

ABSTRACT
Lymphoma is a common malignant tumor globally. Tumor-derived extracellular vesicles (Evs) participate in genetic information exchange between tumor cells. We investigated the role and mechanism of human Burkitt lymphoma cells Raji-derived Evs (Raji-Evs) in lymphoma cells. Effects of Evs on lymphoma cell proliferation, invasion, autophagy, and apoptosis were assessed using Cell Counting Kit-8 method, Transwell assay, laser confocal microscopy, Western blotting, and flow cytometry. microRNA (miR)-106a expression in lymphoma cells was determined using reverse transcription-quantitative polymerase chain reaction and then downregulated in Raji cells and then Evs were isolated (Evs-in-miR-106a) to evaluate its role in lymphoma cell growth. The binding relationship between miR-106a and Beclin1 was verified using RNA pull-down and dual-luciferase assays. Beclin1 was overexpressed in SU-DHL-4 and Farage cells and SU-DHL-4 cell autophagy and apoptosis were detected. The levels of miR-106a and Beclin1 in SU-DHL-4 cells were detected after adding autophagy inhibitors. The tumorigenicity assay in nude mice was performed to validate the effects of Raji-Evs in vivo. Raji-Evs promoted lymphoma cell proliferation and invasion and increased miR-106a. miR-106a knockdown reversed Evs-promoted lymphoma cell proliferation and invasion. miR-106a carried by Raji-Evs targeted Beclin1 expression. Beclin1 overexpression or miR-106a inhibitor reversed the effects of Evs on lymphoma cell autophagy and apoptosis. Autophagy inhibitors elevated miR-106a expression and lowered Beclin1 expression. Raji-Evs-carried miR-106a inhibited Beclin1-dependent autophagy and apoptosis in lymphoma cells, which were further verified in vivo, together with promoted tumor growth. We proved that Raji-Evs inhibited lymphoma cell autophagy and apoptosis and promoted cell growth via the miR-106a/Beclin1 axis.
KEYWORDS: Lymphoma cells, extracellular vesicles, microRNA-106a, Beclin1, autophagy, apoptosis
Introduction
Lymphoma, generally considered as a group of heterogeneous tumors, can affect any organ in the body and manifest with clear clinical and morphological characteristics, together with various symptoms, such as consecutive fever, weight loss and night sweats [1–3]. Lymphoma mainly develops as a consequence of smoking and obesity, in the other hand, genetic factors, infection and inflammation are important contributors [4]. There are two main subtypes of lymphoma: Hodgkin’s lymphoma (HL) (about 10%; classical and non-classical types) and non-Hodgkin’s lymphoma (NHL) (90%; origin of B-cells, T-cells or natural killer cells) [5]. Different lymphomas have greatly-varied clinical treatments; aggressive lymphoma needs immediate treatment, while indolent forms have no requirement for treatment over years, which is largely incurable [5,6]. Currently, chemotherapy is still the standard treatment for aggressive lymphoma; however, a large number of patients (20–30% NHL and 15% HL) experience recurrence after initial treatment [7]. Therefore, it is urgent to find new therapies to improve the prognosis and clinical outcome of lymphoma patients.
In recent years, extracellular vesicles (Evs) are gaining importance given that they show an intrinsic involvement in the intercellular communication and serve as an essential part in tumor development [8,9]. Evs including exosomes and microvesicles are as membranous structure and can be secreted by multiple cell types [10,11]. Among them, tumor-derived Evs are shown to change the tumor microenvironment and regulate tumor development, promising to be a valuable biomarker for cancer diagnosis and prediction [12]. Importantly, tumor-derived Evs have also been highlighted to communicate between lymphoma cells and thus play a role in the pathogenesis of lymphoma [13].
Evs are shown to participate in diverse cell processes by transporting theirs cargo [proteins, microRNAs (miRNAs) and mRNAs] to recipient cells [14]. Mounting evidence has demonstrated that Evs-transported miRNAs make a great contribution to the creation of tumor environment [15]. miRNAs, as non-coding molecules inducing epigenetic regulation, have shown a close relation with substantial solid malignancies; deregulated miRNAs are strongly linked to the pathogenesis of lymphoma [16,17]. miR-106a/b is shown to be indicative of higher mortality of relapsed lymphoma patients and can be a reliable biomarker for recurrent lymphoma [18]. miR-106a/363 clusters are found to be aberrantly upregulated in cutaneous T-cell lymphoma [19]. Evs can regulate the expression of Beclin1 by paracrine [20], and Beclin1 can negatively regulate innate immune signal and lymphoma development [21]. However, little is known about miR-106a/Beclin1 in lymphoma cell-derived Evs.
Therefore, it is reasonable to hypothesize that lymphoma cell-derived Evs may play an underlying role in lymphoma progression through a possible system with the involvement of miR-106a. Consequently, we performed a series of histological and molecular experiments to identify the regulatory role and mechanism of lymphoma cell-derived Evs in lymphoma development, with the purpose to provide some novel insights for lymphoma treatment.
Materials and methods
Ethics statement
This study was supervised by the Ethics Committee of The Affiliated Hospital of Southwest Medical University. Significant efforts were made to minimize both the number of animals and their suffering. All procedures were strictly conducted in accordance with the code of ethics.
Lymphoma cell culture
Human B-lymphoma cell lines SU-DHL-4 and Farage (Shanghai Mingjing Biology Co., Ltd., Shanghai, China) and human Burkitt lymphoma cell-line Raji (Cell Resource Center, Shanghai Institutes for Biological Sciences, the Chinese Academy of Sciences, Shanghai, China) were cultured in RPMI-1640 medium (Gibco Company, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin (Solarbio, Beijing, China) at 37°C with 5% CO2. The cells at logarithmic growth phase were selected for subsequent experiments.
Isolation of Raji cells-derived Evs (Raji-Evs)
The Raji cells were centrifuged (at 300 g; 5 min) and then the medium was replaced with serum-free medium (Umibio Co., Ltd., Shanghai, China) for further culture. After 24 h, the cells successively underwent several centrifugations (at 300 g, 10 min; at 2000 g, 10 min; and at 10,000 g, 30 min) at 4°C to remove cells and cell debris, dead cells and large vesicles. Next, following a centrifugation (at 100,000 g; 70 min), the supernatant was discarded, and the precipitate was re-suspended using phosphate-buffered saline (PBS). Finally, the Evs were obtained after a 70-min centrifugation (at 100,000 g; 4°C) and resuspension using 100 μL PBS. miR-106a inhibitor or its negative control (NC) (Shanghai GenePharma Co., Ltd., Shanghai, China) was transfected into Raji cells (in-miR-106a/in-NC) using Lipofectamine 3000 (Invitrogen Inc., Carlsbad, CA, USA). Then Evs were extracted from Raji cells transfected with in-miR-106a (Evs-in-miR-106a) or in-NC (Evs-in-NC). Bicinchoninic acid (BCA) kit (Solarbio) was utilized to quantify the Ev protein. Evs were characterized utilizing transmission electron microscopy (TEM; JEOL, Ltd., Tokyo, Japan) and the qNano system (Izon Science Ltd., Christchurch, New Zealand). The Ev markers CD63, CD9 and Calnexin were detected using Western blotting (WB). Meanwhile, a GW4869 group [Raji cells were treated with 10 μM GW4869 (Sigma-Aldrich Chemical Company, St Louis, MO, USA), and then the supernatant of culture medium was absorbed] was used as the control.
