

Abstract
Human neutrophil elastase (hNE) is an abundant serine protease that is a major constituent of lung elastolytic activity. However, when secreted in excess, if not properly attenuated by selective inhibitor proteins, it can have detrimental effects on host tissues, leading to chronic lung inflammation and non‐small cell lung cancer. To improve upon the design of inhibitors against hNE for therapeutic applications, here, we report the crystal structure of hNE in complex with an ecotin (ET)‐derived peptide inhibitor. We show that the peptide binds in the nonprime substrate binding site. Unexpectedly, compared with full‐length (FL) ET, we find that our short linear peptides and circular amide backbone–linked peptides of ET are incapable of efficient hNE inhibition. Our structural insights point to a preferred amino acid sequence and the potential benefit of a scaffold for optimal binding and function of the peptide inhibitor, both of which are retained in the FL ET protein. These findings will aid in the development of effective peptide‐based inhibitors against hNE for targeted therapy.
Keywords: elastase, inhibition, peptide, serine protease, structure
Short abstract
PDB Code(s): 7WHU.
1. INTRODUCTION
Neutrophil serine proteases are released in excess at inflammatory sites following lung injury. Under normal circumstances, the host, in response, will also secrete protease inhibitors to protect the host tissue from degradation once the initial injury has abated. However, in chronic inflammatory lung conditions, there is an imbalance in the production of proteases and inhibitors in favor of proteases, with the excess production of these proteases associated with the pathogenesis of inflammatory lung disease and cancer. 1 , 2 , 3
Human neutrophil elastase (hNE; MW 29.5 kDa) is a serine protease belonging to the chymotrypsin family. This glycoprotein, in excess, is frequently linked to chronic lung inflammation and the progression of non‐small cell lung cancer 4 and is now considered as a well‐established biomarker of disease progression. 1 , 2 , 5 At present, only two inhibitors against hNE are in clinical use: the peptide‐based drug, Prolastin, and the non‐peptide drug, Sivelestat. 6 , 7 Both drugs, however, are easily degraded under pathophysiological conditions which limits their efficacy. 2 , 7 In addition, there are two potent third‐ and fifth‐generation small‐molecule inhibitors of hNE: AZD9668 and BAY 85‐8501, respectively. 2 The peptide inhibitors are shown to have several advantages over the small‐molecule inhibitors. The peptides are naturally existing biologics and are, therefore, deemed safer for the host than synthetic inhibitors. 8 Indeed, it has been shown that peptides derived from natural protein inhibitors are preferred over synthetic peptides as they cause less cytotoxicity. 9 Current natural inhibitor peptides, however, are unstable under pathophysiological conditions, show a lack of precise selectivity, and have an ineffective inhibitory potential. 2 Synthetic peptides with modified amino acids are therefore frequently sought and optimized to improve the affinity and inhibitory potential of peptides toward proteases. 10 , 11 Taking all these into consideration, non‐synthetic peptide‐derived inhibitors remain the lofty goal in drug development for therapeutic targeting of proteases in disease.
To date, four peptide‐derived inhibitor complexes of hNE have been reported (PDB codes 1HNE, 1PPG, 2RG3*, 4WVP*, where "*" denotes covalently linked complexes). 12 , 13 , 14 , 15 Complexes 1HNE, 1PPG, and 2RG3 describe a peptide inhibitor interaction with hNE, whereas 4WVP is a peptide inhibitor that is also used to probe hNE activity. The crystal structures of these complexes show that the peptide inhibitors target only the non‐prime sites of hNE to inhibit the enzyme. A recent report from Hochscherf et al. detailed the structure of a small‐molecule inhibitor (1,3‐thiazolidine‐2,4‐dione derivative) in complex with leukocyte elastase (PDB code 6F5M), in which the inhibitor targeted the prime site of the substrate binding site. 16
Recently, we reported the crystal structure of hNE in complex with the serine protease inhibitor protein, ecotin (ET; MW 18.3 kDa) 17 that lead to the identification of two independent binding regions of ET involved in the inhibition of hNE. Further, the comparative analyses revealed how the hNE activity can be inhibited by various inhibitors and the broad specific inhibition property of ET against various proteases.
In our continued efforts to understand the inhibitory mechanisms of proteases, here we sought to explore the inhibitory potential of four peptides (Pep1–Pep4) against hNE. These peptides were designed based on our previous study and literature. 17 , 18 We also report the crystal structure of Pep1 in complex with hNE. Compared with ET, we observe that these inhibitors cannot fully inhibit hNE. We propose that optimal inhibition requires preferred amino acid side chains—passing through the substrate binding site(s)—to engage with subsites for hNE inhibition and surmise that a rigid scaffold that resists proteolytic cleavage by the protease hNE would enhance the binding and inhibitory activity of ET‐derived peptides.
