

Abstract
Context: Rare copy number variants (CNVs) have been associated with the development of severe obesity. However, the potential disease-causing contribution of individual genes within the region of CNVs is often not known.
Objective: Screening of rare variants in genes involved in CNVs in Finnish patients with severe early-onset obesity to find candidate genes linked to severe obesity.
Methods: Custom-made targeted exome sequencing panel to search for rare (minor allele frequency <0.1%) variants in genes affected by previously identified CNVs in 92 subjects (median age 14 years) with early-onset severe obesity (median body mass index (BMI) Z-score + 4.0).
Results: We identified thirteen rare heterozygous variants of unknown significance in eleven subjects in twelve of the CNV genes. Two rare missense variants (p.Pro405Arg and p.Tyr232Cys) were found in SORCS1, a gene highly expressed in the brain and previously linked to diabetes risk. Four rare variants were in genes in the proximal 16p11.2 region (a frameshift variant in TAOK2 and missense variants in SEZ6L2, ALDOA and KIF22) and three rare missense variants were in genes in the 22q11.21 region (AIFM3, ARVCF and KLHL22).
Conclusion: We report several rare variants in CNV genes in subjects with childhood obesity. However, the role of the individual genes in the previously identified rare CNVs to development of obesity remains uncertain. More studies are needed to understand the potential role of the specific genes within obesity associated CNVs.
Keywords: childhood obesity, gene panel, rare variants, candidate genes, copy number variants
Introduction
Copy number variants (CNVs) are structural variants that include duplications or deletions of genomic regions ranging from 50 bp to several megabases (Zarrei et al., 2015). These structural variants can arise by different mutational mechanisms such as replication, repair of double-strand breaks and recombination (Gu et al., 2008; Carvalho and Lupski, 2016). CNVs contribute to human diversity but also to several diseases, including neurodevelopmental disorders and severe obesity (Wang et al., 2010; Wheeler et al., 2013; D'Angelo et al., 2018; Saeed et al., 2020).
Deletion in 16p11.2 region is the most established CNV linked to early-onset severe obesity (Bochukova et al., 2010; Walters et al., 2010). CNVs also contribute to syndromic obesity (Vuillaume et al., 2014; Pettersson et al., 2017; D’Angelo et al., 2018; Selvanayagam et al., 2018; Saeed et al., 2020; Madeo et al., 2021). The identification of rare CNVs in obesity has led to the discovery of several novel candidate genes. However, in many cases the disease-causing gene within the region of CNVs is unclear. Indeed, unraveling the pathogenicity of CNVs remains a major challenge (Nowakowska, 2017). Pathogenic CNVs are usually large and contain genes with high evolutionary copy number conservation. It has been proposed that dosage sensitivity of individual genes within the region of CNVs is a frequent cause of CNV pathogenicity. Haploinsufficiency is commonly used as a model of gene dosage sensitivity and predictions of haploinsufficiency have been used to predict the impact of CNVs (Huang et al., 2010; Lek et al., 2016; Rice and McLysaght, 2017).
We previously identified rare CNVs enriched in subjects with severe childhood obesity (Pettersson et al., 2017). In a case-control study of 90 obese subjects and 67 normal-weight controls, using array comparative genomic hybridization, we identified rare CNVs in 17 obese subjects. However, the pathogenicity of several of the identified CNVs remained unclear and the potential phenotypic contribution of the genes within the CNVs is not known. We hypothesized that rare variants in the genes within the CNVs might contribute to the development of obesity. We designed a custom-made targeted exome sequencing panel, including all genes located in the regions of the rare CNVs identified in our cohort of patients with childhood obesity, in search for rare variants and novel candidate genes.
