

ABSTRACT
SARS-CoV-2 infections have resulted in a very large number of severe cases of COVID-19 and deaths worldwide. However, knowledge of SARS-CoV-2 infection, pathogenesis and therapy remains limited, emphasizing the urgent need for fundamental studies and drug development. Studies have shown that induction of macroautophagy/autophagy and hijacking of the autophagic machinery are essential for the infection and replication of SARS-CoV-2; however, the mechanism of this manipulation and the function of autophagy during SARS-CoV-2 infection remain unclear. In the present study, we identified ORF3a as an inducer of autophagy (in particular reticulophagy) and revealed that ORF3a localizes to the ER and induces RETREG1/FAM134B-related reticulophagy through the HMGB1-BECN1 (beclin 1) pathway. As a consequence, ORF3a induces ER stress and inflammatory responses through reticulophagy and then sensitizes cells to the acquisition of an ER stress-related early apoptotic phenotype and facilitates SARS-CoV-2 infection, suggesting that SARS-CoV-2 ORF3a hijacks reticulophagy and then disrupts ER homeostasis to induce ER stress and inflammatory responses during SARS-CoV-2 infection. These findings reveal the sequential induction of reticulophagy, ER stress and acute inflammatory responses during SARS-CoV-2 infection and imply the therapeutic potential of reticulophagy and ER stress-related drugs for COVID-19.
Abbreviations: CQ: chloroquine; DEGs: differentially expressed genes; ER: endoplasmic reticulum; GSEA: gene set enrichment analysis; HMGB1: high mobility group box 1; HMOX1: heme oxygenase 1; MERS-CoV: Middle East respiratory syndrome coronavirus; RETREG1/FAM134B: reticulophagy regulator 1; RTN4: reticulon 4; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; TN: tunicamycin
KEYWORDS: ER stress, inflammatory response, ORF3a, reticulophagy, SARS-CoV-2
Introduction
The COVID-19 pandemic outbreak has resulted in the infection of hundreds of million people and the death of more than five million people worldwide, and the number of patients and deaths are still rapidly increasing. Effective treatments and therapies are still far from satisfactory; the development of specific drugs, therapies and vaccines are quite urgently needed. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of COVID-19. As an emerging coronavirus associated with severe disease, SARS-CoV-2 is highly homologous to two other such coronaviruses, SARS-CoV-1 and Middle East respiratory syndrome coronavirus (MERS-CoV), which have caused two pandemic outbreaks in the past twenty years [1,2]. In high-risk populations, such as older persons and persons with underlying diseases, SARS-CoV-2 infection often causes severe respiratory failure, sepsis and even multiple organ failure, accompanied by severe acute inflammation and excessive cytokine release known as “cytokine storm” [3–5]. To date, knowledge of SARS-CoV-2 infection and disease remains very limited.
Macroautophagy/autophagy is a conserved physiological process in eukaryotic cells by which intracellular waste, damaged organelles and invasive microorganisms are separated and wrapped into double-membrane vacuoles and eventually degraded in lysosomes [6,7]. Autophagy is finely regulated through many core autophagy-related genes and several upstream signaling cascades that are primarily initiated by amino acid and glucose deprivation, extracellular stimuli, intracellular damage and so on [6]. Autophagy plays essential roles in cell activities and behaviors, mainly maintaining intracellular homeostasis, removing intracellular threats, repairing damaged organelles and protecting cells from death [8,9]. However, excessive autophagy also hastens cell death [10,11]. While autophagy has exhibited great therapeutic potential in many diseases [12], safe and effective approaches for clinical applications still require a lengthy development process.
Autophagy usually eliminates invading viruses and promotes antiviral responses; however, autophagy can also be hijacked by viruses to facilitate infection, replication and pathogenesis [13,14]. Many enveloped viruses often enter, replicate, mature and translocate through autophagy-related vesicles [15–17]. Thus, autophagy inhibition by genetic approaches or inhibitors effectively suppresses viral infection and diseases [18,19]. Therefore, autophagy has become a promising therapeutic target for viral infectious diseases.
Coronaviruses differentially regulate autophagy through distinct mechanisms. For example, several viral proteins of SARS-CoV-1 and MHV induce autophagy to support viral infection and replication [20–23], whereas MERS-CoV Nsp6 inhibits autophagy [22]. However, autophagy inhibition by different drugs and inhibitors effectively blocks viral infection and replication, and coronaviruses likely require a low basal level of autophagy for cell entry and incomplete autophagy for virion assembly and maturation.
Previous studies have shown that the autophagy inhibitors chloroquine (CQ) and hydroxychloroquine significantly suppress SARS-CoV-2 infection and replication [24,25], leading to hopes that COVID-19 might be defeated by modulating autophagy. However, the ineffectiveness of or lack of observed benefit from CQ and its derivatives has been confirmed in clinical trials of COVID-19 therapies [26–28], indicating that autophagy-related therapeutic strategies are still far from clinical application. Importantly, the infection and replication of SARS-CoV-2 requires the activation of autophagy, and autophagy-like double-membrane vesicles are employed for viral RNA export and viral replication [29]. Therefore, elucidating the fine regulation of autophagy by SARS-CoV-2 and the function of autophagy during infection and replication is quite crucial for understanding SARS-CoV-2 infection and pathogenesis and for developing autophagy-related therapies and drugs for COVID-19.
In the present study, we performed a systematic screen and found that SARS-CoV-2 ORF3a induces reticulophagy/ERphagy through the HMGB1-BECN1 (beclin 1) axis, consequently activating endoplasmic reticulum (ER) stress and inflammatory responses through reticulophagy and in turn facilitating SARS-CoV-2 infection and sensitizing cells to ER stress-related cell death. Our findings reveal a novel mechanism of induction of reticulophagy, ER stress and inflammatory responses by SARS-CoV-2 and provide a potential target for COVID-19 treatment and drug development.
Results
A systemic screening identified that ORF3a induces autophagy
To investigate how SARS-CoV-2 regulates autophagy, a systematic screen of the viral protein expression library was performed using the Gaussia luciferase-based autophagy reporter Actin-LC3-DN, which detects cleavage of pro-LC3, a key precursor of the autophagic protein LC3/Atg8 [30]. After two rounds of screening, we identified several viral proteins that may decrease autophagy-dependent pro-LC3 cleavage; in contrast, NSP6, ORF3a and ORF8 increased autophagy-dependent pro-LC3 cleavage in a dose-dependent manner, and ORF3a exhibited the strongest induction effect (Fig. S1A). To confirm that ORF3a serves as an inducer of autophagy, an ORF3a-expressing plasmid was transfected into cells with or without subsequent starvation in HBSS. The conversion of LC3-I to LC3-II was obviously enhanced but the SQSTM1/p62 level was decreased by ORF3a expression; both were augmented under HBSS starvation (Figure 1A). However, its homologue SARS-CoV ORF3a barely affected LC3-I-to-LC3-II conversion (Fig. S1B), indicating that SARS-CoV-2 ORF3a mediates specific induction of autophagy. Furthermore, we evaluated the activation of one key kinase, AKT, which negatively regulates autophagy, and found that AKT phosphorylation was greatly decreased by ORF3a expression, similar to the effect of HBSS starvation (Figure 1B), and the phosphorylation of ULK1 was increased in presence of ORF3a expression without treatment or under HBSS starvation (Figure 1B). To further visualize autophagosomes in the absence or presence of ORF3a expression, the ORF3a-expressing plasmid was co-transfected with the GFP-LC3- or ptfLC3-expressing plasmid. Increased numbers of GFP-LC3 puncta were observed in cells with ORF3a expression compared with cells with empty vector transfection (Figure 1C), and the formation of LC3-positive autophagosomes (yellow puncta) and the maturation of autolysosomes (red puncta) were similarly enhanced by ORF3a expression (Figure 1D). Similarly, by electron microscopy, increased double-membrane autophagic vacuoles were observed in ORF3a-expressing cells, with relatively few autophagic vacuoles observed in empty vector-transfected cells (Figure 1E). These results suggest that ORF3a overexpression significantly induces autophagy.
Figure 1.

ORF3a induces autophagy. (A-B) HEK293T cells were transfected with empty vector or Flag-ORF3a-expressing plasmid for 48 h and then left untreated or treated with HBSS for 2 h. The cells were harvested, and the whole cell extracts were analyzed by Western blots as indicated to detect LC3-I-to-LC3-II conversion and SQSTM1 level (A) or AKT and ULK1 phosphorylation (B). (C) HeLa cells were transfected with empty vector or Flag-ORF3a- and GFP-LC3-expressing plasmid, left untreated or followed by HBSS treatment for 2 h, then fixed and subjected to confocal microscopy analysis. Scale bar: 10 μm. The LC3 puncta per cell were calculated from 20 cells/each from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, by Sidak’s multiple comparisons test. (D) HeLa cells were transfected with ptfLC3 and empty vector or Flag-ORF3a-expressing plasmid. Twenty-four hours post transfection, cells were fixed and subjected to confocal microscopy analysis. Scale bar: 5 μm. The red puncta represent autolysosomes and the yellow puncta indicate autophagosomes. The LC3 puncta per cell were calculated from 20 cells/each from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, by Sidak’s multiple comparisons test. (E) Autophagic vacuoles in control or Flag-ORF3a-expressing HEK293 cells were observed by electron microscopy analysis and representative images are shown. N, nuclei. Scale bar: 1 μm. The arrows indicate autophagosomes.