2. RESULTS AND DISCUSSION
2.1. ET‐derived peptides and the inhibition of hNE
ET is a potent inhibitor of hNE. 19 Using our previous complex structure and guided by literature, 17 , 20 , 21 we designed four ET‐derived peptides (Pep1–Pep4) and tested their inhibitory potential against hNE. Peptide 1 (Pep1) was designed as a linear peptide to interact with the non‐prime S site, whereas the linear peptide 3 (Pep3) was designed to interact with both the non‐prime and prime, S and S′ sites, respectively (See Materials and Methods, Section 3). Pep2 and Pep4 were designed as cyclic modifications of Pep1 and Pep3 respectively, where the N‐ and C‐termini of the peptide were amide cyclized (Figure 1a). Pep2 and Pep4 were designed based on previous suppositions that cyclized peptides offered increased inhibition, specificity, stability, and exposed key interacting residues for binding. Furthermore, cyclized peptides are thought to be more rigid, which provides resistance against protease cleavage. 20 , 21 , 22 , 23 These peptides were used in enzymatic activity assays to understand their efficacy of inhibition. The full‐length (FL) inhibitor protein ET was purified and used as a control.
FIGURE 1.

Design of peptides. Peptides were derived from ecotin, a potent inhibitor of human neutrophil elastase (hNE). Four peptides were designed for the inhibition studies. (a) Pep1 is a linear peptide with the sequence 77VSSPVSTM84. Pep2 is an amide‐cyclized peptide with the same sequence as Pep1. The N and C terminals are covalently linked through an amide bond. The Pep2 was designed to ensure lower protease digestion, expose the residues, and facilitate interaction with hNE. The Pep3 is a longer linear peptide with the sequence 77VSSPVSTMMACPA89. This peptide was designed to occupy the prime subsites beyond S1, taking cues from the previously known structure (7CBK, hNE‐ecotin complex). Similar to Pep2, Pep4 is a cyclized peptide of Pep3 designed to maintain the stability of the peptide for better inhibition. (b) The four peptides (Pep1–Pep4) were used in enzymatic assays to evaluate their inhibitory potencies against wild‐type hNE. The chromogenic substrate, upon cleavage by hNE, releases p‐nitroaniline that is detected at 405–410 nm. The inhibition curves shown here indicate that the full‐length ecotin protein has the best inhibition at 0.7 nM. Out of the four peptides, Pep1 has the best IC50 of 1.9 μM followed by Pep3 with 10.8 μM. Pep2 and Pep4 were designed as amide‐cyclized peptides for better stability and interaction with hNE. However, they seem to have very low inhibitory effect on the activity of the enzyme
FL ET inhibited hNE at an IC50 of 0.7 nM (Figure 1b), similar to a previous report. 19 Linear peptides Pep1 and Pep3 inhibited hNE at 1.9 and 10.8 μM, respectively, and are thus not as effective as FL ET at inhibiting hNE. We surmised that these peptides may undergo partial cleavage by hNE. Surprisingly, Pep2 and Pep4 offered no improvement in inhibition potential. There are two possible reasons for the lower inhibition of these peptides: (1) The conformational features of the linear and cyclized peptides might be unfavorable for hNE inhibition and (2) several amino acid preferences in positions P3–P3′ that are known to play a major role in the inhibition of hNE are missing, particularly in Pep1 and Pep2. 11 , 24
We tested the first hypothesis through de novo folding simulations of Pep1 and Pep3. We found that these peptides do not have a similar structure to that of wild‐type FL ET in the same region (hNE‐ET complex, PDB 7CBK). In the hNE‐ET (7CBK) crystal, the 81VSTMMA86 stretch of amino acids protrudes from the structure as a β‐strand, thereby making this conformation more capable of interacting with hNE. Moreover, this amino acid stretch has a small directional twist in the FL ET protein that is not present in the free N‐ and C‐termini of the peptides; this twist is stabilized by the β‐strand, which forms an antiparallel sheet in the hNE‐ET complex with the proximal His53‐Arg54, Ile42‐Val48 and Glu93‐Thr98 β‐strands of FL ET in the complex. Additionally, it is possible that the cyclic peptides (Pep2 and Pep4) provide steric occlusion due to the rigidity to the crucial amino acids that make them incapable of effective interactions.
In terms of our second hypothesis—that amino acid preferences in positions P3–P3′ play a major role in the inhibition of hNE—we note from previous work that differences in the peptide sequences directly contribute to an enhanced affinity to hNE. In addition, ET from Pseudomonas aeruginosa has been shown to inhibit hNE (and other proteases) with better affinity than its Escherichia coli counterpart. 25 , 26 Additionally, multiple reports have shown that chemical modification of amino acids in the peptide routinely offers better inhibition of hNE. 11 , 13 , 27
2.2. Structure of hNE‐ET peptide complex
We next solved the complex structure of hNE‐ETPep1 (Figure 2) by molecular replacement using hNE as a search model (PDB IHNE). The complex was refined to an R value of 0.252 (R‐free 0.306) at 2.9 Å resolution, with good stereochemical parameters (supplementary table 1). The complex molecule is well‐defined in the electron density map, except for the first 15 residues of hNE. The asymmetric unit consists of a tetramer, and each monomer of hNE is bound with the 8‐aa inhibitor peptide (77VSSPVSTM84).
