

Abstract
TLD1433 is the first ruthenium (Ru)-based photodynamic therapy (PDT) agent to advance to clinical trials and is currently in a Phase II study for treating non-muscle bladder cancer with PDT. Herein, we present a photophysical study of TLD1433 and its derivative TLD1633 using complex, biologically relevant solvents to elucidate the excited state properties that are key for biological activity. The complexes incorporate an imidazo [4,5-f][1,10]phenanthroline (IP) ligand appended to α-ter- or quaterthiophene, respectively, where TLD1433 = [Ru(4,4′-dmb)2(IP-3T)]Cl2 and TLD1633 = [Ru(4,4′-dmb)2(IP-4T)]Cl2 (4,4′-dmb=4,4′-dimethyl-2,2′-bipyridine; 3T=α-terthiophene; 4T= α-quaterthiophene). Time-resolved transient absorption experiments demonstrate that the excited state dynamics of the complexes change upon interaction with biological macromolecules (e.g., DNA). In this case, the accessibility of the lowest energy T1 state is increased at the expense of a higher-lying triplet intraligand charge transfer (3ILCT) state. We attribute this behavior to the increased rigidity of the ligand framework upon binding to DNA, which prolongs the lifetime of the T1 state. This lowest lying state is primarily responsible for O2 sensitization and hence photoinduced cytotoxicity. Therefore, in order to gain a realistic picture of the excited-state kinetics that underlie the photoinduced function of the complexes, it is necessary to interrogate their photophysical dynamics in the presence of biological targets once they are known.
Keywords: TLD1433, photodynamic therapy, ct DNA, excited state relaxation, long-lived triplet states
Graphical Abstract

INTRODUCTION
Photodynamic therapy (PDT) uses light to activate an otherwise nontoxic prodrug in the presence of oxygen to selectively destroy tumors and tumor vasculature, and is even capable of inducing an antitumor immune response.1–5 The PDT reaction that results in damage to biomolecular targets is mediated by reactive oxygen species (ROS) formed via electron (Type I) or energy (Type II) transfer pathways between an excited state photosensitizer (PS) and oxygen. Singlet oxygen (1O2) is thought to be the most important ROS for the PDT effect.
Photofrin was the first PS approved for PDT,6 and most subsequent-generation PSs are based on tetrapyrrolic structures.7 These systems generate cytotoxic 1O2 from 3ππ* excited states. There has been ongoing interest in using transition metal complexes as PSs for PDT in order to improve upon earlier PSs. The structural modularity of transition metal complexes makes it relatively straightforward to tune their chemical and photophysical properties. This has been particularly well-documented for Ru(II) polypyridyl complexes, where a variety of excited states can be accessed by combining the appropriate ligands around the metal8–13: metal-to-ligand charge transfer (MLCT), metal-centered (MC), intraligand (IL), intraligand charge transfer (ILCT), and ligand-to- ligand charge transfer (LLCT).
The photophysical properties of transition metal complexes are generally understood in the context of the archetype [Ru(bpy)3]2+ complex, whereby the excited state dynamics tend to be controlled by the lowest-energy 3MLCT state.14,15 Light absorption first populates singlet states that undergo rapid intersystem crossing to form the lowest-lying 3MLCT state. This state decays with a lifetime of approximately 1 μs in deoxygenated fluid solution16 and 200 ns in the presence of oxygen.17 Much work has been done to extend the triplet lifetimes in these systems for applications that rely on charge separation,18–25 and prolonged lifetimes are likewise advantageous for enhancing 1O2 quantum yields in the area of PS design for PDT. One strategy involves creating bichromophoric systems that spatially isolate a pendant π-expanded organic chromophore from the Ru(II) center.18–25 The resulting Ru metal-organic dyads have additional excited states that are localized to the organic chromophore, including triplet intraligand (3IL) states of significant 3ππ* character. Such states often have long μs lifetimes owing to the reduced intersystem crossing (ISC) rates associated with organic triplets. The 3IL states can thus prolong the lifetimes of 3MLCT states if they are relatively close in energy. If the 3IL energy is substantially lower, then the triplet lifetimes can be even longer and exhibit characteristics of pure 3ππ* states.22 Such states in Ru(II) dyads can increase the 1O2 quantum yields from near 56% for [Ru(bpy)3]2+ to almost unity. We have a longstanding interest in exploiting such systems for PDT applications since the prolonged lifetimes and improved 1O2 yields correlate with better photocytotoxicity.26,27
Studies of the fundamental photophysical properties these Ru PSs (and related metal complexes) for PDT necessarily begin with understanding the excited state dynamics in simple solvents. However, ultimately, we want to unravel these dynamics in the complex cellular environment where factors such as pH, ionic strength, and interactions with biomolecules and cellular targets come into play and may substantially alter the photophysical properties. We have begun to undertake these more complicated studies28,29 in an effort to elucidate the mechanism of action of our TLD1433, which is the only Ru-based PS to advance to clinical trials.30,31
Previous studies on a similar complex, bearing an imidazo [4,5-f][1,10]phenanthroline (IP) ligand appended to a pyrenyl substituent at C2 indicate that deprotonation of the imidazo moiety significantly alters the excited-state dynamics compared to the neutral or protonated species. This behavior is likely related to an increase in the energy gap between 3MLCT and 3IL states upon deprotonation of the ligand.28 We also found that intercalation of this complex into salmon sperm (SS) DNA altered the excited state dynamics by increasing the rate of coplanarization (between the imidizo and pyrene groups) compared to water as solvent.29
Herein we probe the effects of biologically relevant environments on the photophysical dynamics of two Ru dyads that incorporate an IP ligand appended to α-terthiophene (3T) or α-quaterthiophene (4T). Both TLD1433 (Ru-ip-3T) and TLD1633 (Ru-ip-4T) contain two 4,4′-dimethyl-2,2′-bipyridine (4,4′-dmb) as coligands and chloride as the counter ion: [Ru(4,4′-dmb)2(IP-3T)]Cl2 and [Ru(4,4′-dmb)2(IP-4T)]Cl2 (Figure 1). These two compounds are exceptional in vitro PDT agents toward cancer cells as demonstrated previously with the SK-MEL-28 melanoma cell line. The additional thienyl ring significantly impacted the photocytotoxicity, decreasing the EC50 value from 1.9×10−4 μM for Ru-ip-3T to 2.8×10−9 μM for Ru-ip-4T.30 Spectroscopic studies in simple organic solvents and water revealed a complex interplay between 3MLCT and 3ILCT states. The 3ILCT state was localized on the oligothiophene chain and dominated the overall photophysics of the complexes.32 In water the long-lived triplet states were predominantly formed by vibrational cooling of hot 3ILCT states, while the initial excitation populates a mixture of 1MLCT and 1ILCT states.33 With increasing oligothiophene chain length, the lifetime of this state was extended into the range of a few tens of μs.32 This study probes the effect of DNA-binding (although DNA is not the suggested biological target) and SK-MEL-28 cellular lysates30,34,35 on these dynamics to underscore the importance of considering nonspecific biomolecular interactions that may influence the electronics and/or conformation of PSs when assessing the relevant photophysical mechanism(s) for photocytotoxicity.