ORF3a localizes to the ER and induces RETREG1-mediated reticulophagy
To investigate the type of autophagy induced by ORF3a, the subcellular localization of ORF3a was determined by immunofluorescence. We found that ORF3a colocalized with the ER membrane protein CANX (calnexin) in the perinuclear region (Figure 2A), suggesting that ORF3a localizes to the ER. Then, we hypothesized that ORF3a regulates ER-related autophagy, and the reticulophagy receptors RTN3 (reticulon 3) and RETREG1 were evaluated in the presence of ORF3a overexpression; their colocalization with ORF3a was observed in the ER compartment (Figure 2B-C). The expression of RETREG1 was slightly promoted by ORF3a expression (Figure 2D) and SARS-CoV-2 infection (Figure 2E), while the levels of the ER membrane proteins CANX and RTN4 (reticulon 4) were greatly decreased in the presence of ORF3a overexpression or SARS-CoV-2 infection, suggesting that ORF3a induces reticulophagy and the degradation of ER membrane proteins. To determine whether the decrease in the levels of CANX and RTN4 is due to autophagic degradation, the expression of RETREG1 in A549 cells was knocked down by shRNA transduction, and we found that RETREG1 depletion attenuated the degradation of CANX and RTN4 in ORF3a-transfected cells or during SARS-CoV-2 infection (Figure 2D-E), indicating that ORF3a and SARS-CoV-2 infection induce reticulophagy. Interestingly, RETREG1 depletion also greatly attenuated LC3-I-to-LC3-II conversion under ORF3a expression or during SARS-CoV-2 infection, indicating that RETREG1 is important for autophagy induction and that RETREG1-dependent reticulophagy is the main type of autophagy occurring in the context of ORF3a overexpression or during SARS-CoV-2 infection (Figure 2D-E). Furthermore, ORF3a expression augmented LC3-I-to-LC3-II conversion and the degradation of CANX and RTN4 during SARS-CoV-2 infection (Figure 2F), while the SQSTM1 level was slightly affected, probably because of incomplete late autophagy during SARS-CoV-2 infection [31,32]. However, the levels of the mitochondrial membrane proteins TIMM23 and TOMM20 were barely affected by ORF3a expression and/or SARS-CoV-2 infection (Figure 2D and 2F), indicating that mitophagy was not induced. To further confirm that ORF3a induces reticulophagy, the specific reticulophagy reporter SERP1/Ramp4-mCherry-GFP was used to monitor reticulophagy in the presence of ORF3a expression, and the formation of SERP1-positive autophagosomes (yellow puncta) and autolysosomes (red puncta) was similarly enhanced by ORF3a expression (Figure 2G). These results suggest that ORF3a promotes RETREG1-related reticulophagy during SARS-CoV-2 infection.
Figure 2.
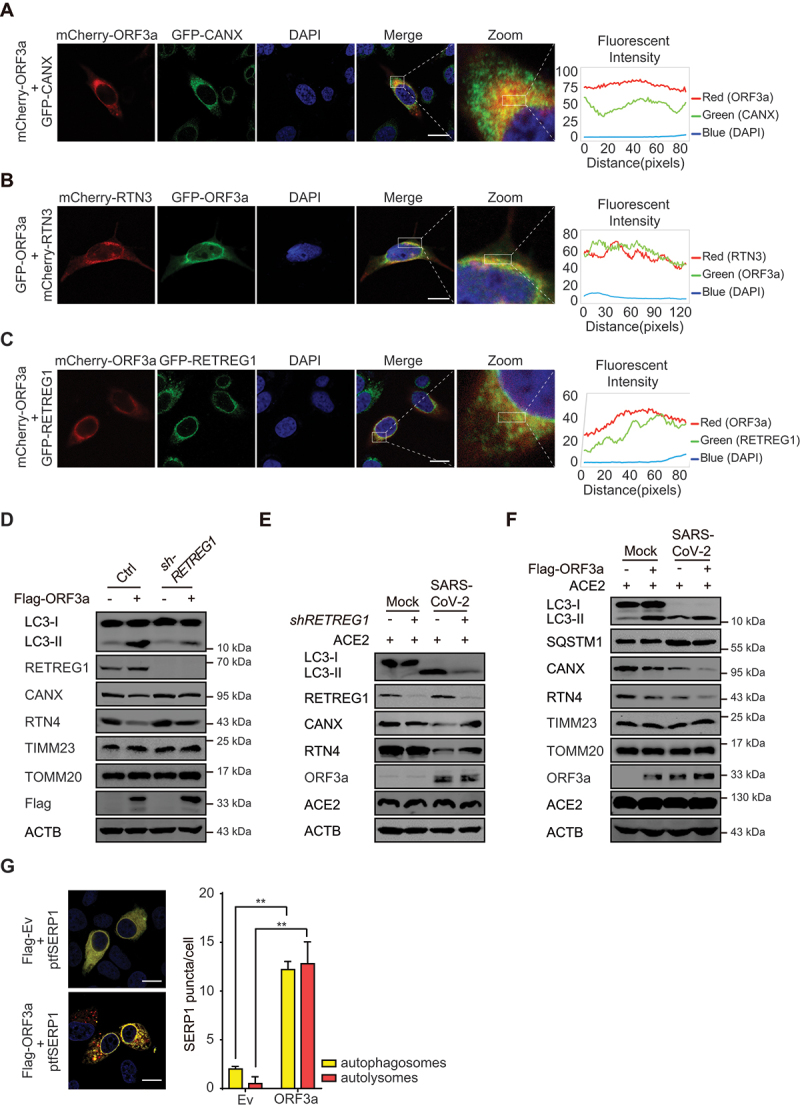
ORF3a localizes to the ER and induces RETREG1-mediated reticulophagy. (A-C) HeLa cells were transfected with mCherry-ORF3a and then stained with ER marker CANX (A), or co-transfected with mCherry-RTN3 and GFP-ORF3a (B), or GFP-RETREG1 and mCherry-ORF3a (C). Twenty-four hours post transfection, cells were fixed, and the images were visualized by confocal microscopy analysis. Scale bar: 5 μm. The coefficient of colocalization was determined by qualitative analysis of the fluorescence intensity of the selected area in merge. (D) Control or RETREG1-knockdown A549 cells were transfected with Flag-ORF3a-expressing plasmid or empty vector for 48 h. The cells were harvested, and the whole cell extracts were immunoblotted with the indicated antibodies. (E) Control or RETREG1-knockdown A549 cells (transfected with ACE2-expressing plasmid for 24 h) were left uninfected or infected with SARS-CoV-2 for 24 h, MOI = 1. the cells were harvested, and the whole cell extracts were analyzed by Western blots to detect the indicated antibodies. (F) A549 cells were transfected with Flag-ORF3a-expressing plasmid or empty vector and ACE2-expressing plasmid for 24 h and then left uninfected or infected with SARS-CoV-2 for 24 h, MOI = 1. The cells were harvested and the whole cell extracts were analyzed. (G) HeLa cells were transfected with SERP1-mCherry-GFP and empty vector or Flag-ORF3a-expressing plasmid. Twenty-four hours post transfection, cells were fixed and subjected to confocal microscopy analysis. The red puncta represent autolysosomes and the yellow puncta indicate autophagosomes. Scale bar: 5 μm. The SERP1 puncta per cell were calculated from 20 cells/each from three independent experiments. *, p < 0.05; **, p < 0.01, by Sidak’s multiple comparisons test.
ORF3a interacts with HMGB1 and induces BECN1-dependent autophagy
To investigate how ORF3a induces autophagy, we investigated public SARS-CoV-2 viral protein-protein interaction network resources and found that several ORF3a-binding proteins are involved in autophagy regulation [33]. Five proteins were depleted by shRNA, and the resulting effects on ORF3a-induced autophagy were carefully evaluated. Since EI24 mainly regulates the basal autophagy pathway and autophagic degradation of aggregated proteins [34,35] and EI24 depletion did not consistently decrease ORF3a-mediated autophagy induction in the repeated experiments, we did not further evaluate whether EI24 is involved in this process. Depletion of HMGB1 (high mobility group box 1) and HMOX1 (heme oxygenase 1) greatly reduced the ability of ORF3a to induce autophagy, whereas VPS11 and VPS39 depletion only weakly affected autophagy induction (Figure 3A) and (Fig. S2A). Furthermore, the interaction between ORF3a and HMGB1 was observed when both were transiently overexpressed in cells (Figure 3B), and the interaction between ORF3a and endogenous HMGB1 was also observed (Figure 3C). As expected, the level of LC3-I-to-LC3-II conversion and degradation of CANX induced by ORF3a were greatly decreased by HMGB1 shRNA in a dose-dependent manner (Figure 3D) and (Fig. S2B), confirming that HMGB1 is required for ORF3a-induced reticulophagy.
Figure 3.