FIGURE 2.
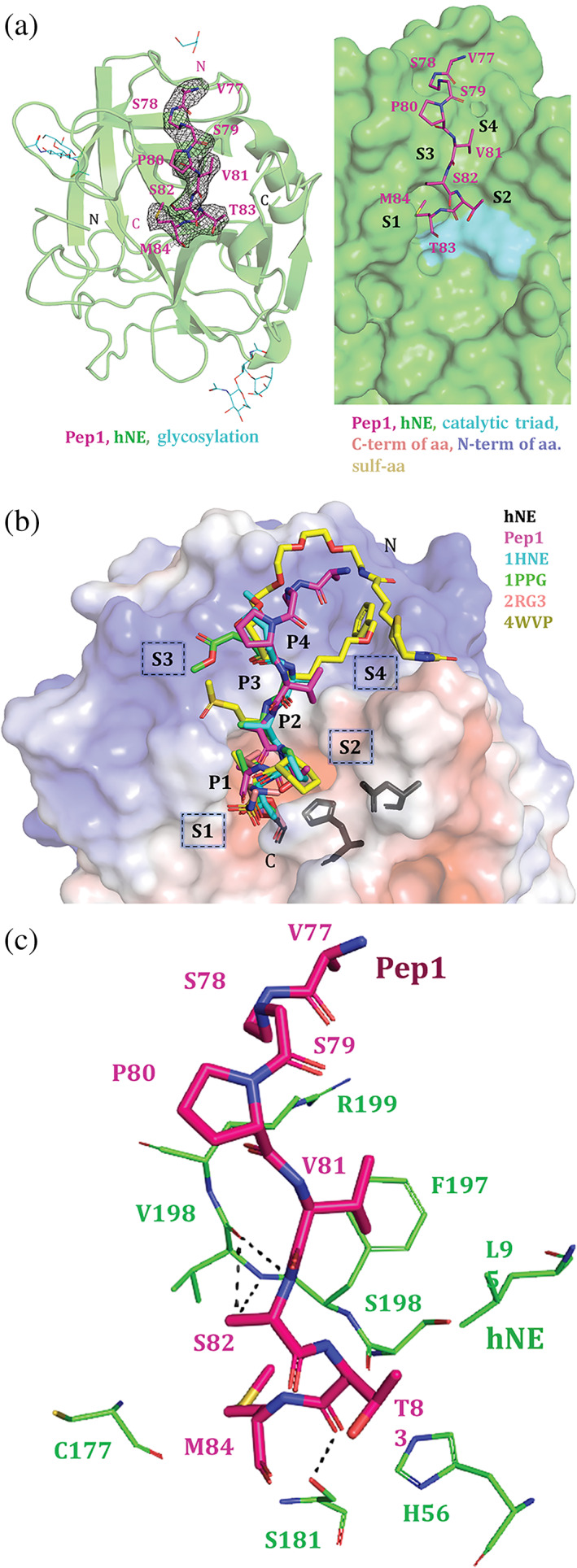
Crystal structure of hNE‐ETPep1. (a) The left panel shows the hNE‐ETPep1 monomer structure and the Pep1 bound to the S subsites of hNE enzyme. The 2Fo − Fc map (gray isomesh) for the bound peptide (magenta) is contoured at 0.9 σ. The hNE (shown in as green cartoon) is glycosylated at two amino acids (shown in cyan)—Asn109 and Asn159. The right panel shows the bound peptide consisting of Met84, Thr83, Ser82, and Val81 which are P1–P4 positions of the inhibitor, and they interact with the S1–S4 subsites of the hNE (shown as green surface representation). The catalytic triad (His57, Asp102, and Ser195) is shown in cyan. (b) Comparison of the peptide orientation from known hNE complex crystal structures. There are four known structures of hNE‐peptide complexes (1HNE, 1PPG, 2RG3, and 4WVP). These peptides in these structures target the same subsites on hNE as Pep1 from our structure. hNE is shown as an electrostatic potential surface with the peptides aligned well. The P1 and P2 target the S1 and S2 subsites which are largely acidic in nature, whereas the P3 and P4 target S3 and S4 subsites which form more basic/alkaline regions in hNE. The superposition of the hNE‐peptide complexes shows that the peptides occupy the same subsites in the hNE (electrostatic surface representation). The catalytic triad residues (shown in dark gray) are close to the S2 subsites. (c) The Pep1 (shown as thick sticks in magenta) interacts with S1–S4 subsites formed by the residues of hNE (thin green sticks). The key hydrogen bonds are shown as thin black dashed line
The 267‐aa‐long hNE adopts a chymotrypsin fold, with the peptidase domain formed by residues 30–247 aa. A catalytic triad (His70, Asp117, and Ser202) is formed by interactions from both the N‐terminal and C‐terminal domains of hNE. Asn109 and Asn159 of hNE are glycosylated in this complex. Glycosylation in the hNE‐ETPep1 structure encompasses two NAG‐NAG‐FUC glycan links (NAG: N‐acetylglucosamine, FUC: fucose), albeit FUC is missing in one monomer and is replaced by BMA in another monomer (beta‐D‐mannose).
