (1 ng per 200 μl of PCR buffer, 3 h, 37.5 °C) or (b) conventional Invader probe DYZ-REF (6 ng, 3 h, 37.5 °C). Fixed isolated interphase nuclei from male bovine kidney cells were incubated with probes in a Tris buffer (20 mM Tris-Cl, 100 mM KCl, pH 8.0) and counterstained with DAPI. Images were obtained by overlaying images from Cy3 (red) and DAPI (blue) channels and adjusting the exposure. Nuclei were viewed at 60X magnification using a Nikon Eclipse Ti-S inverted microscope. Probe concentrations were selected based on initial optimization studies (Fig. S35†).
Lastly, control experiments were performed in which DYZ-NIP was incubated with nuclei from a female bovine endothelial cell line that lacks the DYZ-1 region. Importantly, no signals were produced in nd-FISH experiments (Fig. S36†), demonstrating that nicked Invader probes allow for recognition of mixed-sequence chromosomal DNA region under non-denaturing conditions with excellent binding specificity.
CONCLUSION.
Nicked Invader probes recognize dsDNA targets more efficiently than conventional Invader probes. The distinguishing features of this new probe architecture is that it has three destabilized double-stranded segments and is comprised of four strands, each of which form stabilized duplexes with complementary DNA. It is this stability difference that drives dsDNA-recognition. Appropriately designed nicked Invader probes allow for recognition of long dsDNA targets (~25 base-pairs) under non-denaturing conditions, including chromosomal targets that are refractory to recognition by conventional Invader probes. We expect that this approach can be further elaborated with concatemeric Invader probes that tile along extended chromosomal DNA regions. The use of multiple shorter probe strands rather than fewer longer probe strands is appealing from a synthetic perspective but challenges concerning coordinated delivery will need to be considered. Nonetheless, the development of nicked Invader probes is an important advancement as it allows for highly specific, efficient, and sequence-unrestricted recognition of dsDNA target regions without relying on nucleoproteins or expression vectors. We expect many applications for nicked Invader probes in molecular biology.
EXPERIMENTAL SECTION
Synthesis and purification of ONs.
Modified ONs were synthesized on an automated DNA synthesizer (0.2 μmol scale) using long chain alkyl amine-controlled pore glass (LCAA-CPG) solid support with a pore size of 500 Å. The corresponding protected phosphoramidites of monomer U, A, and, C were prepared as previously described42,43 and incorporated into ONs via hand-couplings (0.05 M in acetonitrile, ~50-fold molar excess) using 0.01 M 4,5-dicyanoimidazole as the activator (15 min) and 0.02 M iodine in THF/H2O/pyridine for extended oxidation (45 s). Cy3-labeling of Invader strands was accomplished by incorporating a commercially available Cy3 phosphoramidite (Glen Research) into ONs by hand-coupling (4,5-dicyanoimidazole, 3 min, anhydrous CH3CN). Subsequent treatment with 32% ammonia (55 °C, 17 h) ensured deprotection and cleavage from the solid support of the crude DMTr-protected ONs, which were purified via ion-pair reverse phase HPLC (XTerra MS C18 column: 0.05 M triethylammonium acetate and acetonitrile gradient) followed by detritylation (80% aq. acetic acid, 20 min) and precipitation (NaOAc, NaClO4, acetone, −18 °C, 16 h). The purities of the synthesized ONs were verified using analytical HPLC (>85% purity, see Figs. S19–S21†), while their identities were verified either by MALDI-MS (using 2,4,6-trihydroxy acetophenone as a matrix) or LC-ESI-MS analysis (Waters/Acquity C18 column; triethylammonium formate and acetonitrile gradient) recorded on a Quadrupole Time-of-Flight (Q-TOF) mass spectrometer (Table S1† and Figs. S1–S18†).
Thermal denaturation experiments.