Methods
Study Subjects
This study included 92 children and adolescents with severe early-onset obesity, defined as height-adjusted weight >60% before 10 years of age according to Finnish growth standards (Saari et al., 2011). The study subjects were recruited through pediatric endocrine clinics at Helsinki University Hospital, Seinäjoki Central Hospital and Oulu University Hospital during years 2011–2017. We have previously investigated the study subjects by targeted exome sequencing for rare variants in 24 genes related to the leptin-melanocortin pathway or hypothalamic development (Loid et al., 2020). Altogether 80% of the patients in this study have also been screened for rare CNVs by array comparative genomic hybridization (a-CGH). In our previous study we identified rare CNVs in 17 obese subjects (Pettersson et al., 2017) and 14 of these 17 subjects with rare CNVs participated in the present study. The study was approved by Research Ethics Committees of Hospital District of Helsinki and Uusimaa, the Pirkanmaa Hospital District and the Northern Ostrobothnia Hospital District. Informed written consents were obtained from all the participants or their parents/guardians. Clinical and growth data were collected from hospital records and anthropometric measurements were obtained during a study visit. Age-and gender specific BMI Z-scores were derived based on the World Health Organization reference values (www.who.int/childgrowth/standards).
Targeted Exome Sequencing
We designed the probes for targeted exome sequencing using SeqCap EZ Choice Library and Nimble Design (Roche NimbleGen, United States). DNA capture and sequencing were performed at Oxford Genomics Centre. Sequence reads were aligned to reference genome GRCh37. In this study, we included 117 genes involved in previously identified rare CNVs (Pettersson et al., 2017). The genes are listed in Supplementary Table S1. We filtered variants using Varaft (version 2.17) (Desvignes et al., 2018). We selected coding and splice site variants with an allele frequency of <0.001 in the Genome Aggregation Database (gnomAD) database (http://gnomad.broadinstitute.org), the 1000 Genomes Project (http://www.internationalgenome.org), and the Sequencing Initiative Suomi database (SISu) (http://www.sisuproject.fi). In a second analysis, we also evaluated coding and splice site variants with an allele frequency of <0.005 to explore whether change of allele frequency threshold would add any potentially interesting discoveries, considering that severe obesity is not an extremely rare phenotype. In silico predictions were performed using Sorting Intolerant From Tolerant (SIFT) (Kumar P. et al., 2009), Polymorphism Phenotyping version 2 (PolyPhen2) (http://genetics.bwh.harvard.edu/pph2/), MutationTaster2 (Schwarz et al., 2014), Combined Annotation Dependent Depletion (CADD) scores (https://cadd.gs.washington.edu/), Mendelian Clinically Applicable Pathogenicity (M-CAP) (http://bejerano.stanford.edu/mcap/), Rare Exome Variant Ensemble Learner (REVEL) score (Ioannidis et al., 2016) and splice AI (https://spliceailookup.broadinstitute.org). We selected rare variants predicted to be pathogenic by several of the abovementioned prediction tools including a CADD score >20. Integrative Genomics Viewer (IGV) was used for visualization of the variants. The identified variants were classified according to the American Collage of Medical Genetics (ACMG) guidelines (Richards et al., 2015) using VarSome tool (Kopanos et al., 2019). Parental and siblings’ samples was obtained from two available families with the identified rare variants and Sanger sequencing was performed to determine inheritance pattern.
Results
Characteristics of Study Subjects
The cohort in this study comprised 92 subjects (51% males) with median age of 13.7 years (interquartile range (IQR) 10.6–16.8 years) and median BMI Z-score of +4.0 (IQR + 3.4 to + 4.9). The study subjects fulfilled our inclusion criteria (height-adjusted weight >60%) at median age of 6.0 years (IQR 4.5–7.0 years). Of the 68 school-aged study subjects for whom information on learning difficulties was available, ten (15%) had learning difficulties. Two study subjects had developmental delay based on previous clinical investigations. In these two patients Prader-Willi syndrome had been ruled out. Ten participants had insulin resistance based on oral glucose tolerance test.