Figure 1.

Chemical structures of (a) TLD1433, [Ru(4,4’-dmb)2(IP-3T)]2+ (Ru-ip-3T) and (b) TLD1633, [Ru(4,4’-dmb)2(IP-4T)]2+ (Ru-ip-4T). 4,4’-dmb=4,4’-dimethyl-2,2’-bipyridine; IP=imidazo [4,5-f][9,10] phenanthroline; 3T=α-terthiophene; 4T=α-quaterthiophene.
METHODS
ct DNA preparation.
ct DNA was obtained from Sigma Aldrich (Merck) and reconstituted in Tris NaCl/HCl buffer (50 mM/5 mM, pH 7.4), sonicated, and frozen until further use. For all measurements in ct DNA, a fixed [DNA]bp / [complex] was used. The concentration of the ct DNA was determined from the UV-vis absorption signal of the ct DNA at 260 nm using OD = εcl, where εbp = 13 200 M−1 cm−1 (Figure S1).
Lysate preparation.
Lysate was prepared from SK-MEL-28 cells at 90% confluence. The cells were detached using 5% trypsin-EDTA, pelleted with centrifugation, and then dissolved in PBS buffer commercially purchased from Merck (1X, without calcium and magnesium, pH 7.0–7.2) with several rounds of ultra-sonication at 30% power. Following sonication the concentrated lysate solution was diluted to 3 mL using Tris NaCl/HCl buffer (50 mM/5 mM, pH 7.4) and added to 50 μL of the complexes. (Figure S1).
Steady state UV-vis absorption and emission spectroscopy.
UV-vis absorption spectra were measured on a Jasco-V760 spectrophotometer in 1 cm quartz cell. Emission spectroscopy was carried out using a FLS980 spectrometer (Edinburgh Instruments) with a spectral resolution in the excitation channel of Δλex.=3 nm and Δλem.=4 nm in the emission channel. The OD of the sample was adjusted to 0.05 at the excitation wavelength for emission spectroscopy.
Time resolved spectroscopy.
The setup for femtosecond time resolved spectroscopy in solution has been described elsewhere.36,37 The maximum delay time window is 2 ns and the samples are excited at 400 nm using the second harmonic of the laser fundamental. Residual light from the fundamental was focused into a CaF2 plate to generate the white light probe. The mutual polarization between pump and probe was set to the magic angle (54.7°). To avoid contributions from coherent artifacts38,39 in the data analysis, data recorded at delay times shorter than 300 fs were omitted from the multi-exponential analysis.
For nanosecond transient absorption experiments, the 410 nm pump pulses are generated from an OPO pumped by the 10 Hz output of a NdYAG laser with a time resolution of 10 ns. The probe light is generated from an arc lamp, and the probe wavelength is stepped in 10 nm increments from 350 to 800 nm. The transient absorption signals are detected by a commercially available system from Princeton Instruments.
Samples.
For ns-TA the OD at the excitation wavelength was set to 0.25 in a 1 cm quartz cell. The nanosecond transient absorption setup was also used for time-resolved emission measurements with the probe light blocked so that only the spontaneous emission was detected at 90° with respect to the pump beam. All experiments were carried out in duplicate.
For fs-TA measurements the OD of the sample in a 1 mm quartz cuvette was adjusted to 0.25 at the pump wavelength except for measurements in the presence of ct DNA and cellular lysate where the OD was adjusted to 0.25 in a 1 cm quartz cell to achieve the desired [DNA]bp/[complex] ratio. When attempting to reach the desired high concentration of ct DNA in a 1 mm cell, precipitation of the DNA was observed. All experiments were performed in duplicate.
Data analysis procedure for femtosecond transient absorption.
The transient absorption data was analyzed by fitting a kinetic model of the form:
In this analysis the probe-wavelength dependent coefficients Ai correspond to the decay-associated spectra (DAS), which are associated with specific characteristic first-order decay constants τi. Any component with a lifetime that exceeds the range of experimentally accessible delay times is summarized in the “Inf.” spectrum in our fit. Thus, the corresponding DAS can be understood as the difference spectrum between the long-lived excited state and the ground state to which it relaxes.
RESULTS AND DISCUSSION
Steady state absorption and emission spectroscopy.
The UV-vis absorption spectrum of Ru-ip-3T (Figure 2a) in water has two major transitions in the visible region, occuring at 420 and 480 nm. The former is assigned to 1ILCT transitions while the latter corresponds to 1MLCT transitions.32 When Ru-ip-3T is mixed with ct DNA, these bands shift bathochromically by 5 nm (Figure 2a). Such spectral shifts are common for binding of Ru(II) complexes to ct DNA,29,40 and similar shifts are observed for Ru-ip-4T (Figure S4). An earlier investigation we carried out on the related [Ru(bpy)2(IP-3T)]2+ complex with ct DNA showed a strong interaction characterized by a binding constant (Kb) on the order of ~108. Since the ionic strength and pH of buffer used in this study is identical to our previous study,41 we anticipate similar Kb values for structurally similar complexes such as [Ru(dmb)2(IP-3T)]2+ and [Ru(bpy)2(IP-3T)]2+. Upon interaction of these complexes with DNA, the long-wavelength absorption shoulder becomes more prominent which could prove convenient for inducing red-light activation of the complex for PDT.42–44 Identical shifts are observed for Ru-ip-3T (Figure 2a) and Ru-ip-4T (Figure S6) in the presence of SK-MEL-28 lysate.