Raji-Ev absorption by human lymphoma cells
Dil solution (4 mg/mL) (Molecular Probes, Eugene, OR, USA) was added to the PBS suspension of Evs. After 30-min incubation, Dil-labeled Evs were obtained by ultracentrifugation and PBS washing, and then incubated with SU-DHL-4 and Farage cells for 3 h. Following the Evs were washed with PBS and fixed with 4% paraformaldehyde, the uptake of Evs was observed under a laser confocal microscope (LSM5, Zeiss Inc., AG, Oberkochen, Germany).
Cell grouping and treatment
SU-DHL-4 and Farage cells were allocated into blank group (treated with 100 μg/mL PBS), GW4869 group (treated with 100 μg/mL supernatant of Raji cells after GW4869 treatment), Evs group [treated with 100 μg/mL Evs [22], Evs-in-NC group (treated with 100 μg/mL Evs-in-NC), Evs-in-miR-106a group (treated with 100 μg/mL Evs-in-miR-106a), Evs + pc-NC and Evs + pc-Beclin1 group. In the Evs + pc-NC and Evs + pc-Beclin1 groups, SU-DHL-4 and Farage cells were treated with 100 μg/mL Evs after transfection of pc-NC or pc-Beclin1 (GenePharma) using Lipofectamine 3000. After 48 h of Ev intervention, the following experiments were performed. SU-DHL-4 cells were treated with 1 nM Bafilamycin A1 (Baf-A1, Sigma-Aldrich) and 20 μM Chloroquine (CQ, Sigma-Aldrich), and DMSO (sigma Aldrich) was used as control.
Cell counting kit-8 (CCK-8) assay
SU-DHL-4 and Farage cells were seeded into 96-well plates (2000 cells/100 μL), respectively. The cell growth at 0 h, 24 h, 48 h, and 72 h after Ev (100 μg/mL) intervention was determined using CCK-8 kits (Solarbio) and expressed as the optical density (OD) value measured at 450 nm using a microplate reader (ELx800, Omega Bio-tek Inc., Norcross, GA, USA).
Transwell assay
Transwell chamber was pre-coated with Matrigel (BD Biosciences, San Jose, CA, USA) and cells were starved for 24 h in serum-free medium. Afterward, the cells were re-suspended in a medium with 1% FBS and the 200 μL cell suspension was seeded into the apical chamber (5 × 105 cells/well). The basolateral chamber was added with 500 μL complete medium containing 10% FBS. After 24 h, the non-invading cells in the apical chamber were gently removed using cotton swabs. The medium in the basolateral chamber was gently mixed, and then 10 μL medium was sucked out and added into the counting plate. The cells were observed and counted under the microscope, and the total number of cells in 4 quadrants (under 100 times microscope) was counted.
Observation of autophagy flow
The sterilized cell slide was put into 24-well plates, and cell suspension added into the plates. When cell coverage rate was about 70%, the cells were transfected with double-labeled green fluorescent protein (GFP)-monomeric red fluorescent protein (mRFP)-LC3 plasmids. After 24 h, the cells were washed 3 times with PBS, 5 min each, and fixed with 4% polymethanol for 30 min. Next, the cell slide was taken out and placed on the cover glass. The red, green and yellow spots in the cells were observed under a laser confocal microscope and then photographed. In the overlapping image, the yellow spots in the cell represented autophagosomes and the red spots represented autolysosomes.
Dual-luciferase reporter gene assay
Starbase (http://starbase.sysu.edu.cn/index.php) was used to predict the binding site between miR-106a and Beclin1. The complementary binding sequence and mutant sequence of miR-106a and Beclin1 were amplified and cloned into pmiR-GLO luciferase vector (Promega Corp., Madison, WI, USA) to construct the wild-type and mutant plasmid of Beclin1 (Beclin1-WT/Beclin1-MUT), which were then co-transfected with mimic-NC or miR-106a mimic into HEK293T cells (Shanghai Institute of Cellular Biology of Chinese Academy of Sciences, Shanghai, China), respectively using Lipofectaminetm 3000 (Invitrogen). After 48 h, luciferase activity was detected using a Dual-Luciferase Reporter Assay System (Promega).
RNA pull-down
miR-106a-biotin, miR-106a-MUT-biotin and the control (NC biotin) were transfected into SU-DHL-4 cells, respectively. After 24 h, the cells were incubated with streptavidin magnetic beads in the lysis (Ambion, Austin, TX, USA) for 2 h, and the RNA binding compound was eluted. Beclin1 abundance was determined using reverse transcription-quantitative polymerase chain reaction (RT-qPCR).
Flow cytometry
The cells were centrifuged (300 g; 10 min) at room temperature, collected and washed with PBS. Fluorescein isothiocyanate Annexin V Apoptosis Detection Kit I (Cat#556,547, BD Biosciences-Pharmingen, Cat#556,547) was used for cell treatments. Finally, the apoptosis was detected using a flow cytometer (CytoFLEX, Beckman Coulter, Inc, Chaska, MN, USA).
Tumorigenicity assay in nude mice
Non-obese diabetic-severe combined immunodeficiency (NOD-SCID) mice [Hunan SJA Laboratory Animal Co., Ltd., Changsha, Hunan, China; SCXK (Xiang) 2016–0002)] were randomly allocated into the NC, Evs, Evs-in-NC and Evs-in-miR-106a groups according to the body weight, with 5 mice in each group. Nude mouse in each group was subcutaneously injected with 5 × 108 SU-DHL-4 cells into the back scapula, following by Evs injection (10 μg) via tail vein [23]. The tumor volume was measured on the 7th, 14th, 21th and 28th days after the experiment, and calculated based on the formula V = 0.5 × a × b [2] (a represents the tumor length; b represents the tumor width). The mice were euthanized by intraperitoneal injection of pentobarbital sodium (800 mg/kg) on the 28th day. The tumor was weighed, frozen in liquid nitrogen and stored at −196°C. RT-qPCR and WB were applied for detection.
WB
Tumor tissues and cell samples were ground in liquid nitrogen, and then the total protein was extracted from tumor tissues and cells using RIPA lysis buffer (Beyotime Biotechnology Co., Ltd., Shanghai, China). The protein concentration was detected using the BCA protein assay kit (Beyotime). After loading, the samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10% separating gel), and then transferred to membranes and blocked with 5% bovine serum albumin. Next, the membranes were incubated with primary antibodies (all from Abcam Inc., Cambridge, MA, USA): rabbit anti-CD9 (ab92726, 1:2000), rabbit anti-CD63 (ab134045, 1:1000), rabbit anti-Calnexin (ab22595, 1 µg/mL), rabbit anti-light chain 3 (LC3) (ab192890, 1: 1000), rabbit anti-p62 (ab109012, 1:1000), rabbit anti-Beclin1 (ab210498, 1:1000), rabbit anti-Cleaved-caspase-3 (ab32042, 1:500), rabbit anti-B-cell lymphoma-2 (Bcl-2) (ab32124, 1:1000), rabbit anti-Beclin1(ab207612, 1:2000) and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (ab181602, 1:10,000), followed by incubation with secondary antibody goat anti-rabbit immunoglobulin G (IgG) (ZB-5301, 1:5000, ZSGB-Bio Co., Ltd., Beijing, China). Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA) was utilized for band exposure and gray value analysis.