Targeted Exome Sequencing
On average 99.3% of the bases in the coding exons were covered by at least 100 reads and the mean read depth of coverage was 750X. We identified 13 rare heterozygous variants in 11 subjects with early-onset obesity. Four of them had learning difficulties and one had developmental delay with autistic features. The variants with allele frequency of <0.001 were in 12 of the genes in the panel, namely AIFM3, ALDOA, ARVCF, BRAF, CHD1L, KIF22, KLHL22, MCTP2, SEZ6L2, SORCS1, TAOK2 and XRN1. Table 1 presents the rare variants with allele frequencies in gnomAD and in silico prediction values. For the rare splicing variant in CHDL1 we also assessed the SpliceAI score, which was 0.990 (donor loss). In one of the genes (SORCS1) we identified two rare variants; p.Pro405Arg and p.Tyr232Cys. Figure 1 presents the segregation of the SORCS1 variants in the two families. In both families the variant was inherited from a parent with overweight. However, also other family members without the variant were overweight or obese.
TABLE 1.
The identified rare variants with minor allele frequencies (MAF) in gnomAD and in silico prediction values.
| Study subject | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BMI (Z-score) | +5.8 | +5.1 | +4.0 | +4.3 | +2.8 | +2.9 | +3.8 | +4.5 | +3.7 | +5.0 | +3.8 | |||
| Associated features | — | insulin resistance, learning difficulties | learning difficulties | — | — | — | learning difficulties | developmental delay, autistic features | learning difficulties | — | — | |||
| Gene | CHD1L | KIF22 | SORCS1 | SEZ6L2 | TAOK2 | BRAF | ALDOA | ARVCF | KLHL22 | SORCS1 | AIFM3 | MCTP2 | XRN1 | |
| Type of variant | Splicing | Missense | Missense | Missense | Frameshift deletion | Missense | Missense | Missense | Missense | Missense | Missense | Missense | Missense | |
| Genotype | Het | Het | Het | Het | Het | Het | Het | Het | Het | Het | Het | Het | Het | |
| Chromosome position (hg19) | 1:146756173G > A | 16:29810594C > T | 10:108466322G > C | 16:29899049G > A | 16:29998896 GGGCTGTG >- | 7:140501243G > A | 16:30080955G > A | 22:19960473T > C | 22:20796725C > T | 10:108589363T > C | 22:21331358G > A | 15:94901832G > A | 3:142136034C > T | |
| DNA change | c.1854 + 1G > A | c.769C > T | c.1214C > G | c.1129C > T | c.3303_3310del | c.829C > T | c.922G > A | c.2525A > G | c.1540G > A | c.695A > G | c.1256G > A | c.1292G > A | c.1384G > A | |
| Amino acid change | — | p.Arg257Trp | p.Pro405Arg | p.Arg377Cys | p.Q1101fs | p.Pro277Ser | p.Val308Ile | p.Asp842Gly | p.Val514Met | p.Tyr232Cys | p.Arg419Gln | p.Arg431His | p.Ala462Thr | |
| rs number | rs371704346 | rs757250411 | — | rs752385379 | rs777383420 | — | rs142759891 | — | rs748490204 | rs201223592 | rs775926412 | rs375821657 | rs1223545973 | |
| MAF gnomAD | 0.0002 | 0.00002 | 0 | 0.0006 | 0.000003 | 0 | 0.00005 | 0 | 0.00003 | 0.00008 | 0.00006 | 0.00002 | 0 | |
| MAF gnomad FIN | 0 | 0 | 0 | 0.00009 | 0.0006 | 0 | 0.0002 | 0 | 0.00017 | 0.0005 | 0.0001 | 0 | 0 | |
| MAF obesity cohort | 0.0054 | 0.0054 | 0.0054 | 0.0054 | 0.0054 | 0.0054 | 0.0054 | 0.0054 | 0.0054 | 0.0054 | 0.0054 | 0.0054 | 0.0054 | |
| CADD | 34 | 26.3 | 28.9 | 23.8 | 33.0 | 27.1 | 23.3 | 22.4 | 33.0 | 27.0 | 25.6 | 24.3 | 27.9 | |