ORF3a interacts with HMGB1 and induces BECN1-dependent reticulophagy. (A) Flag-ORF3a or empty vector was co-transfected into HEK293T cells with scramble or shRNA against EI24, HMGB1, HMOX1, VPS11 or VPS39. Thirty-six hours after transfection, cell pellets were collected, and the whole cell extracts were analyzed by Western blots as indicated to detect LC3-I-to-LC3-II conversion. The densities of LC3-II to ACTB bands were analyzed by a LI-COR Odyssey scanner and quantitated using ImageJ software. The results are shown as the mean ± SD (n = 3), *, p < 0.05; **, p < 0.01; ***, p < 0.001, by Sidak’s multiple comparisons test. (B) GST-tagged HMGB1-expressing plasmid or empty vector was transfected into HEK293T cells with Flag-ORF3a or empty vector for 36 h. Cells were collected, lysed and subjected to immunoprecipitation with GST-affinity beads, and then whole cell lysates and immunoprecipitated complexes were analyzed as indicated. (C) GFP-ORF3a or empty vector was transfected into HEK293T cells for 36 h. Cells were collected, lysed and subjected to immunoprecipitation with GFP-affinity beads, and then whole cell lysates and immunoprecipitated complexes were analyzed by Western blots with anti-HMGB1 antibody as indicated. (D) Different amounts of shRNA against HMGB1 were transfected into HEK293T cells in the presence of Flag-ORF3a-expressing plasmids or empty vector. Thirty-six hours after transfection, cell pellets were collected, and the whole cell extracts were analyzed by Western blots to detect LC3-I-to-LC3-II conversion and CANX. The densities of LC3-II to ACTB were visualized by a LI-COR Odyssey scanner and analyzed using Imagej software. The results are shown as the mean ± SD (n = 3), *, p < 0.05; **, p < 0.01; ***, p < 0.001, by Sidak’s multiple comparisons test. (E) HeLa cells were transfected with empty vector or GFP-ORF3a- and mCherry-HMGB1-expressing plasmid, and then fixed and subjected to confocal microscopy analysis. Scale bar: 10 μm. The coefficient of colocalization was determined by qualitative analysis of the fluorescence intensity of the selected area in merge. (F) Different amounts of Flag-ORF3a-expressing plasmid or empty vector were transfected into HEK293T cells in the presence of GST-HMGB1- and GFP-BECN1-expressing plasmids for 36 h. Cells were collected and lysed, and immunoprecipitation was performed with GST-affinity beads. The whole cell lysates and immunoprecipitated complexes were analyzed as indicated. (G) Flag-ORF3a plasmid or empty vector was transfected into HEK293T cells with GFP-BECN1 plasmid and empty vector or different amounts of GST-HMGB1-expressing plasmid for 36 h. Cells were collected, immunoprecipitation with anti-GFP-affinity beads was performed, and then whole cell lysates and immunoprecipitated complexes were analyzed as indicated. (H) WT and BECN1-KO HEK293T cells were transfected with Flag-ORF3a-expressing plasmid and then left untreated or treated with HBSS for 2 h. Cells were harvested after an additional 50 μM CQ treatment for 4 h, and then cell extracts were detected by Western blots with the indicated antibodies. The densities of LC3, SQSTM1 and ACTB were quantitated and analyzed using ImageJ software. The results are shown as the mean ± SD (n = 3), ****, p < 0.0001, by Sidak’s multiple comparisons test.
In addition, ORF3a interacted with HMOX1 (Fig. S2C), and depletion of HMOX1 moderately attenuated LC3-I to LC3-II conversion in the presence of ORF3a expression, while the degradation of CANX was barely reversed by HMOX1 knockdown (Fig. S2D-E), indicating that HMOX1 is important for ORF3a-induced autophagy but may not be involved in ORF3a-mediated induction of reticulophagy. Given that HMOX1 contributes to oxidative stress and HMGB1 acts as a key regulator of autophagy under oxidative stress, HMOX1 and HMGB1 may cooperate in the regulation of autophagy under oxidative stress in the presence of ORF3a expression.
Importantly, ORF3a promoted HMGB1 translocation from the nucleus to the cytosol, and HMGB1 was then recruited by and colocalized with ORF3a in the endoplasmic reticulum, suggesting that ORF3a induces reticulophagy by regulating HMGB1 translocation and recruiting HMGB1 to the ER (Figure 3E). Mechanistically, HMGB1 associates with BECN1 to enhance autophagy [36,37], and the increased interaction of ORF3a with HMGB1 enhanced the association of BECN1 with HMGB1 (Figure 3F). ORF3a did not directly interact with BECN1, although they were linked via HMGB1 in a dose-dependent manner (Figure 3G), suggesting that ORF3a interacts with HMGB1-BECN1 complexes to enhance the HMGB1-BECN1 association and subsequently induce autophagy. To further detect the role of BECN1 in ORF3a-mediated regulation of autophagy, WT (wild-type) or BECN1-KO cells were transfected with an ORF3a-expressing plasmid, and the ORF3a-induced increase in LC3-I-to-LC3-II conversion and decrease in the SQSTM1 level were found to be abolished in BECN1-KO cells compared with WT cells (Figure 3G). These results suggest that ORF3a induces BECN1-dependent autophagy through the HMGB1-BECN1 pathway.
ORF3a induces ER stress and inflammatory responses through reticulophagy
To further investigate the function of ORF3a-induced autophagy, the transcriptome of ORF3a-expressing A549 cells was analyzed by RNA sequencing. The enrichment of the differentially expressed genes (DEGs) was analyzed by gene set enrichment analysis (GSEA). The enriched DEGs induced by ORF3a expression shared high similarity to the DEGs in SARS-COV-2- and MERS-CoV-infected cells (Fig. S3A). Ninety-seven DEGs were upregulated in both SARS-CoV-2-infected cells and ORF3a-expressing cells (Fig. S3B), and a cluster of genes involved in ER stress and immune and inflammatory responses were highly upregulated by both ORF3a expression and SARS-CoV-2 infection (Fig. S3C), suggesting that ORF3a plays a critical role in the induction of ER stress and inflammatory responses during SARS-CoV-2 infection.
RNA sequencing analysis showed that ER stress-related genes were enriched in the presence of ORF3a expression, indicating that ORF3a activates ER stress (Figure 4A). To confirm that ER stress is induced by ORF3a expression, the expression of ER stress-related genes was evaluated in ORF3a-expressing cells. The expression of HSPA5/BiP/GRP78 and DDIT3/CHOP was gradually increased with increasing ORF3a expression, and the phosphorylation of EIF2A was similarly increased by ORF3a expression (Figure 4B). Notably, the induction of ER stress by ORF3a expression requires autophagy, and the increases in HSPA5 and DDIT3 expression and EIF2A phosphorylation were completely abolished in BECN1-KO cells compared to WT cells (Figure 4C). Furthermore, depletion of RETREG1 greatly attenuated the induction of HSPA5 expression and EIF2A phosphorylation in the presence of ORF3a overexpression (Figure 4D). However, depletion of the ER stress-related gene DDIT3 barely affected autophagy induction mediated by ORF3a expression (Figure 4E), indicating that ORF3a induces autophagy independent of ER stress. To confirm that ORF3a physiologically induces ER stress during SARS-CoV-2 infection, control and ORF3a-overexpressing cells were left uninfected or infected with SARS-CoV-2, and increased HSPA5 expression and EIF2A phosphorylation was observed under ORF3a overexpression or during SARS-CoV-2 infection; moreover, both were augmented by ORF3a in virus-infected cells (Figure 4F). Notably, the increases in HSPA5 expression and EIF2A phosphorylation in SARS-CoV-2-infected cells were attenuated by RETREG1 knockdown (Figure 4G). These results suggest that ORF3a induces ER stress through reticulophagy during SARS-CoV-2 infection, probably because ORF3a-induced reticulophagy reprograms or disrupts ER homeostasis.
Figure 4.
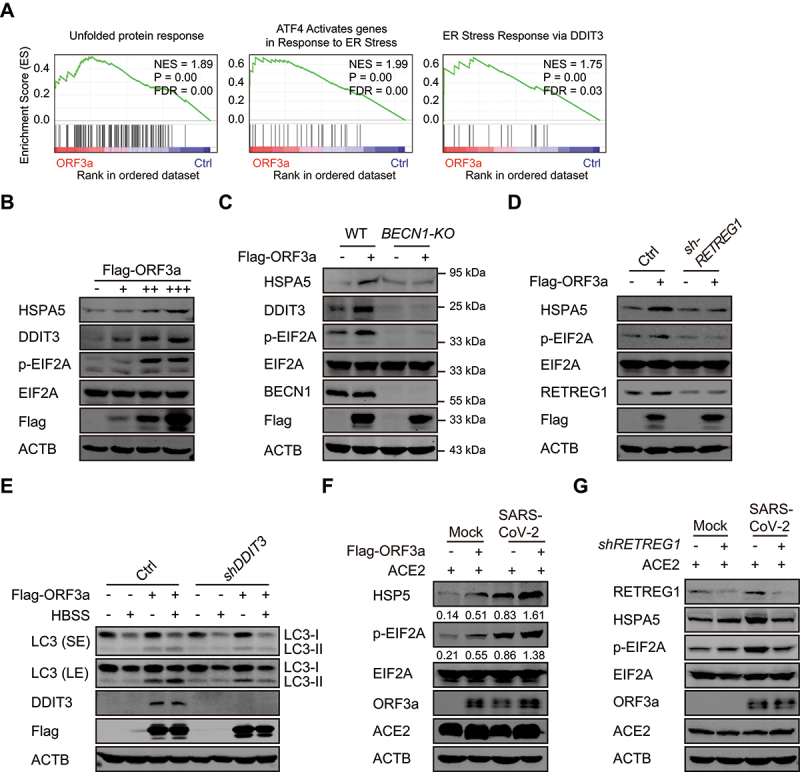
ORF3a activates ER stress through reticulophagy. (A) GSEA analyses of RNA-sequencing data of Flag-ORF3a-overexpressing A549 cells with “Hallmark gene sets” and “Curated gene sets”. NES, normalized enrichment score. FDR, false discovery rate. (B) Flag-ORF3a-expressing plasmid or empty vector was transfected into HEK293T cells. Thirty-six hours after transfection, cell pellets were lysed, and whole cell extracts were analyzed by Western blots with the indicated antibodies. (C) WT or BECN1-KO HEK293T cells were transfected with Flag-ORF3a-expressing plasmid for 36 h, and then lysates were immunoblotted with the indicated antibodies. (D) Control or shRETREG1-transduced A549 cells were transfected with Flag-ORF3a-expressing plasmid for 36 h, and then lysates were immunoblotted with the indicated antibodies. (E) Control and DDIT3 knockdown HEK293T cells were transfected with Flag-ORF3a-expressing plasmid or empty vector for 36 h, cell pellets were harvested after untreated or HBSS treatment for an additional 2 h, and then cell lysates were detected by Western blots with the indicated antibodies. (F) A549 cells were transfected with Flag-ORF3a-expressing plasmid or empty vector and ACE2-expressing plasmid for 24 h and then left uninfected or infected with SARS-CoV-2 for 24 h, MOI = 1. The cells were harvested and the whole cell extracts were analyzed as indicated. The relative ratio of HSPA5/p-EIF2A to ACTB were calculated and shown. (G) Control or RETREG1 knockdown A549 cells (transfected with ACE2-expressing plasmid for 24 h) were left uninfected or infected with SARS-CoV-2 for 24 h, MOI = 1. The cells were harvested, and the whole cell extracts were analyzed by Western blots.