In the current complex structure, Pep1 occupies the same subsites as ET in the hNE‐ET complex previously solved by us (7CBK). 17 Notably, the peptide consists of Val81 (P4), Ser82 (P3), Thr83 (P2), and Met84 (P1) interacting with the S4 (Val198), S3 (Val198), S2 (Ser196, Ser181, and His56), and S1 (Ser181, Gly179, Phe178, Cys177, and His57) subsites of hNE, respectively. Pep1 forms several hydrogen bonds with the hNE protein, the complex further strengthened by numerous hydrophobic interactions. The catalytic triad (His70, Asp117, and Ser202) is located close to the S1/S2 subsite (Figure 2b), as in the previous hNE‐peptide complex structures. 12 , 13 , 14 , 15
A comparison of the hNE‐ETPep1 and hNE‐ET (7CBK) structures indicated good alignment, with a root‐mean‐square deviation of 0.455 Å (for 196 Cα atoms) (Figure 3b). The bound Pep1 is oriented similarly to the equivalent region of ET in the hNE‐ET complex. Structural superposition of hNE‐ETPep1 with each of the other four peptide inhibitor‐hNE complexes (PDB codes 1HNE, 1PPG, 2RG3, and 4WVP) 12 , 13 , 14 , 15 , 27 also indicated good alignment. hNE‐ETPep1 structure has a larger (460 Å2) buried surface area (BSA) than that of other hNE‐peptide complexes (281–345 Å2), which is suggestive of a larger peptide–protein interface and additional interactions in the hNE‐ETPep1 complex. The equivalent region in the hNE‐ET complex, however, has a higher BSA and a higher number of hydrogen bonds between hNE and ET than in our hNE‐ETPep1 complex structure (Figure 3b).
FIGURE 3.

The scaffold of the inhibitor is crucial. (a) The structural superposition of hNE‐ETPep1, hNE‐ET (human neutrophil elastase–ecotin) and the known hNE‐peptide complexes (PDB codes 1HNE, 1PPG, 2RG3, 4WVP) show that the peptides bind to an active site groove on hNE. The groove is shown in different orientations. Left side view, central view and right‐side view are shown here. Primarily, the groove consist of all residues (41CG42, 56HC57, 95NL96, 161LCR163, 176VCFGDSGS183, 195ASFVRGG201 and 206Y) of hNE within 5 Å distance from the inhibitory peptides (shown as sticks in the central view right panel). The ecotin inhibitory region (shown in grey) traverses this hNE groove deeply and therefore responsible for the effective nanomolar inhibition of the enzyme. In the bottom right panel, the surface is shown to be transparent with the Pep1 (magenta sticks) and ecotin (grey ribbon). (b) The comparison of the hNE‐ETPep1, hNE‐ET and the known peptide complexes (PBD codes 1HNE, 1PPG, 2RG3, 4WVP) are shown here. The amino acids at P4 to P3′ positions from different peptides (and FL ecotin) are shown. The superposition and related RMSD (root‐mean‐square deviation) values show that the structures align well. The buried surface area (BSA) is the highest in the ecotin‐hNE complex followed by hNE‐ETPep1 and then other peptides complexes. The number of hydrogen bonds between the inhibitor peptide (or ecotin protein) and hNE protein is shown in the last row. The figures below correspond to the inhibitor present in the respective structure
2.3. Amino acid residue preferences of hNE for peptides targeting the subsites
The previous peptide‐inhibitor complexes (PDB codes 1HNE, 1PPG, 2RG3, and 4WVP) 12 , 13 , 14 , 15 used modified (or unnatural) amino acids, with binding predominantly confined to the non‐prime S (1–4) subsites. Notably, all subsites (1–4) in the hNE‐PK101 complex (4WVP) were occupied by unnatural amino acid groups. 13 In our complex, the P4 position (Val81 in Pep1) of the peptide maintains its interactions with Val198 and Phe197 of hNE, and in all of the other complexes—with the exception of 4WVP—this position in the peptide is occupied by small hydrophobic amino acids such as alanine or valine (Figure 3b). Similar to the P4 position, the P3 position in our complex is also occupied by amino acids with small side chains, like serine or alanine. Ser82 of Pep1 from hNE‐ETPep1 structure is at position P3 and interacts with Val198 of hNE. In 4WVP, P3 has a methionine‐derived “methionine sulfone (OMT)” (also known as Met(O)2) group, which forms additional hydrogen bonds with hNE. The P2 position in our complex is a threonine residue, which makes four hydrogen‐bonding contacts with hNE. However, when substituted by proline (in 1HNE and 1PPG) or by “ L‐ octahydroindole‐2‐carboxylic acid (OIC)” (in 4WVP), these hydrogen bonds are missing.