| MutationTaster2 | Disease causing | Disease causing | Disease causing | Disease causing | NA | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | |
| M-CAP | NA | Damaging | Damaging | Tolerated | NA | Damaging | Damaging | Damaging | Damaging | Damaging | Tolerated | Damaging | Tolerated | |
| REVEL | NA | Benign | Pathogenic | Benign | NA | Pathogenic | Benign | Benign | Pathogenic | Benign | Benign | Benign | Benign | |
| SIFT | NA | Damaging | Damaging | Tolerated | NA | Damaging | Tolerated | Damaging | Damaging | Damaging | Tolerated | Tolerated | Damaging | |
| Polyphen2 | NA | Probably Damaging | Probably Damaging | Benign | NA | Probably Damaging | Benign | Benign | Probably Damaging | Probably Damaging | Probably Damaging | Probably Damaging | Posssibly Damaging | |
| VarSome ACMG Classification (criteria used) | VUS (PP3, BS1) | VUS (PM2, PP3) | VUS (PM2, PP3, BP1) | LB (BS1, BP1, BP4) | VUS (PVS1, PP3, BS1) | LP (PM2, PM1, PP2,PP3) | VUS (PM2, PP3) | VUS (PM2, BP1) | VUS (PVS1, PM2, PP3) | LB (PP3, BS1, BP1) | LB (BS1) | VUS/B (BP1) | VUS (PM2, PP3) | |
MAF gnomad FIN; minor allele frequency in gnomad Finnish population
NA; data not available
ACMG; American College of Medical Genetics and Genomics
VUS; Variant of uncertain significance
B; benign
LB; likely benign
LP; likely pathogenic
FIGURE 1.
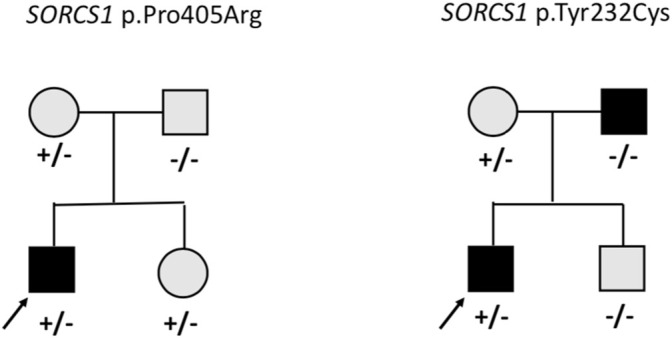
Pedigrees of the families with missense variants in SORCS1. Square = male, circle = female, +/− heterozygote, −/− wildtype. Black square/circle = obesity, gray square/circle = overweight.
Table 2 presents the cytogenic location of the genes with the rare variants. Four of the genes (ALDOA, KIF22, SEZ6L2 and TAOK2) were in the 16p11.2 region and three of the genes (AIFM3, ARVCF, KLHL22) were in the 22q11.21 region. We further evaluated the gnomAD constrained metrics for the genes that were involved in the rare deletions that we previously identified. Table 3 presents the predicted constraint metrics. Three of the genes (BRAF, TAOK2 and XRN1) had a pLI = 1, indicating dosage sensitivity to loss-of-function variants.
TABLE 2.
The cytogenic location for the 12 genes with identified rare variants.
| Cytogenic location | Gene symbol | Gene | Clinical genomic database | Inheritance |
|---|---|---|---|---|
| 16p11.2 | ALDOA | Aldolase, fructose-bisphosphate A | Glycogen storage disease XII | AR |
| 16p11.2 | KIF22 | Kinesin Family Member 22 | Spondyloepimetaphyseal dysplasia with joint laxity, type 2 | AD |
| 16p11.2 | SEZ6L2 | Seizure Related 6 Homolog Like 2 | — | — |
| 16p11.2 | TAOK2 | TAO Kinase 2 | — | — |
| 22q11.21 | AIFM3 | Apoptosis inducing factor mitochondria associated 3 | — | — |
| 22q11.21 | ARVCF | ARVCF Delta Catenin Family Member | — | — |
| 22q11.21 | KLHL22 | Kelch Like Family Member 22 | — | — |
| 7q34 | BRAF | B-Raf Proto-Oncogene, Serine/Threonine Kinase | Noonan syndrome 7, cardiocutaneous syndrome, LEOPARD syndrome 3 | AD |
| 1q21.1 | CHD1L | Chromodomain helicase DNA Binding Protein 1 Like | — | — |
| 15q26.2 | MCTP2 | Multiple C2 And Transmembrane Domain Containing 2 | — | — |
| 10q25.1 | SORCS1 | Sortilin Related VPS10 Domain Containing Receptor 1 | — | — |
| 3q23 | XRN1 | 5′-3′ Exoribonuclease 1 | — | — |
AR; autosomal recessive, AD; autosomal dominant
TABLE 3.
