

Abstract
High-throughput sequencing technologies have generated massive protein sequences, but the annotations of protein sequences highly rely on the low-throughput and expensive biological experiments. Therefore, accurate and fast computational alternatives are needed to infer functional knowledge from protein sequences. The gene ontology (GO) directed acyclic graph (DAG) contains the hierarchical relationships between GO terms but is hard to be integrated into machine learning algorithms for functional predictions. We developed a deep learning system named PANDA2 to predict protein functions, which used the cutting-edge graph neural network to model the topology of the GO DAG and integrated the features generated by transformer protein language models. Compared with the top 10 methods in CAFA3, PANDA2 ranked first in cellular component ontology (CCO), tied first in biological process ontology (BPO) but had a higher coverage rate, and second in molecular function ontology (MFO). Compared with other recently-developed cutting-edge predictors DeepGOPlus, GOLabeler, and DeepText2GO, and benchmarked on another independent dataset, PANDA2 ranked first in CCO, first in BPO, and second in MFO. PANDA2 can be freely accessed from http://dna.cs.miami.edu/PANDA2/.
INTRODUCTION
The number of protein sequences generated by high-throughput experiments far exceeds the number of experimentally annotated proteins. Computational protein function predictors can quickly annotate proteins, which are fast and affordable alternatives to biochemical experiments. The automatic protein function prediction is a multi-label classification problem. A predictor labels a protein from about 45 937 Gene Ontology (GO) terms (1). GO terms are defined in three ontologies: molecular function ontology (MFO), biological process ontology (BPO) and cellular component ontology (CCO). For each ontology, the relationships between GO terms can be represented as a directed acyclic graph (DAG), where a node indicates a GO term, and an edge shows the relationship between two GO terms. Since these hierarchical relationships are hard to be integrated with deep learning algorithms, only a few methods consider the hierarchical structures of GO terms in their algorithms (2,3). To improve performance, DeepGOPlus (4) no longer conducts hierarchical classification as their previous tool DeepGO (2). The recent development of graph neural networks (5,6) provides an approach to naturally integrate the deep learning algorithms and the hierarchical relationships between GO term classes.
According to the keyword analysis of CAFA3 participating teams (7), machine learning and sequence alignment are the most widely used and helpful approaches for protein function prediction. Approaches such as sequence-profile alignment, profile-profile alignment, homolog, sequence properties are also popular and helpful. Alignment-based approaches were heavily used in our previous method, PANDA (8). PANDA includes profile-profile alignments against protein databases, where profiles are position-specific scoring matrices (PSSMs) generated from multiple sequence alignments (MSAs). PANDA also uses PSI-BLAST (9), a profile-sequence alignment approach, to search for homologous proteins against UniProt (10). The protein domain is a structurally or functionally reserved segment. PANDA uses a profile-profile alignment approach to detect protein domains and then integrates a Bayesian statistic model that infers GO terms based on domains. PANDA uses Z-scores to pool and filter all candidate GO terms, then uses an affinity propagation algorithm to cluster the remaining GO terms, and then performs a second round of filtering on the GO term clusters. However, the performances of alignment-based approaches highly rely on homologous sequences having functional annotations. If a protein only has less than 60% global sequence identities with annotated proteins, it is usually labeled as a difficult protein (11). For these difficult proteins, other features (such as sequence properties) are needed to predict their functions. For example, GOLabeler (12), one of the best methods based on the evaluations of CAFA3 (7), uses sequence property features including biophysical properties, domains, and motifs.
Sequence properties can also be learned by deep learning algorithms. The protein-sequence-language models can generate sequence representations directly from a protein sequence without BLAST (9) search. UDSMProt (13), a tool that uses a supervised recurrent neural network (RNN) and modern natural language processing methods to predict protein functions directly from sequences, has achieved competitive performances with the leading methods in CAFA3. Heinzinger et al. retrained the language model ELMo (SeqVec) for modeling unlabeled protein sequences (14). Littmann et al. transferred protein annotations according to the similarity of sequence embeddings from the language models (15) and achieved comparable performance with the best ten CAFA3 methods. Rives et al. used attention networks to learn sequence representations from 250 million protein sequences (16). The representation space of this evolutionary-scale language modeling (ESM) of proteins contains protein structures, functions, and binding information. By adding language modeling features to the state-of-the-art methods, the performances of protein secondary structure prediction and residue-residue contact prediction have been improved (16). In our PANDA2, ESM was used as one of the deep learning features since ESM outperformed SeqVec in terms of secondary structure predictions and long-range residue-residue contact predictions (16).
Machine learning algorithms are the most widely used and successful methods according to the keyword analysis of CAFA3 (7). GOLabeler and DeepGOPlus are two of the best-performed methods on the CAFA3 dataset (4). GOLabeler (12) applied the algorithm of learning to rank (17) on candidate GO terms. DeepGOPlus used a one-dimensional (1D) convolutional neural network (CNN) to capture the associations between motifs and GO terms. UDSMProt (13) used a supervised recurrent neural network (RNN) to predict protein functions from sequences. However, these leading machine learning methods do not take advantage of the hierarchical structure of gene ontology (GO) terms.
Graph network (GN) frameworks process the data represented in graph domains for relational reasoning (6). GNs can be implemented with neural networks or other approaches. Those GNs implemented with fully connected layers are referred to as graph neural networks (GNNs), and those GNs implemented with convolutional layers are referred to as graph convolutional networks (GCNs) (6). Several GN applications are closely related to protein structures and function predictions. GraphQA (18) used GCN to assess the qualities of predicted protein structures (6). DeepFRI (19) and PersGNN (20) used GCN (5) to predict protein functions from residue-residue contact maps. Deepgoa (21) used a correlation matrix of GO terms to represent the hierarchical structure of GO terms and then integrated DeepGOCNN (4) and GCN (5) to predict maize protein functions.
