

Abstract
The RNA helicase DHX15 is widely expressed in immune cells and traditionally thought to be an RNA splicing factor or a viral RNA sensor. However, the role of DHX15 in NK-cell activities has not been studied thus far. Here, we generated Dhx15-floxed mice and found that conditional deletion of Dhx15 in NK cells (Ncr1CreDhx15fl/fl mice) resulted in a marked reduction in NK cells in the periphery and that the remaining Dhx15-deleted NK cells failed to acquire a mature phenotype. As a result, Dhx15-deleted NK cells exhibited profound defects in their cytolytic functions. We also found that deletion of Dhx15 in NK cells abrogated their responsiveness to IL-15, which was associated with inhibition of IL-2/IL-15Rβ (CD122) expression and IL-15R signaling. The defects in Dhx15-deleted NK cells were rescued by ectopic expression of a constitutively active form of STAT5. Mechanistically, DHX15 did not affect CD122 mRNA splicing and stability in NK cells but instead facilitated the surface expression of CD122, likely through interaction with its 3′UTR, which was dependent on the ATPase domain of DHX15 rather than its splicing domain. Collectively, our data identify a key role for DHX15 in regulating NK-cell activities and provide novel mechanistic insights into how DHX15 regulates the IL-15 signaling pathway in NK cells.
Keywords: DEAH-box helicase 15 (DHX15), Innate immunity, Natural killer (NK) cells, Homeostasis, IL-15 signaling pathway
Subject terms: Innate immune cells, Innate immunity, NK cells
Introduction
DHX15 is a member of the DEAH-box family of RNA helicases that is widely expressed by immune cells [1–3] (and data from the Immgen consortium, http://www.immgen.org). It is a motor protein that possesses both ATPase and helicase activities, and by hydrolyzing ATP, DHX15 unwinds complex RNA structures for further processing [4–6]. Traditionally, DHX15 is believed to be a pre-mRNA splicing factor that facilitates mRNA maturation and ribosome biogenesis. DHX15 can also modulate gene expression by regulating CMTR1-dependent mRNA cap formation [7, 8]. Recently, the role of DHX15 in various malignant tumors has attracted considerable attention [9–13]. Wang et al. recently reported that depletion of Dhx15 in gut epithelial cells renders mice susceptible to infections, owing to impaired α-defensin production in the gut [14]. In the immune system, DHX15 is mostly studied as a viral RNA sensor involved in activation of the NF-κB and MAPK signaling pathways in dendritic cells, thus mediating strong protective immunity [2, 3]. Others have reported a role for DHX15 in the survival and proliferation of B cells, as B-cell-specific deletion of Dhx15 results in reduced peripheral B-cell numbers and dysregulated expression of genes linked to humoral immunity [1]. However, the exact role of DHX15 in regulating NK cells has not been studied and remains completely unknown.
NK cells, their survival and maturation, as well as their cytolytic activity are regulated by an intricate network of intrinsic and extrinsic factors [15]. In mice, NK cells are mainly derived from common lymphoid progenitors in the bone marrow (BM), where developing NK cells transition sequentially through various stages of maturation, a process that is tightly controlled by a plethora of transcription factors, including Ikaros, Pu.1, Id2, Ets1, Foxo1, E4BP4, and MEF[15–17]. After acquiring the classical NK-cell marker NK1.1, NK cells can be further divided into three subsets based on the expression of CD27 and the integrin CD11b (i.e., CD11b− CD27+, CD11b+ CD27+, and CD11b+ CD27−), with the CD11b+ CD27− subset being the most differentiated. Mature NK cells also express cytokine receptors and complex stimulatory and inhibitory surface receptors that further regulate their functional attributes [18]. It is generally believed that the terminal differentiation of NK cells is regulated by the transcription factors Tbx21 (T-bet), Eomes, Tox, Gata3, and Irf2 as well as by the metabolic regulators mTORC1/2 [19–21]. The maturation, survival, and effector functions of NK cells are also impacted by extrinsic cytokines, including IL-15, IL-2, IL-12, and IL-18. These cytokines regulate diverse aspects of NK-cell activities, mostly through signaling pathways involving the aforementioned transcription factors, induction of proliferation and survival molecules, and metabolic reprogramming of NK cells [22–24]. From a functional perspective, natural killer (NK) cells are a subset of innate lymphocytes; they have the capacity to readily release inflammatory cytokines (e.g., IFN-γ and TNF-α) and cytolytic granules (perforin and granzymes) in the absence of prior antigen sensitization [25, 26]. As a result, NK cells often provide the first line of defense against pathogens, eliminate stressed and cancerous cells, and help kick-start the adaptive immune response [27, 28]. Recently, NK cells have been proposed as a safe and effective platform for engineering chimeric antigen receptors for cancer immunotherapies [29–32]. Thus, a better understanding of the fundamental mechanisms that govern NK-cell activities is an important and clinically relevant issue.
In the present study, we generated Dhx15-floxed mice and examined the role of DHX15 in regulating NK cells by deleting Dhx15 specifically in NK cells (Ncr1CreDhx15fl/fl mice). We found that conditional deletion of Dhx15 in NK cells had a profound impact on NK cells, in that the numbers of NK cells were greatly reduced, and their cytolytic activity was severely impaired. Interestingly, most of the remaining NK cells in the periphery were functionally immature and unable to transition into the terminally differentiated phenotype, primarily due to a defect in the IL-15 signaling pathway. Importantly, DHX15 appears to be critical in the surface expression of the IL-2/IL-15Rβ chain (CD122) but not in CD122 mRNA splicing and stability.
Materials and methods
Mice, tumor models, and in vivo killing assays
All mice used in this study were bred and maintained under specific pathogen-free conditions at the Houston Methodist Research Institute in accordance with the Institutional Animal Care and Use Committee guidelines. Animals between 6 and 12 weeks of age were used for all studies, and all mice were on the C57BL/6 genetic background. The mouse strains included C57BL/6 (CD45.2; The Jackson Laboratory), B6. SJL (CD45.1; Taconic), Dhx15fl/fl (generated in our laboratory, Fig. S1A), Ncr1Cre [33], Rag2−/−IL-2Rγ−/− (Taconic), and ERCre (The Jackson Laboratory). Ncr1CreDhx15fl/fl and ERCreDhx15fl/fl mice were bred at Houston Methodist Research Institute.
To establish the tumor models, 1 × 106 RMA-S cells in phosphate-buffered saline (PBS) were subcutaneously injected into the shaved flanks of littermate control (Dhx15fl/fl) and Ncr1Cre Dhx15fl/fl mice. Tumor volume was measured using digital calipers and calculated as V = (L × W2)/2. At the endpoint of the study (tumor diameter of 20 mm), all mice were euthanized. To establish the tumor metastasis model, 2 × 105 B16F10 melanoma cells in PBS were injected into mice via the tail vein. Pulmonary metastasis was assessed by comparing lung weights, and liver metastasis was evaluated by comparing the numbers of metastatic nodules on Day 14 after tumor cell injection.
To evaluate NK-cell functions in vivo, a cytotoxicity assay with RMA-S cells was adopted as described previously [34]. Briefly, mice were intraperitoneally injected with a mixture of target cells (106) consisting of NK-cell-sensitive RMA-S cells labeled with CFSE and NK-cell-resistant RMA cells labeled with CTV. Eighteen hours after tumor cell injection, the mice were euthanized, and cells in the peritoneal cavity were collected. The relative percentages of RMA-S and RMA cells were analyzed by flow cytometry. Immunocompromised Rag2−/−IL-2Rγ−/− mice were used as controls. The percentage of RMA-S cytotoxicity was calculated as follows: 100 × (1-[percentage of residual CFSE+ population among the total CFSE+ and CTV+ population in the experimental group/percentage of residual CFSE+ population among the total CFSE+ and CTV+ population in the Rag2−/−IL-2Rγ−/− mouse group]).
Cell isolation, cell lines and culture
NK cells were isolated by negative selection using an EasySep Mouse NK Cell Isolation Kit (STEMCELL Technologies) from spleen (SP), BM and other tissues. For some experiments, such as Western blotting, the isolated NK cells were subjected to FACS. Primary NK cells were cultured in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS) and 50 μM 2-mercaptoethanol (2-ME) in the absence or presence of cytokines (200 U/mL IL-2, 10 ng/mL IL-12, 20 ng/mL IL-15, and 50 ng/mL IL-18). B16F10, NIH3T3, and HEK293T cells (ATCC) were cultured in DMEM containing 10% (v/v) FBS, 100 U/ml penicillin G and 100 µg/mL streptomycin in a humidified 5% CO2 atmosphere at 37 °C. Platinum-E cells (Cell Biolabs) were cultured in DMEM containing 10% (v/v) FBS, 100 U/ml penicillin G, 100 µg/mL streptomycin, 1 µg/mL puromycin, and 10 µg/mL blasticidin in a humidified 5% CO2 atmosphere at 37 °C. OP9 cells (ATCC) were cultured in ribonucleoside- and deoxyribonucleoside-free alpha-MEM supplemented with sodium bicarbonate, 20% (v/v) FBS, 100 U/ml penicillin G and 100 µg/mL streptomycin in a humidified 5% CO2 atmosphere at 37 °C. RMA and RMA-S cells were kind gifts from Dr. Lewis L. Lanier (University of California, San Francisco, CA) [35] and were cultured in RPMI 1640 medium containing 10% (v/v) FBS, 50 μM 2-ME, 100 U/mL penicillin G and 100 µg/mL streptomycin in a humidified 5% CO2 atmosphere at 37 °C.
