

Abstract
Nuclear receptor subfamily 1 group D member 1 (NR1D1, also known as Rev‐erbα) is a nuclear transcription factor that is part of the molecular clock encoding circadian rhythms and may link daily rhythms with metabolism and inflammation. NR1D1, unlike most nuclear receptors, lacks a ligand‐dependent activation function domain 2 and is a constitutive transcriptional repressor. Amyotrophic lateral sclerosis (ALS) is the most common adult‐onset motor neuron disease, caused by the progressive degeneration of motor neurons in the spinal cord, brain stem, and motor cortex. Approximately 10%–20% of familial ALS is caused by a toxic gain‐of‐function induced by mutations of the Cu/Zn superoxide dismutase (SOD1). Dysregulated clock and clock‐controlled gene expression occur in multiple tissues from mutant hSOD1‐linked ALS mouse models. Here we explore NR1D1 dysregulation in the spinal cord of ALS mouse models and its consequences on astrocyte–motor neuron interaction. NR1D1 protein and mRNA expression are significantly downregulated in the spinal cord of symptomatic mice expressing mutant hSOD1, while no changes were observed in age‐matched animals overexpressing wild‐type hSOD1. In addition, NR1D1 downregulation in primary astrocyte cultures induces a pro‐inflammatory phenotype and decreases the survival of cocultured motor neurons. NR1D1 orchestrates the cross talk between physiological pathways identified to be disrupted in ALS (e.g., metabolism, inflammation, redox homeostasis, and circadian rhythms) and we observed that downregulation of NR1D1 alters astrocyte–motor neuron interaction. Our results suggest that NR1D1 could be a potential therapeutic target to prevent astrocyte‐mediated motor neuron toxicity in ALS.
Keywords: amyotrophic lateral sclerosis, astrocytes, inflammation, motor neurons, NR1D1, Rev‐erbα
Abbreviations
- ALS
amyotrophic lateral sclerosis
- NR1D1
nuclear receptor subfamily 1 Group D member 1
- ROR
RAR‐related orphan receptor
- SOD1
superoxide dismutase 1
1. INTRODUCTION
NR1D1 (also known as Rev‐erbα) is a transcription factor that belongs to the nuclear hormone receptor family. NR1D1 is a constitutive transcriptional repressor and was initially classified as an orphan receptor until the discovery of its ligand, heme, in 2007. 1 , 2 NR1D1, unlike most nuclear receptors, lacks a ligand‐dependent activation function domain 2 (AF‐2). 3 NR1D1 binds as a monomer or homodimer to the ROR response element (RORE) or Rev‐DR2 motif, respectively. During promoter binding, nuclear receptor corepressor 1 (NCoR1) and histone deacetylase 3 (HDAC3) are recruited by NR1D1 to repress transcription. 4 , 5
NR1D1 represses the expression of genes involved in numerous physiological functions, including circadian rhythm. Circadian rhythms are genetically encoded by a molecular clock composed of interlocked positive and negative transcriptional/translational feedback loops (TTFL), which induce and repress the transcription of target genes, respectively. The positive arm of the core clock consists of brain and muscle ARNT‐like 1 (Bmal1) and circadian locomotor output cycles protein kaput (Clock), while the negative arm of the core clock consists of Period 1/Period 2 (Per1/2) and Cryptochrome 1/Cryptochrome 2 (Cry1/2). 6 NR1D1 participates in a second interlocking TTFL, where it suppresses the positive arm of the core clock. NR1D1 and RAR‐related orphan receptor α (Rorα) compete for binding to the RORE motif located in the Bmal1 promoter to directly repress or stimulate transcription, respectively. 7 , 8 , 9
In addition to its role in the molecular clock, NR1D1 is also a known repressor of metabolic and inflammatory genes whose expressions also fluctuate in a time‐of‐day‐dependent manner. 10 , 11 , 12 Therefore, NR1D1 can potentially be the link coordinating circadian rhythm, metabolism, and inflammation. In vivo, NR1D1 is not required for circadian rhythm generation 7 ; however, Nr1d1 knockout mice display spontaneous microglial and astrocyte activation and dysregulated NF‐κB signaling. 13
Amyotrophic lateral sclerosis (ALS) is characterized by the progressive loss of motor neurons in the motor cortex, brain stem, and spinal cord. No cure or effective treatment is currently available, although two drugs (riluzole and edaravone) appear to slightly slow disease progression. 14 , 15 Approximately 10% of the ALS cases present with a familial history of the disease (familial ALS (FALS)) and are most frequently linked to dominant mutations. The remaining ALS cases do not have a familial history (sporadic ALS (SALS)) and may result from yet unidentified interaction between environmental and genetic risk factors. 16 The first gene linked to ALS was superoxide dismutase 1 (SOD1) 17 and mice overexpressing ALS‐linked mutant hSOD1 develop an ALS‐like phenotype. 18 The pathophysiological analysis of these mouse models has contributed to a significant portion of our mechanistic understanding of ALS pathology.
In ALS patients and animal models, astrocytes display increased inflammatory markers. 19 , 20 , 21 , 22 Upregulation of inflammatory genes and downregulation of metabolic genes appear to be among the earlier transcriptional changes observed in astrocytes from ALS mice. 23 The molecular mechanism underlying the death of motor neurons in ALS remains uncertain. However, the phenotype acquired by ALS astrocytes likely contributes to motor neuron demise—an observation that has been clearly established in FALS and SALS models. 24 , 25 , 26 , 27 , 28 , 29 , 30
We recently analyzed the expression of clock and clock‐controlled genes in multiple tissues from mutant hSOD1‐linked ALS mouse models. 31 Interestingly, NR1D1 was the only core clock gene that displayed significant expression changes in all tissue types analyzed from early symptomatic mice overexpressing the ALS‐linked hSOD1G93A mutation. Here we further explore NR1D1 dysregulation in the spinal cord of ALS mouse models and its consequences on astrocyte–motor neuron interaction.
2. METHODS AND MATERIALS
2.1. Reagents
All chemicals and reagents were purchased from Sigma‐Aldrich unless otherwise specified. Culture media and supplements were from ThermoFisher Scientific unless otherwise specified. Primers sequences were previously described 31 and were obtained from Integrated DNA Technologies.
2.2. Animals
B6.Cg‐Tg(SOD1*G93A)1Gur/J and B6. Cg‐Tg(SOD1)2Gur/J mice 18 were obtained from The Jackson Laboratory and maintained as hemizygous animals in a C57BL/6J background. hSOD1H46R/H48Qmice were provided by Dr. David Borchelt 32 and have been backcrossed into C57BL/6J pure background for more than 10 generations. Tissue harvesting was performed at the same time of the day for all animals in this study. All animal procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH. The Animal Care and Use Committee of UW‐Madison approved the animal protocol pertinent to the experiments reported in this publication.