Finally, the P1 subsite—regarded as the most crucial amino acid position for effective hNE inhibition—is occupied by either an unnatural or small hydrophobic amino acid in the previous four complexes. In the hNE‐ETPep1 structure, Met84 (of Pep1) is at the P1 position and makes interactions with the S1 subsite. Many other studies report the use of unnatural substitutions in this P1 position to avoid cleavage and degradation by the protease. 12 , 13 , 14 , 15 For example, although multiple interactions between P1 and S1 are facilitated by hydrophobic amino acids, such as methionine, valine, or isoleucine, at this position, these amino acid residues are cleaved by hNE, thereby making the peptide interaction less effective. 24 , 28 , 29 Therefore, many unnatural substitutions were reported in P1 position to avoid cleavage and degradation by the proteases. 12 , 13 , 14 , 15 In the 2RG3 and 4WVP complexes, the P1 position is occupied by synthetic groups, such as “BG1” and “3V2,” which make several hydrogen‐bonding contacts with hNE. This singular difference might explain why the peptides in these two complexes, 2RG3 and 4WVP, bind tighter to hNE than the other peptides, despite their loss of interaction with the S2 subsite, as mentioned before.
2.4. Scaffold of inhibitory peptides
Although FL ET binds and strongly inhibits hNE, the peptides derived from ET are much less effective: This may be attributed to the overall framework of the peptides in addition to the preferred amino acids substitutions at specific subsites. In other words, FL ET possibly provides a better scaffold or framework for interacting with the S subsites of hNE. This could be for two reasons. First, FL ET fits tightly into a large groove formed by 41CG42, 56HC57, 95NL96, 161LCR163, 176VCFGDSGS183, 195ASFVRGG201, and 206Y at 5 Å or less from ET. The bound peptides from the previously solved hNE complex structures also largely target this groove for inhibition, and, in our structure, Pep1 also aligns well with ET but has shorter and fewer interactions. Second, FL ET makes additional interactions with hNE at a secondary site, which involves the 60s loop (Leu64‐Tyr69) of ET and secondary binding site residues of hNE (Gly92, Gln126, Gln233, Phe234, Asn236, Trp237, and Ser240). This accessory interaction may facilitate a more efficient binding and, in turn, inhibition of the protein. 17 , 30
In our investigations—and contrary to many previous reports 20 , 21 , 23 —the cyclized peptides had no enhanced hNE inhibition over the linear peptides, indicating that the amide cyclization of these peptides did not favorably orient the P4–P1 (or P4–P4′ in the case of the larger peptide, Pep3) amino acids to enhance hNE inhibition. We surmise that a larger cyclic peptide might offer an improved binding orientation. Alternatively, various amino acid substitutions at P4′‐P4 positions (to target the S′‐S subsites) could be made to improve the interactions between the inhibitor and enzyme. Moreover, chimeric peptides designed with a linker to engage the secondary binding site of hNE may also improve the inhibition potential of the peptides. Indeed, augmenting the ET peptide with an optimal scaffold design and a linker region to connect the secondary binding site could improve its insertion into the active site groove and avoid cleavage and degradation by the protease.
Notably, the overexpression of hNE in pathogenic state must be regulated but not completely inhibited. 31 The development of peptide inhibitors for hNE has to be focused on ensuring moderate inhibition of hNE and thereby reverse disease phenotype. A small‐molecule inhibitor, Sivelestat, has comparable inhibition values to Pep1 and Pep2 (in micromolar range) and accompanied by improved pulmonary function and general betterment in patients with acute lung injury. This inhibitor (drug) is being permitted to use in Japan and other countries. 7 , 32
In summary, here we have reported the use of natural ET‐derived peptide inhibitors against hNE. Combined with literature, we propose that the following points should be considered to optimize an effective peptide for hNE inhibition. (1) There is a preference for specific amino acid residue side chains that pass through and interact with the hNE subsites, including the active site region. Notably, the P1 position is crucial, and the presence of a modified or an unnatural amino acid side chain in this subsite will avoid degradation of the residue by the enzyme and allow for multiple interactions to enhance binding. (2) A scaffold is required to enhance the stability of the peptide‐enzyme interaction. FL ET possesses a stable β‐sheet in which one β‐strand of this β‐sheet effectively interacts with the hNE residues. An inhibitory peptide abridged with a scaffold will be better able to wedge itself into the hNE groove (i.e., subsites of protein substrate binding pocket) and engage in more interactions at the primary binding site (the groove along the active site region). Furthermore, the use of a linker that connects with the secondary binding site will likely enhance the interaction. Next‐generation peptide inhibitors of hNE that take these factors into consideration should be the focus of future studies.