Antibodies and reagents
Antibodies used for flow cytometry staining, sorting and isolation, including antibodies specific for CD3ε (145-2C11), CD4 (GK1.5), CD122 (TM-β1), CD19 (6D5), B220 (RA3-6B2), TER119 (TER-119), CD8α (53-6.7), TCRβ (H57-597), NK1.1 (PK136), CD49b (DX5), CD27 (LG.3A10), CD11b (M1/70), CD45 (30-F11), CD45.1 (A20), CD45.2 (104), CD107a (1D4B), CD132 (TUGm2), Gr-1 (RB6-8C5), Sca-1 (E13-161.7), c-Kit (2B8), and GFP (FM264G), were purchased from Biolegend. CD49a (Ha31/8) was purchased from BD Biosciences. Intracellular staining for IFN-γ (XMG1.2), Granzyme B (GB11), Perforin (S16009A), T-bet (4B10), Eomes (W17001A) (Biolegend), and Ki67 (SolA15) (eBioscience) was performed with a Foxp3 kit (eBioscience). Fix buffer I and Perm buffer III (BD Biosciences) were used for intracellular staining of phosphorylated STAT5 (p-STAT5, pY694) (BD Biosciences). Anti-mouse antibodies specific for the following proteins were used for Western blotting: DHX15 (Abcam, Cat# ab70454), IL-2Rβ (Santa Cruz, sc-393092), HuR (Abcam, Cat# ab136542), β-actin (Santa Cruz, sc-47778), and p-mTOR (CST, #5536 s). The recombinant cytokines used for NK or BM stem cell culture were as follows: murine IL-2, IL-15, IL-12p70, SCF, IL-3, and IL-6 (all purchased from Peprotech) and murine IL-18/IL-1F4 (purchased from R&D Systems). Restriction enzymes (EcoR1-HF, Not1-HF, and Nhe1), Q5 High-Fidelity DNA Polymerase, T4 DNA ligase, a Q5 Site-Directed Mutagenesis Kit, and NEB Stable Competent E. coli were obtained from NEB Biolabs. CellTrace™ Violet and CellTrace™ CFSE were obtained from Thermo Fisher Scientific.
Generation of bone marrow chimeras
Recipient mice (C57BL/6 J CD45.1±, and CD45.1+CD45.2+ mice) were lethally irradiated at 9 Gy. Donor BM cells were obtained from Dhx15fl/fl or Ncr1CreDhx15fl/fl mice (both CD45.2+) and mixed at a 1:1 ratio with BM cells from wild-type C57BL/6 J CD45.1+ mice. Five million cells of each population were injected i.v. into recipient mice. After 6–8 weeks of BM reconstitution, the spleens were harvested, and the percentages of CD45.2+ and CD45.1+ NK cells as well as NK-cell subsets based on CD11b expression on CD45.2+ NK cells were analyzed by flow cytometry.
Flow cytometry and cell sorting
Single-cell suspensions were first incubated with viability dyes (Zombie Aqua; Biolegend) to identify dead cells and with an anti-FcγRII/III antibody (2.4G2; BD Bioscience) to block nonspecific antibody binding. After washing, the cells were incubated with mixtures of fluorescently labeled antibodies for 20 min at 4 °C. For analysis of CD107a, IFN-γ, Perforin, and Granzyme B, splenic NK cells were isolated using an NK-cell negative selection kit and were then treated with IL-2 (200 U/ml) and IL-18 (50 ng/ml), a plate-bound anti-NK1.1 antibody (10 μg/ml), IL-12 (10 ng/ml), IL-18 (50 ng/ml), and IL-15 (100 ng/ml) for 6 h in the presence of GolgiStop™ protein transport inhibitor. Cells were collected for intracellular staining. After surface staining, cells were fixed using a Foxp3 fixation & permeabilization kit or BD Phosflow buffers according to the manufacturer’s instructions. Samples were acquired using an LSRFortessa™ or LSR II flow cytometer (BD Bioscience) and analyzed using FlowJo software (Tree Star). For sorting of BM and splenic NK cells for qRT–PCR, Western blotting, or RNA sequencing, these cells were enriched with a kit from STEMCELL Technologies (19855), and total NK cells or NK-cell subsets were then sorted using a BD FACSAria cell sorter (BD Bioscience).
For analysis of NK-cell apoptosis, splenic NK cells were stained with an Annexin V kit according to the manufacturer’s instructions (BioLegend). A flow cytometry-based ex vivo NK-cell cytotoxicity assay was adopted to analyze Dhx15-deleted NK-cell function as previously reported [36]. In brief, splenic NK cells were sorted from control and Dhx15-deleted mice. The sorted NK cells (effector cells) were cocultured with RMA-S cells (target cells), which were labeled with EGFP using retroviral vectors (pMYs-EGFP-SV40-puromycin), for 4 h in the presence of 20 ng/ml IL-15 in a 96-well U-bottom plate. At the end of the 4 h incubation, the total cell population was stained with 7-AAD and analyzed by flow cytometry. The percentage of specific cytotoxicity was calculated as [% 7-AAD+ cells among EGFP+ cells] (Effector+Target) well – [%7-AAD+ cells among EGFP+ cells] Target-only well.
Assay for IL-15 responsiveness in vivo
The IL-15/IL-15Rα/Fc complex was prepared using human recombinant IL-15 (R&D Systems) and a recombinant fusion protein consisting of the ectodomain of the mouse IL-15α chain and human IgG1 Fc (IL-15Rα/Fc) (R&D Systems) as previously reported [37]. In brief, both IL-15 and IL-15Rα/Fc were resuspended in PBS solution and were then mixed together at a protein mass ratio of 1:6 (IL-15:IL-15Rα/Fc). The mixture was incubated at 37 °C for 30 min and was then used for in vivo injection. For stimulation of NK cells in vivo in Dhx15fl/fl or Ncr1CreDhx15fl/fl mice, each mouse was i.p. administered 2.5 μg of IL-15/IL-15Rα/Fc complex in 200 μl of PBS, and all analyses of NK-cell expansion were performed 4 days later in the treated mice.
Viral vectors and cell transduction
The DHX15-full-length (FL) (residues 1-795), CD122 and activated STAT5 (a-STAT5b, H298R/S715F) constructs were generated in the pMYs-IRES-GFP retroviral vector (Cell Biolabs). The EGFP expression vector was generated in the pMYs-SV40-puromycin retroviral vector (Cell Biolabs) backbone. The DHX15-FL (residues 1-795), DHX15-E261Q mutation, and DHX15-R222G mutation constructs were generated in the pCDH-MSCV-MCS-EF1α-GFP + Puro vector (System Biosciences) backbone. The CD122 GFP reporters with or without the CD122 3′UTR were generated in the pMYs-IRES-GFP retroviral vector (Cell Biolabs) backbone. Briefly, the extracellular domain of CD122 was replaced with the GFP sequence, and other domains were initially retained. The 3′UTR (Transcript ID: ENSMUST00000089398) was amplified from the mouse genome and fused to the GFP reporter. Retroviral particles were prepared by transfection of vectors into Plat-E packaging cells according to the manufacturer’s recommendations (Cell Biolabs). For transduction of BM cells enriched with hematopoietic stem cells (HSCs, Lin− Sca1+ and/or c-Kit+ cells), BM cells were first activated for 24 h by treatment with the cytokines IL-3 (10 ng/mL), IL-6 (50 ng/mL) and SCF (100 ng/mL) and then transduced with freshly prepared retroviral particles by centrifugation for 2 h at 800 × g and 32 °C in the presence of 10 µg/ml polybrene (Sigma–Aldrich). After centrifugation, the cells were cultured for 4 h at 32 °C, and these BM cells enriched with HSCs were then cultured in complete DMEM in the presence of the same cytokines for an additional 2 days at 37 °C. To increase the transduction efficiency, a second round of transduction by centrifugation was performed after 2 days, and BM cells were transferred into recipient mice after another 24 h of culture. For the production of lentiviral particles, expression vectors were transfected along with the envelope plasmid pMD2.G (Addgene, #12259) and packaging plasmid psPAX2 (Addgene, #12260) into 293 T cells. The procedures used for subsequent transduction of BM cells enriched with HSCs were similar to those used for retroviral particles.
Viral vector-modified BM chimera studies were conducted using age- and sex-matched mice in accordance with approved institutional protocols. Briefly, modified BM cells mixed with (for CD122, a-STAT5 retrovirus and all lentiviruses) or without (for empty retroviral vectors and DHX15-FL retrovirus) 0.4 million CD45.1+ BM cells were transferred into lethally irradiated recipient mice. For the empty (using WT mouse BM cells) or DHX15-FL (using Ncr1CreDhx15fl/fl BM cells) retroviral vector-modified study, the blood or spleen of recipient mice was analyzed after 4 weeks of development. For BM cell transfer (for CD122, a-STAT5 retrovirus and all lentiviruses) from ERCreDhx15fl/fl mice, TMF was administered for 5 consecutive days after 3 weeks of development. Then, the spleens of the recipient mice were analyzed after another 2 weeks. The effect of each DHX15 mutant on NK cells after TMF treated was evaluated as followed formula: [(CD45.2+GFP+ NK cells/CD45.2+GFP+ NK cells + CD45.1+ NK cells) TMF-treated]/ [(CD45.2+GFP+ NK cells/CD45.2+GFP+ NK cells + CD45.1+ NK cells) Control].
Bone marrow-derived NK cells
For the transduction of CD122 GFP reporters, BM-derived NK cells were used as previously reported [38, 39]. In brief, c-kit+ BM cells (ERCre Dhx15fl/fl) were sorted and were then activated by incubation for 24 h with the cytokines IL-7 (0.5 ng/mL), Flt-3L (50 ng/mL) and SCF (60 ng/mL) and transduced with GFP reporters as described above. After 5 or 6 days of culture in the same cytokines, the BM cells were harvested, washed, and replated in complete RPMI 1640 medium containing IL-15 (30 ng/mL) alone into 6-well plates coated with OP9 cells. After an additional 3 days, the cultures were refed with the same medium, and on Day 11 or 12 of the total culture period, the cells were harvested and treated with tamoxifen (TMF) or with DMSO as the control for the indicated time. Then, surface GFP expression was analyzed using an anti-GFP flow cytometry antibody, and total GFP expression was evaluated by flow cytometry in the FITC channel. The relative expression was determined based on the surface GFP expression ratio (gated on GFP+ NK cells) of the With-UTR group to the No-UTR group, which was calculated as follows: {With-UTR group [TMF-treated (surface GFP expression/total GFP expression)/DMSO-treated (surface GFP expression/total GFP expression)]}/{No-UTR group [TMF-treated (surface GFP expression/total GFP expression)/DMSO-treated (surface GFP expression/total GFP expression)]}.