Protein function prediction can also benefit from other types of approaches. DeepText2GO (11) outperformed GOLabeler by integrating relevant citation knowledge and sequence-based features. Protein structure is another key feature that can help predict protein functions. I-TASSER is a structure-based approach, which predicts protein functions by transferring functions from similar templates that have known functions (22). The limitations of using a three-dimensional (3D) structure-based feature are slow computation and large memory usage. To represent protein 3D structures in a memory-efficient way, some groups simplified protein 3D structures as residue–residue contact maps and then applied graph networks to learn the relationship between complex structures and functions (19,20). Moreover, protein-protein interaction (PPI) provides useful information in terms of biological processes (2,23). For example, the authors of (23) inferred protein functions by integrating PPI and retrieving functions from the literature. Furthermore, gene expression is also useful for predicting protein functions by providing closely associated genes with the query protein (24).
We developed and benchmarked a protein function prediction system named PANDA2, which used the cutting-edge graph neural network to model the GO DAG. PANDA2 also used the ESM features, which were generated by the single-sequence protein language models that were trained from 250 million protein sequences using transformers (16). Benchmarked on the CAFA3 dataset, the 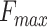 of PANDA2 ranked first in CCO, tied for first in BPO but with a higher coverage rate, and ranked second in MFO when compared with the best 10 CAFA3 methods. Benchmarked on another dataset and compared with the top performed methods GOLabeler, DeepGOPlus and DeepText2GO, the
of PANDA2 ranked first in CCO, tied for first in BPO but with a higher coverage rate, and ranked second in MFO when compared with the best 10 CAFA3 methods. Benchmarked on another dataset and compared with the top performed methods GOLabeler, DeepGOPlus and DeepText2GO, the 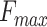 of PANDA2 ranked first in CCO and BPO and ranked second in MFO.
of PANDA2 ranked first in CCO and BPO and ranked second in MFO.
MATERIALS AND METHODS
Datasets
The first dataset we used for training and testing is CAFA3 official training data and target proteins. The data is publicly available at https://www.biofunctionprediction.org/cafa/. Protein sequences are from Swiss-Prot, a manually annotated and reviewed knowledge base (10). Protein annotations are from both Swiss-Prot and gene ontology annotation (GOA) (25). The training and validation datasets contain 66 841 proteins. Each protein has at least one experimentally annotated GO term(s). We used the GO definition released on 2016-01-16. The experimental annotations are linked with experimental codes: EXP, IDA, IMP, IGI, IEP, TAS or IC (26). The blind test dataset contains the target proteins that did not have experimentally annotated GO terms (no knowledge) before the challenge (September 2016) but gained experimental annotations before the benchmark (November 2017).
The second dataset we used to compare PANDA2 with the leading methods is the 2016 dataset provided by DeepGOPlus (4), which was generated using the same time-delayed evaluation method (7) as the one used by GOLabeler (12) and DeepText2GO (11). The proteins with experimental annotations before 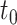 (January 2016) belong to the training dataset. The proteins having no experimentally annotated GO term (no knowledge) before the
(January 2016) belong to the training dataset. The proteins having no experimentally annotated GO term (no knowledge) before the 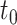 but gaining experimental annotation between the
but gaining experimental annotation between the 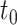 and
and  (October 2016) belong to the testing dataset.
(October 2016) belong to the testing dataset.
The third dataset (27) was only used for blind testing, which contained the hypothetical proteins of pathogenic genera of nine bacterial phyla: Actinobacteria, Bacteroidetes, Chlamydiae, Cyanobacteria, Firmicutes, Fusobacteria, Proteobacteria, Spirochaetes and Tenericutes. Hypothetical proteins (HPs) are proteins whose functions are unknown (28), and the existences of HPs lack experimental evidence at protein, transcript, or homology levels. The proteins with the evidence of existence ‘predicted’ or ‘uncertain’ and with the annotation scores of less than 3 points out of 5 were included in the third dataset. The annotation score measures the annotation content of a protein in UniProt, where 1 point out of 5 indicates a protein with only basic annotation, and 5 points out of 5 denotes having the best annotations.
By cross-referencing the third dataset created in 2019 with the Swiss-Prot dataset released in March 2021, we collected the hypothetical proteins that have been manually reviewed during this period. These recently reviewed proteins in general have limited function annotations, that is, most annotations are inferred from electronic annotation (IEA) that is not an experimental evidence code. Only the hypothetical protein O33285 (putative envelope-preserving system protein Rv2742c) in the third dataset was annotated with the GO:0005886 (plasma membrane) by high throughput direct assay (HDA) that was an experimental evidence code. Our deep learning models were trained using the data that were gathered before 2019, which did not include the annotation of this protein. Therefore, this protein along with its HDA-based GO term was used for benchmarking the prediction accuracy of our system on hypothetical proteins.
We trained PANDA2 using the GO terms that were annotated to at least 50 proteins (4). Propagated annotations were taken into account, which meant that if a GO term was annotated to a protein, then all of its ancestor GO terms would also be annotated to that protein. All types of GO term relationships were considered. Table 1 shows the number of proteins for training, validation and testing and the number of GO terms used for training. In both CAFA3 and 2016 blind test datasets, the 23 target species evaluated in CAFA3 (7) were considered.
Table 1.
The number of proteins and GO terms in the training, validation and testing datasets
| Dataset | training size | validation size | testing size | MFO classes | BPO classes | CCO classes |
|---|---|---|---|---|---|---|
| 2016 | 58 525 | 6503 | 1788 | 652 | 3904 | 545 |
| CAFA3 | 60 157 | 6684 | 3328 | 677 | 3992 | 551 |
The GO terms counted in this table are annotated with at least 50 proteins.
Supplementary Table S1 shows the number of proteins and the number of GO terms (target classes for the graph neural networks) when the GO terms with at least 100 proteins (instead of 50 as in Table 1) were used as the target classes. Supplementary files 1 and 2 list each GO term in the CAFA3 and 2016 datasets, respectively, and the number of training and validation proteins that are annotated with each of the GO terms.
Learning architecture
Overview
Figure 1 shows the neural network architecture of PANDA2. There are three graph neural network blocks (GNNs) in PANDA2, each of which is a basic computational unit that updates edge, node, and global features. In the first and second graph network blocks, we sequentially updated edge features, node features, and global features with all features of the graph neural network. We used a fully connected layer to change the size of ESM features to the number of classes. Then we concatenated node features, the output of the fully connected layer, DIAMOND scores, and priority scores and inputted them into the third graph network block. We used node features of the third graph network block as the final prediction after a sigmoid function.
Figure 1.

The overall architecture of the learning system of PANDA2. Psitop10 denotes PSI-BLAST top 10 scores; Dia denotes DIAMOND scores; Pri denotes priority scores; PAAC denotes the pseudo amino acid composition of sequence; ESM denotes ESM representation; 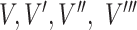 denotes node features;
denotes node features;  denotes edge features;
denotes edge features;  denotes global features;
denotes global features; 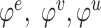 denote feature ‘update’ functions; and
denote feature ‘update’ functions; and  denote feature ‘aggregation’ functions.
denote feature ‘aggregation’ functions.
In PANDA2, we trained one model to predict the GO terms in BPO, CCO, and MFO ontologies at once instead of three different models predicting for the three ontologies. The graph topology that is modeled by the graph neural network of PANDA2 is the GO DAG. For different query proteins, the topology of the input GO term graph or DAG is the same, which contains all of the GO terms that have at least 50 proteins annotated with them, except that the node and global features are different and determined by the specific query protein. In comparison, the graph modeled by the GNN in another research in protein model quality assessment (18) consists of protein residues. By stacking the GNN blocks, the PANDA2 network learns knowledge from neighboring GO terms using the first block and then enlarges to a broader range containing more GO terms in the DAG using the following two blocks.
The features of PANDA2 include the bidirected unweighted graphs of GO terms (GO DAGs), sequence-alignment-based features, and sequence-based features. The nodes in the graph are the GO terms having at least 50 annotations with an experimental evidence code in the training and validation sets. The edges are ‘is_a,’ ‘part_of,’ and ‘regulations’ relationships between GO terms. Each directed edge starts from a ‘sender’ node and ends at a ‘receiver’ node.
The sequence-alignment-based node features contain PSI-BLAST top 10 scores, DIAMOND scores, and priority scores (See the following sections for details). Those scores are engineered based on the sequence alignment results of the query protein. We used the pseudo amino acid composition (PAAC) (29) as the global feature of the graph neural network. Moreover, after a fully connected layer, we merged the ESM features into node features.
Graph neural network
We trained GNN with node features, edge features, and global features. The node feature was a vector of length 12, including PSI-BLAST top 10 scores, a DIAMOND score, and a priority score. The edge feature is a vector with length one that has an initial value of 1 if there is an edge between two GO terms. We tested different values for ‘is_a,’ ‘part_of,’ and ‘regulates,’ e.g. 0.8, 0.6 and 0.5, but our evaluations indicated that having initial values of 1 for all relationships achieved the best performance. The global feature is a vector of amino acid composition with a length of 20 (30).
These features were updated in three GNN blocks, each of which was a basic computation unit in the GNN framework (6). Table 2 shows the step-by-step computations in a GNN block of PANDA2. A GNN block has ‘update’ functions 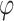 and ‘aggregation’ functions
and ‘aggregation’ functions 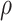 . We used a Linear-ReLU-Linear layer in the ‘update’ functions. In other words, we used fully connected layers in the GNN blocks since a linear layer in Pytorch (31) was implemented as a fully connected layer. These blocks could also be implemented as convolutional layers and recurrent layers.
. We used a Linear-ReLU-Linear layer in the ‘update’ functions. In other words, we used fully connected layers in the GNN blocks since a linear layer in Pytorch (31) was implemented as a fully connected layer. These blocks could also be implemented as convolutional layers and recurrent layers.
Table 2.
Computational steps in a GNN block
| procedure GNNBlock(E, V, u): // Sender: the start node of a directed edge | The major dimensions of features in different blocks | ||
|---|---|---|---|
| // Receiver: the end node of a directed edge | Block 1 | Block 2 | Block 3 |
| // E: input edge features | 1 | 10 | 10 |
| // V: input node features | 12 | 20 | 23 |
| // u: input global features | 20 | 30 | 20 |
| // u_edge: the global features merged to edges | 20 | 30 | 20 |
| // u_node: the global features merged to nodes | 20 | 30 | 20 |
| // v_sender: the node features of all senders | 12 | 20 | 23 |
| // v_receiver: the node features of all receivers | 12 | 20 | 23 |
// step1: update edge features 
|
|||
| E’ ← concatenate(v_sender, v_receiver, E, u_edge) | 45 (E’) | 80 (E’’) | 76 (E’’’) |
| E’ ← linear_transformation(E’) | 45→5 | 80→5 | 76→5 |
| E’ ← ReLU(E’) | 5→5 | 5→5 | 5→5 |
| E’ ← linear_transformation(E’) | 5→10 | 5→10 | 5→10 |
// step2: aggregate edge features per node 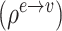
|
|||
| e_v ← concatenate(v_sender, E’) | 22 | 30 | 33 |
| e_v ← linear_transformation(e_v) | 22→15 | 30→20 | 33→23 |
| e_v ← ReLU(e_v) | 15→15 | 20→20 | 23→23 |
| e_v ← linear_transformation(e_v) | 15→20 | 20→25 | 23→10 |
| e_v ← scatter_mean(e_v) //average e_v over receivers | 20→20 | 25→25 | 10→10 |
// step3: update node features 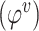
|
|||
| V’ ← concatenate(V, e_v, u_node) | 52 (V’) | 75 (V’’) | 53 (V’’’) |
| V’ ← linear_transformation(V’) | 52→20 | 75→25 | 53→10 |
| V’ ← ReLU(V’) | 20→20 | 25→25 | 10→10 |
| V’ ← linear_transformation(V’) | 20→20 | 25→20 | 10→1 |
// step4: aggregate edge features globally 
|
|||
| e_u ← scatter_mean(E’) //average E’ over receivers | 10→10 | 10→10 | |
| e_u ← scatter_mean(e_u) //average e_u over graphs | 10→10 | 10→10 | |
// step5: aggregate node features globally 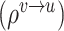
|
|||
| v_u ← scatter_mean(V’) //average V’ over graphs | 20→20 | 20→20 | |
// step6: update global features 
|
|||
| u’ ← concatenate(u, v_u, e_u) | 50 (u’) | 60 (u’’) | |
| u’ ← linear_transformation(u’) | 50→25 | 60→25 | |
| u’ ← ReLU(u’) | 25→25 | 25→25 | |
| u’ ← linear_transformation(u’) | 25→30 | 25→20 | |
| return E', V', u' | |||
| end procedure | |||
Steps 4, 5 and 6 are only applied in blocks 1 and 2.