3. MATERIALS AND METHODS
3.1. Design of ET‐derived peptides
Our previous structure of the hNE‐ET complex (PDB: 7CBK) showed interactions between ET and hNE. Peptides resembling the ET protein were designed to inhibit hNE. The peptides are based on the 77VSSPVSTM84 region of the ET sequence that corresponds to the region of primary interaction between hNE and ET. Specifically, two peptides 77VSSPVSTM84 (Pep1) and 77VSSPVSTMMACPA89 (Pep3) were designed and procured from Biomatik (Wilmington, DE). Apart from this, two cyclic peptides (Pep2 and Pep4) were also designed to maintain stability in the interacting regions.
3.2. Protein expression and purification
hNE (peptidase domain; residues 30–247 with a molecular weight of 23 kDa, Uniprot P08246) was purchased from Elastin Products Company, buffer exchanged (50 mM Tris–HCl pH 8, 100 mM NaCl, and 20 mM CaCl2), and concentrated to 200 μM prior to complex formation. The ET gene was cloned into a modified pET32a vector with an N‐terminal hexa‐histidine tag between the BamHI and XhoI sites and overexpressed in E. coli BL21 (DE3). The primary culture of 100 ml was grown in Luria Bertani media at 37°C overnight and transferred to 1 L of LB media. Both primary and secondary LB media contain ampicillin with final concentration of 100 μg/ml. Cultures were grown at 37°C till they reached an optical density of 0.6–0.8 and induced with 250 μM of isopropyl β‐d‐1‐thiogalactopyranoside at 18°C for 16 hr. The cell cultures were pelleted at 3500 rpm (Beckmann Coulter Centrifuge JLA‐8.1 Rotor). The pellets were resuspended in lysis buffer (50 mM Tris–HCl pH 8, 100 mM NaCl, and 5 mM CaCl2) and lysed by sonication. After lysis, ET was purified from the supernatant by nickel‐nitriloacetic acid affinity chromatography followed by size‐exclusion chromatography using Superdex 75 column. ET has a molecular weight of 16 kDa, and it elutes as a dimer with molecular weight of 32 kDa.
3.3. Enzymatic assay for hNE activity
The elastase activity assay was performed with the peptide substrate similar to previous reports. 11 , 14 , 29 In short, the peptides/protein was incubated with the elastase for 15 min on ice. Master mix (containing chromogenic substrate [Calbiochem]; 1 mM of N‐MeO‐Suc‐Ala‐Pro‐Val‐p‐NA and Tris–HCl, pH 7.5) was added to make up 200 μl assay volume to initiate the reaction. End‐point measurements were made using Tecan microplate reader at 405 nm wavelength. The data points were plotted on GraphPad prism and fitted using regression values to get inhibition curves and IC50.
3.4. Crystallization and structure determination of hNE‐ET peptide complex
We have started with hNE‐ET complex for crystallization; however, we have obtained the crystals of hNE‐ETPep1 (77VSSPVSTM84). The crystallization screening of the complex was carried out using commercially available screens by incubating 1 μl protein‐complex solution (9 mg/ml) with 1 μl crystallization solution in a hanging drop vapor diffusion method. Crystals appeared after 7 days in 0.2 M ammonium sulfate, 0.1 M BIS‐TRIS pH 5.5, and 25% w/v polyethylene glycol 3350 at room temperature (24°C) which were further optimized to obtain diffraction quality crystals. Prior to data collection, the crystals were cryo‐protected using the crystallization solution supplemented with 25% glycerol. The crystal diffracted up to 2.9 Å, and data were collected using Rigaku MicroMax‐007. The data were processed and scaled using HKL2000. 33
The crystals belong to space group P21 with a hetero‐tetrameric complex molecule in the asymmetric unit. The initial phases were obtained using Phenix 34 by molecular replacement with the PHASER program using the search models PDB 1HNE (for neutrophil elastase). 12 Refinement was carried out using Phenix‐Refine, and the remaining model building was performed using COOT. 35 The final model has good stereochemical parameters evaluated with PROCHECK. 36 PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC) was used to prepare all structure‐related figures. 37 The predicted peptide folds for Pep1 and Pep3 were calculated de novo through an online PEP‐FOLD server. 38 , 39
DATA DEPOSITION
The coordinates of the hNE‐ETPep1 structure in the RCSB PDB database under the code 7WHU.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
AUTHOR CONTRIBUTIONS
Tanaya Bagga: Formal analysis (supporting); methodology (supporting); writing – original draft (equal). Su Ning Loh: Data curation (lead); methodology (equal). Sivaraman J: Conceptualization (lead); resources (lead); writing – review and editing (lead). Srihari Shankar: Conceptualization (lead); data curation (lead); formal analysis (lead); methodology (equal); writing – original draft (lead); writing – review and editing (lead).