Figure 2.

(a) Steady state UV-vis absorption spectra of Ru-ip-3T in water32, ct DNA, and lysate of SK-MEL-28 cells. (b) Emission of Ru-ip-3T in water and ct DNA at excitation wavelength of 450 nm. (c) ns-TA of Ru-ip-3T in ct DNA recorded at selected delay times upon excitation at 410 nm. (d) Ground state recovery lifetimes (τTA) associated with T1 state of Ru-ip-3T in water32, ct DNA and lysates of SK-MEL-28 cell under aerated conditions. A [DNA]bp/[complex] of 5:1 was used for all measurements in ct DNA solutions. Both ct DNA and SK-MEL-28 lysate solutions were prepared in Tris-HCl / NaCl (5 mM / 50 mM, pH 7.4) buffer. The kinetics from 630–680 nm were averaged to obtain the resultant kinetics and fitted to obtain the corresponding lifetimes. Note: For data analysis in Figure 2c, the contribution by emission is subtracted from the experimental data in the spectral range between 630 and 680 nm, thereby highlighting the features of the excited-state absorption spectrum (rather than the transient absorption spectrum, which of course contains information on emission). Experimentally, a transient absorption measurement is performed first. In order to account for the contribution by emission from this differential absorption kinetics, an additional measurement is conducted exclusively in only ‘pump on’ mode. This process allows us to separately collect the emission kinetics which is calibrated accordingly to ΔAbs. unit and subtracted from the differential absorption kinetics.
The weak 3MLCT emission from both Ru-ip-3T and Ru-ip-4T is centered at 640 nm (upon excitation at 450 nm)32 and does not shift in the presence of ct DNA. However, DNA enhances the emission by sixfold for Ru-ip-3T and by threefold for Ru-ip-4T (Figure 2b and Figure S4). This increased emission agrees with what has been previously observed for the structurally-related complex [Ru(bpy)2(IP-3T)]2+ bound to DNA41 and has been attributed to the ability of DNA to shield the complex and thereby reduce excited state quenching by water or dissolved oxygen.45,46 Unfortunately, large scattering effects precluded similar emission studies of Ru-ip-3T and Ru-ip-4T in SK-MEL-28 lysate solutions.
Nanosecond transient absorption and emission spectroscopy.
As we previously reported,32 the transient absorption (TA) spectrum obtained for Ru-ip-3T in water reveals an excited-state absorption (ESA) band at 640 nm, which was assigned to the lowest energy 3ILCT state (referred to herein as the T1 state)32 that decays with a lifetime of 1.4 μs in aerated solution (Figure 2d). In addition to the T1 decay, an emissive 3MLCT state decays with a lifetime of 0.3 μs. The distinct lifetimes for T1 and the 3MLCT states indicate that they are not electronically coupled.
The nanosecond transient absorption spectra recorded for Ru-ip-3T in aqueous solution containing ct DNA solution show ESA features identical to those obtained in water. A broad ESA with a maximum at 640 nm represents absorption associated with the T1 state, while a GSB occurs below 500 nm. The positive differential absorption band at 640 nm, reaching a maximum at 3.5 μs with a rise time constant of 0.5 μs (Figure 2c), results from overlap of the ESA with the 3MLCT emission that yields a negative contribution to the differential absorption signal and decays on the sub-μs timescale. T1 decays bi-exponentially with time constants of 4 (50%) and 10 μs (50%) as reflected in the ESA lifetime. The emissive 3MLCT state for Ru-ip-3T in the presence of ct DNA also decays with a bi-exponential lifetime of 0.3 (60%) and 1.1 μs (40%) (Figure S2), with the shorter component being similar to the emissive lifetime in water. Bi-exponential emission kinetics suggests that there are two populations, a fraction of molecules that are unbound or loosely associated with ct DNA (the shorter component) alongside another that is more strongly bound (the longer component).
When Ru-ip-3T is dissolved in SK-MEL-28 cellular lysate, T1 decays bi-exponentially with characteristic lifetimes of 1.4 (60%) and 10 μs (40%) (Figure 2d and Figure S2) observed by TA. The shorter time constant is consistent with that obtained in water, suggesting a population of unbound or loosely-bound complexes in the lysate. It should be noted that there are a variety of biomolecules in the lysate, and thus multiple targets, but the observation of two lifetimes with roughly equal amplitudes suggests two major types of environments. Assuming that the shorter lifetime represents unbound or loosely-associated metal complex, we sought to use lysate not diluted with buffer in order to increase the concentration of DNA and other biomolecules in the solution. However, higher lysate concentrations led to immediate precipitation, precluding any determination on preferential lifetime effects.
We ascribe the increased stability of T1, as reflected in the transient absorption data, to the fact that the biomolecule-bound complexes are less exposed to water and dissolved oxygen quenchers. A similar rational was used to explain the increase in the T1 lifetime in the presence of ct DNA. The 3MLCT lifetime of Ru-ip-3T is also prolonged by the lysate, increasing from 0.3 μs in water32 to 0.5 μs in lysate (Figure S2). While comparable changes in lifetimes are observed for the T1 state of Ru-ip-3T in DNA and lysate solutions, the effect on the MLCT state in DNA (appearance of a 1.1 μs component) is significantly greater than in lysate (0.5 μs). This could be due to stronger binding interactions by Ru-ip-3T in ct DNA solution compared to lysate. However further investigation is required to verify this hypothesis.