Conventional PCR and quantitative RT–PCR analysis
Total RNA of NK cells isolated from the BM or SP was purified using a Direct-zol RNA Microprep kit (Zymo Research) and treated with DNase I, and cDNA was synthesized from 1 μg of RNA using iScript™Reverse Transcription Supermix (Bio–Rad). Cytoplasmic and nuclear RNA of NK cells was purified using an NE-PER™ Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific), and cDNA was synthesized using iScript™ Reverse Transcription Supermix (Bio–Rad). For synthesis of nuclear cDNA, the U6 snRNA-Reverse primer was adopted (Table S1). Real-time quantitative PCRs were set up in duplicate with SsoAdvanced™ Universal SYBR® Green Supermix (Bio–Rad) and carried out in a Bio–Rad CFX 96 Real-Time System (Bio–Rad). All reactions were carried out with amplification of target genes and an internal control (GAPDH or U6) in the same plate. Each sample was normalized using an internal control. Amplification was performed with the following thermal cycling program: initial denaturation at 95 °C for 5 min, followed by 45 cycles of denaturation at 95 °C for 15 s and annealing and elongation at 60 °C for 1 min, with a final cycle for melt curve generation at 55–95 °C. Relative quantitative evaluation of target gene expression was performed by comparing the threshold cycle (CT) values, and samples with CT values ≥ 35 were removed from analysis. Primer specificity was confirmed by melt curve analysis. The primers used in this study are listed in Supplementary Table S1. For analysis of CD122 alternative splicing, total RNA was extracted from Dhx15fl/fl and ERCreDhx15fl/fl mouse NK cells that were treated with TMF or DMSO for 5 days in vitro after confirmation of decreased surface CD122 protein expression. cDNA was synthesized using oligo(dT)18 primers (NEB) and/or random primers. A series of primers (Table S1) spanning different exons were used for analysis of possible longer products, including intron RNA, via normal PCR amplification.
mRNA stability assay
Splenic NK cells sorted from ERCre Dhx15fl/fl mice were cultured with IL-15 (20 ng/ml) with alone or in combination with TMF for 6 d. Then, the NK cells were treated with actinomycin D (5 μg/ml, A9415, Sigma) for the indicated time. Cells without actinomycin D treatment were used as the 0 h control. Cells were collected at the indicated time, and total RNA was extracted using a Direct-zol RNA Microprep Kit (Zymo Research). Total RNA was subjected to reverse transcription using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher) with random or oligo(dT) primers. The mRNA half-life was calculated and plotted in GraphPad Prism using a nonlinear regression curve fitting method (one-phase decay) as previously described by Ratnadiwakara et al. [40]. Primer sequences are listed in Table S1.
RNA-seq analysis
RNA-seq was performed on CD11b- NK cells sorted from Dhx15fl/fl and Ncr1CreDhx15fl/fl mice by Novogene as previously reported [41]. The RNA-seq reads were aligned to the mouse reference genome mm10 using TopHat. Prior to differential gene expression analysis, the read counts for each sequenced library were adjusted with the edgeR package with a single scaling factor for normalization. Differential expression analysis was performed using the DEGSeq R package (1.20.0). All p values were adjusted using the Benjamini & Hochberg method. For samples without biological replicates, differentially expressed genes were determined by the criteria |log2(FoldChange)|> 1 and padj (q value) < 0.005.
Immunoblotting and immunoprecipitation (IP)
Cells were lysed with ice-cold lysis buffer, and proteins were separated on 4–15% SDS–PAGE gels (Bio–Rad). Proteins were then transferred to PVDF membranes and incubated at room temperature in blocking buffer containing 5% BSA or nonfat milk in TBST (Tris-buffered saline, 0.1% Tween 20) for 1 h. The membranes were then incubated overnight with primary antibodies at a dilution of 1:1000 in blocking buffer. The membranes were washed and incubated with anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibodies at a 1:2000 dilution for 1 h. Immunoreactive bands were imaged on a UVP imaging System or using X-ray film with enhanced chemiluminescence reagents (Thermo Fisher Scientific). β-Actin was used as the loading control.
For the immunoprecipitation assay, 48 h after cotransfection of the HA-DHX15 and CD122 plasmids, 293 T cells were lysed with IP lysis buffer (Thermo Fisher Scientific) with Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Cat# 78440) for 10 min. Soluble extracts of cell lysates were collected by centrifugation at 15,000 × g for 10 min. Precleaning was performed by adding protein A beads into the collected lysate and incubating the mixture with constant rotation for 1 h at 4 °C, followed by centrifugation at 15,000 × g for 10 min at 4 °C. CD122 in the supernatant was immunoprecipitated by incubation with anti-HA agarose beads with constant rotation overnight at 4 °C. The precipitates were washed four times with cold IP lysis buffer. After the final wash, the beads were resuspended in sample buffer and boiled for 10 min. Coimmunoprecipitated samples were analyzed by Western blotting with the indicated Abs. The specific bands were visualized using ECL reagents (Thermo Fisher Scientific).
RNA immunoprecipitation (RIP) assay
RNA immunoprecipitation was performed using a Magna RNA-Binding Protein Immunoprecipitation Kit (17-700, Millipore) following the manufacturer’s protocol. Briefly, NK cells were sorted from the spleens of littermate control mice and cultured for 7 days with 20 ng/mL IL-15. Then, NK cells (4 × 107) were collected, lysed, and incubated with 5 μg of magnetic beads labeled with an anti-DHX15 antibody (Cat# ab70454, Abcam) or normal rabbit IgG (PP64B, Millipore) as the negative control at 4 °C overnight with gentle rotation. The immunoprecipitated RNAs were isolated using proteinase K digestion and detected by qRT–PCR. The gene-specific primer sequences used for qRT–PCR of the immunoprecipitates from the RIP assay are listed in the supplemental material (Table S1).
RNA pulldown assay
The RNA pulldown assay was conducted as previously reported [42]: (1) The CD122 3′UTR and antisense 3′UTR sequences were inserted into the pcDNA3.1 (+) vector containing the T7 promoter using the Nhe I and Not I restriction enzymes. The template DNA used for in vitro transcription was generated by PCR using primers (see the supplementary material) that inserted the T7 promoter into the targeting sequence. The CD122 3′UTR and its antisense RNA were in vitro transcribed using a MEGAscript Kit (AM1333, Thermo Fisher) and purified using a MEGAclear™ Transcription Clean-Up Kit (AM1908, Thermo Fisher). Then, the transcribed RNA was labeled with biotin using a Pierce™ RNA 3′ End Desthiobiotinylation Kit (20163, Thermo Fisher) and streptavidin magnetic beads. (2) NIH3T3 cells (2 × 107) were lysed with 1 mL of IP lysis buffer at 4 °C for 30 min. The supernatant was collected by centrifugation (15,000 × g, 4 °C, 10 min), 10 mM ATP was added, and the solution was mixed with the labeled RNA by rotation at 4 °C for 1 h. (3) The precipitated proteins were boiled in 1× reducing sample buffer and subjected to Western blot analysis.
Statistical analysis
Two-tailed unpaired Student’s t test was used to determine the significance of differences. A p value< 0.05 was considered to indicate statistical significance. All statistical analyses were performed and plots were generated in Prism 7 (GraphPad Software).
Results
Deletion of Dhx15 in NK cells impaired NK-cell maturation and functions
To examine the role of DHX15 in NK cells, we generated Dhx15fl/fl mice by inserting two LoxP sites flanking the exon 4 of the Dhx15 locus and crossed these mice with Ncr1Cre mice, which resulted in deletion of Dhx15 in NK cells in the obtained Ncr1CreDhx15fl/fl mice (Fig. S1A). The genotype of the Ncr1CreDhx15fl/fl mice was confirmed by PCR and Western blot analysis (Fig. S1B, C), and these Dhx15-deleted mice were healthy and did not show any gross abnormalities (data not shown). We then analyzed NK cells in the spleen, blood, and BM from Ncr1CreDhx15fl/fl mice by gating on the NK1.1+ CD3ε− population, and compared them with those in age-matched littermate control mice. As shown in Fig. 1A, B, the number of NK cells was markedly reduced in Ncr1CreDhx15fl/fl mice, and the relative percentage of NK cells was reduced by 5- to 8-fold compared to that in control mice. A similar reduction in NK cells was observed in the lymph nodes (LNs), liver and lung in Ncr1CreDhx15fl/fl mice (Fig. S1D). Interestingly, in addition to conventional NK cells (Dx5+ CD49a−), tissue-resident NK cells (Dx5− CD49a+) in the liver were reduced in Ncr1CreDhx15fl/fl mice (Fig. S1E). As expected, the populations of other cell types (CD4+ cells, CD8+cells, B cells, and NKT cells) were comparable between control and Dhx15-deleted mice (Fig. S2A). The absolute numbers of NK cells were similarly reduced in the spleen and BM in the Ncr1CreDhx15fl/fl mice (~7-fold reduction; Fig. 1C, D and Fig. S2B, C). We then gated on the Lin− NK1.1+ population in the spleen and further segregated these cells into different subsets based on CD27 and CD11b expression. As shown in Fig. 1C, deletion of Dhx15 in NK cells prominently affected the CD11b+CD27− subset and the CD11b/CD27 double-positive subset compared to those in control mice. A majority of the NK cells in Dhx15-deleted mice retained the CD11b− and CD27+phenotype (Fig. 1C, D), suggesting that deletion of Dhx15 preferentially affects the terminal differentiation of NK cells.
Fig. 1.

Impaired NK-cell maturation and functions in Dhx15-deleted NK cells. A, B Representative flow cytometry plots and quantification of NK cells (CD45+ CD3ε− NK1.1+) in peripheral blood, spleen, and bone marrow in littermate control (Dhx15fl/fl) and Dhx15-deleted (Ncr1CreDhx15fl/fl) mice (n = 5 each group). C, D Representative flow cytometry plots and quantification of the absolute numbers of NK cells (Lin− NK1.1+) and NK-cell subsets based on CD11b and CD27 expression in the spleen of control (n = 6) and Dhx15-deleted mice (n = 3). E Flow cytometric analysis of CD107a, IFN-γ, granzyme B (GrzB), and perforin (Prf) expression in splenic NK cells (CD3ε− NK1.1+) from control and Dhx15-deleted mice. Cells were pretreated with the IL-15/IL-15RαFc complex for 4 days (n = 3 each group). F, G Histogram of CD107a (F) and IFN-γ (G) expression in splenic NK-cell subsets (CD11b− NK cells and CD11b+ NK cells), as analyzed by flow cytometry, from control and Dhx15-deleted mice. Cells were pretreated with the IL-15/IL15RαFc complex for 4 days (n = 3 each group). H Representative data of the cytoxicity assay performed with sorted control and Dhx15-deleted NK cells against RMA-S target cells at the indicated effector:target ratio (n = 3). I, J Representative images of subcutaneous RMA-S cell tumors from control and Dhx15-deleted hosts on Day 8 (I) and curves showing the average growth of tumors (J) are shown (n = 3 each group). The data shown are representative of at least three independent experiments, and the calculated data are shown as the mean ± standard deviation (s.d.) values. n.s., not statistically significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001
To determine their functionality, we first stimulated NK cells in Ncr1CreDhx15fl/fl and littermate control mice using the IL-15/IL-15RαFc complex in vivo. Then, splenic NK cells were purified and analyzed for expression of CD107a, IFN-γ, granzyme B, and perforin, markers that are typically associated with NK-cell functions. As shown in Fig. 1E, the expression of CD107a in response to IL-2 and IL-18 and to an anti-NK1.1 antibody was significantly decreased in NK cells from Ncr1CreDhx15fl/fl mice, and both the CD11b− and CD11b+ NK-cell subsets were affected (Fig. 1F). Similarly, IFN-γ production induced by stimulation with IL-12 and IL-18 and the anti-NK1.1 antibody was also impaired in Ncr1CreDhx15fl/fl mice (Fig. 1E). Both the CD11b− and CD11b+ NK-cell subsets exhibited impaired IFN-γ production (Fig. 1G). Granzyme B (GrzB) and perforin (Prf) expression induced by stimulation with IL-2 and IL-15 was significantly decreased in Dhx15-deleted mice (Fig. 1E). In addition, the cytotoxicity of Dhx15-deleted NK cells was severely impaired compared with that of control NK cells in vitro (Fig. 1H). These data demonstrate an essential role for DHX15 in regulating NK-cell activities.
Deletion of Dhx15 in NK cells altered tumor surveillance and metastasis in vivo
As NK cells are critical for the control of certain tumor cells in vivo [43, 44], we utilized the B16F10 melanoma model, an extensively characterized model of tumor metastasis that is controlled by NK cells [45], to further determine the function of Dhx15-deleted NK cells. We injected B16F10 cells into control and Ncr1CreDhx15fl/fl mice and then monitored the emergence of tumor nodules in the lungs of recipient mice. As shown in Fig. S2D, deletion of Dhx15 in NK cells resulted in a significant increase in the number of B16 nodules in the lung compared to that in control mice. This increase was also observed in the liver and the abdominal walls of Ncr1CreDhx15fl/fl mice inoculated with B16 melanoma cells (Fig. S2D). In another model, we subcutaneously inoculated control and Ncr1CreDhx15fl/fl mice with RMA-S cells, an NK-cell-sensitive cell line (due to the lack of self-MHC class I expression) [34, 35], and monitored tumor growth. As shown in Fig. 1I, J, deletion of Dhx15 in NK cells significantly accelerated the formation of solid tumors compared to that in control mice. In in vivo cytotoxicity assays with fluorescently labeled RMA-S cells, we confirmed that NK-cell-mediated killing of RMA-S cells was severely impaired in Ncr1CreDhx15fl/fl hosts (Fig. S2E).Collectively, these results demonstrate that deletion of Dhx15 in NK cells impairs their tumor surveillance activities and facilitates tumor growth in vivo.
DHX15 had an NK-cell-intrinsic role in regulating NK-cell survival and development
To gain further insights related to the impaired NK cells in Ncr1CreDhx15fl/fl mice, we first compared the expression of T-bet and Eomes, two key transcription factors in NK-cell maturation [46, 47], in control and Dhx15-deleted NK cells. Analysis by flow cytometry showed that their expression levels were comparable between control and Dhx15-deleted mice (Fig. 2A). Moreover, the expression of other NK-related transcription factors, as assessed by qRT–PCR, showed no marked differences between control and Dhx15-deleted NK cells (Fig. 2B). We also performed RNA-seq analysis to compare the global transcriptional profiles of control and Dhx15-deleted NK cells and again observed no marked differences, especially in the expression of transcripts key to NK-cell development (Fig. 2C). Interestingly, staining for the cell proliferation marker Ki67 showed high levels of expression in Dhx15-deleted NK cells under static conditions (Fig. 2D, Fig. S3A). The survival of Dhx15-deleted NK cells was compromised when these cells were cultured in vitro in the presence of IL-15, although this effect was not obvious in vivo either with or without IL-15/IL-15RaFc treatment (Fig. S3B).
Fig. 2.

DHX15 is intrinsically required for NK-cell survival and development. A Flow cytometric analysis and quantification of mean fluorescence intensity (MFI) representing the expression of T-bet and Eomes in NK cells (Lin− CD45+ NK1.1+), the CD11b− NK-cell subset, and the CD11b+ NK-cell subset in the bone marrow (BM) and spleens (SP) of control and Ncr1CreDhx15fl/fl mice (n = 3 each group). B qRT–PCR analysis of transcription factors in Dhx15-deleted NK cells. NK cells were purified from the BM and spleen of control and Ncr1CreDhx15fl/fl mice and cultured in IL-15 (20 ng/ml) for 7 days. CD11b− NK cells (CD45+CD3ε−NK1.1+CD11b−) were sorted and analyzed for the expression of the indicated transcription factors. C Volcano plot of the RNA-seq analysis results for CD11b− NK cells from control and Ncr1CreDhx15fl/fl mice. A series of genes (total differentially expressed genes, 56; upregulated genes, 47; downregulated genes, 9) was affected in Dhx15-deficient NK cells. D Histogram of Ki67 expression in NK cells (Lin− CD45+ NK1.1+), the CD11b− NK-cell subset, and the CD11b+ NK-cell subset in the spleen and bone marrow of control and Ncr1CreDhx15fl/fl mice (n = 3 each group). E Flow cytometric analysis of adoptively transferred NK cells purified from the spleen of ERCreDhx15fl/fl (CD45.2+) and control mice (CD45.1+) after adoptive cotransfer into C57BL/6 (CD45.1±) hosts (1:1 ratio) and TMF treatment (5 injections). Representative flow cytometry plots (left) and graph (right) showing the percentages of Dhx15-deleted NK cells (CD45.2) and control NK cells (CD45.1) in the spleen and BM of host mice 10 and 28 days post cell transfer (n = 3). F Schematic diagram showing the generation of BM chimeric mice (left): BM cells isolated from CD45.1+ control mice were mixed with either Dhx15fl/fl or Ncr1CreDhx15fl/fl BM cells (CD45.2) at a 1:1 ratio and were then injected into lethally irradiated CD45.1± recipient mice. Representative flow cytometry plots (right) showing the percentages of CD45.1 cells versus CD45.2 cells gated on CD3ε− NK1.1+ cells and CD45.2+ NK-cell subsets based on CD11b expression (n = 3). The data shown are representative of at least three independent experiments and are shown as the mean ± s.d. values. ***, p < 0.001
We further generated CD45.2+ERCreDhx15fl/fl mice, in which deletion of Dhx15 in NK cells is inducible upon administration of TMF [48]. We sorted splenic NK cells from control (CD45.1) and ERCreDhx15fl/fl mice (CD45.2) by FACS, mixed them at a 1:1 ratio, and injected them into CD45.1± hosts (CD45.1+CD45.2+). The host mice were administered TMF for 5 days, and the transferred NK cells were examined 10 and 28 days later. As shown in Fig. 2E, TMF treatment markedly reduced the recovery of CD45.2+ NK cells (transferred from ERCreDhx15fl/fl mice), while the control NK cells (CD45.1) remained in the treated hosts on Days 10 and 28 (Fig. 2E). The reduction in NK cells on D28 was even more prominent, with few CD45.2+ NK cells detected in the treated hosts. Thus, deletion of Dhx15 in mature NK cells clearly compromised their survival. We also created BM chimeric mice; BM cells from control (CD45.1 WT) and Dhx15fl/fl or Ncr1CreDhx15fl/fl mice (CD45.2) were mixed (1:1 ratio) and were then injected into lethally irradiated CD45.1± hosts. NK cells were assessed 6–8 weeks later when the lymphoid compartment was completely reconstituted. As shown in Fig. 2F, the development of CD45.2+ NK cells and the CD11b+ NK-cell subset (Dhx15-deleted) was markedly impaired compared to that of control Dhx15fl/fl NK cells. Collectively, these results further confirm that NK cells intrinsically require DHX15 for their survival and development in vivo.
DHX15 played a key role in NK-cell responsiveness to IL-15
As IL-15 is central to NK-cell survival and homeostasis [49, 50], we determined the responsiveness of NK cells to IL-15 in vivo in control and Ncr1CreDhx15fl/fl mice. For this purpose, we intraperitoneally injected the IL-15/IL-15RαFc complex, which acts as an IL-15 superagonist [51], into littermate control and Ncr1CreDhx15fl/fl mice and assessed the expansion of NK cells 4 days later. As shown in Fig. 3A and B, IL-15/IL-15RαFc administration resulted in a 5–7-fold increase in the NK cell population in the spleen and blood in control mice, and similar expansion was observed in the liver and lungs (Fig. 3A). In stark contrast, the IL-15/IL-15RαFc complex failed to induce expansion of NK cells in Ncr1CreDhx15fl/fl mice (Fig. 3A). Furthermore, analysis of NK-cell subsets based on the expression of CD11b showed that CD11b+ NK cells, especially those in the spleens of Ncr1CreDhx15fl/fl mice, failed to respond to IL-15 (Fig. 3A, B), suggesting that the response to IL-15, especially the differentiation toward mature NK cells, is impaired in Dhx15-deleted NK cells. Staining of IL-15 receptors (IL-2/IL-15Rβ; CD122; and the common γc, CD132) on wild-type NK cells and Dhx15-deleted NK cells showed comparable levels of expression (Fig. S3C, D). However, the phosphorylation level of STAT5 downstream of IL-15R showed a noticeable reduction in Ncr1CreDhx15fl/fl mice, while the level of p-mTOR was comparable between the two groups (Fig. S3F).
Fig. 3.

Deletion of Dhx15 in NK cells abrogates IL-15 responsiveness. A Representative flow cytometry plots showing the responsiveness of NK cells from control and Ncr1CreDhx15fl/fl mice to the IL-15/IL-15RαFc complex (administered i.p.). The percentages of NK cells (top panel) and NK-cell subsets (bottom panel) in the bone marrow, spleen, blood, liver, and lungs of control and Ncr1CreDhx15fl/fl mice treated with or without the IL-15 complex for 4 days. B The histograms show the absolute numbers of NK cells in the bone marrow and spleen and the expansion of the CD11b+ and CD11b− NK-cell subsets in the spleen of control and Ncr1CreDhx15fl/fl mice treated with or without the IL-15/IL-15RαFc complex. The blue numbers in the graph indicate the fold changes in the numbers of NK cells after treatment with the IL-15/IL-15RaFc complex (n = 3 each group). The data shown are representative of at least three independent experiments, and the calculated data are shown as the mean ± s.d. values. **, p < 0.01; ***, p < 0.001
To gain further mechanistic insights, we sorted NK cells from control and ERCreDhx15fl/fl mice by FACS, treated them in vitro with TMF, and assessed IL-2/IL-15Rβ (CD122) and common γc (CD132) expression at different time points (1–5 days). As shown in Fig. 4A and B, we observed significant downregulation of the IL-2/IL-15Rβ chain in Dhx15-deleted NK cells compared to control NK cells, while the expression of common γc was comparable regardless of Dhx15 deletion status (Fig. 4C), and this downregulation became more pronounced on Day 5 after TMF treatment, when Dhx15 deletion was complete. We then treated ERCreDhx15fl/fl mice and their control littermates with TMF for 5 days and examined the expression of the IL-2/IL-15Rβ chain in NK cells in vivo. Again, we observed significant downregulation of the IL-2/IL-15Rβ chain in Dhx15-deleted NK cells (Fig. 4D). These results suggest that the IL-2/IL-15Rβ chain is likely the focal point of regulation by DHX15.
Fig. 4.

Regulation of CD122 expression on NK cells by DHX15 in vitro and in vivo. A Representative flow cytometry plots showing profiles of CD122 expression at different time points (1–5 days) on NK cells isolated from ERCreDhx15fl/fl and control mice. Purified NK cells cultured with 20 ng/ml IL-15 were treated with or without TMF for 5 days in vitro. B The graph shows CD122 expression (MFI) on Day 5 on splenic NK cells treated with or without TMF in vitro. NK cells were purified from control and ERCreDhx15fl/fl mice. C The flow cytometry histogram and plot show the expression of CD132 on Day 5 on splenic NK cells isolated from ERCreDhx15fl/fl mice and treated with or without TMF in vitro. D The flow cytometry histogram and plot show the expression of CD122 on bone marrow NK cells in control and ERCreDhx15fl/fl mice on Day 6 after five consecutive tamoxifen treatments in vivo (100 mg/kg, administered i.p.). E Quantitative RT–PCR (qRT–PCR) analysis of IL-15Rα, CD122 (IL2Rβ), common γ chain (IL2Rγ) and IL-2Rα mRNA expression in NK cells after induced Dhx15 deletion. NK cells sorted from the spleen and bone marrow of control and ERCreDhx15fl/fl mice were treated with TMF or DMSO for 5 days in vitro. Total RNA was extracted from treated NK cells for analysis after confirmation of reduced CD122 surface expression. The data shown are representative of at least three independent experiments and are shown as the mean ± s.d. values. n.s., not (statistically) significant; **, p < 0.01; ***, p < 0.001
Deletion of Dhx15 did not seem to affect CD122 mRNA processing and stability in NK cells
To examine whether DHX15 regulates IL-2/15Rβ mRNA splicing and stability and therefore affects its expression, we sorted NK cells from control and ERCreDhx15fl/fl mice by FACS and cultured them in vitro with or without TMF for 5 days. Then, cDNA was synthesized from total cellular RNA using oligo(dT) and random primers, and the mRNA levels of IL-2Rα, IL-15Rα, CD122, and CD132 were determined by quantitative RT–PCR. As shown in Fig. 4E, we did not observe marked changes in the mRNA levels of CD122 and CD132. However, mRNA of IL-15Ra, which is not expressed in NK cells, was conspicuously absent (Fig. 4E). We then designed specific primer sets that corresponded to defined regions of the coding and noncoding sequences of CD122 mRNA (Fig. 5A) and used these primers for quantitative PCR analysis. Again, we failed to detect marked changes in PCR amplicons in NK cells, either in the cytoplasm or in the nucleus, regardless of Dhx15 deletion status (Fig. 5A). To specifically address CD122 mRNA splicing in NK cells, different primer sets spanning different exons, as well as both random and oligo(dT) primers, were used for reverse transcription. Again, we failed to detect any alternative splicing products of CD122 mRNA regardless of Dhx15 deletion status (Fig. 5B). CD122 mRNA degradation was measured by inhibiting transcription with actinomycin D in ERCreDhx15fl/fl NK cells after treatment with or without TMF for 5 days. As shown in Fig. S4A, CD122 had a longer half-life after induced DHX15 knockout. Taken together, these results indicate that the reduced CD122 expression on Dhx15-deleted NK cells is unlikely to be due to impairment of CD122 mRNA processing and stability.
Fig. 5.
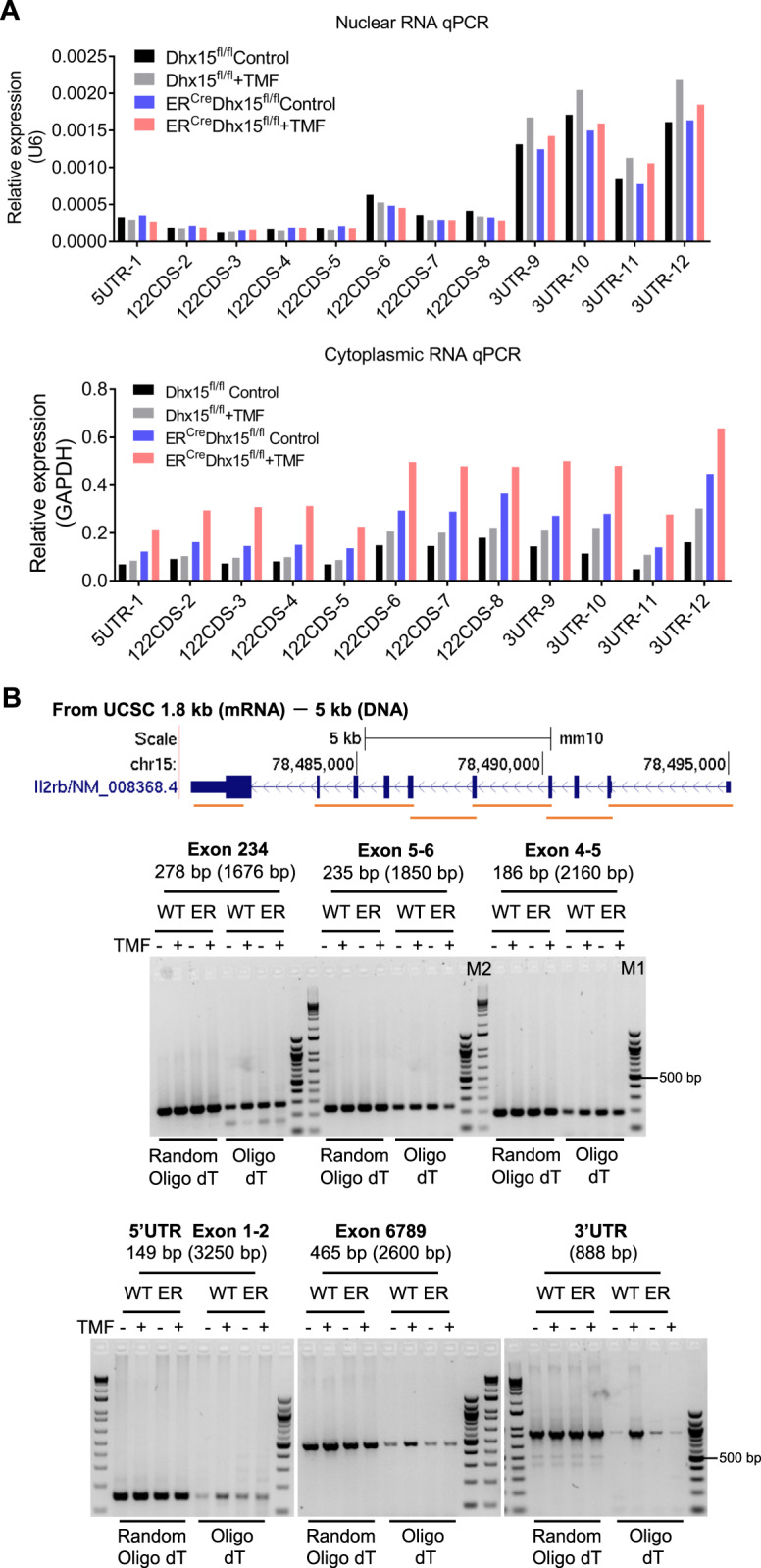
Deletion of Dhx15 does not seem to affect CD122 mRNA processing in NK cells. A Representative plots of qRT–PCR data for CD122 mRNA levels in the cytoplasm and nucleus of Dhx15-deleted NK cells. NK cells sorted from the spleen of control and ERCreDhx15fl/fl mice were treated with TMF or DMSO for 5 days. Cytoplasmic and nuclear RNA were extracted, and qRT–PCR was performed to analyze CD122 mRNA expression using specific primer sets corresponding to defined regions in the coding and noncoding sequences of CD122 mRNA. B Conventional PCR analysis of the effect of Dhx15 deletion on the alternative splicing of CD122 mRNA. Splenic NK cells sorted from control (WT) and ERCreDhx15fl/fl(ER) mice were treated with TMF or DMSO for 5 days in vitro. Total RNA was extracted from treated NK cells, and random and/or oligo(dT) primers were used for cDNA synthesis. A series of primers spanning different exons were used for subsequent PCR amplification. The predicted longer PCR products, including intron RNA, are shown in brackets. The data shown are representative of at least three independent experiments
Mutation of the ATPase domain of DHX15 disrupted NK-cell development
DHX15 consists of multiple domains and has both ATPase and helicase activities (Fig. 6A). The tandem RecA domains (RecA1 and RecA2) form the conserved core, which possesses ATPase activity [52]. To assess whether the ATPase activity of DHX15 (rather than its splicing activity) is involved in regulating NK cells, we generated a DHX15 mutant (i.e., E261Q), and this point mutation selectively abolished the ATPase activity of DHX15 [5]. As a control, we also introduced the R222G mutation into DHX15, which abrogates its helicase function [9, 53] (Fig. 6A).These DHX15 mutants were expressed in ERCreDhx15fl/fl BM stem cells (CD45.2) using lentivirus-mediated gene transfer (marked by GFP expression). We then generated chimeric mice by transferring the transduced BM cells together with CD45.1 control BM cells into lethally irradiated CD45.1± hosts and then treating the host mice with TMF. As shown in Fig. 6A and Fig. S5A, in host mice reconstituted with the E261Q (ATPase-dead) mutant, the development of NK cells was impaired, whereas in those reconstituted with the R222G (helicase-dead) mutant little effect on NK-cell development was observed, further supporting the idea that the helicase activity of DHX15 is dispensable for NK-cell activities.
Fig. 6.

Role of DHX15 in the surface expression of CD122 and its interactions with the CD122 3′UTR. A Schematic diagram (top) showing the domain structure of DHX15 and sites of point mutations. Flow cytometry plots showing the effect of DHX15 mutants (E261Q and R222G) on NK-cell development in vivo in ERCreDhx15fl/fl host mice. BM cells from CD45.2 ERCreDhx15fl/fl mice were spin-infected with lentivirus expressing full-length (FL) Dhx15, the Dhx15 E261Q mutant, and the Dhx15 R222G mutant. The transduced BM cells were mixed with 0.4 million CD45.1+ BM cells and used to reconstitute lethally irradiated CD45.1± host mice. Three weeks later, the host mice were treated with TMF for 5 consecutive days, and NK cells in the host spleens were analyzed 2 weeks later by gating on the CD45.1+ and CD45.2+GFP+ populations (bottom left panel). The graph (bottom right panel) shows the relative percentages of Dhx15-modified NK cells treated with or without TMF (n = 3 each group). B Western blot analysis of CD122 expression on NK cells sorted from control and ERCreDhx15fl/fl mice, cultured in IL-15 (20 ng/ml), and treated with TMF or DMSO for 5 days in vitro. The protein expression of CD122 was analyzed at by Western blotting. C RIP assay showing the binding of DHX15 to various regions of CD122 mRNA. Total mRNA was immunoprecipitated using an anti-DHX15 antibody or normal rabbit IgG as the control. The precipitated RNA was then analyzed by qRT–PCR using a series of primer sets specific for CD122 mRNA (Transcript ID: ENSMUST00000089398). The graph is representative of one experiment showing fold enrichment as measured by RIP-qPCR. D RNA pulldown assay assessing RNA/protein interactions. The CD122 3′UTR or CD122 3′UTR antisense (3′UTR-AS) RNA was used to pulldown protein complexes from NIH3T3 cell lysates, and enrichment of DHX15 in the protein complexes was assessed by Western blotting (left panel). Experiments involving the RNA binding protein HUR were included as a positive control (right panel). E Representative Western blot analysis assessing interactions between DHX15 and CD122. CD122 and HA-tagged DHX15 proteins were coexpressed in 293 T cells, and the cell lysate was subjected to immunoprecipitation with anti-HA antibody-conjugated agarose beads or unconjugated agarose beads as the negative control (NC) and analyzed by immunoblotting with an anti-CD122 antibody. F Schematic diagram showing the GFP reporters (with or without the 3′UTR) of CD122 generated by replacing the extracellular domain of CD122 with GFP. The graph shows the relative surface GFP expression of the GFP reporter with the 3′UTR compared to the GFP reporter without the 3′UTR on bone marrow-derived NK cells treated with TMF for the indicated time. The data shown are representative of at least three independent experiments. n.s., not significant; *, p < 0.05; **, p < 0.01
DHX15 impacted surface expression by interacting with the 3′UTR of CD122
The finding that CD122 mRNA processing and stability were not altered in Dhx15-deleted NK cells in spite of reduced cell surface expression of CD122 was unexpected. The impairment of NK cells induced by the point mutation in the ATPase domain of DHX15 prompted us to examine whether the corresponding DHX15 mutant still interacts with CD122 mRNA, thus indirectly affecting IL-15R signaling. To address this possibility, we sorted NK cells from control and ERCreDhx15fl/fl mice by FACS, extracted total protein from the sorted cells after treatment in vitro with TMF for 5 days, and quantitated total CD122 protein expression by Western blotting. As shown in Fig. 6B, CD122 protein expression was comparable in control and Dhx15-deleted NK cells, suggesting that translation of the mRNA into the CD122 protein is not altered in the absence of DHX15. We performed extensive RIP-qPCR experiments examining the binding of DHX15 to various regions of CD122 mRNA extracted from NK cells. As shown in Fig. 6C, we mapped the DHX15 binding sites and found that DHX15 preferentially bound to the 3′UTR of CD122 mRNA. To further confirm the binding of DHX15 to CD122 mRNA, an RNA pulldown assay was performed in NIH3T3 cells. As shown in Fig. 6D, the 3′UTR of CD122 mRNA could interact with DHX15. We then analyzed whether DHX15 also binds to the CD122 protein using a co-IP assay in 293 T cells. As shown in Fig. 6E, DHX15 failed to interact directly with the CD122 protein. We further evaluated whether the 3′UTR of CD122 mRNA affects surface protein expression In the absence of DHX15 using GFP reporters, which were constructed by replacing the extracellular domain of CD122 with GFP, as shown in Fig. 6F. Bone marrow-derived NK cells (ERCreDhx15fl/fl), which were modified by GFP reporter transfection before differentiation, were treated with TMF for the indicated time. The surface expression level of the With-UTR GFP reporter was significantly decreased on D5 and D7 compared to that of the No-UTR reporter (Fig. 6F and Fig. S4B). These results indicate that DHX15 likely facilitates CD122 surface expression by interacting with its 3′UTR.
Ectopic expression of CD122 or a-STAT5 rescued the defects in Dhx15-deficient NK cells
To determine whether altered surface expression of IL-2/IL-15Rβ affects IL-15R signaling in NK cells, we assessed the status of phosphorylated STAT5 (p-STAT5), an essential signaling molecule downstream of IL15/IL-15R engagement [54]. We sorted NK cells from control and ERCreDhx15fl/fl mice, stimulated them with IL-15 in vitro and treated them with TMF, and then examined the p-STAT5 level. As shown in Fig. 7A, STAT5 was prominently phosphorylated (i.e., p-STAT5) in control NK cells 10 min after IL-15 stimulation, while the p-STAT5 level was significantly decreased in Dhx15-deleted NK cells (treated with TMF). The total STAT5 protein level was not affected (data not shown).
Fig. 7.

Ectopic expression of CD122 or a-STAT5 can rescue the Dhx15-deleted NK-cell phenotype. A The flow cytometry plots and graph show the expression of p-STAT5 in NK cells from control and ERCreDhx15fl/fl mice in vitro. NK cells sorted from ERCreDhx15fl/fl or control mice and cultured in IL-15 (20 ng/ml) were treated with TMF or DMSO for 5 days. After recovery for 6 h without IL-15, NK cells were stimulated with 20 ng/ml IL-15 for 1, 5, or 10 min. The phosphorylation status of STAT5 was analyzed by intracellular staining. B Schematic diagram shows the bone marrow chimera experiment timeline. ERCreDhx15fl/fl mice were treated with 5-FU for 4 days, and BM cells were collected for spin infection with retrovirus. Infected BM cells were then mixed with control CD45.1+ BM cells (0.4 million) and transferred into lethally irradiated CD45.1± recipient mice. After 3 weeks, these mice were treated with TMF for 5 consecutive days (i.p.) to delete Dhx15. Splenic NK cells were analyzed by flow cytometry 2 weeks later. C Representative flow cytometry plots and graphs showing the percentages of CD11b+ NK cells after ectopic expression of CD122 and a-STAT5 (marked by GFP expression) (n = 3). Bone marrow cells from ERCreDhx15fl/fl mice were spin-infected with retrovirus expressing CD122 or a-STAT5 with the GFP marker for reconstitution. The data shown are representative of at least three independent experiments. ***, p < 0.001
We also generated a constitutively active mutant of STAT5 (a-STAT5) and overexpressed a-STAT5 in BM stem cells using retrovirus-mediated gene transfer (Fig. 7B). We then generated BM chimeric mice to determine whether the NK cell defects in Dhx15-deleted mice could be rescued by ectopic expression of a-STAT5. BM cells from ERCreDhx15fl/fl mice transduced with the a-STAT5 vector (marked by GFP expression) were injected into lethally irradiated CD45.1± hosts, which were treated with TMF 3 weeks after reconstitution. After another 2 weeks, we analyzed NK cells in the spleens of host mice by flow cytometry. As shown in Fig. 7C, ectopic expression of a-STAT5 largely rescued the CD11b+ NK cells, as well as the CD11b+CD27− subset. Similar findings in rescuing NK cell defects were observed when CD122 was overexpressed in BM stem cells (Fig. 7C). As expected, ectopic expression of full-length DHX15 using Ncr1CreDhx15fl/fl BM cells completely restored the CD11b+ NK cells and CD11b+CD27− NK-cell subset (Fig. S5B). Collectively, these results further support the role of DHX15 in regulating NK cells via the IL-15R signaling pathway.
Discussion
NK cells are a key cell type in the innate immune system, and their regulation by various transcription factors and cytokines has been extensively studied [15, 16, 22–24]. In the present study, we demonstrated an important role for the RNA helicase DHX15 in regulating NK-cell maturation, survival and cytolytic functions, thus, for the first time, placing the helicases on a growing list of regulators that control NK cells. We found that deletion of Dhx15 in NK cells disrupts their differentiation and survival in the periphery, with the most prominent effects on the terminally differentiated CD11b+CD27− subset, thus resulting in reduced NK-cell numbers and impaired cytolytic functions. We also found that Dhx15-deleted NK cells are defective in their responsiveness to IL-15, a key cytokine mediating the control of NK cells [49, 50], primarily due to impaired IL-2/IL-15Rβ chain (CD122) expression and impaired IL-15R signaling. Mechanistically, we showed that the IL-2/IL-15Rβ chain, which is noticeably downregulated in Dhx15-deleted NK cells, seems to be the focal point of this regulation. We further showed that the effects of DHX15 on NK cells depend on its ATPase domain and not its helicase domain. Hence, our study provides novel mechanistic insights into the regulatory networks that control NK-cell activities and markedly expands the role of RNA helicases in the regulation of innate immune responses.
Clearly, the discovery that DHX15 is an essential regulator of NK cells is novel and significant, as it highlights an additional layer of control of NK-cell activities beyond the known transcription factors, cytokines and metabolic reprogramming events [16, 19, 22]. DHX15 is an RNA binding protein possessing both ATPase and helicase activities [6, 52]. Traditionally, DHX15 is known as an RNA splicing factor involved in unwinding complex RNA structures [6]. Because of its RNA binding capacity, DHX15 also acts as a molecular sensor of RNA viruses to trigger dendritic cell activation [2]. We demonstrated in the current study that DHX15 also plays an indispensable role in NK cells, regulating both the differentiation and survival, as well as the cytolytic functions, of NK cells. In fact, the role of DHX15 in NK cells was consistently demonstrated in mice with conditional Dhx15 deletion, in adult mice with induced Dhx15 deletion, and models involving BM chimeric mice and adoptive transfer of Dhx15-deleted NK cells. Importantly, one of the striking features of Dhx15-deleted NK cells is their impaired responsiveness to IL-15, a finding that is consistent with the key role of IL-15 in the control of NK cells [49, 55, 56]. Indeed, in both IL-15 knockout and IL-15R knockout mice, NK cells are defective [49, 57–59]. Interestingly, this impairment of IL-15 responsiveness appears to be related to altered IL-2/IL-15Rβ expression and, consequently, impaired IL-15R signaling (i.e., STAT5 phosphorylation). This idea is further supported by the finding that ectopic expression of CD122 or a constitutively active form of STAT5 (a-STAT5) largely rescued the defects of Dhx15 deletion in NK cells. In fact, the selective control of the IL-2/IL-15Rβ chain but not the common γc chain or the IL-15Rα chain by DHX15 is highly interesting. Indeed, we did not detect altered splicing or reduced stability of CD122 mRNA in Dhx15-deleted NK cells, and CD122 protein expression was comparable to that in control NK cells. Thus, the reduced surface expression of CD122 on Dhx15-deleted NK cells appears to involve novel mechanisms that warrant further clarification. Our analysis of the DHX15 mutants revealed that the ATPase domain rather than the helicase domain of DHX15 seems to be critical for surface expression of CD122.
Another interesting finding was that DHX15 did not seem to interact directly with the CD122 protein but consistently bound to the 3’UTR of CD122 mRNA, as shown by RIP assays. A recent study suggested that DHX9, another member of the RNA helicase family, inhibits CDK6 expression by binding to its 3’-untranslated region (3’UTR) in cancer cells [42]. Furthermore, others recently showed that an RNA binding protein called HuR specifically regulates surface CD47 expression by interacting with the long 3’UTR of CD47 transcripts in U2OS and HEK293 cells [60]. Our GFP reporter assay indicated that the impairment of surface CD122 expression was regulated through the DHX15-CD122 3′UTR interaction. However, the detailed mechanism by which the DHX15-CD122 3′UTR complex contributes to surface CD122 expression was not completely clarified in the current study. Thus, our data and those of others suggest a novel mechanism involving RNA binding proteins and 3′UTRs in the cell surface expression of signaling receptors.
In our model, deletion of DHX15 did not completely abolish NK-cell development in vivo but instead reduced the number of NK cells in the periphery, with the greatest effect on the mature and terminally differentiated subsets, suggesting that the role of DHX15 in NK-cell regulation is conditional and likely depends on the stage of NK-cell differentiation. Previous studies have demonstrated that NK cells at these stages are also controlled by T-bet, Eomes, and other transcription factors [16, 46, 47]. In addition, their activation and effector functions are also sensitive to IL-15 at these stages [61]. Another pathway of NK cell activation operates through PI3K-mTOR-dependent phosphorylation of ribosomal protein S6 by p70-S6 kinase, as NK cells were found to be defective in mice with either STAT5 or mTOR knockout [19, 54, 62]. Our adoptive transfer experiments further support the idea that the maturation and terminal differentiation of NK cells are more sensitive than other NK-cell properties to Dhx15 deletion. Clearly, the terminal differentiation of NK cells is controlled by diverse regulatory mechanisms, and how the DHX15 pathway interacts with other regulatory pathways in fine tuning NK-cell activities awaits further clarification.
In summary, our study demonstrates that the RNA helicase DHX15 is critically involved in regulating NK cells and that its deletion disrupts NK-cell homeostasis, survival and cytolytic activity. DHX15 plays a particularly important role in the terminal differentiation of NK cells in the periphery, primarily by regulating the IL-15 signaling pathway. Importantly, the role of DHX15 in the control of NK cells depends on its ATPase activity, not its helicase activity, and is most likely mediated via regulation of surface expression of the IL-2/IL-15Rβ chain. Clearly, these findings establish the importance of helicases in regulating NK-cell activities and uncover new therapeutic targets involved in the modulation of NK cells in various clinical settings.
Supplementary information
Acknowledgements
We thank Professor Eric Vivier at the Innate Pharma Research Labs in France for the generous gift of Ncr1Cre mice. We acknowledge the excellent services obtained from the flow cytometry core and Comparative Medicine Program at Houston Methodist Hospital in Houston, Texas. This project was supported by National Institutes of Health grants (R01AI080779 and R01 A1155488).
Author contributions
GW designed and performed the experiments and analyzed the data. XX helped with FACS and mouse irradiation. LM provided operational support. XX, YW, XC, RG, and YD performed some experiments and provided helpful discussions. ZZ helped revise the paper. XCL supervised the studies and wrote the paper.
Competing interests
The authors declare no competing interests.
Supplementary information
The online version contains supplementary material available at 10.1038/s41423-022-00852-7.
References
- 1.Detanico T, Virgen-Slane R, Steen-Fuentes S, Lin WW, Rhode-Kurnow A, Chappell E, et al. Co-expression Networks Identify DHX15 RNA Helicase as a B Cell Regulatory Factor. Front Immunol. 2019;10:2903. doi: 10.3389/fimmu.2019.02903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu H, Lu N, Weng L, Yuan B, Liu YJ, Zhang Z. DHX15 senses double-stranded RNA in myeloid dendritic cells. J Immunol. 2014;193:1364–72. doi: 10.4049/jimmunol.1303322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mosallanejad K, Sekine Y, Ishikura-Kinoshita S, Kumagai K, Nagano T, Matsuzawa A, et al. The DEAH-box RNA helicase DHX15 activates NF-kappaB and MAPK signaling downstream of MAVS during antiviral responses. Sci Signal. 2014;7:ra40. doi: 10.1126/scisignal.2004841. [DOI] [PubMed] [Google Scholar]
- 4.Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem Sci. 2011;36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Memet I, Doebele C, Sloan KE, Bohnsack MT. The G-patch protein NF-kappaB-repressing factor mediates the recruitment of the exonuclease XRN2 and activation of the RNA helicase DHX15 in human ribosome biogenesis. Nucleic Acids Res. 2017;45:5359–74. doi: 10.1093/nar/gkx013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semlow DR, Blanco MR, Walter NG, Staley JP. Spliceosomal DEAH-Box ATPases Remodel Pre-mRNA to Activate Alternative Splice Sites. Cell. 2016;164:985–98. doi: 10.1016/j.cell.2016.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inesta-Vaquera F, Chaugule VK, Galloway A, Chandler L, Rojas-Fernandez A, Weidlich S, et al. DHX15 regulates CMTR1-dependent gene expression and cell proliferation. Life Sci Alliance. 2018;1:e201800092. doi: 10.26508/lsa.201800092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toczydlowska-Socha D, Zielinska MM, Kurkowska M, Astha, Almeida CF, Stefaniak F, et al. Human RNA cap1 methyltransferase CMTr1 cooperates with RNA helicase DHX15 to modify RNAs with highly structured 5’ termini. Philos Trans R Soc Lond B Biol Sci. 2018;373:20180161. [DOI] [PMC free article] [PubMed]
- 9.Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet. 2016;48:1551–6. doi: 10.1038/ng.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ito S, Koso H, Sakamoto K, Watanabe S. RNA helicase DHX15 acts as a tumour suppressor in glioma. Br J Cancer. 2017;117:1349–59. doi: 10.1038/bjc.2017.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jing Y, Nguyen MM, Wang D, Pascal LE, Guo W, Xu Y, et al. DHX15 promotes prostate cancer progression by stimulating Siah2-mediated ubiquitination of androgen receptor. Oncogene. 2018;37:638–50. doi: 10.1038/onc.2017.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen XL, Cai YH, Liu Q, Pan LL, Shi SL, Liu XL, et al. ETS1 and SP1 drive DHX15 expression in acute lymphoblastic leukaemia. J Cell Mol Med. 2018;22:2612–21. doi: 10.1111/jcmm.13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao G, Chen K, Qin Y, Niu Y, Zhang X, Xu S, et al. Long Non-coding RNA JHDM1D-AS1 Interacts with DHX15 Protein to Enhance Non-Small-Cell Lung Cancer Growth and Metastasis. Mol Ther Nucleic Acids. 2019;18:831–40. doi: 10.1016/j.omtn.2019.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, He K, Sheng B, Lei X, Tao W, Zhu X, et al. The RNA helicase Dhx15 mediates Wnt-induced antimicrobial protein expression in Paneth cells. Proc Natl Acad Sci U S A. 2021;118:e2017432118. doi: 10.1073/pnas.2017432118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hesslein DG, Lanier LL. Transcriptional control of natural killer cell development and function. Adv Immunol. 2011;109:45–85. doi: 10.1016/B978-0-12-387664-5.00002-9. [DOI] [PubMed] [Google Scholar]
- 16.Luevano M, Madrigal A, Saudemont A. Transcription factors involved in the regulation of natural killer cell development and function: an update. Front Immunol. 2012;3:319. doi: 10.3389/fimmu.2012.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bi J, Wang X. Molecular Regulation of NK Cell Maturation. Front Immunol. 2020;11:1945. doi: 10.3389/fimmu.2020.01945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood. 2009;113:5488–96. doi: 10.1182/blood-2008-10-187179. [DOI] [PubMed] [Google Scholar]
- 19.Marcais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15:749–57. doi: 10.1038/ni.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang C, Tsaih SW, Lemke A, Flister MJ, Thakar MS, Malarkannan S. mTORC1 and mTORC2 differentially promote natural killer cell development. Elife. 2018;7:e35619. doi: 10.7554/eLife.35619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li D, Wang Y, Yang M, Dong Z. mTORC1 and mTORC2 coordinate early NK cell development by differentially inducing E4BP4 and T-bet. Cell Death Differ. 2021;28:1900–9. doi: 10.1038/s41418-020-00715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y, Tian Z, Wei H. Developmental and Functional Control of Natural Killer Cells by Cytokines. Front Immunol. 2017;8:930. doi: 10.3389/fimmu.2017.00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abel AM, Yang C, Thakar MS, Malarkannan S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol. 2018;9:1869. doi: 10.3389/fimmu.2018.01869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brady J, Carotta S, Thong RP, Chan CJ, Hayakawa Y, Smyth MJ, et al. The interactions of multiple cytokines control NK cell maturation. J Immunol. 2010;185:6679–88. doi: 10.4049/jimmunol.0903354. [DOI] [PubMed] [Google Scholar]
- 25.Cooper MA, Colonna M, Yokoyama WM. Hidden talents of natural killers: NK cells in innate and adaptive immunity. EMBO Rep. 2009;10:1103–10. doi: 10.1038/embor.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, et al. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity. 2007;26:798–811. doi: 10.1016/j.immuni.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 27.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–9. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137:624–36. doi: 10.1182/blood.2020007748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daher M, Rezvani K. Outlook for New CAR-Based Therapies with a Focus on CAR NK Cells: What Lies Beyond CAR-Engineered T Cells in the Race against Cancer. Cancer Discov. 2021;11:45–58. doi: 10.1158/2159-8290.CD-20-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell. 2019;177:1701–13. doi: 10.1016/j.cell.2019.04.041. [DOI] [PubMed] [Google Scholar]
- 32.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med. 2020;382:545–53. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Narni-Mancinelli E, Chaix J, Fenis A, Kerdiles YM, Yessaad N, Reynders A, et al. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc Natl Acad Sci USA. 2011;108:18324–9. doi: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang M, Li D, Chang Z, Yang Z, Tian Z, Dong Z. PDK1 orchestrates early NK cell development through induction of E4BP4 expression and maintenance of IL-15 responsiveness. J Exp Med. 2015;212:253–65. doi: 10.1084/jem.20141703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam VC, Folkersen L, Aguilar OA, Lanier LL. KLF12 Regulates Mouse NK Cell Proliferation. J Immunol. 2019;203:981–9. doi: 10.4049/jimmunol.1900396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong P, Wagner JA, Berrien-Elliott MM, Schappe T, Fehniger TA. Flow cytometry-based ex vivo murine NK cell cytotoxicity assay. STAR Protoc. 2021;2:100262. doi: 10.1016/j.xpro.2020.100262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kroemer A, Xiao X, Degauque N, Edtinger K, Wei H, Demirci G, et al. The innate NK cells, allograft rejection, and a key role for IL-15. J Immunol. 2008;180:7818–26. doi: 10.4049/jimmunol.180.12.7818. [DOI] [PubMed] [Google Scholar]
- 38.Williams NS, Klem J, Puzanov IJ, Sivakumar PV, Bennett M, Kumar V. Differentiation of NK1.1+, Ly49+ NK cells from flt3+ multipotent marrow progenitor cells. J Immunol. 1999;163:2648–56. [PubMed] [Google Scholar]
- 39.Male V, Nisoli I, Kostrzewski T, Allan DS, Carlyle JR, Lord GM, et al. The transcription factor E4bp4/Nfil3 controls commitment to the NK lineage and directly regulates Eomes and Id2 expression. J Exp Med. 2014;211:635–42. doi: 10.1084/jem.20132398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ratnadiwakara M, Anko ML. mRNA Stability Assay Using transcription inhibition by Actinomycin D in Mouse Pluripotent Stem Cells. Bio Protoc. 2018;8:e3072. doi: 10.21769/BioProtoc.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Q, Lou Y, Yang J, Wang J, Feng J, Zhao Y, et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut. 2019;68:2019–31. doi: 10.1136/gutjnl-2019-318912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang YL, Liu JY, Yang JE, Yu XM, Chen ZL, Chen YJ, et al. Lnc-UCID Promotes G1/S Transition and Hepatoma Growth by Preventing DHX9-Mediated CDK6 Down-regulation. Hepatology. 2019;70:259–75. doi: 10.1002/hep.30613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Investig. 2018;128:4654–68. doi: 10.1172/JCI99317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li ZY, Morman RE, Hegermiller E, Sun M, Bartom ET, Maienschein-Cline M, et al. The transcriptional repressor ID2 supports natural killer cell maturation by controlling TCF1 amplitude. J Exp Med. 2021;218:e20202032. doi: 10.1084/jem.20202032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takeda K, Nakayama M, Sakaki M, Hayakawa Y, Imawari M, Ogasawara K, et al. IFN-gamma production by lung NK cells is critical for the natural resistance to pulmonary metastasis of B16 melanoma in mice. J Leukoc Biol. 2011;90:777–85. doi: 10.1189/jlb.0411208. [DOI] [PubMed] [Google Scholar]
- 46.Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36:55–67. doi: 10.1016/j.immuni.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med. 2014;211:563–77. doi: 10.1084/jem.20131560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–18. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 49.Cooper MA, Bush JE, Fehniger TA, VanDeusen JB, Waite RE, Liu Y, et al. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood. 2002;100:3633–8. doi: 10.1182/blood-2001-12-0293. [DOI] [PubMed] [Google Scholar]
- 50.Anton OM, Peterson ME, Hollander MJ, Dorward DW, Arora G, Traba J, et al. Trans-endocytosis of intact IL-15Ralpha-IL-15 complex from presenting cells into NK cells favors signaling for proliferation. Proc Natl Acad Sci USA. 2020;117:522–31. doi: 10.1073/pnas.1911678117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo Y, Luan L, Patil NK, Sherwood ER. Immunobiology of the IL-15/IL-15Ralpha complex as an antitumor and antiviral agent. Cytokine Growth Factor Rev. 2017;38:10–21. doi: 10.1016/j.cytogfr.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Studer MK, Ivanovic L, Weber ME, Marti S, Jonas S. Structural basis for DEAH-helicase activation by G-patch proteins. Proc Natl Acad Sci USA. 2020;117:7159–70. doi: 10.1073/pnas.1913880117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McElderry J, Carrington B, Bishop K, Kim E, Pei W, Chen Z, et al. Splicing factor DHX15 affects tp53 and mdm2 expression via alternate splicing and promoter usage. Hum Mol Genet. 2019;28:4173–85. doi: 10.1093/hmg/ddz261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin JX, Du N, Li P, Kazemian M, Gebregiorgis T, Spolski R, et al. Critical functions for STAT5 tetramers in the maturation and survival of natural killer cells. Nat Commun. 2017;8:1320. doi: 10.1038/s41467-017-01477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pfefferle A, Jacobs B, Haroun-Izquierdo A, Kveberg L, Sohlberg E, Malmberg KJ. Deciphering Natural Killer Cell Homeostasis. Front Immunol. 2020;11:812. doi: 10.3389/fimmu.2020.00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huntington ND, Puthalakath H, Gunn P, Naik E, Michalak EM, Smyth MJ, et al. Interleukin 15-mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat Immunol. 2007;8:856–63. doi: 10.1038/ni1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity. 1995;2:223–38. doi: 10.1016/1074-7613(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 58.Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, et al. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–76. doi: 10.1016/S1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- 59.Suzuki H, Duncan GS, Takimoto H, Mak TW. Abnormal development of intestinal intraepithelial lymphocytes and peripheral natural killer cells in mice lacking the IL-2 receptor beta chain. J Exp Med. 1997;185:499–505. doi: 10.1084/jem.185.3.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berkovits BD, Mayr C. Alternative 3’ UTRs act as scaffolds to regulate membrane protein localization. Nature. 2015;522:363–7. doi: 10.1038/nature14321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nandagopal N, Ali AK, Komal AK, Lee SH. The Critical Role of IL-15-PI3K-mTOR Pathway in Natural Killer Cell Effector Functions. Front Immunol. 2014;5:187. doi: 10.3389/fimmu.2014.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood. 2011;117:1565–73. doi: 10.1182/blood-2010-06-291633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.