Furthermore, we constructed a series of ORF3a constructs (Fig. S4A) and mapped the functional regions of ORF3a. The results showed that the C-terminal domain was responsible for HMGB1 binding, and deletion of the C-terminal domain abolished the ORF3a-HMGB1 interaction and the induction of reticulophagy and ER stress (Fig. S4B-C); however, the C-terminal domain alone failed to exert these functions even though it interacted with HMGB1. An additional transmembrane (TM) domain was required, but the TM domain alone also failed in these functions. These results indicate that ORF3a primarily induces reticulophagy through the ORF3a-HMGB1 interaction and its localization in the ER compartment and consequently induces ER stress, which excludes the possibility that ORF3a directly acts as a misfolded protein and induces the unfolded protein response or ER overload response.
ER dysfunction often triggers innate immune and inflammatory responses. The expression of innate immune and inflammatory genes was increased in the presence of ORF3a expression (Figure 5A); thus, the expression of key cytokines and the proinflammatory factors IFNB/IFNβ, IL1B/IL-1β, IL6 and TNF/TNFα was induced by ORF3a expression (Figure 5B-D). The induction of these factors was dependent on reticulophagy, because BECN1 deficiency in BECN1-KO cells or RETREG1 depletion by shRNA transduction almost abolished the increases in the expression of these cytokines observed in WT cells (Figure 5B-C). Similarly, depletion of DDIT3 expression also greatly attenuated these increases in the presence of ORF3a expression (Figure 5D). These results indicate that ORF3a induces the expression of innate immune and inflammatory genes through reticulophagy and ER stress. Consistent with this hypothesis, expression of the key cytokines IFNB and TNF was induced during SARS-CoV-2 infection, and ORF3a overexpression augmented their expression during viral infection (Fig. S3D). Furthermore, SARS-CoV-2 upregulated the gene expression of IFNB, IFIT2, IL6 and TNF, while the expression of these genes was significantly attenuated by depletion of RETREG1, HMGB1, DDIT3 or ERN1 (Figure 5G and Fig. S3E). These results suggest that ORF3a expression and SARS-CoV-2 infection induce innate immune and inflammatory responses through reticulophagy and subsequent ER stress.
Figure 5.
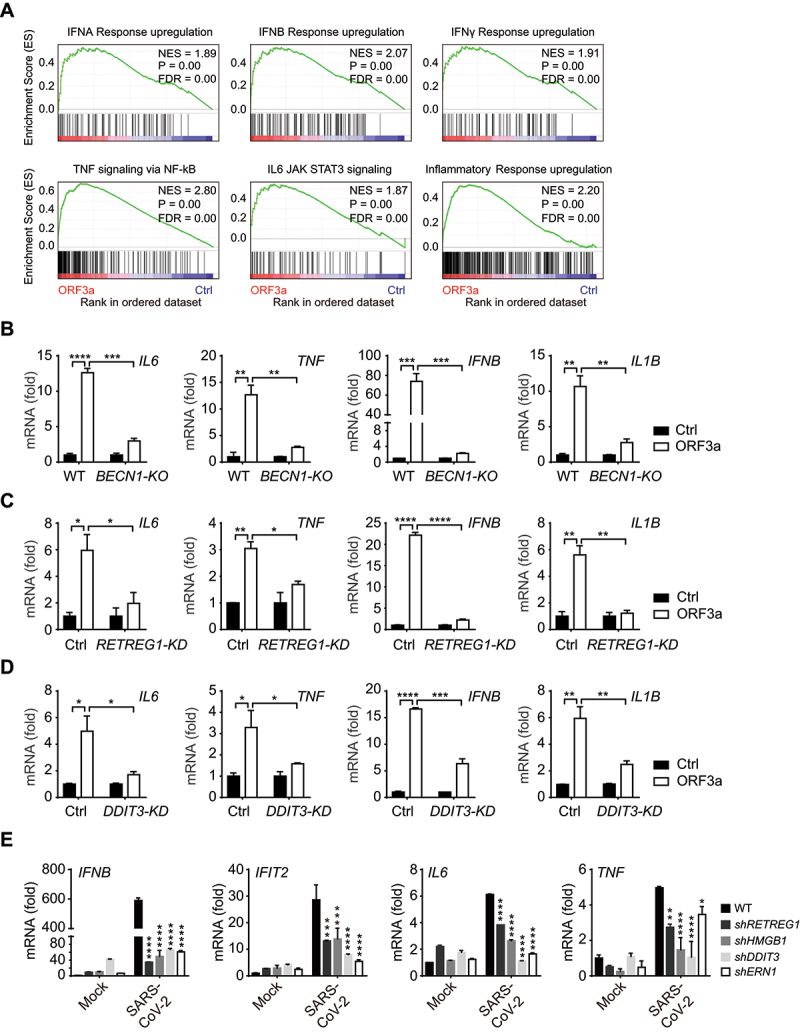
ORF3a and SARS-CoV-2 infection activates inflammatory gene expression through reticulophagy and ER stress. (A) GSEA analyses of RNA-sequencing data of Flag-ORF3a-overexpressing A549 cells with “Hallmark gene sets” and “Curated gene sets”. NES, normalized enrichment score. FDR, false discovery rate. (B-D) WT vs. BECN1-KO HEK293T cells (B), control vs. shRETREG1 (C) and control vs. shDDIT3 A549 cells (D) were transfected with Flag-ORF3a-expressing plasmid or empty vector as a control. Twenty-four hours post transfection, cells were treated with poly(I:C) (5 μg/ml) for 24 h (B). And then total RNA was extracted and subjected to quantitative RT-PCR analysis to detect IL6, TNF, IFNB and IL1B expression. The results are shown as the mean ± SD (n = 3), *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, by Sidak’s multiple comparisons test. (E) Control vs. RETREG1, HMGB1, DDIT3 or ERN1 knockdown A549 cells (transfected with ACE2-expressing plasmid for 24 h) were untreated or infected with SARS-CoV-2 for 24 h, MOI = 1, and then total RNA was extracted and subjected to quantitative RT-PCR analysis to detect IL6, TNF, IFNB and IFIT2 expression. The results are shown as the mean ± SD (n = 3), *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, by Sidak’s multiple comparisons test.
ORF3a-induced reticulophagy and ER stress sensitize cells to ER apoptotic cell death
Considering that ORF3a induces ER stress, we hypothesized that ORF3a disrupts ER homeostasis and promotes cell death in the context of ER dysfunction. SARS-CoV-2 ORF3a-induced apoptosis was investigated using ANXA5/annexin V FITC and propidium iodide/PI double staining plus FACS analysis and was compared between WT and BECN1-KO HEK293T cells and between control and RETREG1 knockdown A549 cells. Under normal culture conditions, the percentage of ANXA5+ PI− early apoptotic cells was significantly increased but that of ANXA5+ PI+ late apoptotic cells were only slightly increased in cells with ectopic ORF3a expression compared with the corresponding control cells, and the increase in the percentage of early apoptotic cells was greatly reversed in BECN1-KO or RETREG1 knockdown cells (Figure 6A-B). These results indicate that ORF3a overexpression induces the early apoptotic phenotype through reticulophagy and ER stress.
Figure 6.