The behavior of Ru-ip-4T with ct DNA and SK-MEL-28 lysate is quantitatively identical to that of Ru-ip-3T (Figure S5). A bi-exponential decay of 1.4 (50%) and 7.4 μs (50%) corresponding to lifetimes of T1 state with ct DNA is observed compared to 1.4 μs in water.32 Also a bi-exponential 3MLCT emission decay, characterized by decay times of 0.1 μs (40%) and 0.9 μs (60%), is observed and is suggestive of two populations (unbound or loosely-associated and bound). The T1 state of Ru-ip-4T shows a bi-exponential decay with lifetimes of 1.4 μs (45%) and 10 μs (55%) in lysate (Figure S6), also suggestive of two environments with the shorter component representing free complex. The increased T1 lifetime in ct DNA solution and lysate would be expected to enhance 1O2 sensitization even under low O2 tension, an important consideration for PDT as many cancer cells thrive under low oxygen tension.29,47
Femtosecond transient absorption spectroscopy.
The fs transient absorption data of Ru-ip-3T along with its excited-state relaxation model, which is derived from femtosecond transient absorption spectra and the DAS spectra obtained from fitting the data, is discussed in our previous study.33 Here we focus on the ultrafast photoinduced relaxation of Ru-ip-3T in a solution of ct DNA. The femtosecond TA spectra of Ru-ip-3T in the presence of ct DNA are characterized by a ground state bleach (GSB) flanked by two regions of excited state absorption (ESA). The ESA includes a weak positive ΔOD signal, which may indicate the presence of a band below 370 nm,s and a strong and broad positive ΔOD band above 550 nm (Figure 3a). Within the first 2.5 ps after photoexcitation, the initial GSB at 480 nm decreases, a negative ΔOD band at 420 nm emerges, and the initially broad and mostly structureless ESA increases. Subsequently, the ESA band sharpens into a peak at 660 nm on a 150 ps timescale. After the build-up of the 660 nm band, the overall signal decays (but not completely) to the ns transient absorption features previously described (Figure 3a).
Figure 3.
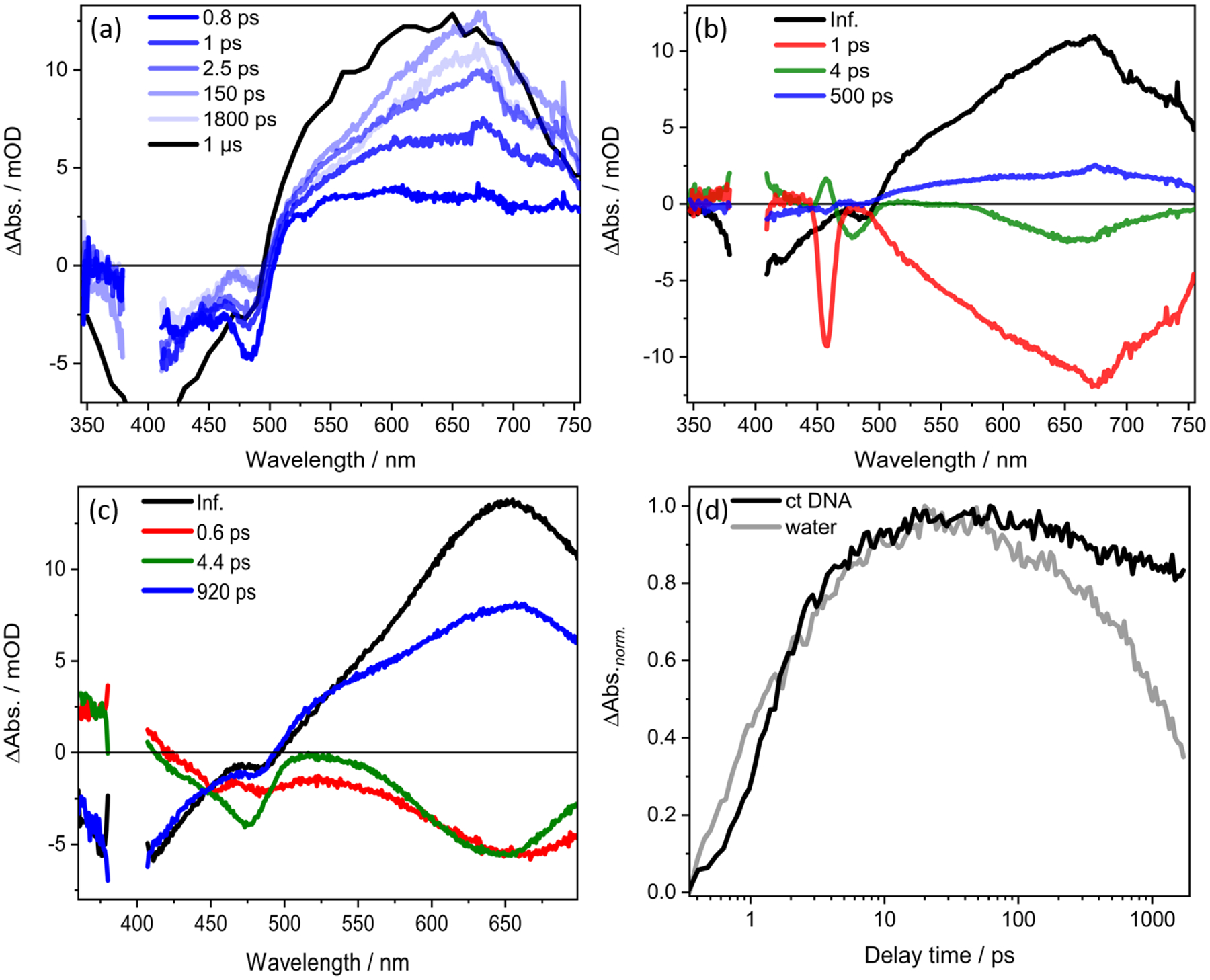
(a) Femtosecond TA spectra with 400-nm excitation of Ru-ip-3T in ct DNA solution. The spectrum at 1 μs is extracted from the ns-TA spectrum of Ru-ip-3T in ct DNA solution with contribution from emission subtracted from the overall spectra. (b) DAS for Ru-ip-3T in ct DNA solution. The intense band at at 460 nm in the 1-ps spectrum is due to Raman scattering by water. (c) DAS for Ru-ip-3T in water. (d) Transient absorption kinetics recorded at 660 nm of Ru-ip-3T in ct DNA solution versus water following excitation at 400 nm. Data for Ru-ip-3T in water were reported elsewhere.33
The data are analyzed quantitatively by fitting a sum of exponentials to give the characteristic decay times: τ1=1 ps, τ2=4 ps, and τ3=500 ps. A long-lived component is also observed, which corresponds to the transients in the ns experiments. We refer to this component as the “infinite component” in the context of the femtosecond transient absorption spectra.