The concentrations of ONs were estimated using the following extinction coefficients (OD260/μmol): G (12.01), A (15.20), T/U (8.40), C (7.05), Cy3 (4.93), and pyrene (22.4)47. Tms of duplexes (0.5 μM final concentration of each strand) were determined using a Cary 100 UV/Vis spectrophotometer equipped with a 12-cell Peltier temperature controller and determined as the maximum of the first derivative of thermal denaturation curves (A260 vs. T) recorded in medium salt buffer (Tm buffer: 100 mM NaCl, 0.2 mM EDTA, and pH 7.0 adjusted with 10 mM Na2HPO4 and 5 mM Na2HPO4). Strands were mixed in quartz optical cells having a path-length of 1.0 cm and annealed by heating to ~90 °C (2 min) followed by cooling to the starting temperature of the experiment. A temperature range from 5–20 °C to 90 °C was typically used, with Tms determined as the average of at least two experiments within ±1.0 °C unless otherwise noted. A temperature ramp of 1 °C/min was used in all experiments.
Electrophoretic mobility shift assay.
Unmodified DNA strands were obtained from commercial sources and used without further purification. Target strands were DIG-labelled using the 2nd generation DIG Gel Shift Kit (Roche Applied Bioscience). Briefly, 11-digoxigenin-ddUTP was incorporated at the 3′-end of the strand (100 pmol) using a recombinant DNA terminal transferase. The reaction mixture was quenched through the addition of EDTA (50 mM), and then diluted to 100 nM in 2X HEPES buffer and used without further processing. The recognition experiments were conducted essentially as previously reported.25 Thus, conventional/toehold/nicked Invader probes (concentration as specified) were annealed (90 °C for 2–3 min, followed by cooling to room temperature) and subsequently incubated with separately pre-annealed DIG-labelled dsDNA targets (50 nM final concentration in 1X HEPES buffer: 50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, pH 7.2, 10% sucrose, 1.44 mM spermine tetrahydrochloride) at 37 °C ± 2 °C for 17 h.
Following incubation, loading dye (6X) was added and the mixtures were then loaded onto 12 % non-denaturing TBE-PAGE gels (45 mM tris-borate, 1 mM EDTA; acrylamide:bisacrylamide (19:1)). Mixtures were resolved via electrophoresis, which was performed using constant voltage (70 V) at ~4 °C. Bands were blotted onto positively charged nylon membranes (100 V, 30 min, ~4 °C) and cross-linked through exposure to UV light (254 nm, 5 × 15 W bulbs, 3 min). The membranes were incubated with anti-digoxigenin alkaline phosphatase Fab fragments as recommended by the manufacturer and transferred to a hybridization jacket. Membranes were incubated with the chemiluminescence substrate (CSPD) for 10 min at 37 °C, and chemiluminescence was captured on X-ray films. Digital images of developed X-ray films were obtained using a BioRad ChemiDoc™ MP Imaging system and used for densitometric quantification of the bands. The percentage of dsDNA recognition was calculated as the intensity ratio between the recognition band relative to the total lane. An average of at least three independent experiments is reported along with standard deviations (±). Electrophoretograms may be composite images from different runs.
Non-linear regression was used to fit data points from dose-response experiments. A script written for the “Solver” module in Microsoft Office Excel,48 was used to fit the following equation to the data points: y = C + A (1 – e−kt) where C, A and k are constants. The resulting equation was used to calculate C30 values by setting y = 30 and solving for t.
Cell culture and nuclei preparation.