Predicted constrained metrics for the genes involved in deletions.
| Cytogenic location | Gene symbol | pLI | LOF Z-score | LOF o/e | LOEUF | Missense Z-score | Missense o/e | %HI |
|---|---|---|---|---|---|---|---|---|
| 16p11.2 | ALDOA | 0 | 2.3 | 0.4 | 0.8 | 0.3 | 1.0 | 11.3 |
| 16p11.2 | KIF22 | 0 | 2.2 | 0.6 | 0.9 | 0.1 | 1.0 | 33.8 |
| 16p11.2 | SEZ6L2 | 0.12 | 4.4 | 0.3 | 0.4 | 1.8 | 0.8 | 34.6 |
| 16p11.2 | TAOK2 | 1 | 6.0 | 0.13 | 0.2 | 3.2 | 0.7 | 27.9 |
| 7q34 | BRAF | 1 | 5.9 | 0.1 | 0.2 | 3.7 | 0.5 | 9 |
| 1q21.1 | CHD1L | 0 | −0.2 | 1.0 | 1.3 | −0.6 | 1.1 | 45.4 |
| 3q23 | XRN1 | 1 | 8.1 | 0.12 | 0.2 | 2.1 | 0.8 | 23.3 |
pLI; an estimate of the probablility of being loss-of-funtion intolerant. pLI closer to 1 indicates that the transcript cannot tolerate protein truncating variation.
LOF; loss of function, Z-score; A greater Z-score indicates more intolerane to the class of variation.
o/e; Observed over expected ratio of variants in the gene. When a gene has a low o/e, it is under strong selection for that class of variation.
LOEUF; loss-of-function observed/expected upper bound fraction.
%HI; haploinsufficiency rank. Genes with lower numbers are more likely to be dosage sensitive.
We also explored coding and splice variants with an allele frequency of <0.005. These variants were in nine genes; ARVCF, C16orf54, C22orf39, ESF1, GNB1L, KLHL22, MCTP2, MVP and TRMT2A (Supplementary Table S2). We did not find any potential pathogenic/likely pathogenic variants or candidate genes in this second analysis.
Furthermore, we evaluated the subjects in our study with the rare CNVs to look for additional rare variants in the specific genes corresponding to the regions of the deletion since a heterozygous deletion can also affect the function of a gene by unmasking a rare pathogenic variant on the other allele following recessive inheritance. No rare variants were found in these genes, thus excluding biallelic presence of damaging variants.
Discussion
CNVs are causing several diseases. However, in many cases it is challenging to determine the pathogenicity and clinical impact of the CNVs and to pinpoint the potential contribution of specific genes to the phenotype. In addition, the clinical relevance of rare CNVs can be difficult to determine because of incomplete penetrance and variable phenotypic expression. We performed targeted exome sequencing to identify rare variants in 117 genes located in previously identified CNV regions in subjects with severe early-onset obesity (Pettersson et al., 2017) in search for potential candidate genes. We found rare heterozygous variants with allele frequencies <0.1% in twelve of the CNV genes.