The present data obtained in ct DNA solution (Figure 5) is interpreted in the context of the photophysical model derived for water as solvent, whereby photoexcitation leads to rapid ISC from the singlet to the triplet manifold that takes place within the temporal resolution of the experiment (120 fs).14,15 The shortest relaxation time constant available from the data presented here, τ1= 1 ps, is associated with hot 3MLCT→3ILCT energy transfer as indicated by a decrease in the MLCT bleach at 480 nm and an increase in the ILCT bleach at 420 nm with a simultaneous increase in the ESA band at 660 nm that is associated with the 3ILCT state (Figure 3a). This is followed by the sharpening of the ESA at 660 nm with a time constant τ2 = 4 ps. The process associated with τ2 leads to a decrease in the ESA below 575 nm and above 700 nm (Figure 3b), with a simultaneous peaking of the band at 660 nm that is indicative of the molecules populating the lowest vibrational quanta of the 3ILCT state. Hence, we associate this process to vibrational cooling from the initially populated hot 3ILCT to form the cooled 3ILCT state. The time constant of 500 ps (τ3) decreases the overall ESA with a partial recovery of the GSB (Figure 3b). Though the DAS associated with the process is comparatively broader than the spectrum of the long-lived (infinite) component, it overall resembles the features of the 3ILCT state. As discussed in our previous work,33 this process is associated with the deactivation of molecules from a thermally-relaxed 3ILCT state which has a molecular conformation different from the T1 state.
Figure 5.

Jablonski diagram to describe the relaxation dynamics of Ru-ip-3T and Ru-ip-4T in biologically complex solvents with 400 nm excitation. The solid bold larger arrow in black indicates that the preferential population of T1 state increases on moving from water to ct DNA solution. The ct DNA solution was 5:1 [DNA]bp/[complex].
Although the overall photophysical processes of Ru-ip-3T on a ps-timescale remain essentially unchanged upon binding to ct DNA, changes associated with τ3 are observed. Interaction with ct DNA leads to a shorter characteristic time constant of 500 ps compared to 920 ps in water.33 Furthermore, the contribution of the process (i.e., amplitude of the respective DAS) is smaller for the ct DNA solution. This can be rationalized by comparing the ratio A where
| (1) |
in the ct DNA solution (Figure 3b) with that in water (Figure 3c). In this equation, the area of τInf. reflects the fraction of molecules populating the long lived T1 state, while the area of τ3 is proportional to the number of molecules decaying to the ground state from a higher lying 3ILCT state.33 In ct DNA solution, the ratio A ≈5, whereas in water the value drops to 1.5. Ru-ip-3T strongly bound to ct DNA may orient the thiophene chain in conformation(s) favoring the population of T1 over the cooled 3ILCT state.
The femtosecond transient absorption profile of Ru-ip-4T bound to ct DNA (Figure 4a) also differs compared to Ru-ip-4T in water. As for Ru-ip-3T the relative weights of the processes associated with τ3 and τInf. change upon DNA binding (Figure 4). The lifetime associated with the decay of the cooled 3ILCT state (τ3) accelerates from 970 ps to 520 ps on moving from water to ct DNA solution. As for Ru-ip-3T the amplitude / contribution of this process to the overall signal is considerably lowered in ct DNA solution relative to water. We rationalize this by comparing the ratio A between ct DNA solution and water (4.5 versus 0.5, respectively), which implies that the population of the T1 state dominates over the cooled 3ILCT channel in ct DNA solution.
Figure 4.

(a) Femtosecond TA spectra for Ru-ip-4T in ct DNA solution with 400-nm excitation. The spectrum at 1 μs is extracted from the ns-TA spectrum for Ru-ip-4T in ct-DNA solution with contribution from emission subtracted from the overall spectra. (b) DAS of Ru-ip-4T in ct DNA solution. The intense band at 460 nm in the 1-ps spectrum is due to Raman scattering by water. (c) DAS for Ru-ip-4T in water. (d) Transient absorption kinetics recorded at 700 nm of Ru-ip-4T in ct DNA solution versus water recorded upon excitation at 400 nm. Data for Ru-ip-4T in water was reported elsewhere.33
The ultrafast transient absorption data for Ru-ip-4T in ct DNA solution (Figure 4a) shows a GSB below 520 nm and an ESA at longer wavelengths. Within the first 15 ps, the GSB at 480 nm decreases with a simultaneous increase in the GSB at 425 nm. The ESA is marked by a peak at 700 nm and a broad shoulder at 550 nm. This ESA band which is primarily associated with a 3ILCT state33 increases within the first 15 ps. Following this delay time, the overall ESA signal decays with a concomitant decrease in the GSB. A sum of tri-exponential fit in addition to a long lived component (infinite) is used to globally fit the kinetics which yields the time constants: τ1=1 ps, τ2=8 ps, and τ3=520 ps (Figure 4b). The excited state model derived for Ru-ip-4T in water33 can be used to account for its overall behavior in ct DNA solution. τ1=1 ps increases the ESA at 700 nm which is associated with 3ILCT state with a simultaneous decrease with the GSB at 480 nm. Hence, we associate this time constant with energy transfer from the hot 3MLCT→hot 3ILCT. τ2=8 ps specifically results in a loss in ESA at 550 nm and 750 nm with an increase of the 700 nm peak, which represents the molecules relaxing to the lowest vibrational quanta of the 3ILCT state. Therefore, the 8 ps time constant is associated with vibrational cooling from the hot 3ILCT→cool 3ILCT state. τ3=520 ps leads to a decrease in the ESA and GSB. As mentioned above, we associate this process with a loss in the spectral features of the thermally relaxed 3ILCT state (Figure 5), which is electronically and a conformationally distinct from the long-lived T1 state.
Our findings regarding the photoinduced dynamics in Ru-ip-3T and Ru-ip-4T when interacting with ct DNA indicate that increasing rigidity imposed on the complex may limit the number of conformations adopted, thereby affecting processes which are dependent on structural reorganization. This may selectively favor one excited-state relaxation pathway over the other.