RT-qPCR
Total RNA was extracted from ground tumor tissues or cell samples using TRIzol regent (Invitrogen) and transcribed into cDNA using PrimeScript RT reagent kit (Takara Biotechnology Co., Ltd., Dalian, China). The qPCR was performed on ABI 7900HT fast PCR real-time system (Applied Biosystems, Foster City, CA, USA) in accordance with the instructions of SYBR® Premix Ex TaqTM II (Takara). GAPDH was used as the internal parameter, and the data were analyzed using 2−ΔΔCt method. The amplified primer sequences of the genes and their primers are shown in Table 1.
Table 1.
Primer sequences for RT-qPCR
| Gene | Primer sequence |
|---|---|
| Beclin1 | F: 5’-CCATGCAGGTGAGCTTCGT-3’ |
| R: 5’-GAATCTGCGAGAGACACCATC-3’ | |
| miR-106a | F: 5’-GATGCTCAAAAAGTGCTTACAGTGCA-3’ |
| R: 5’-TATGGTTGTTCTGCTCTCTGTCTC-3’ | |
| U6 | F: 5’-TTCTTGGGTAGTTTGCAGTT-3’ |
| R: 5’-TTCTTGGGTAGTTTGCAGTT-3’ | |
| GAPDH | F: 5’-GGAGCGAGATCCCTCCAAAAT-3’ |
| R: 5’-GGCTGTTGTCATACTTCTCATGG-3’ |
Statistical analysis
SPSS 21.0 (IBM Corp., Armonk, NY, USA) was used for data analysis. Kolmogorov-SmiRnov test verified that data were in normal distribution. The results were represented as mean ± standard deviation. The comparison between the two groups was analyzed by independent sample t-test. One-way or two-way analysis of variance (ANOVA) was used for comparison analysis among multiple groups, followed by Tukey’s multiple comparisons test. The p value was obtained from a two-tailed test. p < 0.05 indicated statistically significant difference, and p < 0.01 meant highly statistically significant difference.
Results
Raji-Evs promoted human lymphoma cell proliferation and invasion
A study has shown that tumor cells can release Evs to promote tumor cell proliferation [24]. To analyze the effect of Evs on lymphoma cells, we isolated Evs from human Burkitt lymphoma cell line Raji. TEM observed that Evs showed a typical saucer-type bilayer membrane structure (Figure 1a). Nanoparticle tracking analysis found that the particle size of most of Evs was 100 nm-200 nm (Figure 1b). CD63 and CD9 were positive in Evs, and Calnexin was negative, as shown by WB results; after GW4869 intervention, no obvious expressions of CD63 and CD9 were observed in the supernatant of human lymphoma cell culture medium (Figure 1c), indicating that we successfully isolated the Raji-Evs. The Evs were labeled with Dil and incubated with human lymphoma cells SU-DHL-4 and Farage, respectively. The laser confocal microscopy observed that Evs could be captured and phagocytized by SU-DHL-4 and Farage cells (Figure 1d). Compared with GW4869 group, Evs-treated SU-DHL-4 and Farage cells showed obviously increased proliferation and invasive cell numbers, as confirmed by CCK-8 assay and Transwell assay (Figures 1e,f) (all p < 0.01).
Figure 1.
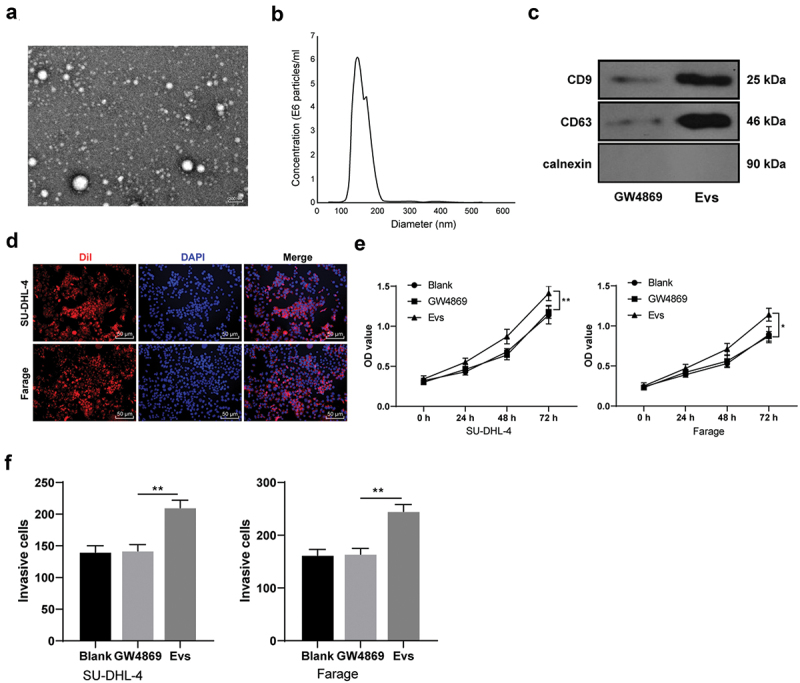
Raji-Evs promoted human lymphoma cell proliferation and invasion. A. The morphology of the Evs was observed using TEM; B. the size distribution of the Evs was detected using qNano system; C. Expressions of positive markers (CD63 and CD9) and negative marker Calnexin of Evs were detected using WB, with the supernatant of Raji cell culture medium after GW4869 intervention as NC; D. Laser confocal microscopy observed the absorption of Dil-labeled Evs by SU-DHL-4 and Farage cells; E. CCK-8 assay was used to detect the proliferation of SU-DHL-4 and Farage cells; F. Transwell assay was used to detect the invasion of SU-DHL-4 and Farage cells in different groups. Three repeated experiments were conducted independently, and the data were expressed as mean ± standard deviation. Data in panels E/F were analyzed using one-way ANOVA, followed by Tukey’s multiple comparisons test. *p < 0.05; **p < 0.01.
Raji-Evs promoted human lymphoma cell proliferation and invasion via carrying miR-106a
As has been previously reported, Evs may promote tumorigenesis via carrying miRNAs and other substances for intercellular genetic material exchange and thus influencing tumor microenvironment [24,25]. miR-106a is highly expressed in human lymphoma tissues [26]. Therefore, we speculated that Raji-Evs may carry miR-106a into lymphoma cells and then promote cell proliferation and invasion. Firstly, miR-106a expression in Evs-treated SU-DHL-4 and Farage cells was detected, and we observed that Ev treatment remarkably upregulated miR-106a expression (Figure 2a) (both p < 0.01), indicating that Raji-Evs may carry miR-106a into human lymphoma cells. To further verify our hypothesis, we transfected miR-106a inhibitor or inhibitor NC into Raji cells (in-miR-106a/in-NC). miR-106a expression in Raji cells and Raji-Evs was detected, and it was found that miR-106a was notably decreased in in-miR-106a-treated Raji cells and Evs-in-miR-106a (Figure 2b) (both p < 0.01). After treatment with Evs-in-miR-106a, SU-DHL-4 and Farage cells showed noticeably reduced miR-106a expression (Figure 2c) (all p < 0.01), and decreased proliferation ability (Figure 2d)(all p < 0.05) and invasive cell numbers (Figure 2e) (both p < 0.01) relative to those treated with Evs. From all above, Raji-Evs promoted human lymphoma cell proliferation and invasion by carrying miR-106a.