Supporting information
Supporting Information.
ACKNOWLEDGMENT
This work was supported by the Ministry of Education, Singapore (MoE AcRF Tier 1) grant R154‐000‐C07‐114. Authors thank Dr. Jobichen Chacko for his help in the data collection and structure determination. Tanaya Bagga is a graduate scholar in receipt of the graduate scholarship, National University of Singapore.
Bagga T, Loh SN, Sivaraman J, Shankar S. Sequence preference and scaffolding requirement for the inhibition of human neutrophil elastase by ecotin peptide. Protein Science. 2022;31:933–941. 10.1002/pro.4274
Funding information Ministry of Education ‐ Singapore, Grant/Award Number: R154‐000‐C07‐114
Contributor Information
J Sivaraman, Email: dbsjayar@nus.edu.sg.
Srihari Shankar, Email: srihari.shankar@u.nus.edu.
REFERENCES
- 1. Stockley R, De Soyza A, Gunawardena K, et al. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir Med. 2013;107:524–533. [DOI] [PubMed] [Google Scholar]
- 2. Von Nussbaum F, Li M‐J. Neutrophil elastase inhibitors for the treatment of (cardio) pulmonary diseases: Into clinical testing with pre‐adaptive pharmacophores. Bioorg Med Chem Lett. 2015;25:4370–4381. [DOI] [PubMed] [Google Scholar]
- 3. Zeiher BG, Matsuoka S, Kawabata K, Repine JE. Neutrophil elastase and acute lung injury: Prospects for sivelestat and other neutrophil elastase inhibitors as therapeutics. Crit Care Med. 2002;30:S281–S287. [DOI] [PubMed] [Google Scholar]
- 4. Yamashita J, Ogawa M, Abe M, et al. Tumor neutrophil elastase is closely associated with the direct extension of non‐small cell lung cancer into the aorta. Chest. 1997;111:885–890. [DOI] [PubMed] [Google Scholar]
- 5. Groutas WC, Dou D, Alliston KR. Neutrophil elastase inhibitors. Expert Opin Ther Pat. 2011;21:339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nita I, Hollander C, Westin U, Janciauskiene S‐M. Prolastin, a pharmaceutical preparation of purified human α1‐antitrypsin, blocks endotoxin‐mediated cytokine release. Respir Res. 2005;6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aikawa N, Kawasaki Y. Clinical utility of the neutrophil elastase inhibitor sivelestat for the treatment of acute respiratory distress syndrome. Ther Clin Risk Manag. 2014;10:621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tarhini M, Fessi H, Greige‐Gerges H, Elaissari A. A potential new strategy for using elastase and its inhibitor as therapeutic agents. J Transl Sci. 2018;6:1–8. [Google Scholar]
- 9. Ahmad S, Saleem M, Riaz N, et al. The natural polypeptides as significant elastase inhibitors. Front Pharmacol. 2020;11:688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Groß A, Hashimoto C, Sticht H, Eichler J. Synthetic peptides as protein mimics. Front Bioeng Biotechnol. 2016;3:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vasconcelos A, Azoia NG, Carvalho AC, Gomes AC, Güebitz G, Cavaco‐Paulo A. Tailoring elastase inhibition with synthetic peptides. Eur J Pharmacol. 2011;666:53–60. [DOI] [PubMed] [Google Scholar]
- 12. Navia MA, McKeever BM, Springer JP, et al. Structure of human neutrophil elastase in complex with a peptide chloromethyl ketone inhibitor at 1.84‐Å resolution. Proc Natl Acad Sci U S A. 1989;86:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lechtenberg BC, Kasperkiewicz P, Robinson H, Drag M, Riedl SJ. The elastase‐PK101 structure: Mechanism of an ultrasensitive activity‐based probe revealed. ACS Chem Biol. 2015;10:945–951. [DOI] [PubMed] [Google Scholar]
- 14. Huang W, Yamamoto Y, Li Y, et al. X‐ray snapshot of the mechanism of inactivation of human neutrophil elastase by 1,2,5‐thiadiazolidin‐3‐one 1,1‐dioxide derivatives. J Med Chem. 2008;51:2003–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. An‐Zhi W, Mayr I, Bode W. The refined 2.3 Å crystal structure of human leukocyte elastase in a complex with a valine chloromethyl ketone inhibitor. FEBS Lett. 1988;234:367–373. [DOI] [PubMed] [Google Scholar]
- 16. Hochscherf J, Pietsch M, Tieu W, et al. Crystal structure of highly glycosylated human leukocyte elastase in complex with an S2′ site binding inhibitor. Acta Crystallogr. 2018;74:480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jobichen C, Prabhakar MT, Loh SN, Sivaraman J. Structural basis for the inhibition mechanism of ecotin against neutrophil elastase by targeting the active site and secondary binding site. Biochemistry. 2020;59:2788–2795. [DOI] [PubMed] [Google Scholar]