CONCLUSIONS
We investigated the excited state dynamics of the photoactive complex Ru-ip-3T in two representative biologically relevant environments, ct DNA and SK-MEL-28 cell lysate. We also explore the related Ru-ip-4T with ct DNA. Binding of the complexes to ct DNA alters their photoinduced dynamics compared to water as solvent. Most importantly, the decay of the oligothiophene-localized 3ILCT state to reform the ground state was accelerated by interaction with the DNA, and the fraction of molecules that populate the long-lived T1 state increases. Both findings hold true both for Ru-ip-3T and Ru-ip-4T. Thus, they indicate that also the excited-state reactivity in the triplet manifold, specifically the partitioning between the 3ILCT and the T1 state, is altered when the complexes interact with DNA. We tentatively ascribe this phenomenon to conformational restriction upon DNA binding, specifically with regard to the ip-nT ligand. Nanosecond transient absorption data show a significantly slower ground state recovery of the T1 state when the complexes are in DNA or cell lysate solutions. The prolonged excited-state lifetime in the presence of biomolecules should enhance singlet oxygen production, an important requirement for PDT. This study highlights the importance of interrogating the photophysical dynamics in solvents which are more biologically relevant than water in order to understand the function-determining excited-state processes in transition metal complexes designed to act as photoactive drugs.
Supplementary Material
Table 1.
Time constants associated with Ru-ip-3T and Ru-ip-4T in solutions under aerated conditions. Time constants, τTA and τem. are associated with the lifetimes of T1 and 3MLCT state respectively. The two time constants for τTA and τem. are due to contributions from unbound (shorter time constant) and bound (longer time constant) forms of the complexes.
| Ru-ip-3T | Ru-ip-4T | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| τ1 / ps | τ2 / ps | τ3 / ps | τTA / μs | τem. / μs | τ1 / ps | τ2 / ps | τ3 / ps | τTA / μs | τem. / μs | |
| Water* | 0.6 | 4.4 | 920 | 1.4 | 0.3 | 0.7 | 9.6 | 970 | 1.4 | 0.3 |
| ct DNA | 1 | 4 | 500 | 4, 10 | 0.3, 1.1 | 1 | 8 | 520 | 1.4, 7.4 | 0.1, 0.9 |
| SK-MEL-28 lysate | - | 4 | 600 | 1.4, 10 | 0.5 | n.d. | n.d. | n.d. | 1.4, 10 | 0.4 |
ACKNOWLEDGEMENTS
K.R.A.S. thanks the Carl-Zeiss-Foundation for financial support. A.C. and B.D.I. thank the DFG (project 395358570) for financial support.
S.A.M. and C.G.C. thank the National Science Foundation (NSF) (award 2102459) and the National Cancer Institute (NCI) of the National Institutes of Health (NIH) (Award R01CA222227) and the for partial support. The content in this review is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information UV-visible spectra of ct DNA and SK-MEL-28 lysate for preparing solutions of requisite DNA concentration, nanosecond transient absorption spectra of Ru-ip-3T in SK-MEL-28 lysate and time resolved emission kinetics of Ru-ip-3T in SK-MEL-28 lysate and ct DNA with 410 nm excitation, femtosecond transient absorption spectra, DAS and kinetics of Ru-ip-3T in SK-MEL-28 lysate with 410 nm excitation, UV-visible absorption and emission spectra of Ru-ip-4T in buffer and ct DNA with 450 nm excitation, nanosecond transient absorption spectra, kinetics and time resolved emission kinetics of Ru-ip-4T in ct DNA with 410 nm excitation, UV-visible, nanosecond transient absorption spectra, kinetics and time resolved emission kinetics of Ru-ip-4T in SK-MEL-28 lysate with 410 nm excitation.
S.A.M. has a potential research conflict of interest due to a financial interest with Theralase Technologies, Inc. and PhotoDynamic, Inc. A management plan has been created to preserve objectivity in research in accordance with UTA policy
REFERENCES
- (1).Bonnett R Chemical Aspects of Photodynamic Therapy; CRC Press: London, 2000. [Google Scholar]
- (2).Photodynamic Therapy: Basic Principles and Clinical Applications; Henderson BW, Dougherty TJ, Eds.; CRC Press: New York, 1992 [Google Scholar]
- (3).Handbook of Photomedicine, 1st ed.; Hamblin MR, Huang Y, Eds.; CRC Press: Boca Raton, FL, 2013. [Google Scholar]
- (4).Hamblin MR; Mroz P Advances in Photodynamic Therapy: Basic, Translational, and Clinical; Engineering in Medicine and Biology; Artech House: Norwood, MA, 2008. [Google Scholar]
- (5).Photodynamic Medicine: From Bench to Clinic, 1st ed.; Kostron H, Hasan T, Eds.; Royal Society of Chemistry: Cambridge, U.K., 2016. [Google Scholar]
- (6).Usuda J; Kato H; Okunaka T; Furukawa K; Tsutsui H; Yamada K; Suga Y; Honda H; Nagatsuka Y; Ohira T Photodynamic Therapy (PDT) for Lung Cancers. J. Thorac. Oncol 2006, 1, 489–493. [PubMed] [Google Scholar]
- (7).Kataoka H; Nishie H; Hayashi N; Tanaka M; Nomoto A; Yano S; Joh T New Photodynamic Therapy with Next-Generation Photosensitizers. Ann. Transl. Med 2017, 5, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Friedman AE; Barton JK; Chambron JC; Sauvage JP; Turro NJ; Barton JK Molecular “Light Switch” for DNA: Ru(bpy)2(dppz) 2+. J. Am. Chem. Soc 1990, 112, 4960–4962. [Google Scholar]