The 16p11.2 deletion is the most common CNV linked to severe obesity. However, the mechanism by which the proximal 16p11.2 deletion causes the phenotype is poorly understood and none of the genes in this region have so far been identified as a solely causative gene for the complex phenotype associated with proximal 16p11.2 CNVs. The patients with 16p11.2 deletions often present with various neurodevelopmental and neurobehavioral conditions, including autism spectrum disorders (ASD) and speech and language problems (Zufferey et al., 2012; Chung et al., 2021). The link between obesity and neurodevelopmental disorders may be due to common genes involved in the neurodevelopment that affect pathways regulating energy balance. Recently, new approaches for fine-mapping the genes in 16p11.2 region have been developed using GWAS and biobank data. Vysotskiy et al. performed an association analysis between imputed gene expression levels and BMI and they found four genes in the 16p11.2 region (TMEM219, SPN, TAOK2 and INO80E) associated with BMI (Vysotskiy et al., 2021). We identified rare heterozygous variants in four genes in the proximal 16p11.2 region; a frameshift variant in TAOK2 and heterozygous missense variants in SEZ6L2, ALDOA and KIF22. These four genes are potential candidate genes for the phenotype in patients with 16p11.2 CNVs. TAOK2 has a pLI of 1, suggesting rare loss-of-function variants may be deleterious (Lek et al., 2016). TAOK2 has been implicated in neurodevelopmental disorders and TAOK2 mutations found in patients with ASD showed impaired dendrite and synaptic development; the BMIs in the affected patients were not reported (Richter et al., 2019).TAOK2 heterozygous and knockout mice show gene brain morphological and behavioral abnormalities (Richter et al., 2019). SEZ6L2 has also been proposed as a candidate gene for ASD because of high expression in the brain (Kumar R. A. et al., 2009; Konyukh et al., 2011). Subcutaneous adipose tissue expression of ALDOA has been correlated with weight regain after dietary intervention (Roumans et al., 2017). ALDOA encodes an enzyme involved in glycolysis and the upregulation of glucose intake may be related to the risk of weight regain (Roumans et al., 2017). Furthermore, analysis of human fetal gene expression data found that KIF22 and ALDOA are significantly enriched in progenitors compared to post-mitotic cells making them candidates for having specific roles in neurogenesis in the developing human fetal cerebral cortex. It has been suggested that neurogenesis is disrupted in 16p11.2 CNVs (Morson et al., 2021). The participants in our study with rare variants in ALDOA, KIF22, TAOK2 and SEZ6L2 did not present with ASD or neurodevelopmental disorders (Table 1). However, the subjects with the missense variants in KIF22 and SEZ6L2 had learning difficulties. To our knowledge, no variants in these four genes have been reported in monogenic obesity. We consider the contribution of the identified rare variants to development of obesity unclear.
Two of the CNVs (16p11.2 deletion and 22q11.21 duplication) that we previously identified in our obese cohort include over 20 genes. In large CNVs, several genes within the locus may contribute to the phenotype. Disruption of multiple genes may be responsible for the complex phenotype seen in patients with 16p11.2 deletions, explaining the difficulty of identifying a single causative gene in the 16p11.2 region. Jensen et al. proposed that haploinsufficiency of certain genes may be altered by haploinsufficiency of other genes in the same CNV region and that these genes interact with each other via shared biological pathways (Jensen and Girirajan, 2019).
Interestingly, we identified heterozygous missense variants in SORCS1 in two subjects. Another study investigating genetic variants in 72 individuals with obesity also found two rare missense SORCS1 variants of unknown significance (Ginete et al., 2021). SORCS1 is highly expressed in the central nervous system and encodes a vacuolar protein sorting 10 domain-containing receptor that binds neuropeptides. SORSC1 has been associated with insulin secretion and diabetes risk (Goodarzi et al., 2007) and loss of both Sorcs1 and Sorcs3 in mutant mice caused increased adiposity and enhanced food intake, but not increased body weight (Subkhangulova et al., 2018). The participants in our study with the rare variants in SORSC1 did not present with insulin resistance or diabetes. In each family the variants were inherited from a parent with overweight, but overweight/obesity was present also in family members without the variant and therefore the contribution of these variants to the phenotype remains uncertain. More studies are needed to investigate the molecular mechanisms of SORCS1 and the possible contribution of this gene to early-onset obesity.