2.3. Primary astrocyte cultures and astrocyte–motor neuron cocultures
Primary astrocyte cultures were prepared from 1‐day‐old pups according to the procedure of Saneto and De Vellis. 33 Briefly, spinal cords were dissociated and incubated in 0.25% trypsin in PBS for 25 min at 37°C. Dissociated cells were passed through a 70‐μm sieve, centrifuged and plated at a density of 1.5 × 106 cells per 25 cm2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, HEPES (3.6 g/L), penicillin (100 IU/ml), and streptomycin (100 μg/ml). When confluent, cultures were shaken for 48 h at 250 rpm at 37°C, followed by a 48‐h treatment with 10 μM cytosine arabinoside. After 48 h, cytosine removal cells were replated at a density of 2 × 106 cells/cm2. Experiments were performed when cultures reach confluency. Control induced pluripotent stem cells (iPSCs) lines were obtained from the NINDS Human Cell and Data Repository or commercial vendors. Details of iPSC lines are as follows: line 1 (iPSC ID# FA0000011, control), line 2 (iPSC ID# ND38555, control), line 3 (XCL‐1, Stemcell #70901, control). iPSCs were differentiated into induced NPCs using an embryoid body formation protocol in the presence of SMAD signaling inhibitors (STEMdiff SMADi Media, Stemcell). Induction was confirmed by an increase in MAP2, PAX6, and NESTIN gene expression and concurrent decrease in SOX2, OCT3, and NANOG expression. Induced NPCs were cultured for 3 weeks in astrocyte differentiation media (STEMdif Astrocyte Differentiation Media, Stemcell), following by 3 weeks in astrocyte maturation media (STEMdif Astrocyte Maturation Media, Stemcell). Astrocyte differentiation was confirmed by assessing GFAP, S100B, and ALDH1L1 gene expression. Following differentiation, iAs were cultured in DMEM‐F12 supplemented with 10% FBS and 0.3% N2 supplement. Motor neuron cultures were prepared from 12.5‐embryonic‐day mouse spinal cords. Motor neurons were purified by a combination of differential centrifugation on BSA cushions and an OptiPrep gradient as previously described. 34 For coculture experiments, motor neurons were plated on primary astrocyte monolayers at a density of 300 cells/cm2 and maintained for 16 or 72 h in L15 medium supplemented with 0.63 mg/ml bicarbonate, 5 μg/ml insulin, 0.1 mg/ml conalbumin, 0.1 mM putrescine, 30 nM sodium selenite, 20 nM progesterone, 20 mM glucose, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 2% horse serum. For coculture experiments with iPSC‐derived astrocytes, motor neurons were plated on astrocyte monolayers at a density of 1500 cells/cm2 and maintained for 72 h in Neurobasal medium supplemented with 1X B27 supplement and GDNF (1 ng/ml). Motor neurons in cocultures with mouse primary astrocytes were identified by immunostaining against βIII‐tubulin (Millipore, 05‐559, lot:2757108), and survival was determined by counting all cells displaying intact neurites longer than 4 cell bodies in diameter. Counts were performed over an area of 0.90 cm2 in 24‐well plates. The cocultures with iPSC‐derived astrocytes were performed with motor neurons isolated from ROSAmT/mG transgenic mice (B6.129(Cg)‐Gt(ROSA)26Sortm4(ACTB‐tdTomato,‐EGFP)Luo/J; Stock No. 007676, The Jackson Laboratory), which express a cell membrane‐localized tdTomato (mT) fluorescent protein. ROSAmT/mG motor neurons in the coculture were identified by immunostaining with a CF594‐conjugated anti‐Red Fluorescent Protein antibody (Biotium, 20422, lot: 20C1003). Motor neuron counting was performed as described above.
2.4. Immunofluorescence
Antigen retrieval and staining in paraffin‐embedded tissues was performed as previously described. 35 Mice lumbar spinal cord sections were stained with anti‐GFAP (Novus, NB300‐141, lot 179‐071616) or anti‐NeuN (Cell Signaling, 24307, lot 2) and anti‐NR1D1 (Abnova, H00009572‐MO2, lot J9051‐4F6) antibodies. Nuclei were counterstained with DAPI (4′,6‐diamidino‐2‐phenylindole dihydrochloride). Images were acquired on an LSM 880 microscope (Zeiss) with Zen Black (v2.3) image software. To determine the percent of NR1D1‐positive nuclei, images were analyzed using ImageJ (version 1.52p). After setting a threshold, individual image channels were converted to binary, overlaid, and then the number of nuclei containing overlapping areas were obtained using the Analyze Particles function. In each group, there were four or five mice and two to four images were analyzed per animal.
2.5. Real‐time PCR and western blot
RNA extraction, RNA retrotranscription, real‐time PCR, and western blot analysis were performed as previously described. 36 Membranes were incubated overnight with anti‐NR1D1 (Novus, H00009572‐M02) and anti‐Actin (Sigma, A5441, lot: 061M4808). Image acquisition was performed in a chemiluminescent western blot scanner (Li‐Cor) or exposed on Kodak BioMax Light film. The western blot image presented in Figure 4A was quantified using ImageJ Software (NIH), all other western blot quantifications were performed using the Image Studio Software (Li‐Cor).
FIGURE 4.
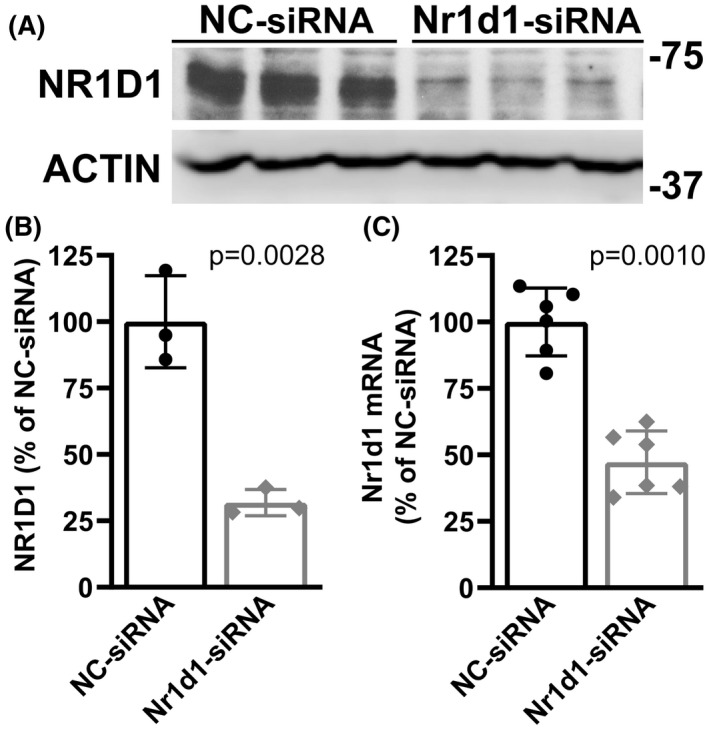
NR1D1 silencing in primary nontransgenic spinal cord astrocytes. Confluent primary astrocyte cultures from nontransgenic mice were treated with a negative control siRNA (NC‐siRNA) or an Nr1d1‐siRNA for 48 h. (A) Western bolt analysis of NR1D1 protein levels in astrocytes following siRNA treatment. (B) Quantification of NR1D1 protein levels shown in A. NR1D1 expression was quantified, normalized by ACTIN levels, and expressed as a percentage of NC‐siRNA‐treated cultures (mean ± SD). (C) Nr1d1 mRNA levels in Nr1d1‐siRNA‐treated astrocytes. Nr1d1 mRNA levels were determined by real‐time PCR and corrected by Actin mRNA levels (mean ± SD)
2.6. Cell treatment and transfections
siRNA transfections with a mouse‐specific Nr1d1‐siRNA (duplex sequence: 5′‐AGAAUAUCCAGUACAAACGGUGUCT‐3′ and 5′‐AGACACCGUUUGUACUGGAUAUUCUGU‐3′) or human‐specific Nr1d1‐siRNA (Horizon, ID: L‐003411‐00‐0005) were performed using Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions. Astrocytes were transfected with 25 nM of a negative control siRNA (NC‐siRNA) or siRNA for Nr1d1 (Nr1d1‐siRNA). After 48 h, astrocytes were harvested for analysis or used in coculture experiments.