The major dimensions of features and the dimensions of inputs and outputs of each fully connected layer are also shown in Table 2. We averaged features over receivers and graphs to aggregate features with the scatter mean function (31). Before aggregating edge features per node, we used the Linear-ReLU-Linear layer to update edge features. The steps in the first and second blocks are the same. The difference between the third block and the first two blocks is that the third block does not need to update global features. Therefore, steps 4, 5 and 6 apply for blocks 1 and 2 only.
Features
PSI-BLAST top 10 scores
The query protein was searched against the training and validation proteins using PSI-BLAST (9). PSI-BLAST top 10 scores were engineered by transferring the annotations of top 10 PSI-BLAST hits. We converted the E-value of an alignment result to a confidence score as:
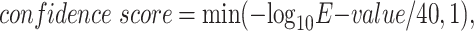 |
where the E-value greater than or equal to 1 was filtered out. For each protein sequence in the top 10 hits, we transferred its experimental annotations to the query protein with a confidence score between 0 and 1. The GO terms associated with the PSI-BLAST top 10 hits along with their confidence scores were input to the GNN as node features.
DIAMOND score
This score was generated by applying a fast and sensitive sequence alignment tool DIAMOND (32) to search against the training and validation proteins. The DIAMOND score was defined in (4):
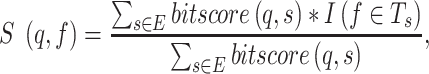 |
where 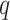 was a query sequence,
was a query sequence, 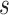 was a set of sequences,
was a set of sequences, 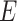 was a set of similar sequences with E-values smaller than 0.001,
was a set of similar sequences with E-values smaller than 0.001,  was a set of experimental annotations for sequence
was a set of experimental annotations for sequence 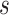 , and
, and 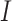 was an identity function that returned 1 if same and 0 otherwise. The GO terms associated with DIAMOND hits (E-values < 0.001) were input into the GNN as node features.
was an identity function that returned 1 if same and 0 otherwise. The GO terms associated with DIAMOND hits (E-values < 0.001) were input into the GNN as node features.
Priority score
We defined a new type of score named priority score that integrated the identity score of PSI-BLAST hits and the number of occurrences of each GO term associated with the top hits. We only considered the hits with E-values <1. The priority score for a GO term was calculated as:
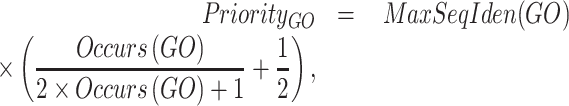 |
where 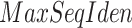 returned the highest sequence identity of the sequences that were annotated with the GO term or its descendants, and
returned the highest sequence identity of the sequences that were annotated with the GO term or its descendants, and  counted the total occurrences of a GO term and its descendants in the annotations of the top hits. The GO terms and their priority scores were input into the GNN.
counted the total occurrences of a GO term and its descendants in the annotations of the top hits. The GO terms and their priority scores were input into the GNN.
Evolutionary-scale language model
PANDA2 was integrated with the evolutionary-scale language model that was trained on 250 million protein sequences covering 66% of publicly available protein sequences (16). We inputted each query protein sequence into the evolutionary-scale language model that outputted a vector of 1280 by the sequence length. The mean was calculated along the sequence length to obtain a fixed-length vector of 1280 by 1. Since the ESM was pre-trained, we only used a fully connected layer or a neural network to change the dimension of the ESM output to the number of GO terms or target classes of our GNN.
Availability of node features
Supplementary Figure S1 shows that most of the proteins in the training and validation sets have at least 10 PSI-BLAST hits. Supplementary Figures S2–S4 show that most of the target GO terms or nodes in the input GO DAG in the first two GNN blocks have non-empty node features. The output of the neural network for ESM features makes all target GO terms or nodes in the GO DAG have non-empty node features for any protein in the CAFA3 and 2016 datasets.
Implementation details
We implemented PANDA2 with the PyTorch library (31) and a graph network framework library (6). The models were trained on an NVIDIA Tesla V100 GPU with a training time of about 40 min. The usage of GPU memory was less than 10GB. The loss function that we used was binary cross-entropy with a log function. Since the negatively labeled GO terms are much more than the positively labeled GO terms, we set a positive class weight of 3 when calculating loss values. We tested different combinations of learning rates, dropout rates, number of GNN blocks, number of channels, and loss functions, and we eventually selected the model with the lowest validation loss. We used an Adam optimizer with a learning rate of 0.01 for the GNN and another Adam optimizer with a learning rate of 0.0001 for the neural network modeling ESM features. These two optimizers updated the parameters of the graph network and the neural network separately.
We implemented a web server for PANDA2 at http://dna.cs.miami.edu/PANDA2/. The model trained on the CAFA3 dataset was used by the web server, which could make predictions for 5220 GO terms. The PANDA2 web server can annotate a protein with 601 amino acids in 1 min and 40 s, including ∼30 s for predicting functions and about 1 min and 10 s for preprocessing and loading models. The results of a query job will be sent back to the user by email. The users need to check their spam folder, as that email sometimes will be mistakenly labeled as a spam email.
Evaluation metrics
We performed protein-centric evaluation measures using the official CAFA assessment tool (7,33) on the CAFA3 and 2016 datasets. The evaluation was performed on propagated predictions and ground truth. A propagated prediction or ground truth contains the GO term annotations and their ancestors. In the benchmarks with the CAFA3 dataset, we compared PANDA2 with the literature results of CAFA top-performing methods, DeepGOPlus (a recently developed leading method), Naïve, and BLAST methods (4,7). The Naïve method predicted GO terms according to the relative frequency of each GO term shown in the Swiss-Prot database. The BLAST method predicted GO terms based on the hits of BLAST (9). In the benchmarks with the 2016 dataset, we compared PANDA2 with the literature results (4,11–13) of UDSMProt, DeepGOPlus, GOLabeler, and DeepText2GO.