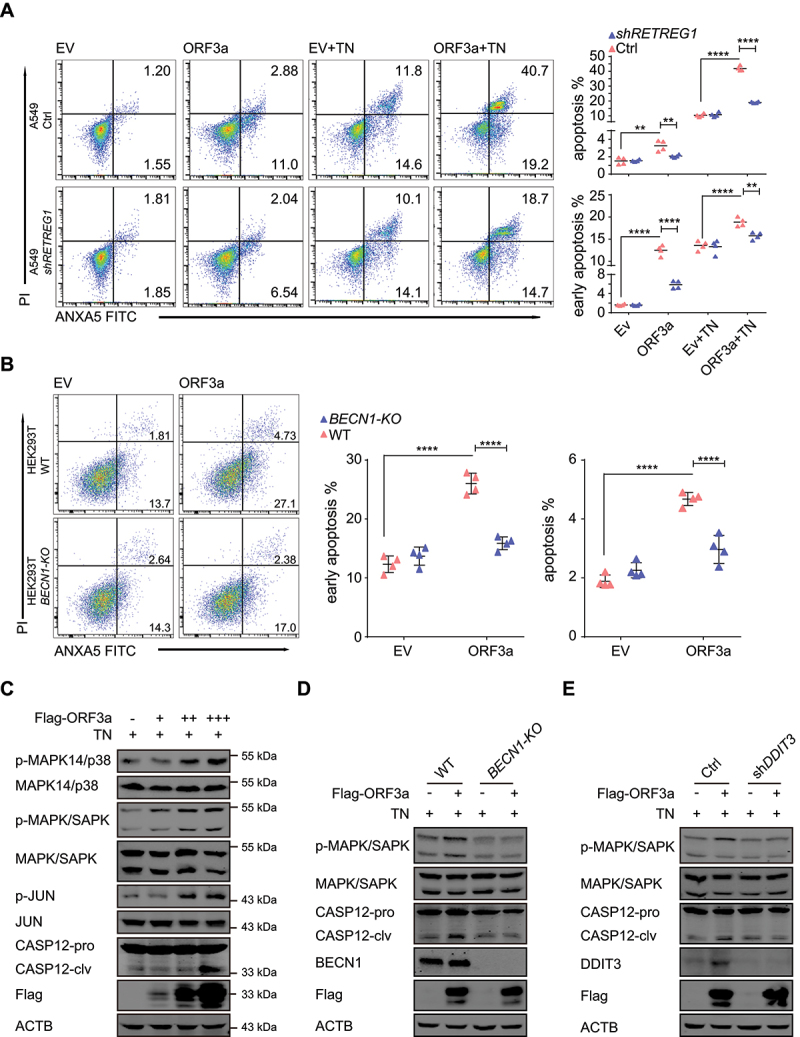
ORF3a enhances ER stress-induced apoptosis through autophagy. (A) WT vs. RETREG1-knockdown A549 cells were transfected with Flag-ORF3a-expressing plasmid or empty vector. After treated with 3 μg/ml TN for 15 h, cells were stained with ANXA5 FITC-PI for flow cytometric analysis, and the representative images and the percentages of apoptotic cells was measured and shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, by Sidak’s multiple comparisons test. (B) WT vs. BECN1-KO HEK293T cells were transfected with Flag-ORF3a-expressing plasmid or empty vector for 48 h. Cells were stained with ANXA5 FITC-PI for flow cytometric analysis, and the representative images and the percentages of apoptotic cells was measured and shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, by Sidak’s multiple comparisons test. (C) Flag-ORF3a-expressing plasmid or empty vector was transfected into HEK293T cells. Twenty-four hours after transfection, cells were treated with 3 μg/ml TN for 15 h, cell pellets were collected, and whole cell extracts were analyzed by Western blots with the indicated antibodies. (D) WT and BECN1-KO HEK293T cells were transfected with Flag-ORF3a-expressing plasmid, cells were harvested after 3 μg/ml TN treatment for 15 h, and then lysates were detected with the indicated antibodies. (E) WT and DDIT3 knockdown HEK293T cells were transfected with Flag-ORF3a-expressing plasmid, cell extracts were prepared after TN (3 μg/ml) treatment for 15 h, and then lysates were detected by Western blots with the indicated antibodies.
To further evaluate ER stress-related cell death, ORF3a-expressing HEK293T cells were treated with tunicamycin (TN), an inducer of ER stress that causes the unfolded protein response and induces ER stress by inhibiting N-linked glycosylation of GlcNAc phosphotransferase. TN-induced apoptosis was also augmented by ORF3a expression in a reticulophagy-dependent manner, and the enhanced apoptotic phenotype was abolished by RETREG1 depletion (Figure 6A). Under TN treatment, phosphorylation of the stress-related kinases MAPK14/p38 and MAPK/SAPK, as well as JUN, was increased in the presence of ORF3a expression (Figure 6C, top). Importantly, cleavage of ER-specific CASP12 (caspase 12) was increased by ORF3a expression under TN treatment (Figure 6C, bottom). These results suggest that ORF3a enhances ER apoptotic cell death under ER stress.
To confirm that the increased cell death is responsible for reticulophagy and ER stress, apoptotic cell death in the ER was further investigated under autophagy-deficient or ER stress-deficient conditions in cells with depletion of BECN1 or DDIT3, respectively. ORF3a-induced phosphorylation of MAPK/SAPK and cleavage of CASP12 were greatly reversed when reticulophagy was blocked in BECN1-KO cells (Figure 6D), indicating that reticulophagy is required for ORF3a-induced ER-related apoptosis. Similarly, blockade of ER stress by DDIT3 depletion also eliminated ER-related apoptosis in ORF3a-expressing cells (Figure 6E). Collectively, our results suggest that ORF3a overexpression triggers ER stress and ER abnormalities through reticulophagy and consequently enhances the sensitivity of SARS-CoV-2-infected cells to ER stress-related drugs.
ORF3a promotes virus replication through reticulophagy and ER stress
Studies have shown that autophagy is essential for SARS-CoV-2 infection and replication [24], and we further investigated whether ORF3a promotes SARS-CoV-2 infection and replication. When A549 cells were transfected with the ORF3a construct and then infected with SARS-CoV-2, the expression levels of the viral genes S, N and RdRp were greatly increased in the presence of ORF3a overexpression (Figure 7A), indicating that ORF3a promotes SARS-CoV-2 infection and replication. To further investigate the role of ORF3a-induced reticulophagy and ER stress in SARS-CoV-2 infection and replication, A549 cells were transfected with shRNAs to block reticulophagy or ER stress and were then infected with SARS-CoV-2. We found that depletion of the reticulophagy-related genes RETREG1 and HMGB1 or the ER stress-related genes ERN1 and DDIT3 significantly downregulated S, N and RdRp expression compared with that in control cells infected with SARS-CoV-2 (Figure 7B), suggesting that reticulophagy and ER stress are required for SARS-CoV-2 infection and replication. In addition, the enhancement of virus replication by ORF3a was greatly abolished in RETREG1 or DDIT3 knockdown cells compared with WT cells (Figure 7A), suggesting that ORF3a promotes SARS-CoV-2 replication through reticulophagy and ER stress. In conclusion, our studies reveal that SARS-CoV-2 hijacks reticulophagy and ER stress through ORF3a to facilitate viral infection and replication.
Figure 7.
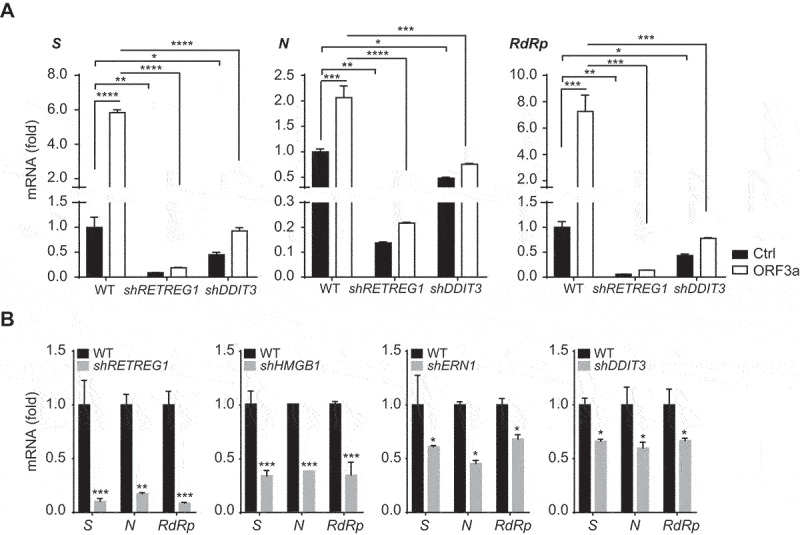
ORF3a induces virus replication through reticulophagy and ER stress. (A) Control vs. RETREG1 or DDIT3 knockdown A549 cells were transfected with empty vector or Flag-ORF3a- and ACE2-expressing plasmid for 24 h and then left untreated or infected with SARS-CoV-2 for 24 h, MOI = 1. The total RNA was extracted and subjected to quantitative RT-PCR analysis to the expression of SARS-CoV-2 S, N and RdRp. The results are shown as the mean ± SD (n = 3), *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, by Sidak’s multiple comparisons test. (B) Control vs. RETREG1, HMGB1, ERN1 or DDIT3 knockdown A549 cells (transfected with ACE2-expressing plasmid for 24 h) were infected with SARS-CoV-2 for 24 h, MOI = 1.The total RNA was extracted and subjected to quantitative RT-PCR analysis to detect the expression of SARS-CoV-2 S, N and RdRp. The results are shown as the mean ± SD (n = 3), *, p < 0.05; **, p < 0.01; ***, p < 0.001, by Sidak’s multiple comparisons test.
Discussion
Studies have confirmed that autophagy plays essential roles in the infection and replication of coronaviruses [38,39]; inhibition of autophagy by genetic approaches or chemical inhibitors effectively suppresses coronavirus infection and might offer a promising therapeutic strategy for treating the related infectious diseases. As SARS-CoV-2 is an emerging coronavirus causing severe disease, the modulation and function of autophagy in its infection and resulting disease remain unclear. In the present study, we revealed that SARS-CoV-2 ORF3a localizes to the ER compartment and induces RETREG1-mediated reticulophagy through the HMGB1-BECN1 pathway and then contributes to ER stress, consequently promoting SARS-CoV-2 infection/replication and activating proinflammatory responses through reticulophagy and ER functional reprogramming (Figure 8). As a consequence, sensitivity to ER stress-related drugs and an early ER apoptotic phenotype were greatly enhanced in ORF3a-expressing cells. This enhancement improves the therapeutic potential of autophagy- and ER stress-related drugs in the treatment of SARS-CoV-2 infection and COVID-19 to suppress severe inflammatory responses and induce the death of SARS-CoV-2-infected cells.
Figure 8.
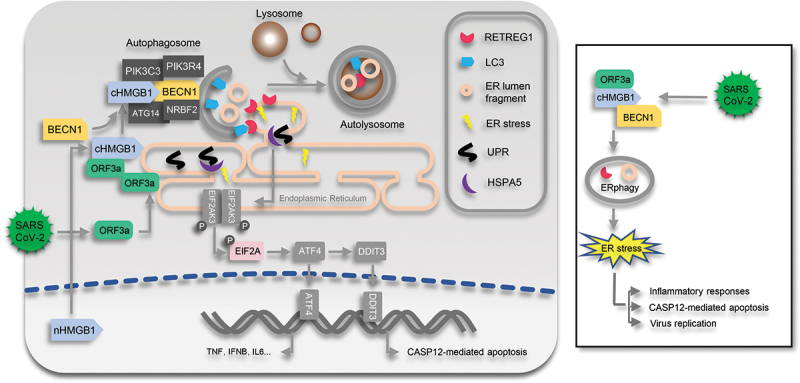
Diagram of ORF3a-induced reticulophagy, ER stress and inflammatory responses. During SARS-CoV-2 infection, ORF3a localizes to ER and interacts with HMGB1 and then enhances the association between HMGB1 and BECN1 to initiate RETREG1-mediated reticulophagy. Consequently, ER stress is induced by ORF3a through reticulophagy to facilitate SARS-CoV-2 infection, trigger proinflammatory responses and enhance the sensitivity of cells to CASP12-mediated ER apoptotic cell death. nHMGB1, nuclear HMGB1; cHMGB1, cytosolic HMGB1.
The autophagy inhibitor CQ and its derivatives suppress SARS-CoV-2 infection and replication [24,25], raising considerable hope for the use of these agents in therapeutic COVID-19 treatment and prevention. Although these agents have exhibited low effectiveness and benefit in clinical trials [26,27], autophagy has been confirmed to play essential roles in the SARS-CoV-2 life cycle. First, coronaviruses enter cells through endocytosis [40] and egress through lysosomes [41], a process that requires fine control of autophagy and lysosomal degradation. Second, a recent study showed that SARS-CoV-2 infection and replication require autophagosome-like double-membrane vacuoles for RNA export and replication [29], indicating the essentiality of autophagy and the autophagic machinery for viral replication and assembly. Finally, the maturation and transport of SARS-CoV-2 virions, like those of other RNA viruses, might also require autophagy or autophagic vacuoles [16]. However, excessive autophagy is not beneficial to either cells or viruses, because the killing of infected cells or clearance of intracellular viruses by strong autophagy induction is an effective antiviral defense mechanism. Therefore, autophagic processes are finely regulated to limit them to the appropriate strength and duration for facilitating cell survival and viral replication. Inhibitory viral proteins or other products of autophagy must exist during SARS-CoV-2 infection, as indicated in our preliminary screening; i.e., several viral proteins can suppress autophagy (Fig. S1). In fact, several viral proteins of coronaviruses induce incomplete autophagy, and replication of coronaviruses utilizes the autophagic machinery while not requiring the complete set of autophagic genes and pathways [23,42]. Thus, viral regulation of autophagy and its function in SARS-CoV-2 infection require further investigation.
Several studies have revealed that ORF3a inhibits the fusion of autophagosomes and lysosomes by binding to VPS39 [31,32,43], resulting in accumulation of LC3-II and autophagosomes; however, our results showed that depletion of VPS11 or VPS39 did not decrease LC3-I-to-LC3-II conversion in the presence of ORF3a expression, while HMOX1 or HMGB1 knockdown did (Figure 3A). This finding can possibly be explained as follows: ORF3a may induce incomplete autophagy, by which it induces the initiation of autophagy at an early stage through BECN1-dependent pathways while inhibiting the formation of autolysosomes and autophagic degradation, consequently resulting in accumulation of autophagosomes and double-membrane vesicles to facilitate SARS-CoV-2 replication.
ORF3a interacts with several proteins that may regulate autophagy and lysosomal degradation [33], and our studies confirmed that ORF3a interacts with HMGB1 and HMOX1, which are involved in the induction of autophagy under oxidative stress; however, HMGB1 but not HMOX1 was found to be essential for reticulophagy in the presence of ORF3a expression. ORF3a altered HMGB1 translocation from the nucleus to the cytosol and recruited it to the ER, consequently initiating reticulophagy through a BECN1-dependent pathway. Our studies revealed that ORF3a interacts with HMGB1-BECN1 complexes and enhances the HMGB1-BECN1 association, although it may not directly bind to BECN1(Figure 3). In addition, the autophagy regulator UVRAG is excluded from BECN1 complexes by ORF3a [44]. These results indicate that ORF3a regulates the formation and activity of BECN1 complexes during the initiation of autophagy by recruiting HMGB1 and excluding UVRAG.
SARS-CoV-2 infection induces RETREG1-mediated reticulophagy, similar to ORF3a overexpression alone under comparable ORF3a expression levels, and ORF3a induces reticulophagy during SARS-CoV-2 infection, while both minimally affect mitophagy (Figure 2F), indicating that ORF3a mainly induces reticulophagy during SARS-CoV-2 infection. However, several viral proteins may activate or inhibit autophagy in different manners [31,32,43], and thus, the possibility that other viral proteins, such as NSP6 and ORF8, exhibit this function cannot be excluded because they can also induce autophagy (Fig. S1A).
The membranes of the ER and mitochondria are the main sources of autophagic membranes, and our studies showed that ORF3a localizes to the ER and induces RETREG1-dependent reticulophagy (Figure 2). The membrane of which ORF3a-induced autophagosomes are formed probably originates mainly from the ER [45] and is then degraded/recycled through reticulophagy. On the other hand, our findings revealed that ORF3a-induced reticulophagy reprograms ER homeostasis and activates ER stress (Figure 4), which also benefits SARS-CoV-2 replication (Figure 7), probably to allow infected cells to tolerate and adapt to the accumulation of viral proteins and recover ER function under virus-induced stresses. Many proteins of coronavirus rapidly translocate to the ER, reprogram ER function [46] and form double-membrane vacuoles from the ER for viral replication [16,29]. We confirmed that ORF3a expression and SARS-CoV-2 infection induce RETREG1-mediated reticulophagy; consequently, both activate ER stress through BECN1- and RETREG1-dependent reticulophagy, and BECN1 deficiency or RETREG1 depletion abolishes the activation of ER stress in the presence of ORF3a expression or during SARS-CoV-2 infection, representing a novel mechanism for the induction of adaptive ER stress through reticulophagy during SARS-CoV-2 infection. Reticulophagy and ER stress are probably activated by SARS-CoV-2 through ORF3a to restrict the inappropriate overload of viral proteins in the ER membrane and lumen, recycle overabundant viral proteins or repair ER damage and maintain ER homeostasis.
As a consequence, reticulophagy and ER stress induced by ORF3a reprogram ER function and activate inflammatory responses. Studies have shown that SARS-CoV-2 infection induces ER stress and activates proinflammatory responses in diverse types of cells [47,48]. Our studies confirmed that inflammatory responses are mainly induced by ORF3a through reticulophagy-mediated ER stress and that blockade of reticulophagy by RETREG1 depletion or of ER stress by DDIT3 depletion greatly eliminates the induction of key cytokines and inflammatory factors under ORF3a expression or during SARS-CoV-2 infection. Notably, ORF3a-mediated induction of inflammatory gene expression was not completely abolished in BECN1-KO or RETREG1-depleted cells (Figure 5B-C), likely because ORF3a associates with other targets, such as HMOX1, and triggers additional inflammatory gene expression through oxidative stress or other mechanisms. This finding provides evidence that reticulophagy and ER stress are the key upstream modulators of inflammatory responses during SARS-CoV-2 infection and may act as the initial precipitating factors of cytokine storm in COVID-19 patients, providing a novel target and approach to alleviate acute inflammatory responses during early SARS-CoV-2 infection through suppression of autophagy and ER stress.