2.7. NF‐κB reporter assay
Adenovirus expressing a firefly luciferase gene under the control of a synthetic promoter that contains direct repeats of the NF‐κB binding site (Ad‐NFkb‐Luc) or a Renilla luciferase under a constitutive promoter (Ad‐pRL‐Luc) were obtained from Vector BioLabs. Adenovirus‐mediated transductions were performed at a multiplicity of infection of 10 and 3, respectively. Seventy‐two hours after viral transduction, cultures were transfected with NC‐siRNA or Nr1d1‐siRNA as described above, and firefly and Renilla luciferase activity were consecutively assayed 48 h later with the Dual‐Glo luciferase system (Promega).
2.8. Statistical analysis
Groups of three or four animals were used for biochemical analysis. Unless otherwise noted, cell culture experiments were repeated in at least three independent primary culture preparations, and values from each independent experiment were combined for data reporting. All data are reported as mean ± SD. Comparisons between two groups were performed with an unpaired t‐test. Multiple group comparisons were performed by one‐way ANOVA with Tukey's posttest. When comparing the effect of combinations of treatments, two‐way ANOVA was used followed by Tukey's posttest. Differences were declared statistically significant if p ≤ .05. All statistical computations were performed using Prism 9.0 (GraphPad Software).
3. RESULTS
We have previously shown evidence of dysregulated clock and clock‐controlled gene expression at mRNA level in early symptomatic animal models of ALS. 31 Here, we examined NR1D1 expression in the spinal cord of overtly symptomatic hSOD1G93A mice (about 140‐day‐old). Compared to nontransgenic controls, hSOD1G93A mice showed significant downregulation in both NR1D1 protein (Figure 1A,B) and mRNA levels (Figure 1C). Likewise, NR1D1 protein and mRNA expression were also significantly downregulated in the spinal cord of symptomatic hSOD1H46R/H48Q mice (about 210‐day‐old, Figure 1D–F). Spinal cords from 7‐month‐old mice overexpressing wild‐type hSOD1, which do not develop overt motor neuron degeneration, showed no difference in NR1D1 expression levels (Figure 1G–I); suggesting that NR1D1 downregulation is linked to the expression of the mutant hSOD1 protein in these mouse models. Consistent with the downregulation in NR1D1 protein expression described in Figure 1, immunostaining in the ventral horn of the spinal cord of hSOD1G93A mice shows that NR1D1 downregulation appears to occur in both neuronal and nonneuronal cells (Figure 2). Moreover, when quantified, we observed a significant decrease in the number of NR1D1‐positive nuclei in the ventral horn of the spinal cord from both hSOD1G93A and hSOD1H46R/H48Q mice (Figure 3).
FIGURE 1.
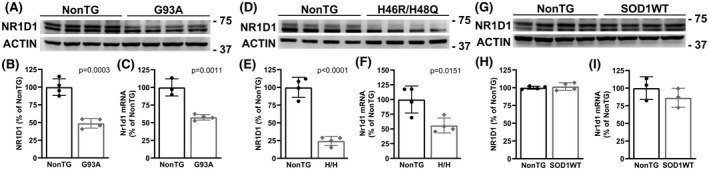
Decreased NR1D1 expression in the spinal cord of mutant hSOD1‐linked mouse models. (A, D, and G) Western blot analysis of NR1D1 expression in the lumbar spinal cord of age‐matched nontransgenic (NonTG) and symptomatic hSOD1G93A (G93A, about 140 days old) mice (A) or symptomatic hSOD1H46R/H48Q (H46R/H48Q, about 210 days old) mice (D) or age‐matched control hSOD1WT mice (about 210 days old) (G). (B, E, and H) Quantification of NR1D1 protein levels shown in A, D, and G, respectively. NR1D1 expression was quantified, normalized by ACTIN levels and expressed as a percentage of age‐matched NonTG mice (each lane corresponds to a different animal, mean ± SD). Both bands shown in the NR1D1 western blot were used for intensity quantification. (C, F, and I) Nr1d1 mRNA levels in the lumbar spinal cord of age‐matched NonTG and symptomatic G93A (C), or symptomatic H46R/H48Q (F), or age‐matched hSOD1WT (I) mice. Nr1d1 mRNA levels were determined by real‐time PCR and corrected by Actin mRNA levels (n = 3 or 4 mice, mean ± SD)
FIGURE 2.
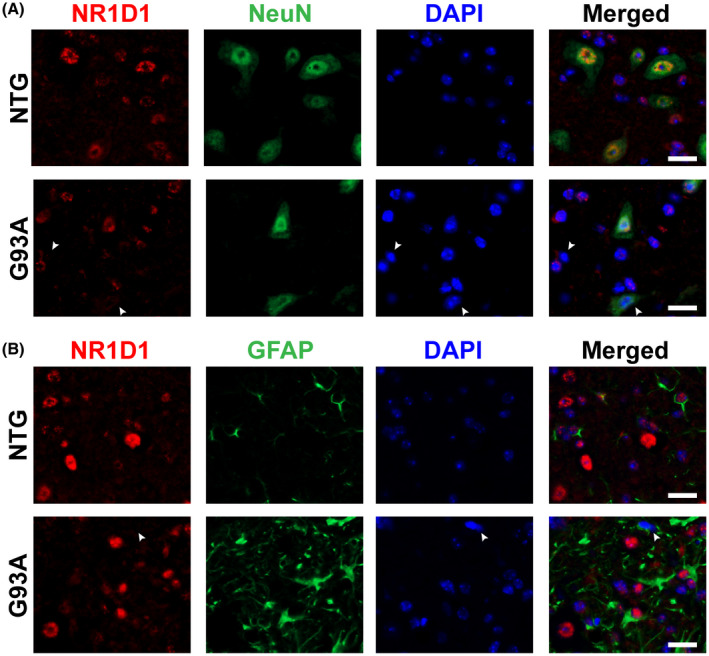
Decreased NR1D1 expression in the spinal cord of symptomatic hSOD1G93A mice. (A) Representative images showing NR1D1 (red) and NeuN (green) immunofluorescence in the ventral horn of the lumbar spinal cord from symptomatic hSOD1G93A (G93A) and age‐matched nontransgenic (NTG) mice. Nuclei were counterstained with DAPI (blue). The arrowheads point to NR1D1‐negative nuclei from NeuN‐negative and NeuN‐positive cells. Scale bar: 20 μm. (B) Representative images showing NR1D1 (red) and GFAP (green) immunofluorescence in the ventral horn of the lumbar spinal cord from symptomatic G93A and age‐matched NonTG mice. Nuclei were counterstained with DAPI (blue). The arrowhead points to a NR1D1‐negative, GFAP‐positive nucleus. Scale bar: 20 μm
FIGURE 3.
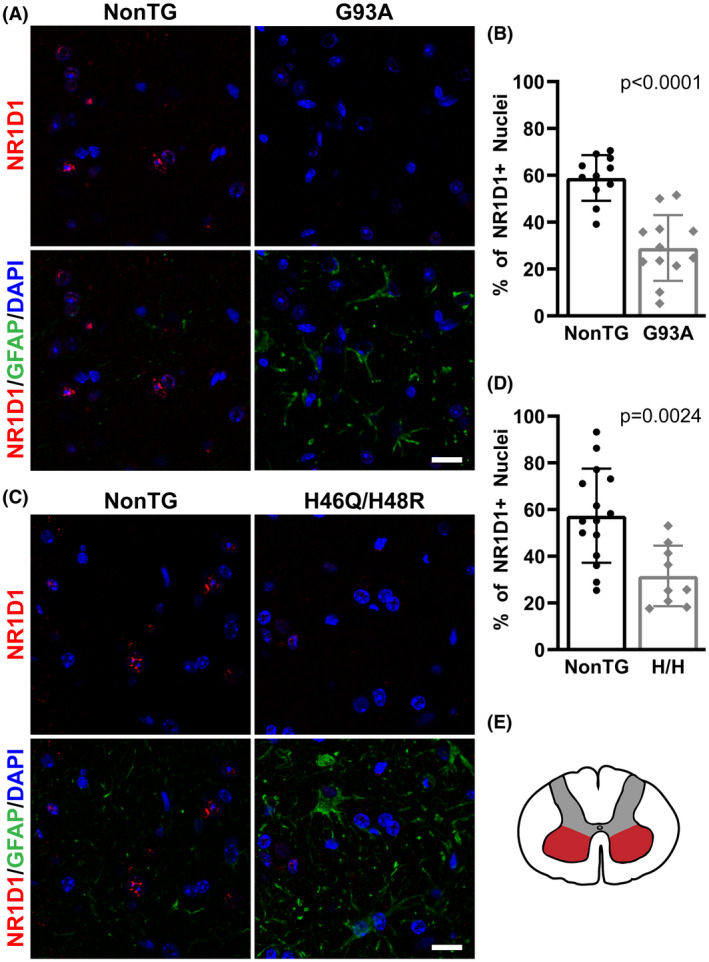
Decreased NR1D1 immunostaining in the spinal cord of mutant hSOD1‐linked mouse models. (A and C) Representative images showing GFAP (green) and NR1D1 (red) immunofluorescence in the ventral horn of the lumbar spinal cord from symptomatic hSOD1G93A (G93A), hSOD1H46R/H48Q (H46R/H48Q), and age‐matched nontransgenic (NonTG) mice. Nuclei were counterstained with DAPI (blue). Scale bar: 20 μm. (B, D) Quantification of NR1D1‐positive nuclei in the ventral horn of the lumbar spinal cord from mice in A and C. Data are expressed as the percentage of NR1D1‐positive nuclei from the total number of nuclei analyzed for each genotype (n = 4 or 5 mice per group and two to four images per animal; mean ± SD). (E) Schematic representation of the spinal cord indicating the area (red) in which the analysis was performed
NR1D1 downregulation in the spinal cord of these two ALS models does not seem to be restricted to a specific cell type. However, since NR1D1 displays altered rhythmicity in iPSC‐derived astrocytes from ALS patients 31 and decreased NR1D1 expression can lead to dysregulated inflammation, 13 , 37 we sought to determine the effects of decreasing NR1D1 expression on the inflammatory profile of spinal cord astrocytes in culture. We transfected primary nontransgenic astrocyte cultures with a negative control siRNA (NC‐siRNA) or an Nr1d1‐specific siRNA (Nr1d1‐siRNA). After 48 h, we observed a significant NR1D1 downregulation in cultures treated with Nr1d1‐siRNA (Figure 4). In addition, we observed that the downregulation of NR1D1 lead to increased luciferase expression from an NF‐κB‐driven promoter (Figure 5A). Accordingly, mRNA of NF‐κB target genes, including Nos2, Il6, Ptgs2, Ccl5, and Cxcl10 were significantly upregulated in astrocytes with decreased NR1D1 expression (Figure 5B), indicating that NR1D1 downregulation induces a pro‐inflammatory phenotype in astrocytes.
FIGURE 5.
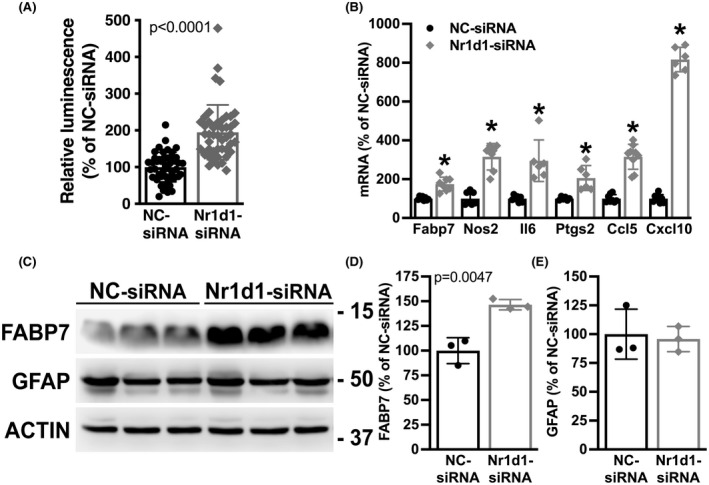
NR1D1 downregulation induces a pro‐inflammatory phenotype in nontransgenic astrocytes. (A) Relative luminescence produced by a firefly luciferase expressed under an NF‐κB‐driven promoter 48 h after NC‐siRNA or Nr1d1‐siRNA treatment of nontransgenic astrocytes. Relative firefly luciferase luminescence was corrected by the amount of Renilla luciferase activity controlled by a constitutive promoter and expressed as a percentage of NC‐siRNA‐treated control cells. (B) Fabp7, Nos2, Il6, Ptgs2, Ccl5, and Cxcl10 mRNA levels in astrocytes 48 h after NC‐siRNA or Nr1d1‐siRNA treatment. mRNA levels were determined by real‐time PCR and corrected by Actin mRNA levels. Data are expressed as a percentage of NC‐siRNA‐treated control cells. (C) Western blot analysis of FABP7 and GFAP expression in astrocytes treated as in A. (D, E) Quantification of the images shown in (C). FABP7 and GFAP protein levels were quantified, corrected by ACTIN levels and expressed as a percentage of NC‐siRNA‐treated control cells. For all graph panels, data are expressed as mean ± SD (*p < .05, significantly different from NC‐siRNA‐treated cells)
It has been previously reported that NR1D1 has a key role in regulating the expression of fatty acid‐binding protein 7 (FABP7) in astrocytes. 38 , 39 Accordingly, we observed that Nr1d1‐siRNA‐treated spinal cord astrocytes displayed upregulated FABP7 expression (Figure 5B–D). We have previously shown that FABP7 upregulation induces a phenotype that is detrimental to the survival of cocultured motor neurons. 40 To examine the effect of NR1D1 silencing on the way astrocytes interact with neighboring neurons, we cocultured motor neurons with astrocytes pretreated with Nr1d1‐siRNA or NC‐siRNA. Downregulation of NR1D1 in astrocytes does not alter the number of motor neurons able to attach to the astrocyte monolayer, since a similar number of motor neurons was observed after 16 h in coculture with Nr1d1‐siRNA or NC‐siRNA (Figure 6A). However, after 3 days in culture, a significant decrease in motor neuron survival was observed in cocultures with astrocytes pretreated with Nr1d1‐siRNA, compared to cocultures with NC‐siRNA‐treated astrocytes (Figure 6B). Moreover, the decrease in motor neuron survival does not appear to be a consequence of decreased trophic support, since the addition of exogenous trophic factors (GDNF 10 ng/ml and BDNF 0.1 ng/ml), capable of supporting motor neuron survival in pure cultures, did not prevent motor neuron loss in cocultures with Nr1d1‐silenced astrocytes (Figure 6B). In transdifferentiated astrocytes obtained from three different human iPSC lines from healthy subjects, silencing of Nr1d1 expression also leads to increased luciferase expression from an NF‐κB‐driven promoter (Figure 7A–C). In addition, a significant decrease in motor neuron survival was also observed in cocultures with transdifferentiated astrocytes pretreated with Nr1d1‐siRNA, compared to cocultures with NC‐siRNA‐treated astrocytes.
FIGURE 6.
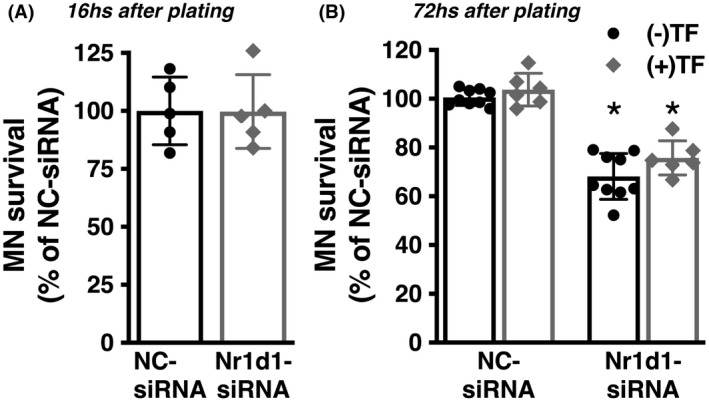
The phenotype induced by NR1D1 downregulation in primary nontransgenic astrocytes is detrimental to cocultured motor neurons. (A) Spinal cord nontransgenic astrocytes were treated with NC‐siRNA or Nr1d1‐siRNA. Forty‐eight hours later embryonic motor neurons were plated on top. Motor neuron (MN) survival was determined 16 h later (data obtained from two independent coculture experiments, two or three replicas per coculture). (B) The same coculture setup as in A but motor neuron survival was determined 72 h later. The addition of trophic factors (TF: GDNF 10 ng/ml and BDNF 0.1 ng/ml) to the coculture does not prevent the motor neuron loss induced by Nr1d1‐siRNA treated astrocytes. Cocultures were treated at the time of motor neuron plating with (+TF) or without (−TF) trophic factors, and motor neuron survival was determined 72 h later. Data were obtained from at least three independent coculture experiments, two or three replicas per coculture. For all panels, data are expressed as percentage of NC‐siRNA control mean ± SD (*p < .05, significantly different from NC‐siRNA‐treated cells)
FIGURE 7.
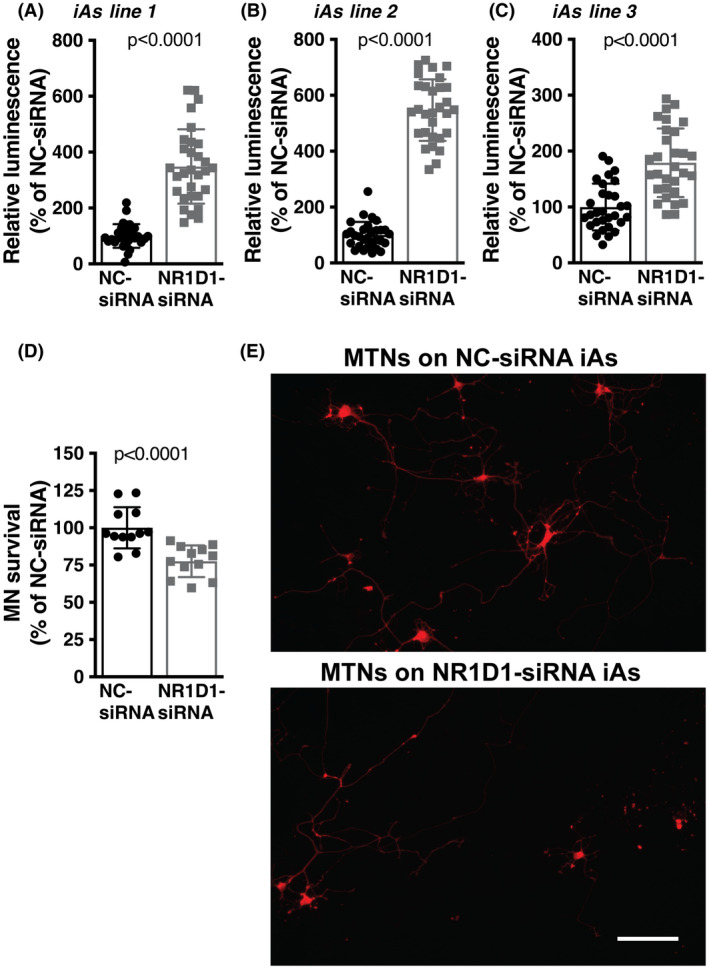
NR1D1 downregulation in transdifferentiated human astrocytes is detrimental to cocultured motor neurons. (A, B, C) Relative luminescence produced by a firefly luciferase expressed under an NF‐κB‐driven promoter 48 h after NC‐siRNA or Nr1d1‐siRNA treatment of three different transdifferentiated human astrocytes (iAs) lines derived from control (nondisease) healthy subjects. Relative firefly luciferase luminescence was corrected by the amount of Renilla luciferase activity controlled by a constitutive promoter and expressed as a percentage of NC‐siRNA‐treated cells. (D) Control iAs (line 1) were treated with NC‐siRNA or Nr1d1‐siRNA. Forty‐eight hours later embryonic motor neurons were plated on top. Motor neuron (MN) survival was determined 72 h later. Data were obtained from four independent coculture experiments, three replicas per coculture. For all panels, data are expressed as percentage of NC‐siRNA control (mean ± SD). (E) Representative images showing motor neurons cultured on the top of NC‐siRNA or Nr1d1‐siRNA‐treated iAs. Scale bar: 20 μm
4. DISCUSSION
Astrocyte‐mediated motor neuron death is a feature observed in many ALS models 24 , 25 , 27 , 29 , 30 , 41 , 42 , 43 , 44 , 46 and strategies aimed at curbing this neurotoxic phenotype have been shown to increase motor neuron survival when translated into ALS mouse models. 35 , 45 , 47 , 48 , 49 Here we show that NR1D1 (also known as Rev‐erbα) expression is significantly downregulated in the spinal cord of symptomatic mice expressing mutant hSOD1 and that this decrease in NR1D1 expression could potentially impact the way astrocytes interact with motor neurons.
In FALS and SALS patients, reactive astrocytes surround both upper and lower degenerating motor neurons, as well as the region where the descending fibers of corticospinal tracts enter the gray matter. 50 , 51 , 52 , 53 The astrocyte reactivity observed in ALS patients is recapitulated in mutant hSOD1 transgenic rodents. 32 , 54 , 55 , 56 , 57 , 58 , 59 Reactive astrocytes in patients and ALS animal models express inflammatory makers such as cyclooxygenase‐2 (Ptgs2) and inducible nitric oxide synthase (Nos2 or iNos). 19 , 20 , 21 , 22 Many of these markers are under the direct control of the transcription factor NF‐κB and activation of the canonical NF‐κB pathway is a prominent feature of activated glia in ALS patients and animal models. 23 , 27 , 60 Moreover, translational profiling of astrocytes from hSOD1G37R‐overexpressing mice identified upregulation of inflammatory genes and downregulation of metabolic genes as the earlier changes observed. 23
Since silencing NR1D1 expression in astrocytes causes an inflammatory profile similar to the one seen in ALS animal models, our data suggest that the decrease in NR1D1 expression observed in symptomatic ALS mice may be contributing to the altered inflammatory profile of ALS astrocytes. Moreover, the consequence of this decrease in NR1D1 expression is likely detrimental, since the phenotype induced by silencing NR1D1 in nontransgenic spinal cord murine astrocytes causes a decrease in the survival of cocultured motor neurons in the absence of an exogenous noxious stimulus. Since motor neurons in coculture with these astrocytes cannot be rescued with the addition of exogenous trophic factors, this observation likely reflects the acquisition of a neurotoxic phenotype in Nr1d1‐silenced astrocytes, rather than a decrease in trophic support. Moreover, this negative effect of decreased NR1D1 expression in astrocyte–neuron interaction is also observed in human transdifferentiated astrocytes, suggesting that it could potentially contribute to motor neuron toxicity in the context of ALS pathology.
NR1D1‐KO mice display spontaneous glial activation in the hippocampus and dysregulated NF‐κB signaling. 13 In addition, primary neurons exposed to conditioned media from mixed glia cultures treated with an Nr1d1‐siRNA are more susceptible to hydrogen peroxide toxicity than neurons cultured in conditioned media from control glia. 13 These results suggest that glia NR1D1 deficiency may impact neuronal health in response to oxidative stress in vitro. On the other hand, pharmacological inhibition or genetic knockdown of NR1D1 stimulates microglial amyloid‐β clearance and reduces amyloid plaque deposition in the 5XFAD mouse model of Alzheimer's disease. 61 While these observations reflect that NR1D1 may likely have a complex role in glia biology, they also point at the potential therapeutic role of NR1D1 manipulation during neurodegeneration.
Circadian rhythm coordinates different physiological functions to recurring daily environmental changes like light/dark cycles and food availability. 62 The ability to anticipate these changes increases the fitness of the organism, while impairment of normal circadian rhythmicity leads to a range of metabolic defects. 63 , 64 , 65 , 66 Beyond progressive motor impairment, patients with ALS suffer from major, yet incompletely characterized, defects in energy metabolism. 67 , 68 Since NR1D1 appears to coordinate metabolism and circadian rhythms, it is plausible that NR1D1 dysregulation contributes to the metabolic and redox impairment observed in ALS patients and mouse models. 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 Moreover, based on the current understanding of the role of astrocytes in circadian rhythm, 83 , 84 , 85 , 86 it would be important to determine the potential effect that circadian rhythms have in astrocyte–neuron metabolic and redox coupling.
Overall, our data suggest that NR1D1 downregulation promotes a pro‐inflammatory response in astrocytes that is ultimately detrimental for motor neuron survival. Thus, modulating NR1D1 expression could be a potential therapeutic approach to prevent astrocyte‐mediated motor neuron toxicity in the context of ALS pathology.
DISCLOSURES
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
Kelby M. Killoy, Benjamin A. Harlan, Mariana Pehar and Marcelo R. Vargas performed the experiments. Kelby M. Killoy, Mariana Pehar, and Marcelo R. Vargas analyzed the data and wrote the paper. All authors reviewed and approved the content of the manuscript.
ACKNOWLEDGMENTS
This study was funded by NIH grants R21NS102599 and R01NS089640. This work used resources and facilities of the William S. Middleton Memorial Veterans Hospital (Madison, WI, USA).
Killoy KM, Harlan BA, Pehar M, Vargas MR. NR1D1 downregulation in astrocytes induces a phenotype that is detrimental to cocultured motor neurons. FASEB J. 2022;36:e22262. doi: 10.1096/fj.202101275R
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the article. The data are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Raghuram S, Stayrook KR, Huang P, et al. Identification of heme as the ligand for the orphan nuclear receptors REV‐ERBalpha and REV‐ERBbeta. Nat Struct Mol Biol. 2007;14:1207‐1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yin L, Wu N, Curtin JC, et al. Rev‐erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318:1786‐1789. [DOI] [PubMed] [Google Scholar]
- 3. Renaud JP, Harris JM, Downes M, Burke LJ, Muscat GE. Structure‐function analysis of the Rev‐erbA and RVR ligand‐binding domains reveals a large hydrophobic surface that mediates corepressor binding and a ligand cavity occupied by side chains. Mol Endocrinol. 2000;14:700‐717. [DOI] [PubMed] [Google Scholar]
- 4. Harding HP, Lazar MA. The orphan receptor Rev‐ErbA alpha activates transcription via a novel response element. Mol Cell Biol. 1993;13:3113‐3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harding HP, Lazar MA. The monomer‐binding orphan receptor Rev‐Erb represses transcription as a dimer on a novel direct repeat. Mol Cell Biol. 1995;15:4791‐4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reppert SM, Weaver DR. Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol. 2001;63:647‐676. [DOI] [PubMed] [Google Scholar]
- 7. Preitner N, Damiola F, Lopez‐Molina L, et al. The orphan nuclear receptor REV‐ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251‐260. [DOI] [PubMed] [Google Scholar]
- 8. Ueda HR, Chen W, Adachi A, et al. A transcription factor response element for gene expression during circadian night. Nature. 2002;418:534‐539. [DOI] [PubMed] [Google Scholar]
- 9. Sato TK, Panda S, Miraglia LJ, et al. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 2004;43:527‐537. [DOI] [PubMed] [Google Scholar]
- 10. Cho H, Zhao X, Hatori M, et al. Regulation of circadian behaviour and metabolism by REV‐ERB‐alpha and REV‐ERB‐beta. Nature. 2012;485:123‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gibbs JE, Blaikley J, Beesley S, et al. The nuclear receptor REV‐ERBalpha mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc Natl Acad Sci USA. 2012;109:582‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Solt LA, Wang Y, Banerjee S, et al. Regulation of circadian behaviour and metabolism by synthetic REV‐ERB agonists. Nature. 2012;485:62‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Griffin P, Dimitry JM, Sheehan PW, et al. Circadian clock protein Rev‐erbalpha regulates neuroinflammation. Proc Natl Acad Sci USA. 2019;116:5102‐5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585‐591. [DOI] [PubMed] [Google Scholar]
- 15. Sawada H. Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin Pharmacother. 2017;18:735‐738. [DOI] [PubMed] [Google Scholar]
- 16. Brown RH, Al‐Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:162‐172. [DOI] [PubMed] [Google Scholar]
- 17. Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59‐62. [DOI] [PubMed] [Google Scholar]
- 18. Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772‐1775. [DOI] [PubMed] [Google Scholar]
- 19. Schiffer D, Fiano V. Astrogliosis in ALS: possible interpretations according to pathogenetic hypotheses. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5:22‐25. [DOI] [PubMed] [Google Scholar]
- 20. Barbeito LH, Pehar M, Cassina P, et al. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res Brain Res Rev. 2004;47:263‐274. [DOI] [PubMed] [Google Scholar]
- 21. Vargas MR, Johnson JA. Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurotherapeutics. 2010;7:471‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 2011;10:253‐263. [DOI] [PubMed] [Google Scholar]
- 23. Sun S, Sun Y, Ling SC, et al. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1‐mediated ALS. Proc Natl Acad Sci USA. 2015;112:E6993‐E7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR‐dependent motor neuron apoptosis. J Neurochem. 2006;97:687‐696. [DOI] [PubMed] [Google Scholar]
- 25. Nagai M, Re DB, Nagata T, et al. Astrocytes expressing ALS‐linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non‐cell autonomous effect of glia on motor neurons in an embryonic stem cell‐based ALS model. Nat Neurosci. 2007;10:608‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haidet‐Phillips AM, Hester ME, Miranda CJ, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meyer K, Ferraiuolo L, Miranda CJ, et al. Direct conversion of patient fibroblasts demonstrates non‐cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci USA. 2014;111:829‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ditsworth D, Maldonado M, McAlonis‐Downes M, et al. Mutant TDP‐43 within motor neurons drives disease onset but not progression in amyotrophic lateral sclerosis. Acta Neuropathol. 2017;133:907‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kia A, McAvoy K, Krishnamurthy K, Trotti D, Pasinelli P. Astrocytes expressing ALS‐linked mutant FUS induce motor neuron death through release of tumor necrosis factor‐alpha. Glia. 2018;66:1016‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Killoy KM, Pehar M, Harlan BA, Vargas MR. Altered expression of clock and clock‐controlled genes in a hSOD1‐linked amyotrophic lateral sclerosis mouse model. FASEB J. 2021;35:e21343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Xu G, Gonzales V, et al. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper‐binding site. Neurobiol Dis. 2002;10:128‐138. [DOI] [PubMed] [Google Scholar]
- 33. Saneto RP, Vellis JD . Neuronal and glial cells: cell culture of the central nervous system. In Neurochemistry: A Practical Approach; 1987:27‐63. [Google Scholar]
- 34. Harlan BA, Pehar M, Sharma DR, Beeson G, Beeson CC, Vargas MR. Enhancing NAD+ salvage pathway reverts the toxicity of primary astrocytes expressing amyotrophic lateral sclerosis‐linked mutant superoxide dismutase 1 (SOD1). J Biol Chem. 2016;291:10836‐10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harlan BA, Killoy KM, Pehar M, Liu L, Auwerx J, Vargas MR. Evaluation of the NAD(+) biosynthetic pathway in ALS patients and effect of modulating NAD(+) levels in hSOD1‐linked ALS mouse models. Exp Neurol. 2020;327:113219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harlan BA, Pehar M, Killoy KM, Vargas MR. Enhanced SIRT6 activity abrogates the neurotoxic phenotype of astrocytes expressing ALS‐linked mutant SOD1. FASEB J. 2019;33:7084‐7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang S, Lin Y, Yuan X, Li F, Guo L, Wu B. REV‐ERBalpha integrates colon clock with experimental colitis through regulation of NF‐kappaB/NLRP3 axis. Nat Commun. 2018;9:4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schnell A, Chappuis S, Schmutz I, et al. The nuclear receptor REV‐ERBalpha regulates Fabp7 and modulates adult hippocampal neurogenesis. PLoS One. 2014;9:e99883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vanderheyden WM, Fang B, Flores CC, Jager J, Gerstner JR. The transcriptional repressor Rev‐erbalpha regulates circadian expression of the astrocyte Fabp7 mRNA. Curr Res Neurobiol. 2021;2:100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Killoy KM, Harlan BA, Pehar M, Vargas MR. FABP7 upregulation induces a neurotoxic phenotype in astrocytes. Glia. 2020;68:2693‐2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen H, Qian K, Chen W, et al. Human‐derived neural progenitors functionally replace astrocytes in adult mice. J Clin Investig. 2015;125:1033‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qian K, Huang H, Peterson A, et al. Sporadic ALS astrocytes induce neuronal degeneration in vivo. Stem Cell Reports. 2017;8:843‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamanaka K, Chun SJ, Boillee S, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang L, Gutmann DH, Roos RP. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum Mol Genet. 2011;20:286‐293. [DOI] [PubMed] [Google Scholar]
- 45. Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28:13574‐13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pehar M, Harlan BA, Killoy KM, Vargas MR. Role and therapeutic potential of astrocytes in amyotrophic lateral sclerosis. Curr Pharm Des. 2017;23:5010‐5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. de Boer AS, Koszka K, Kiskinis E, Suzuki N, Davis‐Dusenbery BN, Eggan K. Genetic validation of a therapeutic target in a mouse model of ALS. Sci Transl Med. 2014;6:248ra104. [DOI] [PubMed] [Google Scholar]
- 48. Miquel E, Cassina A, Martinez‐Palma L, et al. Neuroprotective effects of the mitochondria‐targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic Biol Med. 2014;70:204‐213. [DOI] [PubMed] [Google Scholar]
- 49. Song S, Miranda CJ, Braun L, et al. Major histocompatibility complex class I molecules protect motor neurons from astrocyte‐induced toxicity in amyotrophic lateral sclerosis. Nat Med. 2016;22:397‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kushner PD, Stephenson DT, Wright S. Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. J Neuropathol Exp Neurol. 1991;50:263‐277. [DOI] [PubMed] [Google Scholar]
- 51. Nagy D, Kato T, Kushner PD. Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J Neurosci Res. 1994;38:336‐347. [DOI] [PubMed] [Google Scholar]
- 52. O'Reilly SA, Roedica J, Nagy D, et al. Motor neuron‐astrocyte interactions and levels of Cu, Zn superoxide dismutase in sporadic amyotrophic lateral sclerosis. Exp Neurol. 1995;131:203‐210. [DOI] [PubMed] [Google Scholar]
- 53. Schiffer D, Cordera S, Cavalla P, Migheli A. Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci. 1996;139(suppl):27‐33. [DOI] [PubMed] [Google Scholar]
- 54. Wong PC, Pardo CA, Borchelt DR, et al. An adverse property of a familial ALS‐linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105‐1116. [DOI] [PubMed] [Google Scholar]
- 55. Bruijn LI, Becher MW, Lee MK, et al. ALS‐linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1‐containing inclusions. Neuron. 1997;18:327‐338. [DOI] [PubMed] [Google Scholar]
- 56. Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23:249‐256. [DOI] [PubMed] [Google Scholar]
- 57. Nagai M, Aoki M, Miyoshi I, et al. Rats expressing human cytosolic copper‐zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246‐9254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Howland DS, Liu J, She Y, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant‐mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA. 2002;99:1604‐1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Arnold ES, Ling SC, Huelga SC, et al. ALS‐linked TDP‐43 mutations produce aberrant RNA splicing and adult‐onset motor neuron disease without aggregation or loss of nuclear TDP‐43. Proc Natl Acad Sci USA. 2013;110:E736‐E745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Swarup V, Phaneuf D, Dupre N, et al. Deregulation of TDP‐43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB‐mediated pathogenic pathways. J Exp Med. 2011;208:2429‐2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lee J, Kim DE, Griffin P, et al. Inhibition of REV‐ERBs stimulates microglial amyloid‐beta clearance and reduces amyloid plaque deposition in the 5XFAD mouse model of Alzheimer's disease. Aging Cell. 2020;19:e13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schibler U, Gotic I, Saini C, et al. Clock‐talk: interactions between central and peripheral circadian oscillators in mammals. Cold Spring Harb Symp Quant Biol. 2015;80:223‐232. [DOI] [PubMed] [Google Scholar]
- 63. Maury E, Hong HK, Bass J. Circadian disruption in the pathogenesis of metabolic syndrome. Diabet Metab. 2014;40:338‐346. [DOI] [PubMed] [Google Scholar]
- 64. Asher G, Sassone‐Corsi P. Time for food: the intimate interplay between nutrition, metabolism, and the circadian clock. Cell. 2015;161:84‐92. [DOI] [PubMed] [Google Scholar]
- 65. Bass J. Circadian topology of metabolism. Nature. 2012;491:348‐356. [DOI] [PubMed] [Google Scholar]
- 66. Stubblefield JJ, Terrien J, Green CB. Nocturnin: at the crossroads of clocks and metabolism. Trend Endocrinol Metab. 2012;23:326‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dupuis L, Pradat PF, Ludolph AC, Loeffler JP. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011;10:75‐82. [DOI] [PubMed] [Google Scholar]
- 68. Gray E, Larkin JR, Claridge TD, Talbot K, Sibson NR, Turner MR. The longitudinal cerebrospinal fluid metabolomic profile of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Front Degener. 2015;16:456‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Desport JC, Preux PM, Truong TC, Vallat JM, Sautereau D, Couratier P. Nutritional status is a prognostic factor for survival in ALS patients. Neurology. 1999;53:1059‐1063. [DOI] [PubMed] [Google Scholar]
- 70. Vargas MR, Pehar M, Cassina P, et al. Fibroblast growth factor‐1 induces heme oxygenase‐1 via nuclear factor erythroid 2‐related factor 2 (Nrf2) in spinal cord astrocytes: consequences for motor neuron survival. J Biol Chem. 2005;280:25571‐25579. [DOI] [PubMed] [Google Scholar]
- 71. Dupuis L, Corcia P, Fergani A, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology. 2008;70:1004‐1009. [DOI] [PubMed] [Google Scholar]
- 72. Bouteloup C, Desport JC, Clavelou P, et al. Hypermetabolism in ALS patients: an early and persistent phenomenon. J Neurol. 2009;256:1236‐1242. [DOI] [PubMed] [Google Scholar]
- 73. Pradat PF, Bruneteau G, Gordon PH, et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11:166‐171. [DOI] [PubMed] [Google Scholar]
- 74. Vargas MR, Johnson DA, Johnson JA. Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS‐linked hSOD1(G93A) mice model. Neurobiol Dis. 2011;43:543‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Paganoni S, Deng J, Jaffa M, Cudkowicz ME, Wills AM. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve. 2011;44:20‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lawton KA, Cudkowicz ME, Brown MV, et al. Biochemical alterations associated with ALS. Amyotroph Lateral Scler. 2012;13:110‐118. [DOI] [PubMed] [Google Scholar]
- 77. Gallo V, Wark PA, Jenab M, et al. Prediagnostic body fat and risk of death from amyotrophic lateral sclerosis: the EPIC cohort. Neurology. 2013;80:829‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vargas MR, Burton NC, Kutzke J, et al. Absence of Nrf2 or its selective overexpression in neurons and muscle does not affect survival in ALS‐linked mutant hSOD1 mouse models. PLoS One. 2013;8:e56625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. O'Reilly EJ, Wang H, Weisskopf MG, et al. Premorbid body mass index and risk of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:205‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pehar M, Beeson G, Beeson CC, Johnson JA, Vargas MR. Mitochondria‐targeted catalase reverts the neurotoxicity of hSOD1G(9)(3)A astrocytes without extending the survival of ALS‐linked mutant hSOD1 mice. PLoS One. 2014;9:e103438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Huisman MH, Seelen M, van Doormaal PT, et al. Effect of presymptomatic body mass index and consumption of fat and alcohol on amyotrophic lateral sclerosis. JAMA Neurol. 2015;72:1155‐1162. [DOI] [PubMed] [Google Scholar]
- 82. Marin B, Arcuti S, Jesus P, et al. Population‐based evidence that survival in amyotrophic lateral sclerosis is related to weight loss at diagnosis. Neurodegener Dis. 2016;16:225‐234. [DOI] [PubMed] [Google Scholar]
- 83. Tso CF, Simon T, Greenlaw AC, Puri T, Mieda M, Herzog ED. Astrocytes regulate daily rhythms in the suprachiasmatic nucleus and behavior. Curr Biol. 2017;27:1055‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Brancaccio M, Patton AP, Chesham JE, Maywood ES, Hastings MH. Astrocytes control circadian timekeeping in the suprachiasmatic nucleus via glutamatergic signaling. Neuron. 2017;93:1420‐1435.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Barca‐Mayo O, Pons‐Espinal M, Follert P, Armirotti A, Berdondini L, De Pietri Tonelli D. Astrocyte deletion of Bmal1 alters daily locomotor activity and cognitive functions via GABA signalling. Nat Commun. 2017;8:14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. McKee CA, Lananna BV, Musiek ES. Circadian regulation of astrocyte function: implications for Alzheimer's disease. Cell Mol Life Sci. 2020;77:1049‐1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article. The data are available from the corresponding author upon reasonable request.