The evaluation metrics that we used included the maximum F-measure (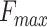 ), the minimum semantic distance (
), the minimum semantic distance ( ), and the area under the precision-recall curve (AUPR). The
), and the area under the precision-recall curve (AUPR). The  and the
and the  are the official measures in CAFA1-3 (7,26,33). The
are the official measures in CAFA1-3 (7,26,33). The  score is the maximum F-measure over confidence score thresholds
score is the maximum F-measure over confidence score thresholds 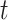 . F-measure is a combination of precision and recall. Precision and recall of a protein were calculated based on the predicted GO term set
. F-measure is a combination of precision and recall. Precision and recall of a protein were calculated based on the predicted GO term set 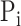 that had confidence score greater than or equal to
that had confidence score greater than or equal to 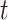 and true GO term set
and true GO term set 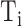 . Only when
. Only when 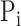 had at least one GO term, we calculated precision and recall for a protein. The precision and recall for
had at least one GO term, we calculated precision and recall for a protein. The precision and recall for 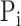 with confidence score threshold
with confidence score threshold  were calculated as:
were calculated as:
 |
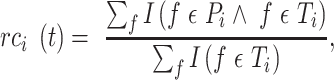 |
where 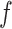 was a GO term, and
was a GO term, and 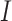 was an indicator function.
was an indicator function.
Precision and recall for all testing proteins over threshold  were averaged as:
were averaged as:
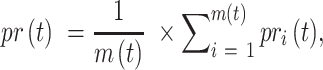 |
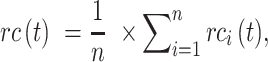 |
where  was the number of proteins with at least one GO term having a confidence score greater than or equal to
was the number of proteins with at least one GO term having a confidence score greater than or equal to 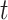 , and
, and  was the number of total testing proteins.
was the number of total testing proteins.
The best F-measure was calculated as:
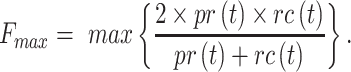 |
The  score was the metric that considered the unbalanced information content (IC) of GO terms. The IC of a GO term f was calculated as:
score was the metric that considered the unbalanced information content (IC) of GO terms. The IC of a GO term f was calculated as:
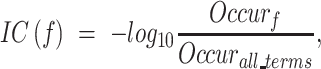 |
where  was a GO term,
was a GO term, 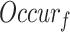 indicated the number of occurrences of
indicated the number of occurrences of  and its descendants in the GO DAG, and
and its descendants in the GO DAG, and 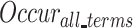 was the total occurrence of all GO terms.
was the total occurrence of all GO terms.
The remaining uncertainty and misinformation were calculated as:
 |
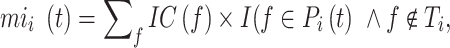 |
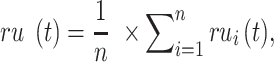 |
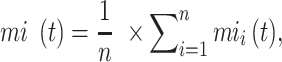 |
where 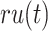 and
and  were the average of remaining uncertainty
were the average of remaining uncertainty  and misinformation
and misinformation 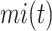 . The
. The  was calculated as:
was calculated as:
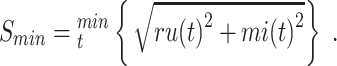 |
RESULTS
Comparison with the leading methods in CAFA3
We trained PANDA2 on the CAFA3 training dataset and then performed the protein-centric evaluation on the CAFA3 testing dataset using the official CAFA assessment tool (7,33). Figure 2 shows the evaluation results of PANDA2, CAFA3 top 10 methods, and DeepGOPlus. PANDA2 achieved the  scores of MFO: 0.5849, BPO: 0.3964 and CCO: 0.6374, respectively. The performance of CAFA3 top-performing methods, DeepGOPlus, and Naïve and BLAST methods were downloaded from (4,7). According to the
scores of MFO: 0.5849, BPO: 0.3964 and CCO: 0.6374, respectively. The performance of CAFA3 top-performing methods, DeepGOPlus, and Naïve and BLAST methods were downloaded from (4,7). According to the 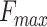 scores, PANDA2 ranked first in cellular component, tied for first in biological process but with higher coverage rate, and ranked second in molecular function.
scores, PANDA2 ranked first in cellular component, tied for first in biological process but with higher coverage rate, and ranked second in molecular function.
Figure 2.

Performance of PANDA2, CAFA3 top10 methods and DeepGOPlus. The evaluation was performed based on molecular function, biological process and cellular component. C (coverage) denotes the percentage of testing proteins that have predictions made by a predictor.
Comparison with the state-of-the-art methods
We further trained and benchmarked PANDA2 on the 2016 dataset and then collected the results from the literature (4,11–13) for Naïve, DIAMONDBlast, DeepGOPlus, UDSMProt, GOLabeler, and DeepText2GO on the same dataset. The benchmark of our previous method, PANDA, was performed using the predictions of the PANDA web server on the 2016 testing dataset. Table 3 shows the performance of these methods in terms of 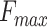 ,
, 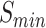 and the area under the precision-recall curve (AUPR). PANDA2 achieved the best
and the area under the precision-recall curve (AUPR). PANDA2 achieved the best 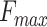 and AUPR in BPO and CCO and the second best
and AUPR in BPO and CCO and the second best 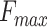 and AUPR in MFO. By using the graph network and new features, PANDA2 outperformed our previously developed method PANDA in terms of
and AUPR in MFO. By using the graph network and new features, PANDA2 outperformed our previously developed method PANDA in terms of 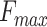 ,
, 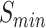 and AUPR. The semantic distance between PANDA2 prediction and the ground truth is larger than some of the other predictors, which is something we could improve in the future.
and AUPR. The semantic distance between PANDA2 prediction and the ground truth is larger than some of the other predictors, which is something we could improve in the future.
Table 3.
The performance of PANDA2 and selected top-performed predictors on the 2016 dataset. The highest 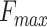 , smallest
, smallest 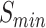 , and highest AUPR are in bold and italic
, and highest AUPR are in bold and italic
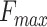
|

|
AUPR | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Method | MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO |
| Naive | 0.306 | 0.318 | 0.605 | 12.105 | 38.890 | 9.646 | 0.150 | 0.219 | 0.512 |
| DIAMONDBlast | 0.525 | 0.436 | 0.591 | 9.291 | 39.544 | 8.721 | 0.101 | 0.070 | 0.089 |
| UDSMProt | 0.582 | 0.475 | 0.697 | 8.787 | 33.615 | 7.618 | 0.548 | 0.422 | 0.728 |
| DeepText2GO | 0.627 | 0.441 | 0.694 | 5.240 | 17.713 | 4.531 | 0.605 | 0.336 | 0.729 |
| GOLabeler | 0.586 | 0.372 | 0.691 | 5.032 | 15.050 | 5.479 | 0.549 | 0.236 | 0.697 |
| DeepGOPlus | 0.585 | 0.474 | 0.699 | 8.824 | 33.576 | 7.693 | 0.536 | 0.407 | 0.726 |
| PANDA | 0.486 | 0.367 | 0.520 | 11.751 | 45.096 | 12.723 | 0.396 | 0.289 | 0.394 |
| PANDA2 | 0.598 | 0.478 | 0.709 | 9.670 | 40.229 | 9.558 | 0.564 | 0.436 | 0.744 |
Feature importance
To benchmark the importance of network components and features, we trained and evaluated PANDA2 by removing different parts of the networks or features. Table 4 shows the results evaluated on the 2016 dataset. For example, PANDA2 rm GNN (PANDA2 with GNN removed) denotes the performance solely based on ESM with a fully connected layer. According to the validation loss, we found the following importance of network components and features: GNN > ESM > PSI-BLAST top 10 scores > DIAMOND scores > the pseudo amino acid composition (PAAC) > priority scores. This ranking indicates that graph neural networks and ESM features are the two most important components and features of the prediction of PANDA2.
Table 4.
The performance of PANDA2 and different versions of PANDA2 after removing GNN or other features. PANDA2 rm GNN denotes the system after removing GNN, which only contains the ESM features and the fully connected layer (or neural network) related to them. The PANDA2 rm NN denotes only using GNN and related features. The PANDA2 rm PAAC denotes the PANDA2 after removing the global (PAAC) features. The PANDA2 rm Psitop10 denotes the PANDA2 after removing PSI-BLAST top 10 features. PANDA2 rm Prior denotes the PANDA2 after removing priority score features. The PANDA2 rm Dia denotes the PANDA2 after removing DIAMOND score features. The highest  , smallest
, smallest 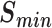 , highest AUPR, and smallest validation loss are in bold
, highest AUPR, and smallest validation loss are in bold

|

|
AUPR | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Method | MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | Validation loss |
| PANDA2 rm GNN | 0.566 | 0.432 | 0.687 | 10.273 | 42.554 | 10.289 | 0.521 | 0.386 | 0.714 | 0.07100 |
| PANDA2 rm NN | 0.561 | 0.456 | 0.651 | 9.860 | 40.913 | 10.188 | 0.530 | 0.400 | 0.668 | 0.07563 |
| PANDA2 rm PAAC | 0.593 | 0.475 | 0.709 | 9.727 | 40.476 | 9.523 | 0.558 | 0.434 | 0.750 | 0.06676 |
| PANDA2 rm Psitop10 | 0.597 | 0.476 | 0.708 | 9.782 | 40.443 | 9.577 | 0.552 | 0.434 | 0.748 | 0.06686 |
| PANDA2 rm Prior | 0.596 | 0.478 | 0.709 | 9.720 | 40.270 | 9.604 | 0.563 | 0.437 | 0.751 | 0.06649 |
| PANDA2 rm Dia | 0.592 | 0.472 | 0.702 | 9.663 | 40.826 | 9.656 | 0.563 | 0.428 | 0.745 | 0.06681 |
| PANDA2 | 0.598 | 0.478 | 0.709 | 9.670 | 40.229 | 9.558 | 0.564 | 0.436 | 0.744 | 0.06632 |
Supplementary Table S2 shows the results of the same type of evaluations as in Table 4 except that the target classes of PANDA2 in Table S2 are the GO terms each of which has at least 100 proteins annotated with it. This causes a decrease in the number of target classes or GO terms that could be predicted by the model. The performances of the models trained with at least 100 annotated proteins were much lower than those trained with at least 50 proteins. Therefore, the final version of PANDA2 was trained with ≥50 as the threshold of the number of annotated proteins.
Figure 3 shows the 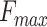 bar plots and precision-recall curves, demonstrating the importance of GNN and other features. We added the performance of DeepGOPlus as a baseline in Figure 3 so that one can compare that leading method with different versions of PANDA2 with GNN or other features removed.
bar plots and precision-recall curves, demonstrating the importance of GNN and other features. We added the performance of DeepGOPlus as a baseline in Figure 3 so that one can compare that leading method with different versions of PANDA2 with GNN or other features removed.
Figure 3.

The 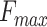 and precision-recall curves of different versions of PANDA2. The evaluation was conducted on the 2016 dataset. A–C show the bar plots of
and precision-recall curves of different versions of PANDA2. The evaluation was conducted on the 2016 dataset. A–C show the bar plots of  in terms of BPO, CCO and MFO, respectively. The empirical bootstrap was applied to estimate the 95% confidence interval with 10 000 resamples on the benchmark dataset (36). The empirical bootstrap was not applied to the literature result of DeepGOPlus. D–F show the precision-recall curves for three ontologies. The highlighted dots on the precision-recall curves indicate the precisions and recalls where
in terms of BPO, CCO and MFO, respectively. The empirical bootstrap was applied to estimate the 95% confidence interval with 10 000 resamples on the benchmark dataset (36). The empirical bootstrap was not applied to the literature result of DeepGOPlus. D–F show the precision-recall curves for three ontologies. The highlighted dots on the precision-recall curves indicate the precisions and recalls where 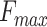 scores were found.
scores were found.
Evaluation of PANDA2 on hypothetical proteins
Mishra's method (34), a deep learning method of protein function prediction, was specially designed to predict the MFO terms of hypothetical proteins. Since the lack of enough experimental annotations, it is hard to evaluate PANDA2 and other methods on all hypothetical proteins. However, we calculated the functional similarities between the predictions of PANDA2 and Mishra's method on all hypothetical proteins in the third dataset and plotted the histogram of similarity scores in Supplementary Figure S5. It can be found that the highest similarity score is around 0.648 and the peak is at around 0.33. Supplementary file 3 contains all PANDA2 predictions for the hypothetical proteins dataset.
Table 5 shows the top 10 CCO terms predicted by PANDA2 for the hypothetical protein O33285, which is the only protein that we have found having been annotated with a GO term based on experimental evidence. The semantic similarities of predicted GO terms and the experimentally determined GO term GO:0005886 were calculated by GOGO (35). Table 5 also shows the depths of predicted CCO GO terms in the GO DAG. It can be found that the sixth predicted GO term from PANDA2 is exactly the experimentally annotated GO term.
Table 5.
Top 10 CCO terms predicted by PANDA2 for the hypothetical protein O33285. GO terms were ranked by confidence scores generated by PANDA2. Depth 0 indicates that the predicted GO term is the root term in the CCO DAG. GO:0005886 (plasma membrane) in bold is the only GO term that is annotated with an experimental evidence code
| Predicted GO term | Confidence score from PANDA2 | Semantic similarity between the predicted GO term and GO:0005886 | Depth of the GO term in the CCO DAG |
|---|---|---|---|
| GO:0044464 | 0.62 | 0.574 | 1 |
| GO:0005575 | 0.61 | 0.313 | 0 |
| GO:0005623 | 0.56 | 0.43 | 1 |
| GO:0071944 | 0.43 | 0.685 | 2 |
| GO:0016020 | 0.39 | 0.49 | 1 |
| GO:0005886 | 0.37 | 1 | 2 |
| GO:0005622 | 0.27 | 0.344 | 2 |
| GO:0005618 | 0.23 | 0.414 | 3 |
| GO:0044424 | 0.2 | 0.315 | 2 |
| GO:0030312 | 0.2 | 0.527 | 2 |
DISCUSSION
The 45 937 currently available GO terms are organized in directed acyclic graphs (DAGs) with ‘is_a’, ‘part_of’, and ‘regulations’ relationships between GO terms. It would be very hard or almost impossible for traditional deep learning algorithms to model this type of network topology. However, the graph neural networks provide a way to naturally model this graph architecture between GO terms and then use it to predict protein functions. The results from this research prove that graph neural networks indeed can be used to model the GO DAG topology and predict protein functions in the format of GO terms.
The graph neural network has edge features, node features, and global features, and in each block of the graph neural network, the edge features are updated and aggregated with node and global features and similarly for the node features and global features. In this research, we model GO DAG as a graph and define the ‘is_a’, ‘part_of’, and ‘regulations’ relationships between GO terms as edges. Moreover, we define the confidence scores of each GO term generated from the homologs of the query protein as node features and the pseudo amino acid composition as the global feature. In this way, the relationships between thousands of GO terms, which is something not directly associated with the query protein but in general for all GO terms, are naturally aggregated with the knowledge of the query protein.
Instead of taking the amino acid sequence of the query protein as input, PANDA2 attempts to use the evolutionary-scale language modeling (ESM) representation of the query protein sequence. This ESM-based sequence representation was trained by other scientists based on 250 million protein sequences, and other studies have proven that this model can provide much more knowledge for the query protein sequence compared to only using the amino acid sequence of the query protein. Since it is enriched with knowledge about the query protein, we use a fully connected layer to change its data shape to the number of possible GO terms and then directly input it to the last block of the graph network. In this way, this ESM-based feature will have a higher weight or impact on the final prediction.
One may argue that since the ESM model was trained on 250 million protein sequences, the blind test proteins used in our benchmarking may already be exposed. However, we want to argue and point out that the ESM model was trained in an unsupervised way, which was nothing related to GO terms but only based on protein sequences. Therefore, using the ESM model to get sequence representation of the query protein does not introduce the data leakage problem to the machine learning architecture.
Since the node, edge and global features are updated and aggregated in each block of the graph neural network, when we stack multiple blocks, it can extract knowledge from increasing neighborhoods or receptive fields. Previous research has taken advantage of that by applying graph networks onto protein residues, in which the graph network blocks can learn, for example, the knowledge regarding α-helices and β-sheets using the first block, domains for the second block, and then the arrangement of domains for the third or fourth blocks (18). For PANDA2, instead of applying this increasing inceptive manner onto the protein residues, we apply it onto the GO terms, that is, it can model or learn knowledge for neighboring GO terms using the first block and then enlarge to a wider range containing more GO terms using the next two blocks. Moreover, our design is not only modeling the query-protein-independent relationships between GO terms but also with the sequence and homologous information of the query protein integrated as the node features. All of this knowledge is integrated naturally by taking advantage of graph neural networks.
DATA AVAILABILITY
The web server of PANDA2 can be freely accessed from http://dna.cs.miami.edu/PANDA2/.
Supplementary Material
Contributor Information
Chenguang Zhao, Department of Computer Science, University of Miami, 1365 Memorial Drive, Coral Gables, FL 33124, USA.
Tong Liu, Department of Computer Science, University of Miami, 1365 Memorial Drive, Coral Gables, FL 33124, USA.
Zheng Wang, Department of Computer Science, University of Miami, 1365 Memorial Drive, Coral Gables, FL 33124, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
National Institutes of Health grant [1R35GM137974 to Z.W., in part].
Conflict of interest statement. None Declared.
REFERENCES
- 1. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T.. Gene ontology: tool for the unification of biology. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kulmanov M., Khan M.A., Hoehndorf R.J.B.. DeepGO: predicting protein functions from sequence and interactions using a deep ontology-aware classifier. Bioinformatics. 2018; 34:660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Deng L., Lin W., Wang J., Zhang J.. DeepciRGO: functional prediction of circular RNAs through hierarchical deep neural networks using heterogeneous network features. BMC Bioinformatics. 2020; 21:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kulmanov M., Hoehndorf R.. DeepGOPlus: improved protein function prediction from sequence. Bioinformatics. 2020; 36:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kipf T.N., Welling M.. Semi-supervised classification with graph convolutional networks. 2016; arXiv doi:22 February 2017, preprint: not peer reviewedhttps://arxiv.org/abs/1609.02907.
- 6. Battaglia P.W., Hamrick J.B., Bapst V., Sanchez-Gonzalez A., Zambaldi V., Malinowski M., Tacchetti A., Raposo D., Santoro A., Faulkner R.. Relational inductive biases, deep learning, and graph networks. 2018; arXiv doi:17 October 2018, preprint: not peer reviewedhttps://arxiv.org/abs/1806.01261.
- 7. Zhou N., Jiang Y., Bergquist T.R., Lee A.J., Kacsoh B.Z., Crocker A.W., Lewis K.A., Georghiou G., Nguyen H.N., Hamid M.N.. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Gen. Biol. 2019; 20:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Z., Zhao C., Wang Y., Sun Z., Wang N.. PANDA: protein function prediction using domain architecture and affinity propagation. Scientific Rep. 2018; 8:3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Altschul S., Madden T., Schaffer A., Zhang J., Zhang Z., Miller W., Lipman D. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:2465–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Apweiler R., Bairoch A., Wu C.H., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M.. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004; 32:D115–D119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. You R., Huang X., Zhu S.. DeepText2GO: improving large-scale protein function prediction with deep semantic text representation. Methods. 2018; 145:82–90. [DOI] [PubMed] [Google Scholar]
- 12. You R., Zhang Z., Xiong Y., Sun F., Mamitsuka H., Zhu S.. GOLabeler: improving sequence-based large-scale protein function prediction by learning to rank. Bioinformatics. 2018; 34:2465–2473. [DOI] [PubMed] [Google Scholar]
- 13. Strodthoff N., Wagner P., Wenzel M., Samek W.. UDSMProt: universal deep sequence models for protein classification. Bioinformatics. 2020; 36:2401–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heinzinger M., Elnaggar A., Wang Y., Dallago C., Nechaev D., Matthes F., Rost B.. Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinformatics. 2019; 20:723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Littmann M., Heinzinger M., Dallago C., Olenyi T., Rost B.. Embeddings from deep learning transfer GO annotations beyond homology. Scientific Rep. 2021; 11:1160–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rives A., Meier J., Sercu T., Goyal S., Lin Z., Liu J., Guo D., Ott M., Zitnick C.L., Ma J.. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li H. A short introduction to learning to rank. IEICE Trans. Inform. Syst. 2011; 94:1854–1862. [Google Scholar]
- 18. Baldassarre F., Menéndez Hurtado D., Elofsson A., Azizpour H.. GraphQA: protein model quality assessment using graph convolutional networks. Bioinformatics. 2020; 37:360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gligorijevic V., Renfrew P.D., Kosciolek T., Leman J.K., Berenberg D., Vatanen T., Chandler C., Taylor B.C., Fisk I.M., Vlamakis H.. Structure-based function prediction using graph convolutional networks. Nat. Commun. 2021; 12:3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Swenson N., Krishnapriyan A.S., Buluc A., Morozov D., Yelick K.. PersGNN: applying topological data analysis and geometric deep learning to structure-based protein function prediction. 2020; arXiv doi:30 October 2020, preprint: not peer reviewedhttps://arxiv.org/abs/2010.16027.
- 21. Zhou G., Wang J., Zhang X., Yu G.. Predicting functions of maize proteins using graph convolutional network. BMC bioinformatics. 2019; 21:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y.. The I-TASSER suite: protein structure and function prediction. Nat.Methods. 2015; 12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jaeger S., Gaudan S., Leser U., Rebholz-Schuhmann D. Integrating protein-protein interactions and text mining for protein function prediction. BMC Bioinformatics. 2008; 9:S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walker M.G., Volkmuth W., Sprinzak E., Hodgson D., Klingler T.. Prediction of gene function by genome-scale expression analysis: prostate cancer-associated genes. Genome Research. 1999; 9:1198–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huntley R.P., Sawford T., Mutowo-Meullenet P., Shypitsyna A., Bonilla C., Martin M.J., O’Donovan C. The GOA database: gene ontology annotation updates for 2015. Nucleic Acids Res. 2015; 43:D1057–D1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Radivojac P., Clark W.T., Oron T.R., Schnoes A.M., Wittkop T., Sokolov A., Graim K., Funk C., Verspoor K., Ben-Hur A.. A large-scale evaluation of computational protein function prediction. Nat. Methods. 2013; 10:221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mishra S., Rastogi Y.P., Jabin S., Kaur P., Amir M., Khatoon S.. A bacterial phyla dataset for protein function prediction. Data Brief. 2020; 28:105002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lubec G., Afjehi-Sadat L., Yang J.-W., John J.P.P.. Searching for hypothetical proteins: theory and practice based upon original data and literature. Prog. Neurobiol. 2005; 77:90–127. [DOI] [PubMed] [Google Scholar]
- 29. Shen H.-B., Chou K.-C.. PseAAC: a flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008; 373:386–388. [DOI] [PubMed] [Google Scholar]
- 30. Cao D.S., Xu Q.S., Liang Y.Z.. propy: a tool to generate various modes of chou's PseAAC. Bioinformatics. 2013; 29:960–962. [DOI] [PubMed] [Google Scholar]
- 31. Paszke A., Gross S., Massa F., Lerer A., Bradbury J., Chanan G., Killeen T., Lin Z., Gimelshein N., Antiga L.. Pytorch: an imperative style, high-performance deep learning library. Advances in neural information processing system. 2019; 32:8026–8037. [Google Scholar]
- 32. Buchfink B., Xie C., Huson D.H.. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015; 12:59–60. [DOI] [PubMed] [Google Scholar]
- 33. Jiang Y., Oron T.R., Clark W.T., Bankapur A.R., D’Andrea D., Lepore R., Funk C.S., Kahanda I., Verspoor K.M., Ben-Hur A. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 2016; 17:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mishra S., Rastogi Y.P., Jabin S., Kaur P., Amir M., Khatun S.. A deep learning ensemble for function prediction of hypothetical proteins from pathogenic bacterial species. Comput. Biol. Chem. 2019; 83:107147. [DOI] [PubMed] [Google Scholar]
- 35. Zhao C., Wang Z.. GOGO: an improved algorithm to measure the semantic similarity between gene ontology terms. Scientific Reports. 2018; 8:15107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Efron B. Nonparametric estimates of standard error: the jackknife, the bootstrap and other methods. Biometrika. 1981; 68:589–599. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The web server of PANDA2 can be freely accessed from http://dna.cs.miami.edu/PANDA2/.