Finally, considering that ER stress is activated and ER function is reprogrammed by ORF3a expression, we hypothesized that ORF3a may enhance the sensitivity of SARS-CoV-2-infected cells to ER stress-related drugs. Indeed, ORF3a-expressing cells exhibit RETREG1- and DDIT3-related early apoptotic phenotypes and are sensitive to treatment with the ER stress-inducing drug TN. The stress-related kinases MAPK14/p38 and MAPK/SAPK were activated, and cleavage of ER-specific apoptotic CASP12 was enhanced by ORF3a under TN treatment. Correspondingly, these effects were abolished in BECN1-KO cells and DDIT3-depleted cells, indicating that ORF3a enhances ER apoptotic cell death through excessive reticulophagy and ER stress. These findings also support the therapeutic potential of reticulophagy and ER stress in COVID-19, consistent with the hypothesis that ER stress is increased in COVID-19 patients and may benefit treatment [49–51].
Interestingly, this finding is different from what is known for other RNA viruses, such as Zika virus. SARS-CoV-2 replicates in autophagosome-like double-membrane vacuoles [29], while Zika replicates within ER membrane-bound compartments [52], named viral replication organelles. Zika virus suppresses reticulophagy and reprograms the ER machinery to establish ER-localized replication compartments [53,54], while SARS-CoV-2 induces reticulophagy and the formation of double-membrane vacuoles, likely from the ER, through an autophagy-like mechanism. Thus, reticulophagy-mediated ER turnover should be differentially regulated and exhibit the opposite effects in response to different viruses.
In summary, our studies identified a novel viral inducer of reticulophagy during SARS-CoV-2 infection. We found that ORF3a localizes to the ER and triggers reticulophagy through the HMGB1-BECN1 pathway, consequently inducing ER stress and promoting SARS-CoV-2 infection and inflammatory responses. Our findings revealed the sequential induction of reticulophagy, ER stress and inflammatory responses by SARS-CoV-2 infection and ORF3a expression, facilitating insight into the hijacking of autophagy, reprogramming of ER functions and activation of acute inflammatory responses during SARS-CoV-2 infection and pathogenesis. Therefore, our studies reveal the regulation, requirement and importance of reticulophagy and its consequences during SARS-CoV-2 infection and replication and provide a novel therapeutic approach for COVID-19 by suppressing autophagy and modulating ER stress.
Materials and methods
Cells, antibodies and chemicals
HeLa (ATCC, CCL-2), HEK293 (ATCC, CL-0001), HEK293T (ATCC, CRL-11268) and BECN1-KO HEK293T (kindly provided by Dr Yang Du, Sun Yat-Sen University) cells were cultured in DMEM (Gibco, 12,800,017) supplemented with 10% fetal bovine serum (FBS; Biological Industries, 04–001-1A); human adenocarcinoma lung tissue-derived epithelial (A549; ATCC, CL-0016) cells were cultured in RPMI 1640 (Gibco, 31,800,022) medium with 10% FBS.
The following antibodies were used in this study: anti-LC3B (3868), BECN1 (3495), AKT (4691), p-AKT (Thr308; 2965), p-AKT (Ser473; 4060), HSPA5 (3183), DDIT3 (2895), p-MAPK/SAPK (Thr183/Tyr185; 4668), MAPK/SAPK (9252), p-MAPK14/p38 (Thr180/Tyr182; 4511), MAPK14/p38 (8690), p-EIF2A (3398), EIF2A (5324), p-JUN (Ser73; 3270) and JUN (9165) were purchased from Cell Signaling Technology; anti-SQSTM1 (P0067) and Flag (F3165) were purchased from Sigma-Aldrich; anti-CASP12 (55,238-1-AP), CANX (10,427-2-AP) and RTN4 (10,740-1-AP) were purchased from Proteintech Group; anti-ACTB/β-actin (ab8227), GST (ab181652) and SARS-CoV-2-ORF3a (ab280953) were purchased from Abcam; anti-TIMM23 (DF12052), p-ULK1 (Ser555; AF7148) and RETREG1 (DF12997) were purchased from Affinity Biosciences; anti-ULK1 (ET1704-63), TOMM20 (ET1609-25) and HMGB1 (ET1601-2) were purchased from HuaAn Biotechnology; anti-GFP (AE012) and HA (AE008) were purchased from ABclonal; goat anti-mouse IRDye680RD (C90710-09) and goat anti-rabbit IRDye800CW (C80925-05) were purchased from Li-COR; tunicamycin (HY-A0098) was purchased from MCE; FITC Annexin V Apoptosis Detection kit (559,763) was purchased from BD Biosciences.
Plasmids
The Flag-ORF3a- and Flag-ACE2-expressing plasmid were kindly and respectively provided by the Pei-Hui Wang laboratory (Shandong University) and Hui Zhang laboratory (Sun Yat-sen University). Plasmids for ORF3a mutations were constructed and subcloned into the pCMV-3Tag-1A vector (Agilent Technologies, 240,195). Plasmids for GFP-ORF3a, GFP-BECN1 and GFP-RETREG1 were constructed and subcloned into the pEGFP-C2 vector (EK-Bioscience, MY1108). Plasmids for mCherry-HMGB1, mCherry-ORF3a and mCherry-RTN3 were constructed and subcloned into the p-mCherry-C2 vector (modified from pEGFP-C2 vector). Plasmids for HA-HMOX1 and Actin-LC3-DN were constructed and subcloned into the pcDNA3.1 vector (EK-Bioscience, MY1012). Plasmids for GFP-LC3 and ptfLC3 were purchased from Addgene (21,073 and 21,074; deposited by Tamotsu Yoshimori). Plasmids for SERP1-mCherry-GFP was modified from ptfLC3. Plasmids for GST-HMGB1 was constructed and subcloned into the pEBG vector (Addgene, 22,227; deposited by David Baltimore). To generate gene knockdown, shRNA target sequences were subcloned into the pLKO.1 vector (EK-Bioscience, MY1413) between EcoRI and AgeI sites. The sequences of shRNAs are listed in Table S1A.
Immunoprecipitation and Western blotting analysis
In brief, one 10-cm dish of cells was transfected with 10–15 μg plasmid for 48 h. The cells were then collected and lysed in the presence of a protease inhibitor cocktail (Roche,6,538,282,001) and phosphatase inhibitors (Sigma-Aldrich, 4,906,845,001). For immunoprecipitation, the same amounts of cell lysates were precleaned and incubated with antibodies at 4°C overnight, and then protein complexes were precipitated with protein G-agarose (Sigma-Aldrich, 11,243,233,001), or the precleaned cell lysates were incubated with GST affinity beads (Cytiva, 17,075,601) at 4°C overnight. After washing five times, the immunoprecipitated complexes were separated by SDS-PAGE and subjected to immunoblotting analysis. For Western blotting, 40–60 μg protein of whole cell extracts per lane was separated by SDS-PAGE and transferred to membranes. The membranes were blocked in 5% dry milk, incubated with primary antibodies at 4°C overnight, and subsequently incubated with species-matched IRDye680- or IRDye800-labeled secondary antibodies for 2 h at room temperature. After 5 washes, images were visualized using a LI-COR Odyssey system.
Real-time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen, 15,596–026) and reverse-transcribed using HiScript® III RT SuperMix (Vazyme, R223-01). Real-time PCR was performed with a SYBR Green I Master Mix kit (Roche, 4,887,352,001) and LightCycler® 480 system. The primer pairs are listed in supplementary Table S1B.
Immunofluorescence staining
The cells were fixed with 4% formaldehyde in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4; BOSTER, AR0030) for 30 min, permeabilized with methanol for 10 min, and blocked with 2% bovine serum albumin in PBS for 30 min. Then, the cells were incubated with primary antibody overnight. After three washes with PBS containing 0.1% Triton X-100 (Beyotime, ST795), the cells were incubated with Alexa Fluor 488- or 555-labeled anti-rabbit IgG antibodies (Invitrogen, A11034 and A27039) for 1 h. The cells were counterstained with 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, D9542) followed by three additional washes. The cells were mounted in antifade agent on glass slides and visualized with a confocal fluorescence microscope (Zeiss LSM800 microscopy with a 64× NA oil-immersion objective).
Luciferase assays
A Gaussia luciferase-based reporter to detect pre-LC3 cleavage was used for the detection of autophagy activation30. Briefly, the Actin-LC3-DN reporter was transfected into HEK293T cells with either empty vector or Flag-ORF3a-expressing plasmid in a 96-well plate. Twenty-four hours later, the cell supernatants were collected, and the secretion of Gaussia luciferase was measured using the reagent coelenterazine (CTZ; MCE, HY-18743) in a TriStar multimode reader.
Electron microscopy
Flag-ORF3a-expressing or control HEK293 cells were scraped, resuspended in DMEM and centrifuged in 1.5-ml centrifuge tubes. The medium was removed, and the cell pellets were fixed in 3% glutaraldehyde overnight at 4°C. The pellets were gently collected and placed in a specimen bottle containing 3% glutaraldehyde for fixation for another 24 h. After the fixed solution was discarded, the cells were rinsed with 0.1 M phosphate buffer for 1 h, washed 3–4 times, and fixed in 1% osmium tetroxide for 1 h. This was followed by another 3 × 10 min washes in 0.1 M phosphate buffer, pH 7.0 for 1 h. After dehydration through a series of ethanol washes from 30% to 2 washes in 100%, the cells were incubated in a mixture of Epon (EMCN, YB-AA-0855) and propylene oxide (3:1, v: v) for 90 min, embedded in a mold of 100% plastic, placed in a 70°C oven and allowed to harden overnight (Epon 812). The cells were cut using a diamond knife to obtain ultrathin slices; the slices were placed on a hexadecimal copper grid (Electron Microscopy Science, H200-Cu). After drying for 2 h, transmission electron microscopy was performed on these grids using an FEI Tecnai G2 Spirit Twin electron microscope (13,500×).