- (9).Howerton BS; Heidary DK; Glazer EC Strained Ruthenium Complexes Are Potent Light-Activated Anticancer Agents. J. Am. Chem. Soc 2012, 134, 8324–8327. [DOI] [PubMed] [Google Scholar]
- (10).Majewski MB; Tacconi NRD; MacDonnell FM; Wolf MO Ligand-Triplet-Fueled Long-Lived Charge Separation in Ruthenium(II) Complexes with Bithienyl-Functionalized Ligands. Inorg. Chem 2011, 50, 9939–9941. [DOI] [PubMed] [Google Scholar]
- (11).Sun Y; Joyce LE; Dickson NM; Turro C Efficient DNA Photocleavage by [Ru(bpy)2(dppn)]2+ with Visible Light. Chem. Commun 2010, 46, 2426–2428. [DOI] [PubMed] [Google Scholar]
- (12).Hissler M; Harriman A; Khatyr A; Ziessel R Intramolecular Triplet Energy Transfer in Pyrene - Metal Polypyridine Dyads: A Strategy for Extending the Triplet Lifetime of the Metal Complex. Chem. - A Eur. J 1999, 5, 3366–3381. [Google Scholar]
- (13).Swavey S; Brewer KJ Visible Light Induced Photocleavage of DNA by a Mixed-Metal Supramolecular Complex: [{(bpy)2Ru(dpp)}2RhCl2]5+. Inorg. Chem 2002, 41, 6196–6198. [DOI] [PubMed] [Google Scholar]
- (14).Bhasikuttan AC; Suzuki M; Nakashima S; Okada T Ultrafast Fluorescence Detection in Tris(2,2′-bipyridine)ruthenium(II) Complex in Solution: Relaxation Dynamics Involving Higher Excited States. J. Am. Chem. Soc 2002, 124, 8398–8405. [DOI] [PubMed] [Google Scholar]
- (15).Cannizzo A; Van Mourik F; Gawelda W; Zgrablic G; Bressler C; Chergui M Broadband Femtosecond Fluorescence Spectroscopy of [Ru(bpy)3]2+. Angew. Chemie - Int. Ed 2006, 45, 3174–3176. [DOI] [PubMed] [Google Scholar]
- (16).Juris A; Balzani V; Barigelletti F; Campagna S; Belser P; von Zelewsky A Ru (II) Polypyridine Complexes: Photophysics, Photochemistry, Eletrochemistry, and Chemiluminescence. Coord. Chem. Rev 1988, 84, 85–277. [Google Scholar]
- (17).Vögtle F; Plevoets M; Nieger M; Azzellini GC; Credi A; De Cola L; De Marchis V; Venturi M; Balzani V Dendrimers with a Photoactive and Redox-Active [Ru(bpy)3]2+-Type Core: Photophysical Properties, Electrochemical Behavior, and Excited-State Electron-Transfer Reactions. J. Am. Chem. Soc 1999, 121, 6290–6298. [Google Scholar]
- (18).Ford WE; Rodgers MAJ Reversible Triplet-Triplet Energy Transfer within a Covalently Linked Bichromophoric Molecule. J. Phys. Chem 1992, 96 (7), 2917–2920. [Google Scholar]
- (19).Goze C; Kozlov DV; Tyson DS; Ziessel R; Castellano FN Synthesis and Photophysics of Ruthenium (II) Complexes with Multiple Pyrenylethynylene Subunits. New J. Chem 2003, 27, 1679–1683. [Google Scholar]
- (20).Kozlov DV; Tyson DS; Goze C; Ziessel R; Castellano FN Room Temperature Phosphorescence from Ruthenium (II) Complexes Bearing Conjugated Pyrenylethynylene Subunits. Inorg. Chem 2004, 43, 6083–6092. [DOI] [PubMed] [Google Scholar]
- (21).Tyson DS; Henbest KB; Bialecki J; Castellano FN Excited State Processes in Ruthenium (II)/Pyrenyl Complexes Displaying Extended Lifetimes. J. Phys. Chem. A 2001, 105, 8154–8161. [Google Scholar]
- (22).McClenaghan ND; Leydet Y; Maubert B; Indelli MT; Campagna S Excited-State Equilibration: A Process Leading to Long-Lived Metal-to-Ligand Charge Transfer Luminescence in Supramolecular Systems. Coord. Chem. Rev 2005, 249, 1336–1350. [Google Scholar]
- (23).Harriman A; Hissler M; Khatyr A; Ziessel R A Ruthenium (II) Tris (2,2′-bipyridine) Derivative Possessing a Triplet Lifetime of 42 μs. Chem. Commun 1999, 735–736. [Google Scholar]
- (24).Simon JA; Curry SL; Schmehl RH; Schatz TR; Piotrowiak P; Jin X; Thummel RP Intramolecular Electronic Energy Transfer in Ruthenium (II) Diimine Donor/Pyrene Acceptor Complexes Linked by a Single C− C Bond. J. Am. Chem. Soc 1997, 119, 11012–11022. [Google Scholar]
- (25).Harriman A; Khatyr A; Ziessel R Extending the Luminescence Lifetime of Ruthenium (II) Poly (Pyridine) Complexes in Solution at Ambient Temperature. Dalt. Trans 2003, 2061–2068. [Google Scholar]
- (26).Monro S; Scott J; Chouai A; Lincoln R; Zong R; Thummel RP; McFarland SA Photobiological Activity of Ru (II) Dyads Based on (Pyren-1-yl) ethynyl Derivatives of 1, 10-Phenanthroline. Inorg. Chem 2010, 49, 2889–2900. [DOI] [PubMed] [Google Scholar]
- (27).Lincoln R; Kohler L; Monro S; Yin H; Stephenson M; Zong R; Chouai A; Dorsey C; Hennigar R; Thummel RP Exploitation of Long-Lived 3IL Excited States for Metal–Organic Photodynamic Therapy: Verification in a Metastatic Melanoma Model. J. Am. Chem. Soc 2013, 135, 17161–17175. [DOI] [PubMed] [Google Scholar]
- (28).Reichardt C; Sainuddin T; Wächtler M; Monro S; Kupfer S; Guthmuller J; Gräfe S; McFarland S; Dietzek B Influence of Protonation State on the Excited State Dynamics of a Photobiologically Active Ru(II) Dyad. J. Phys. Chem. A 2016, 120, 6379–6388. [DOI] [PubMed] [Google Scholar]
- (29).Reichardt C; Schneider KRA; Sainuddin T; Wächtler M; McFarland SA; Dietzek B Excited State Dynamics of a Photobiologically Active Ru(II) Dyad Are Altered in Biologically Relevant Environments. J. Phys. Chem. A 2017, 121, 5635–5644. [DOI] [PubMed] [Google Scholar]
- (30).Monro S; Colón KL; Yin H; Roque J; Konda P; Gujar S; Thummel RP; Lilge L; Cameron CG; McFarland SA Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev 2019, 119, 797–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).McFarland SA; Mandel A; Dumoulin-White R; Gasser G Metal-Based Photosensitizers for Photodynamic Therapy: The Future of Multimodal Oncology? Curr. Opin. Chem. Biol 2020, 56, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chettri A; Roque JA; Schneider KRA; Cole HD; Cameron CG; McFarland SA; Dietzek B It Takes Three to Tango: The Length of the Oligothiophene Chain Determines the Nature of the Long-Lived Excited State and the Resulting Photocytotoxicity of a Ruthenium(II) Photodrug. ChemPhotoChem 2021, 5, 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chettri A; Schneider KRA; Cole HD; Roque III JA; Cameron CG; McFarland SA; Dietzek B String-Attached Oligothiophene Substituents Determine the Fate of Excited States in Ruthenium Complexes for Photodynamic Therapy. J. Phys. Chem. A 2021, 125, 6985–6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lifshits LM; Roque III JA; Konda P; Monro S; Cole HD; von Dohlen D; Kim S; Deep G; Thummel RP; Cameron CG et al. Near-Infrared Absorbing Ru(II) Complexes Act as Immunoprotective Photodynamic Therapy (PDT) Agents against Aggressive Melanoma. Chem. Sci 2020, 11, 11740–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Roque JA; Barrett PC; Cole HD; Lifshits LM; Shi G; Monro S; Von Dohlen D; Kim S; Russo N; Deep G et al. Breaking the Barrier: An Osmium Photosensitizer with Unprecedented Hypoxic Phototoxicity for Real World Photodynamic Therapy. Chem. Sci 2020, 11, 9784–9806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Karnahl M; Kuhnt C; Ma F; Yartsev A; Schmitt M; Dietzek B; Rau S; Popp J Tuning of Photocatalytic Hydrogen Production and Photoinduced Intramolecular Electron Transfer Rates by Regioselective Bridging Ligand Substitution. ChemPhysChem 2011, 12, 2101–2109. [DOI] [PubMed] [Google Scholar]
- (37).Siebert R; Akimov D; Schmitt M; Winter A; Schubert US; Dietzek B; Popp J Spectroscopic Investigation of the Ultrafast Photoinduced Dynamics in π-Conjugated Terpyridines. ChemPhysChem 2009, 10, 910–919. [DOI] [PubMed] [Google Scholar]
- (38).Kovalenko SA; Dobryakov AL; Ruthmann J; Ernsting NP Femtosecond Spectroscopy of Condensed Phases with Chirped Supercontinuum Probing. Phys. Rev. A - At. Mol. Opt. Phys 1999, 59, 2369–2384. [Google Scholar]
- (39).Dietzek B; Pascher T; Sundström V; Yartsev A Appearance of Coherent Artifact Signals in Femtosecond Transient Absorption Spectroscopy in Dependence on Detector Design. Laser Phys. Lett 2007, 4, 38–43. [Google Scholar]
- (40).Sirajuddin M; Ali S; Badshah A Drug-DNA Interactions and Their Study by UV-Visible, Fluorescence Spectroscopies and Cyclic Voltametry. J. Photochem. Photobiol. B Biol 2013, 124, 1–19. [DOI] [PubMed] [Google Scholar]
- (41).Shi G; Monro S; Hennigar R; Colpitts J; Fong J; Kasimova K; Yin H; DeCoste R; Spencer C; Chamberlain L et al. Ru(II) Dyads Derived from α-Oligothiophenes: A New Class of Potent and Versatile Photosensitizers for PDT. Coord. Chem. Rev 2015, 282–283, 127–138. [Google Scholar]
- (42).McKenzie LK; Sazanovich IV; Baggaley E; Bonneau M; Guerchais V; Williams JAG; Weinstein JA; Bryant HE Metal Complexes for Two-Photon Photodynamic Therapy: A Cyclometallated Iridium Complex Induces Two-Photon Photosensitization of Cancer Cells under Near-IR Light. Chem. - A Eur. J 2017, 23, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Al-Afyouni MH; Rohrabaugh TN; Al-Afyouni KF; Turro C New Ru(II) Photocages Operative with near-IR Light: New Platform for Drug Delivery in the PDT Window. Chem. Sci 2018, 9, 6711–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhang P; Wang Y; Qiu K; Zhao Z; Hu R; He C; Zhang Q; Chao H A NIR Phosphorescent Osmium(II) Complex as a Lysosome Tracking Reagent and Photodynamic Therapeutic Agent. Chem. Commun 2017, 53, 12341–12344. [DOI] [PubMed] [Google Scholar]
- (45).Tan LF; Wang F; Chao H; Zhou YF; Weng C Ruthenium(II) Mixed-Ligand Complex Containing 2-(4′-benzyloxy-phenyl)imidazo[4,5-f][1,10]phenanthroline: Synthesis, DNA-Binding and Photocleavage Studies. J. Inorg. Biochem 2007, 101 (4), 700–708. [DOI] [PubMed] [Google Scholar]
- (46).Wu JZ; Yuan L Synthesis and DNA Interaction Studies of a Binuclear Ruthenium(II) Complex with 2,9-bis(2-imidazo[4,5-f][1,10]phenanthroline)-1,10-phenanthroline as Bridging and Intercalating Ligand. J. Inorg. Biochem 2004, 98, 41–45. [DOI] [PubMed] [Google Scholar]
- (47).Reichardt C; Monro S; Sobotta FH; Colón KL; Sainuddin T; Stephenson M; Sampson E; Roque J; Yin H; Brendel JC et al. Predictive Strength of Photophysical Measurements for in Vitro Photobiological Activity in a Series of Ru(II) Polypyridyl Complexes Derived from π-Extended Ligands. Inorg. Chem 2019, 58, 3156–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.