Figure 2.
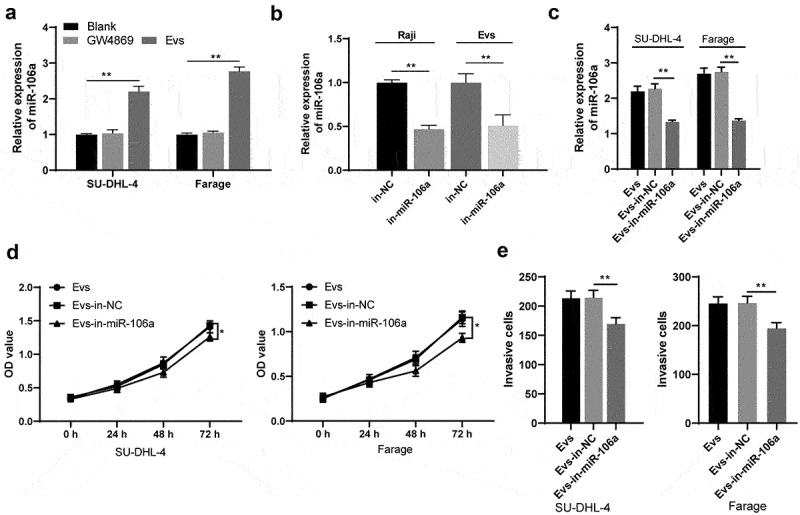
Raji-Evs promoted human lymphoma cell proliferation and invasion via carrying miR-106a. A. RT-qPCR was used to detect the expression of miR-106a in SU-DHL-4 and Farage cells after Evs treatment; B. RT-qPCR was used to detect miR-106a expression in Raji cells and Raji-Evs after transfection of miR-106a inhibitor and its NC; C. RT-qPCR was used to detect the expression of miR-106a in SU-DHL-4 and Farage cells treated with Evs-in-miR-106a; D. CCK-8 assay was used to detect the proliferation of SU-DHL-4 and Farage cells; E. Transwell assay was used to detect the invasion of SU-DHL-4 and Farage cells. Three repeated experiments were conducted independently, and the data were expressed as mean ± standard deviation. Data in panels A/C/D/E were analyzed using one-way ANOVA, followed by Tukey’s multiple comparisons test. *p < 0.05; **p < 0.01.
Raji-Evs carried miR-106a into lymphoma cells to inhibit Beclin1 expression
To further explore the downstream mechanism of Raji-Evs-carried miR-106a in lymphoma cells, we predicted the target binding sites between miR-106a and Beclin1 through Starbase (http://starbase.sysu.edu.cn/index.php). Beclin1 is poorly expressed in some lymphoma patients, which may be a valuable prognostic factor for lymphoma [27]. Dual-luciferase reporter gene assay and RNA pull-down assay verified the targeting relationship between miR-106a and Beclin1 (Figures 3a,b) (all p < 0.01). Meanwhile, Beclin1 mRNA and protein levels in lymphoma cells were detected. We observed that Beclin1 levels were remarkably reduced in Evs-treated lymphoma cells, which were reversed by Evs-in-miR-106a treatment (Figurers 3c,d) (all p < 0.05). Briefly, Raji-Evs transferred miR-106a to inhibit Beclin1 expression in lymphoma cells, thus promoting the proliferation and invasion of lymphoma cells.
Figure 3.
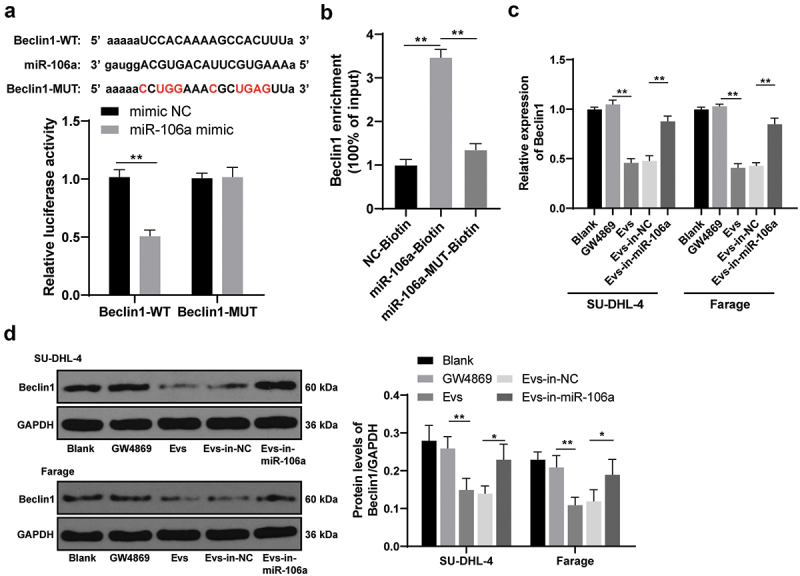
Raji-Evs carried miR-106a into lymphoma cells to inhibit Beclin1 expression. A/B. The binding relationship between miR-106a and Beclin1 was verified using dual luciferase reporter gene assay and RNA pull-down assay; C. Beclin1 expression in SU-DHL-4 and Farage cells was detected using RT-qPCR; D. Beclin1 protein level in SU-DHL-4 and Farage cells was detected using WB. Three repeated experiments were conducted independently, and the data were expressed as mean ± standard deviation. Data in panels A-D were analyzed using one-way ANOVA, followed by Tukey’s multiple comparisons test. *p < 0.05; **p < 0.01.
Beclin1 overexpression reversed Raji-Evs-promoted proliferation and invasion of lymphoma cells
To confirm that Raji-Evs inhibited Beclin1 expression in lymphoma cells via transferring miR-106a, thus promoting lymphoma cell proliferation and invasion, we co-transfected pc-Beclin1 or pc-NC with Evs into SU-DHL-4 and Farage cells (Evs + pc-NC/Evs + pc-Beclin1). Lymphoma cells treated with Evs + pc-Beclin1 showed noticeably elevated Beclin1 expression relative to those with Evs + pc-NC treatment (Figure 4a,b), as well as obviously reduced proliferation ability (Figure 4c) and invasive cells (Figure 4d) (all p < 0.01), suggesting that Beclin1 overexpression reversed the promotion of Raji-Evs on lymphoma cell proliferation and invasion. We confirmed that Raji-Evs inhibited Beclin1 expression in lymphoma cells via transferring miR-106a, thereby promoting lymphoma cell proliferation and invasion.
Figure 4.
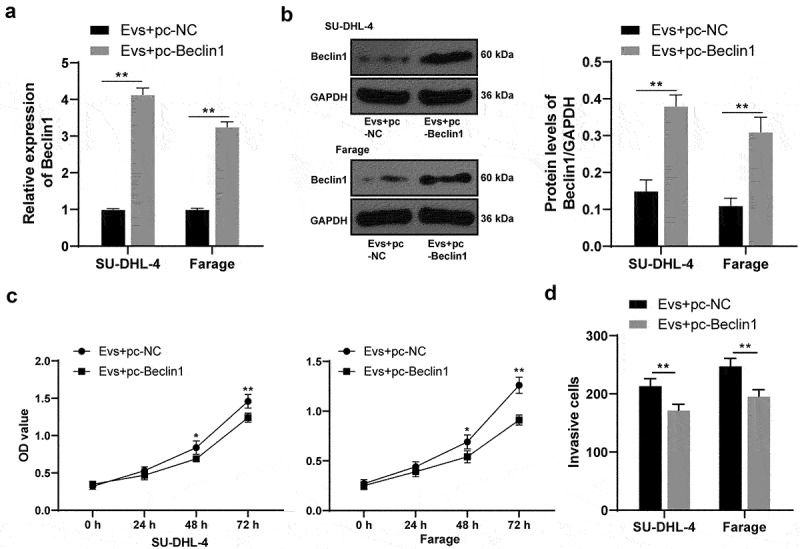
Beclin1 overexpression reversed Raji-Evs-promoted proliferation and invasion of lymphoma cells. A. RT-qPCR was used to detect Beclin1 mRNA expression; B. WB was used to detect Beclin1 protein level; C. CCK-8 assay was used to detect the proliferation ability of lymphoma cells; D. Transwell assay was used to detect the invasion ability of lymphoma cells. Three repeated experiments were conducted independently, and the data were expressed as mean ± standard deviation. Data were analyzed using the t test. *p < 0.05; **p < 0.01.
Raji-Evs inhibited Beclin1-dependent autophagy and apoptosis in lymphoma cells via transferring miR-106a
Beclin1 can affect cell growth by regulating autophagy and apoptosis [28]. Therefore, we speculated that Raji-Evs may regulate autophagy and apoptosis of lymphoma cells via the miR-106a/Beclin1 axis, thereby promoting lymphoma progression. SU-DHL-4 cells were selected for further study. The autophagy level changes in SU-DHL-4 cells after Evs intervention were detected. TEM observed that Evs treatment decreased autophagosome and autolysosome numbers in SU-DHL-4 cells, which were then increased after miR-106a inhibition in Evs (Figure 5a). Similar results were also found using GFP-mRFP-LC3, with remarkably reduced autophagosome and autolysosome densities in Evs-treated SU-DHL-4 cells, and the opposite trends were observed after inhibiting miR-106a in Evs (Figure 5b) (all p < 0.01). Furthermore, the levels of autophagy-related proteins LC3-II, LC3-I and p62 were detected using WB. Evs treatment resulted in notably downregulated LC3-II/LC3-I and clearly upregulated p62; the Evs-in-miR-106 group showed reduced p62 expression and increased LC3-II/LC3-I expression (Figure 5c) (all p < 0.05). These results revealed that Raji-Evs inhibited Beclin1-dependent autophagy in SU-DHL-4 cells.
Figure 5.
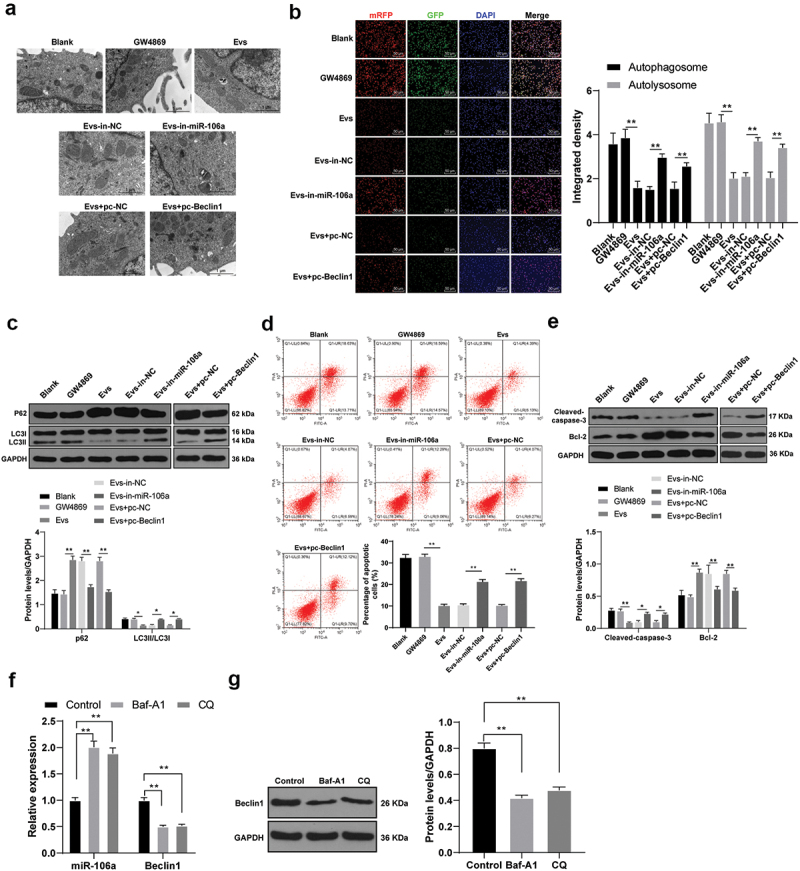
Raji-Evs inhibited autophagy and apoptosis of lymphoma cells via the miR-106a/Beclin1 axis. A. The autophagosomes and autolysosomes in SU-DHL-4 cells in each group were observed using TEM; B. The autophagy flow of SU-DHL-4 cells was detected using GFP-mRFP-LC3; C. Levels of autophagy-related proteins LC3-II, LC3-I and p62 in SU-DHL-4 cells were detected using WB; D. Apoptosis of SU-DHL-4 cells was detected using flow cytometry; E. Protein levels of Bcl-2 and Cleaved-caspase-3 in SU-DHL-4 cells were detected using WB; Autophagy inhibitors 1 nM Baf-A1 and 20 μM CQ were added to the cells. F. RT-qPCR was used to detect the expression of miR-106a and Beclin1 in SU-DHL-4 cells; (g) WB was used to detect the protein expression of Beclin1 in SU-DHL-4 cells. Three repeated experiments were conducted independently, and the data were expressed as mean ± standard deviation. Data in panels B-G were analyzed using one-way ANOVA, followed by Tukey’s multiple comparisons test. *p < 0.05; **p < 0.01.
Evs-treated SU-DHL-4 cells also showed a markedly decreased apoptosis rate, which was then increased after inhibiting miR-106a in Evs (Figure 5d) (all p < 0.01). As indicated by WB results, Evs-treated SU-DHL-4 cells had notably elevated Bcl-2 and reduced Cleaved-caspase-3 expression, and Evs-in-miR-106a treatment caused opposite results (Figure 5e) (all p < 0.05). To further verify the relationship between miR-106a/Beclin1 and autophagy in SU-DHL-4 cells, we added autophagy inhibitors 1 nM Baf-A1 and 20 μM CQ [29,30] to SU-DHL-4 cells, and detected the expression of miR-106a and Beclin1 by RT-qPCR and WB. Compared with control group, the expression of miR-106a was increased in Baf-A1 group and CQ group, while the expression of Beclin1 was decreased (figures 5f,g, p < 0.01). From above, Raji-Evs inhibited autophagy and apoptosis of SU-DHL-4 cells. Based on our previous experimental results, we identified that Raji-Evs inhibited autophagy and apoptosis of lymphoma cells via the miR-106a/Beclin1 axis.
Raji-Evs promoted tumor growth and inhibited autophagy and apoptosis of lymphoma cells in vivo
To further verify the effect of Raji-Evs on the growth of lymphoma cells in vivo, we injected SU-DHL-4 cells subcutaneously into NOD-SCID mice that were grouped into NC, Evs, Evs-in-NC and Evs-in-miR-106a. The tumor changes were detected, and it was found that mice with Ev treatment had notably increased tumor growth rate and weight, which were then remarkably decreased after miR-106a inhibition in Evs (Figures 6a,b) (all p < 0.01). According to RT-qPCR results, miR-106a was noticeably upregulated and Beclin1 was downregulated after Evs treatment, which could be reversed by miR-106a inhibition in Evs (Figure 6c) (all p < 0.05). Evs-treated mice showed markedly reduced LC3-II/LC3-I and Cleaved-caspase-3 expression and obviously increased p62 expression, and Evs-in-miR-106a-treated mice showed the opposite trends; the apoptosis-related protein Cleaved-caspase-3 was notably downregulated in Evs-treated mice, which was markedly upregulated after miR-106a inhibition in Evs (Figure 6d) (all p < 0.01).
Figure 6.
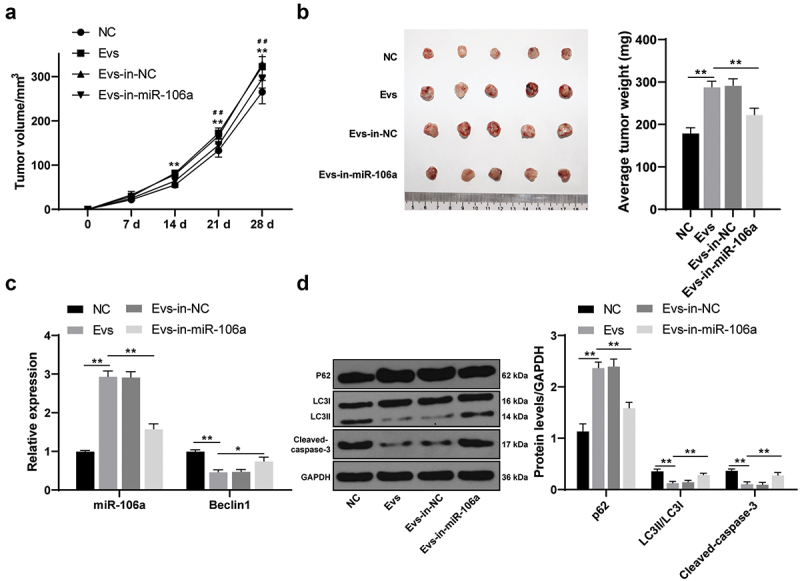
Raji-Evs promoted mouse tumor growth and inhibited autophagy and apoptosis of lymphoma cells. A. The changes of tumor volume in each group were measured; B. The changes of tumor weight in each group were measured; C. mRNA expressions of miR-106a and Beclin1 in each group were detected using RT-qPCR; D. Levels of LC3-II, LC3-I, p62 and Cleaved-caspase-3 in mouse tumor of each group was detected using WB. N = 5. Data were expressed as mean ± standard deviation. Data in panels A-D were analyzed using one-way ANOVA, followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01; #p < 0.05; ##p < 0.01. In panel A, *comparison between the Evs group and the NC group; #comparison between the Evs-in-miR-106a group and the Evs group.
Discussion
Lymphoma affects the survival of patients and is difficult to treat [31]. Tumor cells can secret a great deal of Evs that can be naturally absorbed by host cells in the tumor microenvironment [32]. Ev exchanges between tumor cells and recipient cells are closely related to tumor proliferation and invasion [33–36]. It is validated that tumor-derived Evs contribute to facilitating tumor progression and metastasis [34]. In this study, we identified that Raji-Evs-shuttled miR-106a inhibited Beclin1-dependent autophagy and apoptosis in lymphoma cells, and then promoted lymphoma cell proliferation and invasion.
E. In this study, Raji-Evs could be captured and phagocytized by SU-DHL-4 and Farage cells, and then cause obviously increased proliferation and invasion of lymphoma cells. Consistently, Hodgkin lymphoma cells-isolated Evs can be taken up by fibroblasts in the tumor microenvironment and then help to trigger tumor migration and support tumor growth in HL [35]. Diffuse large B-cell lymphoma-derived Evs display a noticeable role in stimulating tumor proliferation and invasion [36].
It has been well established that miRNAs can be released from cells by encapsulation in Evs and spread to target cells, including tumor cells where Evs-contained miRNAs can promote the malignancy via modulating mRNA and protein expressions [9,37]. miR-106a~363 cluster (including miR-106a, miR-18b, miR-20b, miR-19b, miR-92a, miR-363) is implicated in lymphomas with its aberrant expression [38]. According to our observations, miR-106a expression in Evs-treated SU-DHL-4 and Farage cells was remarkably upregulated, which was then notably decreased after silencing miR-106a in Raji cells, accompanied by decreased proliferation and invasion ability in SU-DHL-4 and Farage cells. Consistent with our study, miR-106a has been demonstrated to be intrinsically related to lymphoma progression, as manifested by the promoted tumor proliferation and suppressed apoptosis with its overexpression [39]. Inhibition of exosome-encapsulated miR-106a helps to hinder hepatocellular carcinoma cell proliferation and invasion [40]. From all above, we concluded that Raji-Evs promoted human lymphoma cell proliferation and invasion via carrying miR-106a.
We then explored the downstream mechanism of Raji-Evs-carried miR-106a in lymphoma cells. Growing studies have pointed out that miR-106a exerts inhibitory effects on tumor cell proliferation and invasion via the interaction of Bcl-2 [41,42]. Beclin1 is validated as a Bcl-2-interacting protein, and the binding relationship between Beclin1 and Bcl-2 has been demonstrated to be crucial in cellular homeostasis [43,44]. Furthermore, Beclin1 is a therapeutic target for lymphoma [45]. We observed that Beclin1 expression was remarkably reduced in Raji-Evs-treated lymphoma cells, which was then reversed after inhibiting miR-106a in Evs. Consistently, Beclin1 is a reliable independent prognostic factor and is indicative of good clinical outcome in NHL [27]. Upregulation of Beclin1 is intimately bound up with the suppressed lymphoma cell proliferation [46]. Beclin1 overexpression reversed Raji-Evs-promoted lymphoma cell proliferation and invasion. Furthermore, Beclin1 is shown to inhibit osteosarcoma cell migration and invasion via a miRNA-mediated regulation [47]. Nevertheless, the interaction between miR-106a and Beclin1 and their combined role in lymphoma have not been elucidated, which, on the other hand, reflects the innovation of this paper. Briefly, our research showed that Raji-Evs transferred miR-106a to inhibit Beclin1 expression in lymphoma cells, thus promoting the proliferation and invasion of lymphoma cells. By the way, miR-106a has been reported to regulate autophagy by targeting ATG7 [48], ULK1 [49] and LKB1 [50], which will become our future research directions.
Autophagy and apoptosis are greatly implicated in the pathogenesis of lymphoma [51,52]. Beclin1 plays an essential role in modulating autophagy and apoptosis that are two processes indispensable for lymphoma malignant behaviors [27]. The induced autophagy can be evaluated by elevated LC3-II/LC3-I and Beclin1 expression, as well as reduced p62 expression [53]. The abrogated Bcl-2 expression and augmented Cleaved-caspase-3 expression are indicative of induced apoptosis [54]. Our results revealed that Raji-Evs decreased autophagosomes and autolysosomes, downregulated LC3-II/LC3-I and upregulated p62 expression, as well as decreased apoptosis rate and Cleaved-caspase-3 expression, and elevated Bcl-2 expression in lymphoma cells; while miR-106a knockdown or Beclin1 overexpression reversed the above results. The role of miR-106a in autophagy and apoptosis of lymphoma cells has rarely studied. The effect of Raji-EVs on lymphoma cells was further verified in NOD-SCID mice in vivo, as manifested by promoted tumor growth and inhibited autophagy and apoptosis of lymphoma cells. All in all, Raji-Evs inhibited autophagy and apoptosis of lymphoma cells via the miR-106a/Beclin1 axis.
Collectively, our study supported that Raji-Evs inhibited lymphoma cell autophagy and apoptosis, and promoted lymphoma cell development via the miR-106a/Beclin1 axis. These results discovered a novel tumor-derived Evs-based therapy for lymphoma patients, and promoting autophagy and apoptosis of lymphoma cells might be developed as a promising therapeutic approach for lymphoma. Although the present study provide therapeutic value for lymphoma treatment, the experiment results and clinical application need to be further verified.
Funding Statement
This work was supported by Sichuan Science and Technology Program (No. 2019YFS0301); Doctor Research Foundation of The Affiliated Hospital of Southwest Medical University (No. 19032; No. 19079); The Key Research Project from Health and Family Planning Commission of Sichuan Province (No. 18ZD014);Applied Basic Research Project of Luzhou Science and Technology Bureau(2019LZXNYDJ54; 2020LZXNYDJ48; 2020-JYJ-50).
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Armitage JO, Gascoyne RD, Lunning MA, et al. Non-Hodgkin lymphoma. Lancet. 2017;390(10091):298–310. [DOI] [PubMed] [Google Scholar]
- [2].Ghattamaneni S, Guttikonda VR, Yeluri S, et al. Early diagnosis of an isolated primary peripheral T-cell lymphoma masquerading as massive gingival enlargement in a pediatric patient. J Oral Maxillofac Pathol. 2017;21(3):421–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yoo KH, Lee H, Suh C, et al. Lymphoma epidemiology in Korea and the real clinical field including the consortium for improving survival of lymphoma (CISL) trial. Int J Hematol. 2018;107(4):395–404. [DOI] [PubMed] [Google Scholar]
- [4].Lewis WD, Lilly S, Jones KL.. Lymphoma: diagnosis and treatment. Am Fam Physician. 2020;101(1):34–41. [PubMed] [Google Scholar]
- [5].Mugnaini EN, Ghosh N.. Lymphoma. Prim Care. 2016;43(4):661–675. [DOI] [PubMed] [Google Scholar]
- [6].Horn H, Staiger AM, Ott G. New targeted therapies for malignant lymphoma based on molecular heterogeneity. Expert Rev Hematol. 2017;10(1):39–51. [DOI] [PubMed] [Google Scholar]
- [7].Zahid U, Akbar F, Amaraneni A, et al. A review of autologous stem cell transplantation in lymphoma. Curr Hematol Malig Rep. 2017;12(3):217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hansen HP, Paes Leme AF, Hallek M. Role of ADAM10 as a CD30 sheddase in classical Hodgkin lymphoma. Front Immunol. 2020;11:398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kosaka N, Yoshioka Y, Fujita Y, et al. Versatile roles of extracellular vesicles in cancer. J Clin Invest. 2016;126(4):1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hill AF. Extracellular vesicles and neurodegenerative diseases. J Neurosci. 2019;39(47):9269–9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Szatanek R, Baj-Krzyworzeka M, Zimoch J, et al. The methods of choice for extracellular vesicles (EVs) characterization. Int J Mol Sci. 2017;18(6);1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tai YL, Chu PY, Lee BH, et al. Basics and applications of tumor-derived extracellular vesicles. J Biomed Sci. 2019;26(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rutherford SC, Fachel AA, Li S, et al. Extracellular vesicles in DLBCL provide abundant clues to aberrant transcriptional programming and genomic alterations. Blood. 2018;132(7):e13–e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Withrow J, Murphy C, Liu Y, et al. Extracellular vesicles in the pathogenesis of rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. 2016;18(1):286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rahbarghazi R, Jabbari N, Sani NA, et al. Tumor-derived extracellular vesicles: reliable tools for Cancer diagnosis and clinical applications. Cell Commun Signal. 2019;17(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Martinez-Escala ME, Choi J. Are microRNAs key to developing biomarkers for cutaneous T-cell lymphoma? J Invest Dermatol. 2018;138(9):1906–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zheng Z, Sun R, Zhao HJ, et al. MiR155 sensitized B-lymphoma cells to anti-PD-L1 antibody via PD-1/PD-L1-mediated lymphoma cell interaction with CD8+T cells. Mol Cancer. 2019;18(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Khare D, Goldschmidt N, Bardugo A, et al. Plasma microRNA profiling: exploring better biomarkers for lymphoma surveillance. PLoS One. 2017;12(11):e0187722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ralfkiaer U, Lindahl LM, Litman T, et al. MicroRNA expression in early mycosis fungoides is distinctly different from atopic dermatitis and advanced cutaneous T-cell lymphoma. Anticancer Res. 2014;34(12):7207–7217. [PubMed] [Google Scholar]
- [20].Yang Y, Li Y, Chen X, et al. Exosomal transfer of miR-30a between cardiomyocytes regulates autophagy after hypoxia. J Mol Med (Berl). 2016;94(6):711–724. [DOI] [PubMed] [Google Scholar]
- [21].Zhu M, Deng G, Tan P, et al. Beclin 2 negatively regulates innate immune signaling and tumor development. J Clin Invest. 2020;130(10):5349–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Huang Y, Liu W, He B, et al. Exosomes derived from bone marrow mesenchymal stem cells promote osteosarcoma development by activating oncogenic autophagy. J Bone Oncol. 2020;21:100280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen Z, You L, Wang L, et al. Dual effect of DLBCL-derived EXOs in lymphoma to improve DC vaccine efficacy in vitro while favor tumorgenesis in vivo. J Exp Clin Cancer Res. 2018;37(1):190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl). 2013;91(4):431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Valadi H, Ekstrom K, Bossios A, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659. [DOI] [PubMed] [Google Scholar]
- [26].Fassina A, Marino F, Siri M, et al. The miR-17-92 microRNA cluster: a novel diagnostic tool in large B-cell malignancies. Lab Invest. 2012;92(11):1574–1582. [DOI] [PubMed] [Google Scholar]
- [27].Nicotra G, Mercalli F, Peracchio C, et al. Autophagy-active beclin-1 correlates with favourable clinical outcome in non-Hodgkin lymphomas. Mod Pathol. 2010;23(7):937–950. [DOI] [PubMed] [Google Scholar]
- [28].Zhang L, Feng Q, Wang Z, et al. Progesterone receptor antagonist provides palliative effects for uterine leiomyoma through a Bcl-2/Beclin1-dependent mechanism. Biosci Rep. 2019;39(7) :BSR20190094. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [29].Yuan N, Song L, Zhang S, et al. Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica. 2015;100(3):345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Masud Alam M, Kariya R, Kawaguchi A, et al. Inhibition of autophagy by chloroquine induces apoptosis in primary effusion lymphoma in vitro and in vivo through induction of endoplasmic reticulum stress. Apoptosis. 2016;21(10):1191–1201. [DOI] [PubMed] [Google Scholar]
- [31].Enblad G, Karlsson H, Loskog AS. CAR T-cell therapy: the role of physical barriers and immunosuppression in lymphoma. Hum Gene Ther. 2015;26(8):498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vu LT, Peng B, Zhang DX, et al. Tumor-secreted extracellular vesicles promote the activation of cancer-associated fibroblasts via the transfer of microRNA-125b. J Extracell Vesicles. 2019;8(1):1599680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hung Y, Wang YL, Lin YZ, et al. The exosomal compartment protects epidermal growth factor receptor from small molecule inhibitors. Biochem Biophys Res Commun. 2019;510(1):42–47. [DOI] [PubMed] [Google Scholar]
- [34].Gangadaran P, Ahn BC. In vivo tracking of tumor-derived bioluminescent extracellular vesicles in mice. Methods Mol Biol. 2020;2081:203–210. [DOI] [PubMed] [Google Scholar]
- [35].Dorsam B, Bosl T, Reiners KS, et al. Hodgkin lymphoma-derived extracellular vesicles change the secretome of fibroblasts toward a CAF phenotype. Front Immunol. 2018;9:1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu W, Zhu M, Wang H, et al. Diffuse large B cell lymphoma-derived extracellular vesicles educate macrophages to promote tumours progression by increasing PGC-1beta. Scand J Immunol. 2020;91(2):e12841. [DOI] [PubMed] [Google Scholar]
- [37].Bayraktar R, Van Roosbroeck K, Calin GA. Cell-to-cell communication: microRNAs as hormones. Mol Oncol. 2017;11(12):1673–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kuppers DA, Schmitt TM, Hwang HC, et al. The miR-106a~363(Xpcl1) miRNA cluster induces murine T cell lymphoma despite transcriptional activation of the p27(Kip1) cell cycle inhibitor. Oncotarget. 2017;8(31):50680–50691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Battistella M, Romero M, Castro-Vega LJ, et al. The high expression of the microRNA 17-92 cluster and its paralogs, and the downregulation of the target gene PTEN, is associated with primary cutaneous B-cell lymphoma progression. J Invest Dermatol. 2015;135(6):1659–1667. [DOI] [PubMed] [Google Scholar]
- [40].Xue X, Zhao Y, Wang X, et al. Development and validation of serum exosomal microRNAs as diagnostic and prognostic biomarkers for hepatocellular carcinoma. J Cell Biochem. 2019;120(1):135–142. [DOI] [PubMed] [Google Scholar]
- [41].Tang W, Li J, Liu H, et al. MiR-106a promotes tumor growth, migration, and invasion by targeting BCL2L11 in human endometrial adenocarcinoma. Am J Transl Res. 2017;9(11):4984–4993. [PMC free article] [PubMed] [Google Scholar]
- [42].You F, Luan H, Sun D, et al. miRNA-106a promotes breast cancer cell proliferation, clonogenicity, migration, and invasion through inhibiting apoptosis and chemosensitivity. DNA Cell Biol. 2019;38(2):198–207. [DOI] [PubMed] [Google Scholar]
- [43].Chiang WC, Wei Y, Kuo YC, et al. High-throughput screens to identify autophagy inducers that function by disrupting beclin 1/Bcl-2 binding. ACS Chem Biol. 2018;13(8):2247–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xu HD, Qin ZH. Beclin 1, Bcl-2 and autophagy. Adv Exp Med Biol. 2019;1206:109–126. [DOI] [PubMed] [Google Scholar]
- [45].Tan P, He L, Xing C, et al. Myeloid loss of Beclin 1 promotes PD-L1hi precursor B cell lymphoma development. J Clin Invest. 2019;129(12):5261–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Meng L, Wen Y, Zhou M, et al. Ouabain induces apoptosis and autophagy in Burkitt’s lymphoma Raji cells. Biomed Pharmacother. 2016;84:1841–1848. [DOI] [PubMed] [Google Scholar]
- [47].Ming He J, Liu PY, Wang J. MicroRNA-17-5p regulates the growth, migration and invasion of the human osteosarcoma cells by modulating the expression of PTEN. J BUON. 2020;25(2):1028–1034. [PubMed] [Google Scholar]
- [48].Hao H, Xia G, Wang C, et al. miR-106a suppresses tumor cells death in colorectal cancer through targeting ATG7. Med Mol Morphol. 2017;50(2):76–85. [DOI] [PubMed] [Google Scholar]
- [49].Rothschild SI, Gautschi O, Batliner J, et al. MicroRNA-106a targets autophagy and enhances sensitivity of lung cancer cells to Src inhibitors. Lung Cancer. 2017;107:73–83. [DOI] [PubMed] [Google Scholar]
- [50].Cui X, Wang X, Zhou X, et al. miR-106a regulates cell proliferation and autophagy by targeting LKB1 in HPV-16-associated cervical cancer. Mol Cancer Res. 2020;18(8):1129–1141. [DOI] [PubMed] [Google Scholar]
- [51].Granato M, Rizzello C, Gilardini Montani MS, et al. Quercetin induces apoptosis and autophagy in primary effusion lymphoma cells by inhibiting PI3K/AKT/mTOR and STAT3 signaling pathways. J Nutr Biochem. 2017;41:124–136. [DOI] [PubMed] [Google Scholar]
- [52].Tan KT, Lin MX, Lin SC, et al. Sinensetin induces apoptosis and autophagy in the treatment of human T-cell lymphoma. Anticancer Drugs. 2019;30(5):485–494. [DOI] [PubMed] [Google Scholar]
- [53].Sun B, Ou H, Ren F, et al. Propofol inhibited autophagy through Ca(2+)/CaMKKbeta/AMPK/mTOR pathway in OGD/R-induced neuron injury. Mol Med. 2018;24(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gu YY, Chen MH, May BH, et al. Matrine induces apoptosis in multiple colorectal cancer cell lines in vitro and inhibits tumour growth with minimum side effects in vivo via Bcl-2 and caspase-3. Phytomedicine. 2018;51:214–225. [DOI] [PubMed] [Google Scholar]