Apoptosis assays
Apoptosis was assessed using the FITC Annexin V Apoptosis Detection kit (ANXA5 FITC, propidium iodide/PI solution and ANXA5 binding buffer). The cells were stained with ANXA5 (a phospholipid-binding protein that binds to disrupted cell membranes) in combination with PI (a vital dye that binds to DNA penetrating into apoptotic cells). Flow cytometric analysis (FACS) was performed to determine the percentage of cells that were undergoing early apoptosis (ANXA5+ PI−) or apoptosis (ANXA5+ PI+).
RNA sequencing analysis and data availability
A549 cells were transfected with Flag-ORF3a or empty vector for 36 h, and total RNA was extracted with TRIzol following manufacturer procedures. Approximately 5 μg of total RNA was subjected to deep RNA sequencing. Libraries were constructed and sequenced on an Illumina platform, and 150 bp paired-end reads were generated. Data analysis was performed with Molecule Annotation System 3.0 (Annoroad Gene Technology Co., Beijing, China). The raw sequencing datasets generated in this study are available on the NCBI Gene Expression Omnibus server under the accession number GSE158484. The RNA-sequencing data of SARS-CoV-2-infected cells were downloaded under the accession number GSE147507 from previously published analysis [55].
GSEA
The results from the RNA sequencing analysis were filtered to yield a list of genes that displayed a ≥ 1.5-fold change in expression across experimental samples and reached a significance level with a corrected p value of ≤ 0.05. Gene set enrichment analysis was performed using GSEA software (https://www.gsea-msigdb.org/gsea/index.jsp) with number of permutations = 1000, permutation type = gene_set, enrichment statistic = weighted and metric for ranking genes = Signal2Noise. In addition, GSEA evaluates a query microarray dataset by using a collection of gene sets called MSigDB (Molecular Signatures Database), consisting of eight major collections. We acquired outputs on ER stress and inflammatory responses with two gene sets, “Hallmark” (specific well-defined biological states or processes and coherent expression): the dataset references included GSE27038, GSE8322, GSE11819, GSE21060, GSE2639 and GSE16193; “Curated” (online pathway databases, publications in PubMed, and knowledge of domain experts): the dataset references included GSE2082, GSE56192 and GSE147507. Finally, enrichment outputs were considered significant and selected when NES ≥ 1, p < 0.05 and FDR < 0.25.
Statistical analyses
All statistical analysis was performed in Prism 7 software (GraphPad Software, La Jolla, CA, USA). One-way or Two-way ANOVA with Sidak adjustments was performed for multiple comparisons. Statistical significance was defined as p value<0.05.
Supplementary Material
Acknowledgments
We thank all the members of our laboratory for their critical assistance and helpful discussions. This work is supported by grants from the National Natural Science Foundation of China (81871643 and 32061143008) to E.K. and the National Natural Science Foundation of China (81971928) to X.Li.
Funding Statement
This work was supported by the National Natural Science Foundation of China [81871643]; National Natural Science Foundation of China [81971928]; National Natural Science Foundation of China [32061143008].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Zhou P, Yang X-L, Wang X-G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhu N, Zhang D, Wang W, et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fehr AR, Channappanavar R, Perlman S.. Middle east respiratory syndrome: emergence of a pathogenic human coronavirus. Annu Rev Med. 2017;68(1):387–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Channappanavar R, Fehr AR, Vijay R, et al. Dysregulated type i interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-INFECTED mice. Cell Host Microbe. 2016;19(2):181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92(4):418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dj K. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–937. [DOI] [PubMed] [Google Scholar]
- [8].Scott RC, Juhász G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. [DOI] [PubMed] [Google Scholar]
- [10].Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115(10):2679–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125(1):14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Orvedahl A, Alexander D, Talloczy Z, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the beclin 1 autophagy protein. Cell Host Microbe. 2007;1(1):23–35. [DOI] [PubMed] [Google Scholar]
- [14].Ding B, Zhang G, Yang X, et al. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe. 2014;15(5):564–577. [DOI] [PubMed] [Google Scholar]
- [15].Chen D, Zheng Q, Sun L, et al. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev Cell. 2021. 6; 56(23): 3250–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wolff G, Melia CE, Snijder EJ, et al. Double-membrane vesicles as platforms for viral replication. Trends Microbiol. 2020;28(12):1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gosert R, Kanjanahaluethai A, Egger D, et al. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J Virol. 2002;76(8):3697–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee JS, Li Q, Lee JY, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11(11):1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yue Y, Nabar NR, Shi CS, et al. SARS-Coronavirus Open Reading Frame-3a drives multimodal necrotic cell death. Cell Death Dis. 2018;9(9):904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shi CS, Nabar NR, Huang NN, et al. SARS-Coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019;5(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cottam EM, Maier HJ, Manifava M, et al. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 2011;7(11):1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Prentice E, Jerome WG, Yoshimori T, et al. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem. 2004;279(11):10136–10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu J, Cao R, Xu M, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020;6(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tang W, Cao Z, Han M, et al. Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: open label, randomised controlled trial. BMJ. 2020;369:m1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Borba MGS, Val FFA, Sampaio VS, et al. Effect of high vs low doses of chloroquine diphosphate as adjunctive therapy for patients hospitalized with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Infection: a randomized clinical trial. JAMA Network Open. 2020;3(4):e208857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schluenz LA, Ramos-Otero GP, Nawarskas JJ. Chloroquine or hydroxychloroquine for management of coronavirus disease 2019: friend or foe? Cardiol Rev. 2020;28(5):266–271. [DOI] [PubMed] [Google Scholar]
- [29].Wolff G, Limpens R, Zevenhoven-Dobbe JC, et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science. 2020;369(6509):1395–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ketteler R, Sun Z, Kovacs KF, et al. A pathway sensor for genome-wide screens of intracellular proteolytic cleavage. Genome Biol. 2008;9(4):R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang Y, Sun H, Pei R, et al. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021;7(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Miao G, Zhao H, Li Y, et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell. 2021;56(4):427–42 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhao YG, Zhao H, Miao L, et al. The p53-induced gene Ei24 is an essential component of the basal autophagy pathway. J Biol Chem. 2012;287(50):42053–42063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Devkota S, Jeong H, Kim Y, et al. Functional characterization of EI24-induced autophagy in the degradation of RING-domain E3 ligases. Autophagy. 2016;12(11):2038–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tang D, Kang R, Cheh CW, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29(38):5299–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Miller K, McGrath ME, Hu Z, et al. Coronavirus interactions with the cellular autophagy machinery. Autophagy. 2020;16(12):2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Maier HJ, Britton P. Involvement of autophagy in coronavirus replication. Viruses. 2012;4(12):3440–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bonam SR, Muller S, Bayry J, et al. Autophagy as an emerging target for COVID-19: lessons from an old friend, chloroquine. Autophagy. 2020;16(12):2260–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ghosh S, Dellibovi-Ragheb TA, Kerviel A, et al. β-Coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell. 2020;183(6):1520–35 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhao Z, Thackray LB, Miller BC, et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy. 2007;3(6):581–585. [DOI] [PubMed] [Google Scholar]
- [43].Hayn M, Hirschenberger M, Koepke L, et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021;35(7):109126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Qu Y, Wang X, Zhu Y, et al. ORF3a-Mediated incomplete autophagy facilitates severe acute respiratory syndrome coronavirus-2 replication[J]. Front Cell Dev Biol. 2021;9:716208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hayashi-Nishino M, Fujita N, Noda T, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11(12):1433–1437. [DOI] [PubMed] [Google Scholar]
- [46].Santerre M, Arjona SP, Allen CN, et al. Why do SARS-CoV-2 NSPs rush to the ER? J Neurol. 2021;268(6):2013–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhu L, Yang P, Zhao Y, et al. Single-cell sequencing of peripheral mononuclear cells reveals distinct immune response landscapes of Covid-19 and influenza patients. Immunity. 2020;53(3):685–96 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vanderheiden A, Ralfs P, Chirkova T, et al. Type I and type iii interferons restrict SARS-COV-2 infection of human airway epithelial cultures. J Virol. 2020;94(19):e00985–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Banerjee A, Czinn SJ, Reiter RJ, et al. Crosstalk between endoplasmic reticulum stress and anti-viral activities: a novel therapeutic target for COVID-19. Life Sci. 2020;255:117842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Koseler A, Sabirli R, Goren T, et al. Endoplasmic reticulum stress markers in SARS-COV-2 infection and pneumonia: case-control study. Vivo. 2020;34(3 suppl):1645–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shabbir S, Hafeez A, Rafiq MA, et al. Estrogen shields women from COVID-19 complications by reducing ER stress. Med Hypotheses. 2020;143:110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Echavarria-Consuegra L, Smit JM, Reggiori F. Role of autophagy during the replication and pathogenesis of common mosquito-borne flavi- and alphaviruses. Open Biol. 2019;9(3):190009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lennemann NJ, Coyne CB. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy. 2017;13(2):322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Evans AS, Lennemann NJ, Coyne CB. BPIFB3 regulates endoplasmic reticulum morphology to facilitate flavivirus replication. J Virol. 2020;94(9). DOI: 10.1128/JVI.00029-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Blanco-Melo D, Nilsson-Payant BE, Liu WC, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181(5):1036–45 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.