Male bovine kidney cells (MDBK, ATCC: CCL-22, Bethesda, MD) were maintained in DMEM with GlutaMax (Gibco, 10569–010) and 10% fetal bovine serum (Invitrogen). Female bovine endothelial cells (CPAE, ATCC: CCL-209) were maintained in Eagle’s Minimum Essential Medium (ATTC, 30–2003) and 20% fetal bovine serum (Invitrogen). The cells were cultured in separate 25 mL or 75 mL flasks at 38.5 °C in a 5% CO2 atmosphere for 72–96 h to achieve 70–80% confluency. At this point, KaryoMax colcemid (Gibco, 15210–040) (65 μL per 5 mL of growth media) was added and the cells were incubated at 37 °C and 5% CO2 for an additional 20 min. At this point, the medium was replaced with pre-warmed 0.05% Trypsin-EDTA in DMEM to detach adherent cells (37 °C, up to 8 min). The cell suspension was transferred to a tube and centrifuged (10 min, 1000 rpm). The supernatant was discarded and the dislodged cell pellet incubated with a hypotonic KCl solution (5–8 mL, 75 mM, 20 min), followed by addition of fixative (10 drops, MeOH:AcOH, 3:1) and further incubation with gentle mixing (10 min, room temperature). The suspension was centrifuged (1000 rpm, 10 min), the supernatant discarded, and additional fixative solution (5–8 mL) added to the suspension of nuclei. This was followed by gentle mixing and incubation (30 min, room temperature). The centrifugation/resuspension/incubation with fixative solution steps were repeated three additional times. The final pellet – containing somatic nuclei – was resuspended in methanol and glacial acetic acid (3:1, v/v) and stored at −20 °C until use.
Preparation of slides for FISH assays.
The nuclei suspension was warmed to room temperature and resuspended in fresh fixative solution. Glass microscope slides were dipped in distilled water to create a uniform water layer across the slide. An aliquot of the nuclei suspension (3–5 μL or enough to cover the slide) was dropped onto the slide, while holding the slide at a 45 ° angle which allowed the suspension to run down the length of the slide. Slides were then allowed to dry at a ~20 ° angle in an environmental chamber at 28 °C and a relative humidity of 38%.
Fluorescence in situ hybridization experiments.
An aliquot of labelling buffer (~200 μL of a solution containing 1–30 ng of Cy3-labeled probes in 1X PCR buffer (10 mM Tris, 50 mM KCl, pH 8.0)) was placed on each slide. Preliminary assay optimization studies revealed that a probe amount of ~1 ng per 200 μL of labeling buffer resulted in the most suitable signal-to-background for nicked Invader probes under non-denaturing conditions (Fig. S35†). Slides with labelling buffer were placed in a glass culture disk, covered with a lid, and incubated in an oven (3 h, 37.5 °C). Slides were subsequently washed (3 min, 37.5 °C) in a chamber with TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0) and allowed to dry at room temperature. Once dried, Gold SlowFade plus DAPI (3 μL, Invitrogen) was placed directly on each slide and a round glass coverslip was mounted for fluorescence imaging.
A Nikon Eclipse Ti-S Inverted Microscope, equipped with a SOLA SMII LED light source system and Cy3 and DAPI filter sets, was used to visualize nuclei at 60X magnification to capture many nuclei in one image. Images of fluorescently labelled nuclei were captured using a 14-bit CoolSNAP HQ2 cooled CCD camera and processed with the NISElements BR 4.20 software. The percentage of nuclei presenting representative signals (i.e., labelling efficiency) was estimated by evaluating >50 nuclei per Invader probe.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Caroline P. Shepard (Univ. Idaho) for preparing DYZ-REF, Dr. Lee Deobald (Murdock Mass Spectrometry Center, Univ. Idaho) for assistance with mass spectrometric analysis, and Ms. Michaela Brown (Univ. Idaho) for proof-reading the manuscript. This study was supported by NIH grant no. GM088697 (National Institute of General Medical Sciences) and the Higher Education Research Council, Idaho State Board of Education [awards IF13-001, IF14-012].
Footnotes
CONFLICTS OF INTERESTS
P. J. H. is an inventor on patents pertaining to Invader probes, which have been issued to the University Idaho.
NOTES AND REFERENCES
- 1).Hsu PD, Lander ES and Zhang F, Cell, 2014, 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Komor AC, Badran AH and Liu DR, Cell, 2017, 168, 20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Duca M, Vekhoff P, Oussedik K, Halby L and Arimondo PB, Nucleic Acids Res, 2008, 36, 5123–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Hari Y, Obika S and Imanishi T, Eur. J. Org. Chem, 2012, 2012, 2875–2887. [Google Scholar]
- 5).Kaihatsu K, Janowski BA and Corey DR, Chem. Biol, 2004, 11, 749–758. [DOI] [PubMed] [Google Scholar]
- 6).Li W, Shi H, Dong B, Nie K, Liu Z and H N, Curr. Med. Chem, 2016, 23, 4681–4705. [DOI] [PubMed] [Google Scholar]
- 7).Brodyagin N, Katkevics M, Kotikam V, Ryan CA and Rozners E, Beilstein J. Org. Chem, 2021, 17, 1641–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Dervan PB and Edelson BS, Curr. Opin. Struct. Biol, 2003, 13, 284–299. [DOI] [PubMed] [Google Scholar]
- 9).Kawamoto Y, Bando T and Sugiyama H, Bioorg. Med. Chem, 2018, 26, 1393–1411. [DOI] [PubMed] [Google Scholar]
- 10).Bahal R, Sahu B, Rapireddy S, Lee C-M and Ly DH, ChemBioChem, 2012, 13, 56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Shigi N, Mizuno Y, Kunifuda H, Matsumura K and Komiyama M, Bull. Chem. Soc. Jpn, 2019, 92, 330–335. [Google Scholar]
- 12).Zheng H, Botos I, Clausse V, Nikolayevskiy H, Rastede EE, Fouz MF, Mazur SJ and Appella DH, Nucleic Acids Res, 2021, 49, 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Zaghloul EM, Madsen AS, Moreno PMD, Oprea II, El-Andaloussi S, Bestas B, Gupta P, Pedersen EB, Lundin KE, Wengel J and Smith CIE, Nucleic Acids Res, 2011, 39, 1142–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Kutyavin IV, Rhinehart RL, Lukhtanov EA, Gorn VV, Meyer RB and Gamper HB, Biochemistry, 1996, 35, 11170–11176. [DOI] [PubMed] [Google Scholar]
- 15).Lohse J, Dahl O and Nielsen PE, Proc. Natl. Acad. Sci, 1999, 96, 11804–11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Aiba Y, Honda Y and Komiyama M, Chem. Eur. J, 2015, 21, 4021–4026. [DOI] [PubMed] [Google Scholar]
- 17).Hibino M, Aiba Y, Watanabe Y and Shoji O, ChemBioChem, 2018, 19, 1601–1604. [DOI] [PubMed] [Google Scholar]
- 18).Bryld T, Højland T and Wengel J, Chem. Commun, 2004, 1064–1065. [DOI] [PubMed] [Google Scholar]
- 19).Filichev VV, Christensen UB, Pedersen EB, Babu BR and Wengel J, ChemBioChem, 2004, 5, 1673–1679. [DOI] [PubMed] [Google Scholar]
- 20).Asanuma H, Niwa R, Akahane M, Murayama K, Kashida H and Kamiya Y, Bioorg. Med. Chem, 2016, 24, 4129–4137. [DOI] [PubMed] [Google Scholar]
- 21).Filichev VV, Vester B, Hansen LH and Pedersen EB, Nucleic Acids Res, 2005, 33, 7129–7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Bohländer PR, Vilaivan T and Wagenknecht H-A, Org. Biomol. Chem, 2015, 13, 9223–9230. [DOI] [PubMed] [Google Scholar]
- 23).Nakamura S, Kawabata H and Fujimoto Kenzo, Chem. Commun, 2017, 53, 7616–7619. [DOI] [PubMed] [Google Scholar]
- 24).Hrdlicka PJ, Kumar TS and Wengel J, Chem. Commun, 2005, 4279–4281. [DOI] [PubMed] [Google Scholar]
- 25).Guenther DC, Anderson GH, Karmakar S, Anderson BA, Didion BA, Guo W, Verstegen JP and Hrdlicka PJ, Chem. Sci, 2015, 6, 5006–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26). For a definition of interstrand zipper nomenclature, see ESI†.
- 27).Crothers DM, Biopolymers, 1968, 6, 575–584. [DOI] [PubMed] [Google Scholar]
- 28).Sau SP, Madsen AS, Podbevsek P, Andersen NK, Kumar TS, Andersen S, Rathje RL, Anderson BA, Guenther DC, Karmakar S, Kumar P, Plavec J, Wengel J and Hrdlicka PJ, J. Org. Chem, 2013, 78, 9560–9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Karmakar S, Madsen AS, Guenther DC, Gibbons BC and Hrdlicka PJ, Org. Biomol. Chem, 2014, 12, 7758–7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Jain SC, Tsai C and Sobell HM, J. Mol. Biol, 1977, 114, 317–331. [DOI] [PubMed] [Google Scholar]
- 31).Williams LD, Egli M, Gao Q and Rich A, in Structure and Function: Nucleic Acids, ed. Sarma RH and Sarma MH, Adenine Press, 1992, vol. 1, pp. 107–125. [Google Scholar]
- 32).Ihmels H and Otto D, Top. Curr. Chem, 2005, 258, 161–204. [Google Scholar]
- 33). A related dsDNA-targeting approach (see reference 21), which is based on DNA duplexes with adjacent incorporations of pyrene-modified non-nucleotide monomers, appeared in the scientific literature following our original study (reference 24).
- 34).Denn B, Karmakar S, Guenther DC and Hrdlicka PJ, Chem. Commun, 2013, 49, 9851–9853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Emehiser RG, Hall E, Guenther DC, Karmakar S and Hrdlicka PJ, Org. Biomol. Chem, 2019, 18, 56–65. [DOI] [PubMed] [Google Scholar]
- 36).Karmakar S, Guenther DC, Gibbons BC and Hrdlicka PJ, Org. Biomol. Chem, 2017, 15, 9362–9371 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Guenther DC, Karmakar S and Hrdlicka PJ, Chem. Commun, 2015, 51, 15051–15054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Anderson BA and Hrdlicka PJ, J. Org. Chem, 2016, 81, 3335–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Emehiser RG and Hrdlicka PJ, Org. Biomol. Chem, 2020, 18, 1359–1368. [DOI] [PubMed] [Google Scholar]
- 40).Adhikari SP, Vukelich P, Guenther DC, Karmakar S and Hrdlicka PJ, Org. Biomol. Chem, 2021, 19, 9276–9290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41).Kaur H, Babu BR and Maiti S, Chem. Rev, 2007, 107, 4672–4697. [DOI] [PubMed] [Google Scholar]
- 42).Karmakar S, Anderson BA, Rathje RL, Andersen S, Jensen TB, Nielsen P and Hrdlicka PJ, J. Org. Chem, 2011, 76, 7119–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Karmakar S, Guenther DC and Hrdlicka PJ, J. Org. Chem, 2013, 78, 12040–12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Demidov VV, Protozanova E, Izvolsky KI, Price C, Nielsen PE and Frank-Kamenetskii MD, Proc. Natl. Acad. Sci. U.S.A, 2002, 99, 5953–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Chen SX, Zhang DY and Seelig G, Nat. Chem, 2013, 5, 782–789.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Perret J, Shia Y-C, Fries R, Vassart G and Georges M, Genomics, 1990, 6, 482–490. [DOI] [PubMed] [Google Scholar]
- 47).Dioubankova NN, Malakhov AD, Stetsenko DA, Gait MJ, Volynsky PE, Efremov RG and Korshun VA, ChemBioChem, 2003, 4, 841–847. [DOI] [PubMed] [Google Scholar]
- 48).Brown AM, Comput. Methods Programs Biomed, 2001, 65, 191–200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.