- 18. Tian S, Swedberg JE, Yi Li C, Craik DJ, de Veer SJ. Iterative optimization of the cyclic peptide SFTI‐1 yields potent inhibitors of neutrophil proteinase 3. ACS Med Chem Lett. 2019;10:1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mccrudden MTC, Ryan LA, Turkington P, Timson DJ. The contribution of key hydrophobic residues in ecotin to enzyme‐inhibitor complex stability: The contribution of key hydrophobic residues in ecotin to enzyme‐inhibitor complex stability. J Enzyme Inhib Med Chem. 2009;24:1207–1210. [DOI] [PubMed] [Google Scholar]
- 20. Joo SH. Cyclic peptides as therapeutic agents and biochemical tools. Biomol Ther (Seoul). 2012;20:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roxin Á, Zheng G. Flexible or fixed: A comparative review of linear and cyclic cancer‐targeting peptides. Futur Med Chem. 2012;4:1601–1618. [DOI] [PubMed] [Google Scholar]
- 22. Horton DA, Bourne GT, Smythe ML. Exploring privileged structures: The combinatorial synthesis of cyclic peptides. J Comput Aided Mol Des. 2002;16:415–431. [DOI] [PubMed] [Google Scholar]
- 23. Choi J‐S, Joo SH. Recent trends in cyclic peptides as therapeutic agents and biochemical tools. Biomol Ther (Seoul). 2020;28:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zimmerman M, Ashe BM. Substrate specificity of the elastase and the chymotrypsin‐like enzyme of the human granulocyte. Biochim Biophys Acta Enzymol. 1977;480:241–245. [DOI] [PubMed] [Google Scholar]
- 25. Eggers CT, Murray IA, Delmar VA, Day AG, Craik CS. The periplasmic serine protease inhibitor ecotin protects bacteria against neutrophil elastase. Biochem J. 2004;379:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Attila Nagy Z, Szakács D, Boros E, et al. Ecotin, a microbial inhibitor of serine proteases, blocks multiple complement dependent and independent microbicidal activities of human serum. PLoS Pathog. 2019;15:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kasperkiewicz P, Poreba M, Snipas SJ, et al. Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc Natl Acad Sci U S A. 2014;111:2518–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McRae E, Nakajima K, Travis J, Powers JC. Studies on reactivity of human leukocyte elastase, cathepsin G, and porcine pancreatic elastase toward peptides including sequences related to the reactive site of alpha 1‐protease inhibitor (alpha 1‐antitrypsin). Biochemistry. 1980;19:3973–3978. [DOI] [PubMed] [Google Scholar]
- 29. Korkmaz B, Attucci S, Hazouard E, et al. Discriminating between the activities of human neutrophil elastase and proteinase 3 using serpin‐derived fluorogenic substrates. J Biol Chem. 2002;277:39074–39081. [DOI] [PubMed] [Google Scholar]
- 30. Mcgrath ME, Erpel T, Bystroff C, Fletterick RJ. Macromolecular chelation as an improved mechanism of protease inhibition: Structure of the ecotin‐trypsin complex. EMBO J. 1994;13:1502–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feng L, Liu X, Zhu W, et al. Inhibition of human neutrophil elastase by pentacyclic triterpenes. PLoS One. 2013;8:82794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pu S, Wang D, Liu D, et al. Effect of sivelestat sodium in patients with acute lung injury or acute respiratory distress syndrome: A meta‐analysis of randomized controlled trials. BMC Pulm Med. 2017;17:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Otwinowski Z, Minor W. Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. [DOI] [PubMed] [Google Scholar]
- 34. Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: A comprehensive python‐based system for macromolecular structure solution. Acta Cryst D. 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emsley P, Cowtan K. Coot: Model‐building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 36. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 37. DeLano WL. PyMOL: A communications tool for computational models. Abstr Pap Am Chem Soc. 2005;230:U1371–U1372. [Google Scholar]
- 38. Venet PT, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P. PEP‐FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012;40:288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shen Y, Maupetit J, Derreumaux P, Tufféry PT. Improved PEP‐FOLD approach for peptide and miniprotein structure prediction. J Chem Theory Comput. 2014;10:4745–4758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.