Another gene of interest is XRN1 located in 3q23 region. XRN1 is highly constrained for both loss-of-function and missense variants. This gene encodes 5′-3′ Exoribonuclease 1, which is involved in replication-dependent histone mRNA degradation. No XRN1 variants in humans have been associated with obesity, but a recent study investigating forebrain-specific Xrn1 knockout mice found that lack of Xrn1 in neurons led to obesity, hyperphagia, leptin resistance and hyperglycemia. The researchers proposed that Xrn1 in the hypothalamus is important for the mRNA degradation in regulating gene expression in the leptin signaling pathway (Takaoka et al., 2021). In our study, we identified a heterozygous missense variant in XRN1 of uncertain significance in a participant with isolated obesity. Further studies are required to explore the clinical relevance of this variant and biological function of XRN1.
Our study was limited by the lack of a control group of normal-weight subjects. However, we compared the allele frequencies of the variants we found in this study with the Finnish population in gnomAD and SISu project, with genotype data of more than 10,000 Finns. Because of the methodological limitation of targeted exome sequencing we may have missed variants in non-coding parts of the selected genes and variants in novel genes that were not included in the panel but may contribute to obesity. We may also have missed more common variants potentially contributing to the phenotype. Other limitations of this study were unavailability of parental DNA samples in part of the cohort and the lack of functional evaluation of the genetic variants. Further studies are needed to explore the possible role of the identified variants in the development of obesity.
In conclusion, the genes and mechanisms mediating the obesity phenotype in the previously identified CNVs remains uncertain and more studies are needed to provide insight to the potential contribution of the rare CNVs and the specific genes to severe obesity.
Acknowledgments
We thank all the study subjects and their family members for participating in the study. We thank RN Päivi Turunen for help with data collection.
Data Availability Statement
The datasets presented in this article are not readily available because Data cannot be shared publicly because the data consists sensitive patient data. More specifically the data consists of individual clinical data and individual genotypes for young children. Data are available from the Helsinki University Hospital’s Institutional Data Access/Ethics Committee for researchers who meet the criteria for access to confidential data. Requests to access the datasets should be directed to Outi Mäkitie MD, Ph.D., outi.makitie@helsinki.fi.
Ethics Statement
The studies involving human participants were reviewed and approved by the Research Ethics Committees of Hospital District of Helsinki and Uusimaa, the Pirkanmaa Hospital District and the Northern Ostrobothnia Hospital District. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
PL, TM, HV, MP, and OM: study design. PL, TM, HV, and PT: data collection. PL, MP, and OM: data analysis and data interpretation. PL, MP, and OM: drafting of manuscript. All authors contributed to manuscript revision and approved the final version.
Funding
Academy of Finland; Sigrid Jusélius Foundation; Foundation for Pediatric Research; Folkhälsan Research Foundation; Päivikki ja Sakari Sohlberg Foundation; Stiftelsen Dorothea Olivia, Karl Walter och Jarl Walter Perkléns minne; Finska Läkaresällskapet; Swedish Research Council; Novo Nordisk Foundation; University of Helsinki through the Doctoral Program in Clinical Research; Helsinki University Hospital research funds.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.839349/full#supplementary-material
References
- Bochukova E. G., Huang N., Keogh J., Henning E., Purmann C., Blaszczyk K., et al. (2010). Large, Rare Chromosomal Deletions Associated with Severe Early-Onset Obesity. Nature 463 (7281), 666–670. 10.1038/nature08689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho C. M. B., Lupski J. R. (2016). Mechanisms Underlying Structural Variant Formation in Genomic Disorders. Nat. Rev. Genet. 17 (4), 224–238. 10.1038/nrg.2015.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W. K., Roberts T. P., Sherr E. H., Snyder L. G., Spiro J. E. (2021). 16p11.2 Deletion Syndrome. Curr. Opin. Genet. