

Abstract
Self-assembly of metallointercalators into DNA nanocages is a rapid and facile approach to synthesizing discrete bioinorganic host:guest structures with a high load of metal complexes. Turberfield’s DNA tetrahedron can accommodate one intercalator for every two base pairs, which corresponds to 48 metallointercalators per DNA tetrahedron. The affinity of the metallointercalator for the DNA tetrahedron is a function of both the structure of the intercalating ligand and the overall charge of the complex, with a trend in affinity [Ru(bpy)2(dppz)]2+ > [Tb-DOTAm-Phen]3+ >> Tb-DOTA-Phen. Intercalation of the metal complex stabilizes the DNA tetrahedron resulting in an increase of its melting temperature and, importantly, a significant increase in its stability in the presence of serum. [Ru(bpy)2(dppz)]2+, which has a greater affinity for DNA than [Tb-DOTAm-Phen]3+, increases the melting point and decreases degradation in serum to a greater extent than the TbIII complex. In the presence of lipofectamine, the metallointercalator@DNA nanocage assemblies substantially increases the cell uptake of their respective metal complex. Altogether, the facile incorporation of a large number of metal complex per assembly, the higher stability in serum, and the increased cell penetration of metallointercalator@DNA make these self-assemblies well-suited as metallodrugs.
Keywords: DNA tetrahedron, metallointercalator, supramolecular self-assembly, serum stability, cell uptake
Graphical Abstract

The ability to control the organization of molecules into networks of well-defined size, shape, and dimensionality is key to developing the next generation of functional materials.1–4 Of these, nanostructures with empty but ordered space are particularly attractive since both the structures and their cavities can hold and organize smaller guest molecules in a manner inspired by biological systems.5 Such templates can not only guide the autonomous organization of their guests, but also subjects them to geometrical constrains that often modify and enhance the biological, chemical and physical properties of the assemblies.5 Recent examples abound and include, for instance, aligning fluorophores in a supramolecular matrix to optimize the charge-transfer and increase the light harvesting properties of the final materials.6,7
Of the many templates investigated in supramolecular chemistry, DNA offers numerous advantages. Its ability to form predictable and programmable duplexes and other secondary structures enables the hierarchical assembly of higher ordered supramolecular three-dimensional constructs such as DNA self-assembled nanostrucutres.8 These factors have contributed greatly to the development of DNA nanotechnology and its applications in a variety of fields such as drug delivery and sensing.9 Many of these applications require incorporation of drugs or imaging agents, either covalently or via supramolecular interactions. Of these two binding modes, the latter is favored as it enables rapid and facile synthesis of a final material that can incorporate a greater number of guests.
Self-assemblies of guests on DNA nanocages have thus far primarily employed organic molecules such as dyes, drugs, or polymers. On the other hand, the synthesis of well-defined structures incorporating metal complexes self-assembled within a DNA nanocages remain under-explored. Metal complexes have been incorporated in DNA self-assembled structures, but mainly via direct conjugation of the ligand to the oligonucleotides such as, for instance, via ssDNA sequences end-modified with terminal coordinating ligands or via site-specific insertion of ligands into the backbone of the DNA.10 In cases where only the ligand is conjugated, subsequent loading with the metal ions affords the final metallated DNA structure.11–15 Unfortunately, such covalent conjugation of the metal complex to the DNA does not take advantage of the benefits afforded by supramolecular chemistry, including the use of non-modified and readily available nucleic acids, the ease of synthesis of each building block, and the self-correcting nature of self-assemblies. The requirements of covalent modification also limit the number of metal complexes that can be incorporated into the DNA structure to at most a few units.
Nonetheless, supramolecular recognition of DNA with metal complexes is a well-established field.16 Minor and major groove binders,17–19 metallointercalators,20,21 metalloinsertors22,23 and helicates24–27 that self-assemble with dsDNA, three-way junctions, or G-quadruplexes28,29 are well known. Some of those are selective for a specific sequence or mismatch.20 Employing these strategies to assemble coordination complexes into DNA nanocages would not only enable rapid and simple functionalization of these nanostructures; it also offers the opportunity to increment and maximize the number of metal complex per hosts. Indeed, crystal structures of dsDNA with both metallointercalators and metalloinsertors indicate that a high load of metal complexes, up to one complex for every two base-pairs, can self-assemble on a dsDNA template.22,30 When applied to DNA nanostructures such as DNA self-assembled nanocages, high loading of metal complexes offers the potential to yield well-defined assemblies with properties that do not necessarily mimic those observed with the DNA template. Functionalization via noncovalent interactions also allows for the formation of reversible systems or dynamic materials in which the DNA template can release its inorganic guests if subjected to an external stimulus such as light, temperatures or changes of ionic strength.
In view of achieving these long-term goals it is first necessary to understand better how the behavior of a DNA nanocage template is affected by a metal complex guest. With this in mind, we synthesized and characterized two different metallointercalators@DNA nanocages that incorporate two different metal complexes. [Ru(bpy)2(dppz)]2+ (Figure 1) is a well-known light-switch metallointercalator whose luminescence increases substantially upon incorporation in the DNA helix. The metal complex unwinds and enters the double helix via the major groove resulting in a doubling of the rise but otherwise minimal distortion of the DNA structure. [Tb-DOTAm-Phen]3+ behaves similarly except that intercalation of the phenanthridine moiety favors photoelectron transfer from the phenanthridine to either guanine or adenosine, which results in significant Tb-centered phosphorescence quenching. Both metallointercalators were previously successfully employed to self-assemble DNA@Au nanoparticles structures.31 Given the high positive charges of both complexes that increase their affinity for dsDNA, we postulated that both complexes could self-assemble in a DNA nanocage such as Turberfield’s DNA tetrahedron with a high and precise metal complex guest to DNA structure host ratio (Figure 2). Moreover, consistent with the decrease of the overall charge of the assembly caused by the positive charge of the complexes, we hypothesized that the metallointercalators@DNA assemblies would display decreased degradation in serum and increased uptake of the metal complex by cells.
Figure 1.

Chemical structures of [Ru(bpy)2(dppz)]2+, [Tb-DOTAm-Phen]3+ and Tb-DOTA-Phen.
Figure 2.

Supramolecular functionalization of DNA tetrahedron structures with metallointercalators.
RESULTS AND DISCUSSION
Turberfield’s DNA tetrahedron was chosen for this study due to its simplicity and ease of synthesis.32 Advantageously, this three-dimensional DNA cage is also is biocompatible, and amenable to further functionalization with chemical tags or other biomolecules of interest as needed for eventual biomedical applications.33–36 The nanocage was self-assembled in one-step from four 55 base-long oligonucleotides, leading to a structure that contains 17 base pairs per edge. Of note, the synthesis of the DNA tetrahedron is facilitated by high concentration of Mg2+ ions in the buffer. Those high concentration of Mg2+, which stabilizes the structure via interaction with the DNA phosphate backbone, disfavors intercalation of positively charged metallointercalators. It is therefore necessary to exchange the buffer used for the synthesis of the DNA tetrahedron to one with lower salt concentrations (10 mM Tris + 5 mM MgCl2) prior to assembling the DNA-metallointercalator host:guest complexes.
As shown in Figure 3, the interaction of both [Ru(bpy)2(dppz)]2+ and [Tb-DOTAm-Phen]3+ with the DNA tetrahedron can be readily monitored by luminescence spectroscopy. In water, the RuII-centered luminescence of [Ru(bpy)2(dppz)]2+ is nearly completely quenched due to hydrogen-bonding between H2O and the nitrogen atoms of the dipyrido[3,2-a:2′,3′-c]phenazine (dppz) ligand that causes the 3MLCT state of [Ru(bpy)2(dppz)]2+, which is primarily phenazine in character, to favor a nonradiative decay process. Intercalation of the dppz ligand in the base pair stacks of the dsDNA shields the dppz from hydrogen bonding to the solvent. As a result, the 3MLCT state, now higher in energy and more bpy in character, favors luminescence. This phenomenon is classically referred to as the light-switch effect.37–41 This effect results in a 16-fold increase in fluorescence intensity as the DNA tetrahedron is titrated to a solution of [Ru(bpy)2(dppz)]2+ (Figure 3a and c).
Figure 3.

(a) Fluorescence spectra of [Ru(bpy)2(dppz)]2+ upon addition of increasing concentrations of DNA tetrahedron. (b) Time-delayed phosphorescence spectra of [Tb-DOTAm-Phen]3+ upon addition of increasing concentrations of DNA tetrahedron. (c) Normalized luminescence intensity as a function of the ratio of DNA base pair (b.p.) to metallointercalator (ML). Error bars represent ±1 standard deviation (n = 3). I = integrated luminescence intensity of the metallointercalator upon addition of increasing amounts of DNA tetrahedron, I0 = integrated luminescence intensity of the metallointercalator in the absence of DNA tetrahedron. Experimental conditions: [metallointercalator] = 10 μM in buffer ([Tris] = 10 mM, [MgCl2] = 5 mM, pH 7.4); T = 20 °C. For [Ru(bpy)2(dppz)]2+: λex = 440 nm, fluorescence is integrated from λem= 500 nm to 800 nm, excitation slit width = 10 nm and emission slit width = 10 nm. For [Tb-DOTAm-Phen]3+: λex = 345 nm, time-delayed luminesce is integrated from λem= 450 nm to 650 nm, delay time = 0.1 ms, gate time = 5 ms, excitation slit width = 5 nm and emission slit width = 5 nm.
On the other hand, intercalation of [Tb-DOTAm-Phen]3+ in the DNA nanocage results in a substantial decrease in TbIII-centered phosphorescence when excited at 345 nm. The origin of the response of [Tb-DOTAm-Phen]3+ is different than that of [Ru(bpy)2(dppz)]2+. In the absence of DNA, excitation of the phenanthridine antenna to its singlet state followed by intersystem crossing to its triplet state enables further energy transfer to the 5D excited state of TbIII and subsequent lanthanide-centered phosphorescence emission.42 Intercalation in dsDNA, however, enables photoelectron transfer from the phenanthridine antenna to either guanosine or adenosine, which prevents sensitization and emission of the TbIII centers.43 As a result, the phosphorescence of the metallointercalator decreases upon titration of the DNA tetrahedron (Figure 3b and c).
The number of DNA binding sites per nanostructure (n) and the average association constants per site (Ka) can both be determined from the spectrofluorometric titrations of the DNA tetrahedron with the metallointercalators (Figure 3c). The binding isotherms of the metallointercalator@DNA nanocage were thus analyzed according to the independent site model and compared to results from a linear 55-base pair long dsDNA (Table 1, Figures S21 and S22).44,45 Importantly, this model assumes that there is at most one metal complex per DNA base pair and that there is no cooperativity between binding sites. The affinity constants calculated for [Ru(bpy)2(dppz)]2+ (Table 1) indicate that the metal complex has an equally high affinity for the DNA tetrahedron and for the linear 55 base pair DNA duplex, which, in both cases, is more than ten orders of magnitude higher than that of the classic organic DNA intercalator ethidium bromide (Ka= 1.23 + 0.07 × 105 M−1).46 Comparison of the Ka values reveals that the affinity of the ruthenium-based metallointercalator for dsDNA is not a function of its three-dimensional structure. [Ru(bpy)2(dppz)]2+ has similar affinity for linear, 1-dimensional dsDNA than for the DNA tetrahedron. However, on a per base pair ratio, the three-dimensional DNA tetrahedron accommodates more metallointercalators than the linear, one-dimensional DNA. This increased loading capacity of the DNA nanocage might be due to the rigidity of the three-dimensional system. Of note, similar experiments were carried out with [Tb-DOTAm-Phen]3+. Unfortunately, an accurate estimation of the affinity constants was not possible as the terbium complex has a weaker affinity for dsDNA and does not appear to behave solely as an intercalator. Treatment of the spectrofluorometric data using the same model thus does not necessarily provide reliable results.
Table 1.
Binding constants for [Ru(bpy)2(dppz)]2+ with DNA tetrahedron and linear dsDNA as determined by fluorescence titrations.
| Ka (M−1) | n | metallointercalator: base pairs ratio | |
|---|---|---|---|
| [Ru(bpy)2(dppz)]2+@DNA tetrahedron | 2.0 ± 0.5 × 106 | 44 ± 2 | 1:2 |
| [Ru(bpy)2(dppz)]2+@linear DNA | 2.7 ± 0.5 × 106 | 16.5 ± 0.5 | 1:3 |
Experimental conditions: 10 mM Tris (aq), 5 mM MgCl2 (aq), pH 7.4
The stoichiometry between the metallointercalator guest and the DNA tetrahedron host can also be determined by the method of continuous variation. The Job’s plot for [Ru(bpy)2(dppz)]2+ titration with Turberfield’s tetrahedron (Figure 4) indicates that a maximum in luminescence intensity is observed for a mole fraction, χ[Ru(bpy)2(dppz)]2+=0.33. The Jobs’ plot for [Tb-DOTAm-Phen]3+ (Figure S18) also has a vertex at χ[Tb(DOTAm-Phen)]3+=0.33. These observations indicate that, regardless of the metallointercalator, the DNA tetrahedron host can accommodate up to one metallointercalator guest for every two DNA base pairs. Each edge of the DNA tetrahedron contains 17 base pairs. Therefore, when completely loaded, Turberfield’s DNA tetrahedron contains 8 metallointercalators per edge, corresponding to 48 metal complexes per DNA nanocage. These observations align with prior observations that demonstrated that Turberfield’s DNA tetrahedron also accommodates 48 organic intercalating dye per assembly.47 The crystal structure of [Ru(bpy)2(dppz)]2+ with a linear (one-dimensional) dsDNA demonstrate that up to 5 metal complexes can intercalate or insert in a 12-mer oligonucleotide; precisely one every two base pair.30 The metallointercalators@DNA self-assemblies thus follow the well-established neighbor-exclusion principle that states that binding of small planar intercalators is anti-cooperative at adjacent sites. In other words, since the two neighboring sites of an occupied intercalation site in dsDNA must remain unoccupied, the metal complex can only intercalate every second base pair.
Figure 4.

Job’s plot of the DNA tetrahedron responsive [Ru(bpy)2(dppz)]2+. I=integrated luminescence intensity from 500 nm to 800 nm. I0 = integrated luminescence intensity in the absence of the DNA tetrahedron from 500 nm to 800 nm. Experimental conditions: Total concentration of [Ru(bpy)2(dppz)]2+ + DNA base pair (b.p) = 10 μM in buffer ([Tris] = 10 mM, [MgCl2] = 5 mM, pH 7.4); T = 20 °C; λex = 440 nm, excitation slit width = 10 nm; emission slit width = 10 nm.
Although both [Ru(bpy)2(dppz)]2+ and [Tb-DOTAm-Phen]3+ form metallointercalators@DNA host:guest assemblies with the same stoichiometry, they do not both have the same affinity for the DNA tetrahedron. The affinity of a metal complex for its intercalating site in the DNA is primarily governed by two factors: the structure of the intercalating ligand and the charge of the complex. The former define how well it can penetrate and stack with its neighboring bases while the latter governs the electrostatic component of the interaction between the positively charged complex and the negatively charged DNA structure. The higher positive charge of [Tb-DOTAm-Phen]3+ contributes to its higher affinity for dsDNA. However, the intercalating ligand of the ruthenium(II) complex, dppz, is more extended and is expected to enable more efficient intercalation and stacking with neighboring base pairs within the DNA base stack.
In order to determine which of the two metallointercalators has the highest affinity for the DNA tetrahedron, a competition experiment was run. In this experiment, the DNA tetrahedron fully loaded with 48 equivalents of [Ru(bpy)2(dppz)]2+ (1 RuII complex for every 2 DNA base pairs) was gradually challenged with increasing concentrations of [Tb-DOTAm-Phen]3+. Advantageously, the significant decrease in RuII emission upon displacement from the DNA structure by the TbIII complex enables us to monitor the competition directly by luminescence spectroscopy. Kinetic studies indicated that the metallointercalators@DNA assembly is dynamic, with guest exchange occurring rapidly, typically in less than 5 min at room temperature. As shown in Figure 5, in the absence of competing [Tb-DOTAm-Phen]3+, the RuII complex intercalates in the DNA tetrahedron resulting in a 13-fold increase in luminescence intensity. Progressive addition of [Tb-DOTAm-Phen]3+ decreases the RuII-centered emission, indicative of the gradual displacement of the RuII metallointercalators. Approximately 5 equivalents of [Tb-DOTAm-Phen]3+ are needed to displace 45 percent of the [Ru(bpy)2(dppz)]2+ (decrease I/I0 from 13.2 to 7.3). This indicates that [Tb-DOTAm-Phen]3+ has slightly lower affinity for the DNA tetrahedron than [Ru(bpy)2(dppz)]2+ despite having a higher charge. This difference can be attributed to the more extended structure of dppz that enables better π-stacking with neighboring bases compared to phenanthridine.
Figure 5.

Displacement of [Ru(bpy)2(dppz)]2+ from the DNA tetrahedron upon addition of competing terbium-based metallointercalators. [Tb-L]= concentration of corresponding [Tb-DOTAm-Phen]3+ and [Tb-DOTA-Phen]. I = integrated luminescence intensity of [Ru(bpy)2(dppz)]+2@DNA upon addition of increasing amounts of competing metallointercalator, I0 = integrated luminescence intensity of [Ru(bpy)2(dppz)]+2@DNA tetrahedron in absence of competing metallointercalator. Error bars represent ±1 standard deviation (n = 3). Experimental conditions: [Ru(bpy)2(dppz)]+2 (10 μM) and DNA tetrahedron (20 μM b.p) in M in buffer ([Tris] = 10 mM, [MgCl2] = 5 mM, pH 7.4); T = 20 °C. Fluorescence integrated from λem = 500 nm to 800 nm, λex = 440 nm, excitation slit width = 10 nm; emission slit width = 10 nm.
Electrostatic forces nonetheless play an important role in the interaction of metallointercalator with dsDNA structures. The affinity of macrocyclic lanthanide complexes with a phenanthridine antenna for nucleotides was previously demonstrated to be highly dependent on the charge of the complex.48 The +3 charged triamide complex [Tb-DOTAm-Phen]3+ has higher affinity for nucleotides than its neutral tricarboxylate analog Tb-DOTA-Phen (Figure 1). Conversely, the higher the negative charge of the nucleotide, the higher its affinity for the positively charged [Tb-DOTAm-Phen]3+.49 These conclusions still holds true for intercalation in dsDNA. Whereas the positively charged [Tb-DOTAm-Phen]3+ can displace most [Ru(bpy)2(dppz)]2+ from the DNA tetrahedron, 5 equivalents of the neutral Tb-DOTA-Phen can barely displace 20% of the RuII complex (Figure 5).
The difference in the affinity of [Ru(bpy)2(dppz)]2+ and [Tb-DOTAm-Phen]3+ for the DNA tetrahedron is also reflected in the melting point of the assemblies. When monitored by UV spectroscopy (λ= 260 nm), the melting point of the DNA tetrahedron increases by 6 °C when fully intercalated with [Ru(bpy)2(dppz)]2+ compared to 4 °C when intercalated [Tb-DOTAm-Phen]3+ (Figure 6). An increase in melting temperature is associated with an increase in the stability of the annealed DNA.50 These data confirm that the higher affinity of [Ru(bpy)2(dppz)]2+ results from a greater stabilization of the metallointercalator@DNA self-assembly with the ruthenium(II) complex compared to the terbium(III) complex. Of note, the melting point of the metallo:DNA host:guest complexes can also be determined by monitoring the RuII-centered luminescence as a function of temperature. As shown in Figure 6, denaturation of the DNA upon heating releases the RuII metallointercalator, resulting in a significant decrease in its luminescence. The melting point for the [Ru(bpy)2(dppz)]2+@DNA tetrahedron assembly as determined by luminescence (69 °C) is comparable to that determined by UV spectroscopy.
Figure 6.

Representative melting curves of DNA tetrahedron and metallointercalator@DNA tetrahedron as measured by UV-spectroscopy and fluorescence. In all cases melting point was calculated as the average of three different measurements. Experimental conditions for UV-melting curves: Absorbance recorded at 260 nm. [DNA tetrahedron] = 150 nM, [metallointercalator] = 7.7 μM in buffer ([Tris] = 10 mM, [MgCl2] = 5 mM, pH 7.4), Heating rate = 1 °C/min. Experimental conditions for fluorescence-melting curve: λem = 618 nm, λex = 440 nm, I= luminescence intensity of [Ru(bpy)2(dppz)]2+@DNA, I0 = luminescence intensity of [Ru(bpy)2(dppz)]2+ in absence of DNA at the same temperature. [DNA tetrahedron] = 30 nM, [ [Ru(bpy)2(dppz)]2+ ] = 1.5 μM in buffer ([Tris] = 10 mM, [MgCl2] = 5 mM, pH 7.4), excitation slit width = 10 nm, emission slit width = 10 nm. Heating rate = 1 °C/min.
Importantly, intercalation of the metal complex does not affect the three-dimensional structure of the DNA tetrahedron. Formation of the parent DNA tetrahedron was initially confirmed via non-denaturing agarose gel electrophoresis where the DNA nanocage exhibited a reduced mobility in comparison to the single stranded DNA oligomers used as starting material (Supplementary Figure S19). Notably, the metallointercalator@DNA tetrahedron nanocages exhibited electrophoretic mobility identical to that of the naked DNA tetrahedron in agarose 2%. Moreover, given their luminescent behavior, the metallointercalator@DNA tetrahedron assemblies can be visualized without the use of any additional nucleic acid stain. The band corresponding to the bioinorganic assembly luminesces either red (for [Ru(bpy)2(dppz)]2+) or green ([Tb-DOTAm-Phen]3+) upon irradiation with UV-light (Figure S23). The fact that metal complexes remain bound to the DNA cage during electrophoresis facilitates detection of the nanocage. More importantly, it corroborates our assertion that the DNA nanostructures are preserved after non-covalent functionalization with metal complexes.
In order to provide further evidence of the formation of the DNA nanostructures, naked and functionalized DNA assemblies were imaged by atomic force microscopy on dry mica (AFM). In the dry state (Figure 7), naked DNA tetrahedron particles are observed as collapsed 2D objects with triangular shapes with about 2-3 nm height, which is consistent with previous reports from the literature.51,52 Electrostatic interactions, as well as the dehydrating conditions, as described by Mao, are likely the cause of such collapse.53 Importantly, the metallointercalators@DNA tetrahedron assemblies also appear as well-dispersed particles with triangular shapes and similar height in the AFM images. These observations strongly indicate that intercalation of the metal complex does not affect the three-dimensional structure of the DNA tetrahedron.
Figure 7.

Atomic force microscopy images of DNA tetrahedron with or without metal complex. a) DNA tetrahedron, b) [Ru(bpy)2(dppz)]2+@DNA assembly, c) [Tb-DOTAm-Phen]3+@DNA assembly.
When fully loaded, 48 metal complexes can intercalate in the DNA tetrahedron. At this ratio, the host:guest assemblies are nearly a third metal complex by mass with an overall charge half (for [Ru(bpy)2(dppz)]2+) or a quarter (for [Tb-DOTAm-Phen]3+) that of the original tetrahedron. Given the propensity of both metallointercalators to stabilize the DNA tetrahedron and the different physical properties of the host:guest complexes, the two components of the supramolecular assemblies were anticipated to affect the behavior of the other one in biological systems. Rapid degradation in serum and other biological media is one of the factors limiting the potential clinical translation of DNA nanostructures as drug delivery or imaging agents. Compact DNA nanocages, although more resistant than ssDNA, remain particularly prone to biodegradation due to nuclease activity.54 The lower concentration of Mg2+ ion in serum (˂1 mM) compared to that in the buffer for the tetrahedron synthesis (50 mM) would also affect stability of the assembly in vivo.55,56 Several studies have looked at increasing the stability of DNA nanostructures in serum via enzymatic ligation of free termini and internal nicks,36,57 or by using peptoids and proteins,58,59 and polymers60,61 as protective coatings.
As shown in Figure 8a, both metallointercalators@DNA self-assemblies substantially slow degradation of the DNA tetrahedron in serum. The native DNA tetrahedron decomposes partially after 1 hour and completely in < 3 hours of incubation in 10% freshly thawed fetal bovine serum (FBS) in Tris buffer supplemented with 5 mM MgCl2. Reports from the literature indicate that the stability of Turberfield’s tetrahedron in FBS vary between 4 to almost 24 hours depending on the proportion of FBS in the incubation media and on the freshness of the serum.36,62–64 Nuclease activity in FBS is known to decrease rapidly with time.55,65 In fresh serum with high nuclease activity, the stability of the DNA tetrahedron increases considerably when intercalated with a metal complex. The [Ru(bpy)2(dppz)]2+@DNA tetrahedron assembly shows minimal degradation even after 7 hours of incubation. The DNA structure does eventually degrade before 24 hours, likely due to the reversible nature of the intercalation. As discussed above, the lanthanide complex [Tb-DOTAm-Phen]3+ has lower affinity for the DNA tetrahedron and does not stabilize the nanocage as much as [Ru(bpy)2(dppz)]2+. The ability of the phenanthridine-based intercalator to protect the DNA tetrahedron from nuclease degradation in serum is therefore also weaker than that of the dppz-based one. This difference is apparent in the faster loss of total band intensity in the agarose gel [Tb-DOTAm-Phen]3+@DNA tetrahedron compared to [Ru(bpy)2(dppz)]2+@DNA tetrahedron.
Figure 8.

Stability of DNA tetrahedron and metallointercalator@DNA tetrahedron assemblies in: a) 10% fetal bovine serum (FBS), b) 10% mouse serum. Percentage of intact DNA tetrahedron was estimated by non-denaturing agarose gel electrophoresis 2% in TBE 1X, visualized with SYBR Safe staining, and plotted as a function of time. Error bars represent ±1 standard deviation (n = 3).
This protective effect could be attributed to the ability of both [Ru(bpy)2(dppz)]2+ and [Tb-DOTAm-Phen]3+ to restrict access of DNases to the DNA. DNase 1, a major serum nuclease,66,67 is a minor groove binder whose cutting rate is significantly affected by the flexibility of the DNA structure.68 Although [Ru(bpy)2(dppz)]2+ is a classic major groove intercalator, it alters and rigidifies the structure of the DNA, which likely affects the activity of DNase-1. Moreover, the positive charges of the metallointercalators mask the negative charge of the sugar-phosphate backbone, which would reduce the electrostatic interactions between DNAse 1 and the DNA tetrahedron. In this regard, the ability of the metallointercalators to decrease dsDNA degradation in serum is unsurprising.22,30,69
Previous studies from Bathe demonstrated that degradation of a wireframed DNA two-helix pentagonal bipyramid occurred almost eight times faster (3 hours) in mouse serum than in FBS (>24 hours) due to higher nuclease activity.70 As stated by Bathe, these experimental observations highlight the importance of accounting for the difference in species-specific nuclease activity in different media when evaluating the stability of DNA nanostructures. To account for this, the protective effect conferred by the metallointercalator on the DNA structure was further explored by evaluating the degradation of the naked DNA tetrahedron and the metallointercalators@DNA tetrahedron assemblies in 10% mouse serum (Figure 8b). Under these conditions, the non-functionalized DNA tetrahedron cage decomposes completely under 30 minutes. At the same time point, almost 40 percent of the [Tb-DOTAm-Phen]3+@DNA tetrahedron assembly remains intact. Predictively, the [Ru(bpy)2(dppz)]2+@DNA tetrahedron assembly exhibits even better stability with almost eighty percent of the DNA nanocage intact after the first 30 minutes. Regardless of the species, our results indicate that addition of metallointercalators to DNA tetrahedron nanocages do considerably reduce the rate of decomposition in serum. In mouse serum, addition of [Tb-DOTAm-Phen]3+ increases the lifetime by a factor of 2, whereas [Ru(bpy)2(dppz)]2+ increases it for up to 3 hours. Altogether, these results corroborate that [Ru(bpy)2(dppz)]2+ provides a better coating effect, presumably by restricting the access of the enzyme to specific regions of the duplex DNA, as well as by modulating the electrostatic interactions between the DNA and enzymes.
Given the ability of metallointercalators to stabilize the DNA nanocage in serum, the supramolecular structure was also anticipated to affect cell uptake of the metal complexes. Neither [Ru(bpy)2(dppz)]2+ nor [Tb-DOTAm-Phen]3+ are noticeably uptaken by cell, a behavior that limits their translation to potent anticancer drugs or cellular probes, respectively. These observations are in agreement with viability assays of [Ru(bpy)2(dppz)]2+ and [Tb-DOTAm-Phen]3+ cells demonstrated that neither complex have notable cytotoxicity, with IC50 values for HEK-293 of 192 μM and >300 μM, respectively (Figure S20). Reports of cell uptake of DNA nanocages in the literature are more inconclusive. The negative charges of the surfaces of cell membranes electrostatically repel and prevent the cell uptake of short ssDNA and dsDNA. Literature reports on cellular uptake of DNA nanostructures vary greatly, but many indicate that the nanostructures of DNA nanocages can overcome the limitations of linear DNA and lead to higher cellular internalization without the aid of transfection agents.63,71,72 Other, however, report minimal cellular association or uptake. Cellular uptake is known to be affected by several parameters, including the shape, size and sequence of the DNA self-assembly as well as functionalization with polymers, targeting vectors, or other moieties intended to stabilize the structure of the DNA in serum. Some reports, unfortunately, do not distinguish conclusively between cellular uptake of the intact fluorescently-labeled DNA nanostructures and uptake of the released fluorophore or fluorescently-labeled degradation products.63
The cellular association of the two fully loaded metallointercalators@DNA assemblies was investigated with both L6 rat myoblast cells and human embryonic kidney cells HEK-293 and monitored by mass cytometry. Mass cytometry, a technique similar to flow cytometry that detects probes labeled with heavy metal isotopes by inductively coupled plasma and time of flight mass spectrometry (ICP-MS) of single cells, is notably suited to monitor the association of the metallointercalators with cells. Sm-DDD, which contains a hydrophilic polar head and two membrane-penetrating hydrophobic tails (Figure 9a), was used to label the cells for recognition in the instrument instead of the traditional iridium DNA intercalator that would interfere with the metallointercalators@DNA assemblies.
Figure 9.
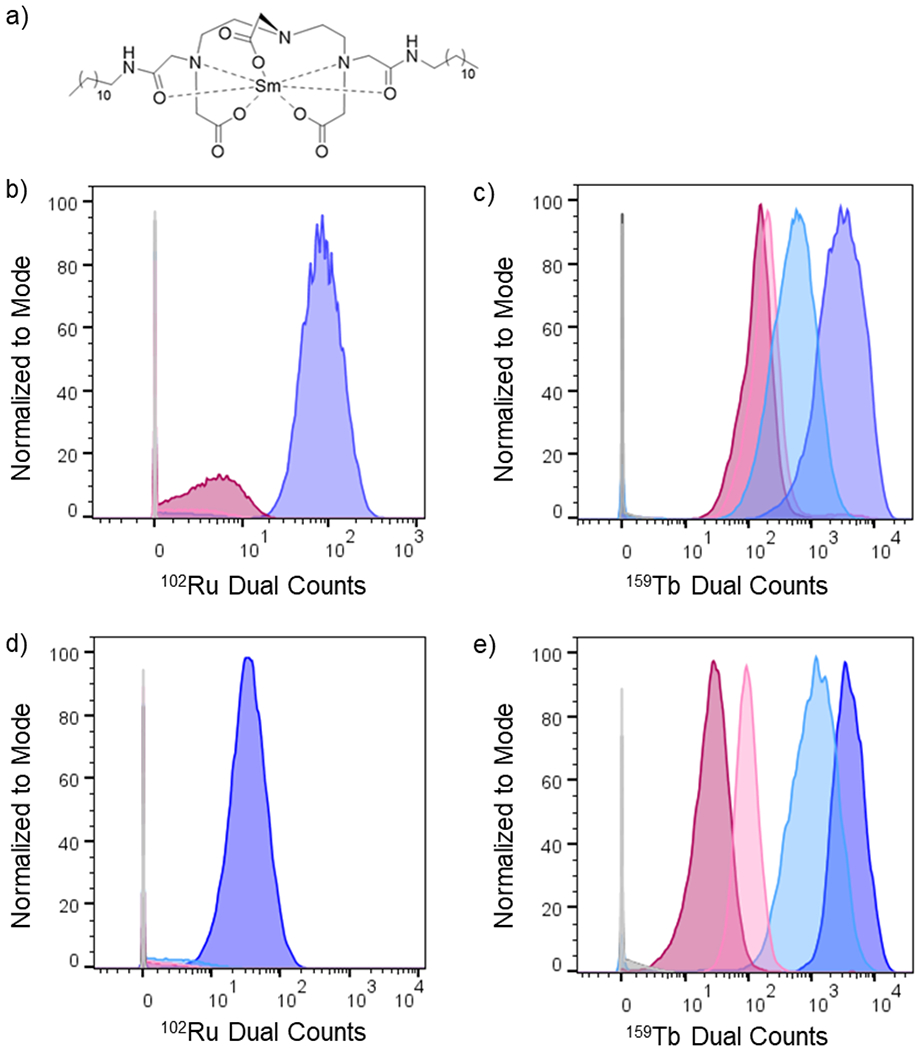
a) Chemical structure Sm-DDD probe for cell labeling by cytometry analysis. b) Histogram showing 102Ru dual count intensity distribution for L6 cells treated for 4 hours with [Ru(bpy)2(dppz)]2+, Lipofectamine + [Ru(bpy)2(dppz)]2+, [Ru(bpy)2(dppz)]2+@DNA, Lipofectamine + [Ru(bpy)2(dppz)]2+@DNA, and controls. c) Histogram showing 159Tb dual count intensity distribution for L6 cells treated for 4 hours with [Tb-DOTAm-Phen]3+, Lipofectamine + [Tb-DOTAm-Phen]3+, [Tb-DOTAm-Phen]3+@DNA, Lipofectamine + [Tb-DOTAm-Phen]3+@DNA and controls. d) Histogram showing 102Ru dual count intensity distribution for HEK-293 cells treated for 4 hours with [Ru(bpy)2(dppz)]2+, Lipofectamine + [Ru(bpy)2(dppz)]2+, [Ru(bpy)2(dppz)]2+@DNA, Lipofectamine + [Ru(bpy)2(dppz)]2+@DNA, and controls. c) Histogram showing 159Tb dual count intensity distribution for HEK-293 cells treated for 4 hours with [Tb-DOTAm-Phen]3+, Lipofectamine + [Tb-DOTAm-Phen]3+, [Tb-DOTAm-Phen]3+@DNA, Lipofectamine + [Tb-DOTAm-Phen]3+@DNA and controls. Legend for samples colors is indicated in table 2.
As shown in Table 2, [Ru(bpy)2(dppz)]2+ does not associate with either L6 or HEK-293 cells after 4 hours of incubation. These observations are in agreement with prior reports describing the poor cell uptake of this ruthenium-based metal complex.38,73 In contrast, [Tb-DOTAm-Phen]3+, some, but still low, uptake were observed in both L6 cells and HEK-293 cells (Table 2 and Figure 9). The addition of Lipofectamine does not increase the cell uptake of the ruthenium metallointercalator in either cell lines. On the contrary, Lipofectamine does increase somewhat the uptake of the terbium complex in L6 cells (2.8 fold increase) and significantly more in HEK-293 cells (11-fold). Interestingly, no cell uptake of [Ru(bpy)2(dppz)]2+@DNA tetrahedron was observed in either cell line. For [Tb-DOTAm-Phen]3+, the DNA self-assembly actually displays lower cell uptake than the metallointercalator alone: less 159Tb were observed per cell for [Tb-DOTAm-Phen]3+@DNA tetrahedron than for [Tb-DOTAm-Phen]3+.
Table 2.
Median dual count intensity values for mass cytometry samples
| L6 Cells | HEK-293 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| Sample | Cell Count | Median Dual Count 152Sm | Median Dual Count 159Tb | Median Dual Count 102Ru | Cell Count | Median Dual Count 152Sm | Median Dual Count 159Tb | Median Dual Count 102Ru | |

|
Media Buffer | 67866 | 139 | n/a | 7.63×10−3 | 46650 | 370 | n/a | ** |

|
Lipofectamine Buffer | 44854 | 163 | n/a | 7.63×10−3 | 18868 | 357 | n/a | ** |

|
[Ru(bpy)2(dppz)]2+ | 42628 | 177 | n/a | 0.91 | 48746 | 310 | n/a | ** |

|
Lipofectamine + [Ru(bpy)2(dppz)]2+ | 38225 | 158 | n/a | 0.055 | 50327 | 261 | n/a | 1.11 |

|
[Ru(bpy)2(dppz)]2+@DNA | 46093 | 150 | n/a | 3.85 | 28821 | 413 | n/a | ** |

|
Lipofectamine + [Ru(bpy)2(dppz)]2+@DNA | 46534 | 188 | n/a | 81.1 | 20759 | 375 | n/a | 32.9 |
|
| |||||||||
|
|
[Tb-DOTAm-Phen]3+ | 37971 | 189 | 185 | n/a | 10524 | 335 | 92.6 | n/a |
|
|
Lipofectamine + [Tb-DOTAm-Phen]3+ | 39089 | 237 | 521 | n/a | 16530 | 321 | 1070 | n/a |
|
|
[Tb-DOTAm-Phen]3+@DNA | 47327 | 134 | 137 | n/a | 38532 | 285 | 26.4 | n/a |
|
|
Lipofectamine + [Tb-DOTAm-Phen]3+@DNA | 36997 | 222 | 2883 | n/a | 15187 | 380 | 3679 | n/a |
Value reported by instrument was below zero due to software randomization indicating no detection of that metal in the sample
limit of detection was found using solution mode resulting in 152Sm 0.0749 pM or 11.4 dual counts, 159Tb 0.129 pM or 20.2 dual counts and 102Ru 0.234 pM or 4.84 dual counts.
Although the metallointercalators and the bioinorganic self-assemblies are poorly taken up by cells, the trend can be reversed upon addition of Lipofectamine. In the presence of Lipofectamine, a significant amount of [Ru(bpy)2(dppz)]2+@DNA is taken up by both L6 and HEK-293 cells (Figure 9b and 9d, purple). Similarly, the addition of Lipofectamine increases the uptake of [Tb-DOTAm-Phen]3+ in L6 and HEK-293 cells by 21-fold and 139-fold, respectively (Figure 9c and 9e, purple). It is clear from these observations that the metallointercalators@DNA tetrahedron are poorly taken up by cells, whereas the addition Lipofectamine to the bioinorganic assembly substantially increases the cellular delivery of the metal.
These results appear to be in part inconsistent with previous literature reports indicating significant cell uptake of various tetrahedral DNA structures. Turberfield, for instance, reported that a similar elongated DNA tetrahedron internalize efficiently in Human embryonic kidney (HEK) cells both with and without the aid of Lipofectin.74 Similar results with a smaller DNA tetrahedron and Human primary glioblastoma cells have also been reported.75 In our case, significant cell uptake is observed only in the presence of Lipofecamine. However, our results are not necessarily contradictory. Prior results were obtained on DNA nanostructures whose functionalization, such as covalent conjugation of a dye, did not significantly affect their charge. Maximum loading of metallointercalators in the DNA tetrahedron (one +2 or +3 charged metal complex for every 2 base pairs) significantly decreases the charge of the assembly to either half (for [Ru(bpy)2(dppz)]2+) or a quarter (for [Tb-DOTAm-Phen]3+) that of the original tetrahedron. The poor cellular association of both metallointercalator@DNA assemblies could be due to their significantly smaller charge and their more rigid structure. On the other hand, the significant cell uptake observed for the metallointercalators@DNA tetrahedron assemblies in the presence of Lipofectamine is in agreement with the mode of action of the transfecting agent and bodes well for further studies of DNA self-assemblies for delivery of metallodrugs.
CONCLUSIONS
Self-assembly of metallointercalators into self-assembled DNA nanocages is a rapid and facile approach to synthesizing discrete bioinorganic host:guest structures with a high loading of metal complexes. Turberfield’s DNA tetrahedron can accommodate at most one intercalator for every two base pairs, which corresponds to 48 metallointercalator guests on a single DNA tetrahedron. This observation is in agreement with the neighbor-exclusion principle. This supramolecular approach thus represents a major advantage over standard bioconjugation since, in comparison, covalent functionalization of oligonucleotides enables the incorporation of at most a few metal complexes into the ssDNA. The affinity of the metallointercalator for the DNA tetrahedron is a function primarily of the structure of the intercalating ligand and the overall charge of the complex. The dppz ligand is a better intercalator than phenanthridine, and as such [Ru(bpy)2(dppz)]2+ has higher affinity for the DNA tetrahedron than [Tb-DOTAm-Phen]3+. The charge of the metallointercalator also influences the affinity of the metal complex for the DNA structure, as evidenced by the much lower affinity of the neutral Tb-DOTA-Phen compared to its positively charged analogue [Tb-DOTAm-Phen]3+. Intercalation stabilizes the DNA resulting in an increase in melting temperature and, importantly, a significant increase in the stability of the DNA construct in the presence of serum. [Ru(bpy)2(dppz)]2+, which has a greater affinity for DNA than [Tb-DOTAm-Phen]3+, increases the melting point and prevents serum degradation to a greater extent than the Tb complex. The significant decrease in the overall charge of the assembly upon intercalation of the metal complexes correlates with very low levels of cell association of the metallointercalator@DNA assemblies. Negligible (for [Ru(bpy)2(dppz)]2+) or very low (for [Tb-DOTAm-Phen]3+) levels of heavy metals are observed per cell by mass cytometry. The addition of Lipofectamine reverses this observation, with significant cell uptake of metallointercalator@DNA assemblies observed in either L6 cells or HEK-293 cells after 4 hours of incubation in the presence of the transfecting agent. Altogether, these observations highlight the potential of metallointercalators@DNA supramolecular structures for biomedical application. In particular, although these assemblies are promising candidates for delivery of intercalating metallodrugs that otherwise have poor cell penetration. Their high loading of metal complexes per DNA tetrahedron, their stability in serum and their high cell penetration in the presence of Lipofectamine suggest that they deserve further investigation as carriers of metallodrugs.
METHODS
Materials.
Unless otherwise noted, starting materials were obtained from commercial suppliers and used without further purification. DNA oligonucleotides were purchased from Integrated DNA Technologies. Deuterated solvents were obtained from Cambridge Isotope Laboratories (Tewskbury, MA, USA). Deionized water was further purified by a Millipore Simplicity UV system (resistivity 18 × 106Ω). Preparative column chromatography was performed using a Teledyne Isco CombiFlash Rf purification system utilizing reversed phase silica gel pre-column load cartridges and gold high performance columns. 1H and 13C NMR spectra were recorded on a Bruker Advance III 400 spectrometer at 400 MHz and 100 MHz, respectively. Chemical shifts are are referenced internally to TMS or the residual solvent peak. Mass spectra were obtained on a Bruker BioTOF II ESI/TOF-MS instrument at the Waters Center for Innovation in Mass Spectrometry of the Department of Chemistry at the University of Minnesota. UV-visible spectra were recorded on a Varian Cary 100 Bio Spectrometer or a Cary 3500 (DNA denaturation analysis) with 1 cm quartz cuvettes in Tris(aq) buffer (10 mM) with MgCl2 (5 mM) at pH 7.4. Luminescence spectra were acquired on a Varian Cary Eclipse fluorescence spectrophotometer using a quartz cell with a path length of 1 cm. Time-gated luminescent spectra were recorded with a time delay of 0.1 ms and a gate time of 5 ms. HPLC analysis was performed on a Varian Prostar 210 (Agilent, Santa Clara, CA) equipped with a Varian ProStar 335 diode array detector and an Agilent Zorbax Eclipse XDB-C18 column (5 μm pore size, 9.4 × 250 mm) using a gradient (15% CH3CN/85% H2O from 0 to 2 minutes, followed with a linear gradient to 100% CH3CN from 2 to 23 minutes, and 15% CH3CN/85% H2O from 30 to 32 minutes) at a flow rate of 1 mL/min. All pH measurements were performed using Thermo Scientific Ag/AgCl refillable probe and a Thermo Orion 3 Benchtop pH meter. Mass cytometry samples were run on a CyTOF2 mass cytometer (Fluidigm, South San Francisco, CA) at the Waters Center for Innovation in Mass Spectrometry at the University of Minnesota. Thermal assembly of non-modified DNA nanostructures was performed in an Eppendorf® Mastercycler Personal. Agarose Gel Electrophoresis (AGE) experiments were carried out on a 7 X 10 cm horizontal Bio-Rad Mini-Sub Cell GT Cell electrophoresis unit.
DOTA-Phen and [Tb-DOTAm-Phen]3+ were synthesized according to previous synthetic protocols developed in our research group.49,77 Likewise, [Ru(bpy)2(dppz)]2PF6 was synthesized according to reports from Barton and Marvi.41,78
Tb-DOTA-Phen.
To a stirred solution of DOTA-Phen (20.0 mg, 33.6 μmol) in mQ water (3 mL) was added TbCl3·6H2O (12.5 mg, 33.5 μmol). The pH of the reaction mixture was adjusted to 8 with 1 M LiOH(aq) and heated at reflux for 3 days. The aqueous reaction mixture was concentrated under reduced pressure and the product was precipitated with diethyl ether. The resulting precipitate was washed with diethyl ether (3 × 20 mL), dissolved in mQ water (5 mL) and lyophilized to afford complex the final Tb-DOTA-Phen as colorless powder (21.0 mg, 84%). See Figure S9 for 1H NMR. ESI HR-MS. Calcd for C30H36N6O7Tb [M + H]+: m/z 751.1893. Found: m/z 751.1922.
Sm-didodecyl-DTPA (Sm-DDD).
Didodecyl-DTPA was synthesized as reported in the literature.79 Its samarium(III) complex was synthesized following a method previously reported for the synthesis of the analog Tb-didodecyl-DTPA.80 Briefly, didodecyl-DTPA (307 mg, 387 μmol) was added to a solution of SmCl3 (148 mg, 576 μmol) in mQ water (20 mL). The resulting reaction mixture was stirred at room temperature for 4 hours then lyophilized. The crude off-white product was dissolved in chloroform (15 mL), filtered, and the solvent removed under reduce pressure to yield Sm-DDD as an off-white solid (270 mg, 80%). See Figure S16 for 1H NMR. ESI HR-MS. Calcd for C38H70ClN5O8Sm [M + Cl]−: m/z 911.4111. Found: m/z 911.4078.
Self-assembly of DNA tetrahedron.
DNA tetrahedron was assembled from the following four oligonucleotides following a slightly modified procedure originally reported by Turberfield.32 Briefly, 2 μL of each single stranded DNA (50 μM stock solutions) were combined in 42 μL of an aqueous solution of Tris buffer (10 mM) and MgCl2 (50 mM) at pH 8. The temperature of the reaction mixture was slowly raised to 95 °C over 10 minutes then cooled to and incubated at 4 °C for 30 minutes. The DNA tetrahedron were then concentrated by ultracentrifugation using 30K MWCO Sartorious vivaspin 500 centrifugal concentrators and re-suspended in a solution of Tris buffer (10 mM) and MgCl2 (5 mM) at pH 7.4.
Oligo-1: 5’-TATCACCAGGCAGTTGACAGTGTAGCAAGCTGTAATAGATGCGAGGGTCCAATAC-3’
Oligo-2: 5’-TCAACTGCCTGGTGATAAAACGACACTACGTGGGAATCTACTATGGCGGCTCTTC-3’
Oligo-3: 5’-TTCAGACTTAGGAATGTGCTTCCCACGTAGTGTCGTTTGTATTGGACCCTC GCAT-3’
Oligo-4: 5’-ACATTCCTAAGTCTGAAACATTACAGCTTGCTACACGAGAAGAGCCGCCATAGTA-3’
Self-assembly of 55 b.p linear DNA:
The linear 55 b.p DNA duplex was assembled by annealing Oligo-1 to its complementary strand (sequence showed below). Briefly, 2 μL of each ssDNA (50 μM stock solutions) were combined in 42 μL of an aqueous solution of Tris buffer (10 mM) and MgCl2 (5 mM) at pH 7.4. The oligomers were hybridized using the same thermal treatment used for the synthesis of the DNA tetrahedron. The resulting linear DNA was concentrated by ultracentrifugation using 30K MWCO Sartorious vivaspin 500 centrifugal filters.
Complementary Oligo-1 Strand: 5’-GTATTGGACCCTCGCATCTATTACAGCTTGCTACACTGT CAA CTG CCT GGT GATA-3’
Native agarose gel electrophoresis.
Oligomers and self-assembled DNA tetrahedron structures were separated using 2 % agarose gel in 1X TBE buffer at 100 V for 100 minutes in an ice bath. Gels were stained by soaking them for 30 minutes with gentil agitation in SYBR safe 1X. Gel images were captured using a Typhoon FLA 9000 Gel Imaging Scanner System from GE Healthcare.
AMF imaging.
Freshly cleaved mica was treated with 0.1% (v/v) APTES ((3-Aminopropyl) triethoxysilane) aqueous solution for 10 min, and then washed with 2 ml ultrapure water and dried under nitrogen gas. 20 μl of 2 μM DNA tetrahedron structure with 102 μM of metal complexes ([Ru(bpy)2(dppz)]2+ or [Tb-DOTAm-Phen]3+) in TRIS buffer 10 mM MgCl2 5 mM pH 7.38 was dropped onto the mica surface and incubated for 10 min before washed by ultra-pure water for 10s and dried under N2 for 5s. The sample was imaged with an Asylum Cypher AFM equipped with a silicon nitride DNP-S probe (Bruker) with a spring constant of 0.35 N/m in AC Molecule tapping mode. Images were recorded at 1.95 Hz and 512 samples per line.
Luminescence titrations.
The affinities of the metallointercalators for the DNA tetrahedron were determined by luminescence spectroscopy by monitoring the change in the luminescence intensity of [Ru(bpy)2(dppz)]2+ or [Tb-DOTAm-Phen]3+ (10 μM) upon gradual addition of concentrated DNA tetrahedron in Tris buffer (10 mM) and MgCl2 (5 mM) at pH 7.4 at 20 °C. Luminescence spectra were recorded 5 minutes after mixing so as to ensure that thermodynamic equilibrium was reached. Prior kinetic studies indicated that thermodynamic equilibrium is reached in < 5 min. Titrations with [Ru(bpy)2(dppz)]2+ were recorded as follow λexc = 440 nm, I = integrated emission from λem = 550 to 800 nm, slit width = 20 nm. Titrations with [Tb-DOTAm-Phen]3+ were recorded as follows λexc = 345 nm, I = integrated emission from λem = 450 to 650 nm, time delay = 0.1 ms, slit width = 5 nm. In both cases, I0 is the integrated luminescence of [Ru(bpy)2(dppz)]2+ or [Tb-DOTAm-Phen]3+ in the absence of DNA tetrahedron. Fitting of the titration data to the theoretical independent site model according to the methods of Vilar45 and Aldrich44 enables determination of both the number of binding sites and the binding constants of the metallointercalators with DNA.
Competition experiments were performed by monitoring the change in luminescence intensity of [Ru(bpy)2(dppz)]2+@DNA (10 μM Ru-complex, 20 μM DNA b.p) upon addition of increasing amounts of the competing metallointercalator [Tb-DOTAm-Phen]3+ or Tb-DOTA-Phen in Tris buffer (10 mM) and MgCl2 (5 mM) at pH 7.4, at 20 °C. Luminescence spectra were recorded 5 minutes after mixing. λexc = 440 nm, I = integrated emission from λem = 500 to 800 nm, slit width = 20 nm. In both cases, I0 is the integrated luminescence of [Ru(bpy)2(dppz)]2+@DNA before addition of the competing metallointercalator.
Stoichiometry of self-assemblies.
The stoichiometries of the host:guest self-assemblies formed with the DNA tetrahedron and the metallointercalators were determined by the method of continuous variation following published procedures.81 Equimolar stock solutions of DNA tetrahedron (10 μM base-pair) and the corresponding metallointercalator (10 μM) were prepared in Tris buffer (10 mM) with MgCl2 (5 mM) at pH 7.4. Samples were prepared by mixing different volumes of DNA tetrahedron stock solution and metallointercalator so as to vary the mole ratio of the metallointercalator between 0.0 and 1.0 while maintaining the total concentration of metallointercalator+DNA tetrahedron constant. Samples were incubated at room temperature for 30 minutes before collecting luminescence spectra to ensure that thermodynamic equilibrium was reached. Prior kinetic studies indicated that, for this system, thermodynamic equilibrium is reached in < 5 min.
Melting points of DNA and DNA assemblies.
The melting temperatures of the DNA tetrahedron and the host:guest DNA tetrahedron@metallointercalator self-assemblies were determined by monitoring the absorbance at 260 nm while increasing the temperature at a rate of 1 °C/min using solution of DNA tetrahedron (150 nM) in Tris buffer (10 mM) and MgCl2 (5 mM) at pH 7.4 in the presence and absence of metallointercalator (7.7 μM, which corresponds to 1 eq. of metal complex per 2 base-pairs). The melting temperatures were calculated from the first derivative of the resulting curve.
The melting temperatures of the DNA tetrahedron@metallointercalator self-assemblies were also determined by monitoring the luminescence intensity of the metallointercalator ([Ru(bpy)2(dppz)]2+: λexc = 440 nm, λem = 618 nm; [Tb-DOTAm-Phen]3+: λexc = 345 nm, λem = 545 nm, time delay = 0.1 ms) while increasing the temperature at a rate of 1 °C/min using solution of DNA tetrahedron (30 nM) in Tris buffer (10 mM) and MgCl2 (5 mM) at pH 7.4 in the presence of metallointercalator (1.53 μM, which corresponds to 1 eq. of metal complex per 2 base-pairs). The melting temperatures were calculated from the first derivative of the resulting I/I0 curve where I is the luminescence intensity of the metallointercalator at a given temperature and I0 is the luminescence intensity of the same metaloointercalator at 20 °C.
Cell Culture.
L6 rat myoblast cells were purchased from ATCC (ATCC CRL-1458). HEK293 cells were donated by The Institute for Therapeutics Discovery and Development from the University of Minnesota. L6 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin while HEK293 cells were cultured in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 ug/mL streptomycin. Cells were incubated at 37 °C with a 5% CO2 atmosphere and seeded into new flask prior to addition of treatments. All cell handling and protocols followed Biosafety Level 1 Procedures.
Incubation of DNA Structures.
Treatment metallointercalator@DNA tetrahedron assemblies and controls, [Ru(bpy)2(dppz)]2+@DNA, [Tb-DOTAm-Phen]3+@DNA, [Ru(bpy)2(dppz)]2+, [Tb-DOTAm-Phen]3+ were prepared in Tris 10 mM MgCl2 5 mM pH 7.4 buffer. Metallointercalators@DNA tetrahedron samples were prepared by adding 50 equivalents of metal complex to 1 equivalent of DNA nanostructure (i.e., 1 equivalent of metal complex per 2 base pairs) and allowing them to equilibrate at room temperature at least 15 minutes before adding to cells. L6 cells were seeded at 0.7 million cells per 25 cm2 flask in complete DMEM 25 hours before treatment. Next, treatment metallointercalator@DNA tetrahedron assemblies and controls were added to the respective culture flasks for a final concentration of 5 μM metal complex or 0.1 μM DNA structure. Cultures were then allowed to incubate for 4 hours at 37 °C. HEK293 cells, on the other hand, were seeded at 1.5 million cells per 25 cm2 flask in complete EMEM 24 hours before treatment. The next day treatment DNA assemblies and controls noted above were added to EMEM media for a final concentration of 5 μM metal complex or 0.1 μM DNA structure. The old media in the cultures was replaced with 5 mL of the treated media and allowed to incubate for 4 hours at 37 °C.
Transfection of DNA Structures in L6 Cells.
Lipofectamine 3000 was diluted in Opti-MEM following manufacture’s guidelines. Treatment metallointercalator@DNA tetrahedron assemblies and controls, [Ru(bpy)2(dppz)]2+@DNA, [Tb-DOTAm-Phen]3+@DNA, [Ru(bpy)2(dppz)]2+, [Tb-DOTAm-Phen]3+, and Tris 10 mM MgCl2 5 mM pH 7.4 buffer, were diluted in Opti-MEM. Equal volumes of the lipofectamine solution and treatment solutions were mixed to form the working solutions and allowed to incubate at room temperature for 10 mins. Media from the cell culture flasks was taken out and replaced with 4.35 mL of DMEM with no supplements. After the 10 min incubation, the working solutions were added to the cells for a final concentration of 5 μM metal complex or 0.1 μM DNA structure and incubated at 37 °C for 4 hours.
Transfection of DNA Structures in HEK293 Cells.
Cells were seeded at 1.5 million cells per 25 cm2 flask in complete EMEM 24 hours before the addition of treatments. Treatment complexes and controls as described above were diluted into Opti-MEM. Lipofectamine 3000 was diluted in Opti-MEM following manufacturer’s guidelines and incubated with treatment solutions for 10 mins at RT to create working solutions. Old media was removed from flasks and replaced with EMEM without supplements. Working solutions were added to the flasks for a final concentration of 4.1 μM metal complex or 0.08 μM DNA structure and placed back in the incubator at 37 °C for 4 hours.
Preparation of Cell Samples for Mass Cytometry.
After incubation time, cells were lifted using either Acutase or trypsin, counted on a Hausser Scientific Ultra Plane hemocytometer and spun down (600 x g for 10 mins for L6 or 130 x g for 6 mins for HEK293) at 4 °C. Pellets were resuspended in 1 mL phosphate buffer solution (PBS) with 0.5% Tween-20 (TPBS), spun down and resuspended in PBS to 1 million cells per mL and 1 mL was aliquoted for next steps. 1 μL of 5mM Cell-ID™ cisplatin was added to each tube, incubated for 1 min then 5 mL of Maxpar cell staining buffer (CSB) was added to neutralize the reaction. Cells were pelleted, the supernatant was remove then resuspended in 1 mL 4% formaldehyde and place on an end over end mixer for 30 mins at room temperature. Cells were pelleted and washed once in 1 mL CSB then resuspended to 1 million cells in 50 μL for L6 cells or 100 μL for HEK293 cells of PBS. 50 μL for L6 cells or 100 μL for HEK293 cells of 2 μM Sm-DDD was added to each sample, for a final concentration of 1 μM, and mixed end over end for 30 mins at room temperature. Cells were washed twice in 1 mL TPBS with a centrifuge speed of 800 x g for 6 mins for L6 cells or 400 x g for 6 mins for HEK-293 cells then brought up in 1 mL Maxpar Fix and Perm buffer and incubated on end over end mixer overnight. The following morning cells were washed once with 1 mL CSB and twice with Millipore water. The cell suspension was counted before the last spin down.
Mass Cytometry.
Cells were brought up in 1X EQ Four Element Calibration Bead beads in HPLC grade water to a concentration below 500,000 cells per mL. Samples were run through the CyTOF2 with noise reduction on and event length from 10-150. FCS files were normalized using the EQ beads with CyTOF software 6.7.1014. Further data processing and analysis was done using FlowJo 10.5.3 software.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge support from the National Science Foundation under award CHE-2107427. Portions of this work were conducted in the Minnesota Nano Center, which is supported by the National Science Foundation through the National Nano Coordinated Infrastructure Network (NNCI) under Award Number ECCS-2025124. ZM was supported by the NIH Chemical Biology Training Grant T32GM132029 and the NIH Functional Proteomics in Aging Training Grant 5T32AG029796-14. We also thank Kristen John for the generous donation of HEK-293 cells and valuable discussion.
Footnotes
Supporting Information
Characterization data including 1H NMR of metal complexes; HR-ESI-MS of ligands and metal complexes; HPLC chromatograms of metallointercalators; UV-visible spectra of metallointercalators; luminescence spectra of Tb-DOTA-Phen; Jobs plot of DNA tetrahedron responsive [Tb-DOTAm-Phen]3+; agarose gel electrophoresis of DNA tetrahedron; cytotoxicity data for [Tb-DOTAm-Phen]3+ and [Ru(bpy)2(dppz)]2+; DNA binding isotherms; melting curves of DNA tetrahedron and metallointercalator@DNA tetrahedron assemblies; mass cytometry data for the cell uptake experiments (PDF).
The authors declare no competing financial interest.
REFERENCES
- (1).Simoncelli S; Li Y; Cortés E; Maier SA Nanoscale Control of Molecular Self-Assembly Induced by Plasmonic Hot-Electron Dynamics. ACS Nano. 2018, 12, 2184–2192. [DOI] [PubMed] [Google Scholar]
- (2).Whitesides GM; Mathias JP; Seto CT Molecular Self-Assembly and Nanochemistry: A Chemical Strategy for the Synthesis of Nanostructures. Science. 1991, 254, 1312–1319. [DOI] [PubMed] [Google Scholar]
- (3).Ariga K; Nishikawa M; Mori T; Takeya J; Shrestha LK; Hill JP Self-Assembly As a Key Player for Materials Nanoarchitectonics. Sci. Technol. Adv. Mater 2019, 20, 51–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhang S Fabrication of Novel Biomaterials Through Molecular Self-Assembly. Nat. Biotechnol 2003, 21, 1171–1178. [DOI] [PubMed] [Google Scholar]
- (5).Tabacchi G Supramolecular Organization in Confined Nanospaces. ChemPhysChem. 2018, 19, 1249–1297. [DOI] [PubMed] [Google Scholar]
- (6).Woller JG; Hannestad JK; Albinsson B Self-Assembled Nanoscale DNA–Porphyrin Complex for Artificial Light Harvesting. J. Am. Chem. Soc 2013, 135, 2759–2768. [DOI] [PubMed] [Google Scholar]
- (7).Duan P; Yanai N; Nagatomi H; Kimizuka N Photon Upconversion in Supramolecular Gel Matrixes: Spontaneous Accumulation of Light-Harvesting Donor–Acceptor Arrays in Nanofibers and Acquired Air Stability. J. Am. Chem. Soc 2015, 137, 1887–1894. [DOI] [PubMed] [Google Scholar]
- (8).Madsen M; Gothelf KV Chemistries for DNA Nanotechnology. Chem. Rev 2019, 119, 6384–6458. [DOI] [PubMed] [Google Scholar]
- (9).Seeman NC; Sleiman HF DNA Nanotechnology. Nat. Rev. Mater 2017, 3, 17068. [Google Scholar]
- (10).McLaughlin CK; Hamblin GD; Sleiman HF Supramolecular DNA Assembly. Chem. Soc. Rev 2011, 40, 5647–5656. [DOI] [PubMed] [Google Scholar]
- (11).Pathak P; Yao W; Hook KD; Vik R; Winnerdy FR; Brown JQ; Gibb BC; Pursell ZF; Phan AT; Jayawickramarajah J Bright G-Quadruplex Nanostructures Functionalized with Porphyrin Lanterns. J. Am. Chem. Soc 2019, 141, 12582–12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yang H; Sleiman HF Templated Synthesis of Highly Stable, Electroactive, and Dynamic Metal–DNA Branched Junctions. Angew. Chemie Int. Ed 2008, 47, 2443–2446. [DOI] [PubMed] [Google Scholar]
- (13).Gothelf KV; Thomsen A; Nielsen M; Cló E; Brown RS Modular DNA-Programmed Assembly of Linear and Branched Conjugated Nanostructures. J. Am. Chem. Soc 2004, 126, 1044–1046. [DOI] [PubMed] [Google Scholar]
- (14).Yang H; McLaughlin CK; Aldaye FA; Hamblin GD; Rys AZ; Rouiller I; Sleiman HF Metal–Nucleic Acid Cages. Nat. Chem 2009, 1, 390–396. [DOI] [PubMed] [Google Scholar]
- (15).Engelhard DM; Meyer A; Berndhäuser A; Schiemann O; Clever GH Di-Copper(II) DNA G-Quadruplexes As EPR Distance Rulers. Chem. Commun 2018, 54, 7455–7458. [DOI] [PubMed] [Google Scholar]
- (16).Zeglis BM; Pierre VC; Barton JK Metallo-Intercalators and Metallo-Insertors. Chem. Commun 2007, 4565–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sánchez MI; Rama G; Calo-Lapido R; Ucar K; Lincoln P; López MV; Melle-Franco M; Mascareñas JL; Vázquez ME Canonical DNA Minor Groove Insertion of Bisbenzamidine–Ru(II) Complexes with Chiral Selectivity. Chem. Sci 2019, 10, 8668–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Holmlin RE; Stemp EDA; Barton JK Ru(phen)2dppz2+ Luminescence: Dependence on DNA Sequences and Groove-Binding Agents. Inorg. Chem 1998, 37, 29–34. [DOI] [PubMed] [Google Scholar]
- (19).Molphy Z; Montagner D; Bhat SS; Slator C; Long C; Erxleben A; Kellett A A Phosphate-Targeted Dinuclear Cu(II) Complex Combining Major Groove Binding and Oxidative DNA Cleavage. Nucleic Acids Res. 2018, 46, 9918–9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Erkkila KE; Odom DT; Barton JK Recognition and Reaction of Metallointercalators with DNA. Chem. Rev 1999, 99, 2777–2796. [DOI] [PubMed] [Google Scholar]
- (21).Sitlani A; Long EC; Pyle AM; Barton JK DNA Photocleavage by Phenanthrenequinone Diimine Complexes of Rhodium(III): Shape-Selective Recognition and Reaction. J. Am. Chem. Soc 1992, 114, 2303–2312. [Google Scholar]
- (22).Pierre VC; Kaiser JT; Barton JK Insights Into Finding a Mismatch Through the Structure of a Mispaired DNA Bound by a Rhodium Intercalator. Proc. Natl. Acad. Sci 2007, 104, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cordier C; Pierre VC; Barton JK Insertion of a Bulky Rhodium Complex Into a DNA Cytosine–Cytosine Mismatch: An NMR Solution Study. J. Am. Chem. Soc 2007, 129, 12287–12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hooper CAJ; Cardo L; Craig JS; Melidis L; Garai A; Egan RT; Sadovnikova V; Burkert F; Male L; Hodges NJ; Browning DF; Rosas R; Liu F; Rocha FV; Lima MA; Liu S; Bardelang D; Hannon MJ Rotaxanating Metallo-Supramolecular Nano-Cylinder Helicates to Switch DNA Junction Binding. J. Am. Chem. Soc 2020, 142, 20651–20660. [DOI] [PubMed] [Google Scholar]
- (25).Oleksi A; Blanco AG; Boer R; Usón I; Aymamí J; Rodger A; Hannon MJ; Coll M Molecular Recognition of a Three-Way DNA Junction by a Metallosupramolecular Helicate. Angew. Chemie Int. Ed 2006, 45, 1227–1231. [DOI] [PubMed] [Google Scholar]
- (26).Hannon MJ; Moreno V; Prieto MJ; Moldrheim E; Sletten E; Meistermann I; Isaac CJ; Sanders KJ; Rodger A Intramolecular DNA Coiling Mediated by a Metallo-Supramolecular Cylinder. Angew. Chemie Int. Ed 2001, 40, 879–884. [DOI] [PubMed] [Google Scholar]
- (27).Zhao C; Wu L; Ren J; Xu Y; Qu X Targeting Human Telomeric Higher-Order DNA: Dimeric G-Quadruplex Units Serve As Preferred Binding Site. J. Am. Chem. Soc 2013, 135, 18786–18789. [DOI] [PubMed] [Google Scholar]
- (28).Campbell NH; Karim NHA; Parkinson GN; Gunaratnam M; Petrucci V; Todd AK; Vilar R; Neidle S Molecular Basis of Structure–Activity Relationships Between Salphen Metal Complexes and Human Telomeric DNA Quadruplexes. J. Med. Chem 2012, 55, 209–222. [DOI] [PubMed] [Google Scholar]
- (29).Wilson T; Costa PJ; Félix V; Williamson MP; Thomas JA Structural Studies on Dinuclear Ruthenium(II) Complexes That Bind Diastereoselectively to an Antiparallel Folded Human Telomere Sequence. J. Med. Chem 2013, 56, 8674–8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Song H; Kaiser JT; Barton JK Crystal Structure of Δ-[Ru(bpy)2dppz]2+ Bound to Mismatched DNA Reveals Side-by-Side Metalloinsertion and Intercalation. Nat. Chem 2012, 4, 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Smolensky ED; Peterson KL; Weitz EA; Lewandowski C; Pierre VC Magnetoluminescent Light Switches – Dual Modality in DNA Detection. J. Am. Chem. Soc 2013, 135, 8966–8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Goodman RP; Berry RM; Turberfield AJ The Single-Step Synthesis of a DNA Tetrahedron. Chem. Commun 2004, 1372–1373. [DOI] [PubMed] [Google Scholar]
- (33).Jia R; He X; Ma W; Lei Y; Cheng H; Sun H; Huang J; Wang K Aptamer-Functionalized Activatable DNA Tetrahedron Nanoprobe for PIWI-Interacting RNA Imaging and Regulating in Cancer Cells. Anal. Chem 2019, 91, 15107–15113. [DOI] [PubMed] [Google Scholar]
- (34).Ge Z; Gu H; Li Q; Fan C Concept and Development of Framework Nucleic Acids. J. Am. Chem. Soc 2018, 140, 17808–17819. [DOI] [PubMed] [Google Scholar]
- (35).He L; Lu D; Liang H; Xie S; Zhang X; Liu Q; Yuan Q; Tan W mRNA-Initiated, Three-Dimensional DNA Amplifier Able to Function Inside Living Cells. J. Am. Chem. Soc 2018, 140, 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Keum J-W; Bermudez H Enhanced Resistance of DNA Nanostructures to Enzymatic Digestion. Chem. Commun 2009, 7036–7038. [DOI] [PubMed] [Google Scholar]
- (37).Friedman AE; Chambron JC; Sauvage JP; Turro NJ; Barton JK A Molecular Light Switch for DNA: Ru(bpy)2(dppz)2+. J. Am. Chem. Soc 1990, 112, 4960–4962. [Google Scholar]
- (38).Puckett CA; Barton JK Methods to Explore Cellular Uptake of Ruthenium Complexes. J. Am. Chem. Soc 2007, 129, 46–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lim MH; Song H; Olmon ED; Dervan EE; Barton JK Sensitivity of Ru(bpy)2dppz2+ Luminescence to DNA Defects. Inorg. Chem 2009, 48, 5392–5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Brennaman MK; Alstrum-Acevedo JH; Fleming CN; Jang P; Meyer TJ; Papanikolas JM Turning the [Ru(bpy)2dppz]2+ Light-Switch On and Off with Temperature. J. Am. Chem. Soc 2002, 124, 15094–15098. [DOI] [PubMed] [Google Scholar]
- (41).McConnell AJ; Lim MH; Olmon ED; Song H; Dervan EE; Barton JK Luminescent Properties of Ruthenium(II) Complexes with Sterically Expansive Ligands Bound to DNA Defects. Inorg. Chem 2012, 51, 12511–12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Thibon A; Pierre VC Principles of Responsive Lanthanide-Based Luminescent Probes for Cellular Imaging. Anal. Bioanal. Chem 2009, 394, 107–120. [DOI] [PubMed] [Google Scholar]
- (43).Bobba G; Dickins RS; Kean SD; Mathieu CE; Parker D; Peacock RD; Siligardi G; Smith MJ; Gareth Williams JA; Geraldes CFGC Chiroptical, ESMS and NMR Spectroscopic Study of the Interaction of Enantiopure Lanthanide Complexes with Selected Self-Complementary Dodecamer Oligonucleotides. J. Chem. Soc. Perkin Trans 2 2001, 1729–1737. [Google Scholar]
- (44).Stootman FH; Fisher DM; Rodger A; Aldrich-Wright JR Improved Curve Fitting Procedures to Determine Equilibrium Binding Constants. Analyst. 2006, 131, 1145–1151. [DOI] [PubMed] [Google Scholar]
- (45).Bandeira S; Gonzalez-Garcia J; Pensa E; Albrecht T; Vilar R A Redox-Activated G-Quadruplex DNA Binder Based on a Platinum(IV)–Salphen Complex. Angew. Chemie Int. Ed 2018, 57, 310–313. [DOI] [PubMed] [Google Scholar]
- (46).Garbett NC; Hammond NB; Graves DE Influence of the Amino Substituents in the Interaction of Ethidium Bromide with DNA. Biophys. J 2004, 87, 3974–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Özhalıcı-Ünal H; Armitage BA Fluorescent DNA Nanotags Based On a Self-Assembled DNA Tetrahedron. ACS Nano. 2009, 3, 425–433. [DOI] [PubMed] [Google Scholar]
- (48).Weitz EA; Chang JY; Rosenfield AH; Pierre VC A Selective Luminescent Probe for the Direct Time-Gated Detection of Adenosine Triphosphate. J. Am. Chem. Soc 2012, 134, 16099–16102. [DOI] [PubMed] [Google Scholar]
- (49).Weitz EA; Chang JY; Rosenfield AH; Morrow EA; Pierre VC The Basis for the Molecular Recognition and the Selective Time-Gated Luminescence Detection of ATP and GTP by a Lanthanide Complex. Chem. Sci 2013, 4, 4052–4060. [Google Scholar]
- (50).Wilson WD; Tanious F; Fernandez-Saiz M; Rigl T Evaluation of Drug-Nucleic Acid Interactions by Thermal Melting Curves. In Drug-DNA Interaction Protocols; Fox K, Ed.; Humana Press: Totowa, New Jersey, 1997; pp 219–240. [DOI] [PubMed] [Google Scholar]
- (51).Kim K-R; Jegal H; Kim J; Ahn D-R A Self-Assembled DNA Tetrahedron As a Carrier for In Vivo Liver-Specific Delivery of siRNA. Biomater. Sci 2020, 8, 586–590. [DOI] [PubMed] [Google Scholar]
- (52).Han X; Jiang Y; Li S; Zhang Y; Ma X; Wu Z; Wu Z; Qi X Multivalent Aptamer-Modified Tetrahedral DNA Nanocage Demonstrates High Selectivity and Safety for Anti-Tumor Therapy. Nanoscale. 2019, 11, 339–347. [DOI] [PubMed] [Google Scholar]
- (53).He Y; Ye T; Su M; Zhang C; Ribbe AE; Jiang W; Mao C Hierarchical Self-Assembly of DNA Into Symmetric Supramolecular Polyhedra. Nature. 2008, 452, 198–201. [DOI] [PubMed] [Google Scholar]
- (54).Chandrasekaran AR Nuclease Resistance of DNA Nanostructures. Nat. Rev. Chem 2021, 5, 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Hahn J; Wickham SFJ; Shih WM; Perrault SD Addressing the Instability of DNA Nanostructures in Tissue Culture. ACS Nano. 2014, 8, 8765–8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Dey S; Fan C; Gothelf KV; Li J; Lin C; Liu L; Liu N; Nijenhuis MAD; Saccà B; Simmel FC; Yan H; Zhan P DNA Origami. Nat. Rev. Methods Prim 2021, 1, 13. [Google Scholar]
- (57).Conway JW; McLaughlin CK; Castor KJ; Sleiman H DNA Nanostructure Serum Stability: Greater Than the Sum of Its Parts. Chem. Commun 2013, 49, 1172–1174. [DOI] [PubMed] [Google Scholar]
- (58).Wang S-T; Gray MA; Xuan S; Lin Y; Byrnes J; Nguyen AI; Todorova N; Stevens MM; Bertozzi CR; Zuckermann RN; Gang O DNA Origami Protection and Molecular Interfacing Through Engineered Sequence-Defined Peptoids. Proc. Natl. Acad. Sci 2020, 117, 6339 – 6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Auvinen H; Zhang H; Nonappa; Kopilow A; Niemelä EH; Nummelin S; Correia A; Santos HA; Linko V; Kostiainen MA Protein Coating of DNA Nanostructures for Enhanced Stability and Immunocompatibility. Adv. Healthcare Mater 2017, 6, 1700692. [DOI] [PubMed] [Google Scholar]
- (60).Ahmadi Y; De Llano E; Barišić I (Poly)Cation-Induced Protection of Conventional and Wireframe DNA Origami Nanostructures. Nanoscale. 2018, 10, 7494–7504. [DOI] [PubMed] [Google Scholar]
- (61).Ponnuswamy N; Bastings MMC; Nathwani B; Ryu JH; Chou LYT; Vinther M; Li WA; Anastassacos FM; Mooney DJ; Shih WM Oligolysine-Based Coating Protects DNA Nanostructures from Low-Salt Denaturation and Nuclease Degradation. Nat. Commun 2017, 8, 15654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Keum J-W; Ahn J-H; Bermudez H Design, Assembly, and Activity of Antisense DNA Nanostructures. Small. 2011, 7, 3529–3535. [DOI] [PubMed] [Google Scholar]
- (63).Lacroix A; Vengut-Climent E; de Rochambeau D; Sleiman HF Uptake and Fate of Fluorescently Labeled DNA Nanostructures in Cellular Environments: A Cautionary Tale. ACS Cent. Sci 2019, 5, 882–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Charoenphol P; Bermudez H Aptamer-Targeted DNA Nanostructures for Therapeutic Delivery. Mol. Pharmaceutics. 2014, 11, 1721–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Green CM; Mathur D; Medintz IL Understanding the Fate of DNA Nanostructures Inside the Cell. J. Mater. Chem. B 2020, 8, 6170–6178. [DOI] [PubMed] [Google Scholar]
- (66).Napirei M; Ludwig S; Mezrhab J; Klöckl T; Mannherz HG Murine Serum Nucleases – Contrasting Effects of Plasmin and Heparin on the Activities of DNase1 and DNase1-like 3 (DNase1l3). FEBS J 2009, 276, 1059–1073. [DOI] [PubMed] [Google Scholar]
- (67).Napirei M; Ricken A; Eulitz D; Knoop H; Mannherz HG Expression Pattern of the Deoxyribonuclease 1 Gene: Lessons from the Dnase1 Knockout Mouse. Biochem. J 2004, 380, 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Suck D DNA Recognition by DNase I. J. Mol. Recognit 1994, 7, 65–70. [DOI] [PubMed] [Google Scholar]
- (69).Li G; Sun L; Ji L; Chao H Ruthenium(II) Complexes with dppz: From Molecular Photoswitch to Biological Applications. Dalt. Trans 2016, 45, 13261–13276. [DOI] [PubMed] [Google Scholar]
- (70).Wamhoff E-C; Huang H; Read BJ; Ginsburg E; Schief WR; Farrell N; Irvine DJ; Bathe M Controlling Wireframe DNA Origami Nuclease Degradation with Minor Groove Binders. 2020, 2020.05.24.110783. bioRxiv 10.1101/2020.05.24.110783 (accessed January 18th, 2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Bastings MMC; Anastassacos FM; Ponnuswamy N; Leifer FG; Cuneo G; Lin C; Ingber DE; Ryu JH; Shih WM Modulation of the Cellular Uptake of DNA Origami Through Control Over Mass and Shape. Nano Lett. 2018, 18, 3557–3564. [DOI] [PubMed] [Google Scholar]
- (72).Wang P; Rahman MA; Zhao Z; Weiss K; Zhang C; Chen Z; Hurwitz SJ; Chen ZG; Shin DM; Ke Y Visualization of the Cellular Uptake and Trafficking of DNA Origami Nanostructures in Cancer Cells. J. Am. Chem. Soc 2018, 140, 2478–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Peterson KL; Dang JV; Weitz EA; Lewandowski C; Pierre VC Effect of Lanthanide Complex Structure on Cell Viability and Association. Inorg. Chem 2014, 53, 6013–6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Walsh AS; Yin H; Erben CM; Wood MJA; Turberfield AJ DNA Cage Delivery to Mammalian Cells. ACS Nano. 2011, 5, 5427–5432. [DOI] [PubMed] [Google Scholar]
- (75).Xia Z; Wang P; Liu X; Liu T; Yan Y; Yan J; Zhong J; Sun G; He D Tumor-Penetrating Peptide-Modified DNA Tetrahedron for Targeting Drug Delivery. Biochemistry. 2016, 55, 1326–1331. [DOI] [PubMed] [Google Scholar]
- (76).Son KK; Patel DH; Tkach D; Park A Cationic Liposome and Plasmid DNA Complexes Formed in Serum-Free Medium Under Optimum Transfection Condition Are Negatively Charged. Biochim. Biophys. Acta - Biomembr 2000, 1466, 11–15. [DOI] [PubMed] [Google Scholar]
- (77).Weitz EA; Chang JY; Rosenfield AH; Pierre VC A Selective Luminescent Probe for the Direct Time-Gated Detection of Adenosine Triphosphate. J. Am. Chem. Soc 2012, 134, 16099–16102. [DOI] [PubMed] [Google Scholar]
- (78).Cook NP; Ozbil M; Katsampes C; Prabhakar R; Martí AA Unraveling the Photoluminescence Response of Light-Switching Ruthenium(II) Complexes Bound to Amyloid-β. J. Am. Chem. Soc 2013, 135, 10810–10816. [DOI] [PubMed] [Google Scholar]
- (79).Leipold MD; Ornatsky O; Baranov V; Whitfield C; Nitz M Development of Mass Cytometry Methods for Bacterial Discrimination. Anal. Biochem 2011, 419, 1–8. [DOI] [PubMed] [Google Scholar]
- (80).Brown HMG; Arriaga EA Quantifying Heterogeneity of Individual Organelles in Mixed Populations via Mass Cytometry. Anal. Chem 2018, 90, 13315–13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Brglez J; Nikolov P; Angelin A; Niemeyer CM Designed Intercalators for Modification of DNA Origami Surface Properties. Chem. – Eur. J 2015, 21, 9440–9446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.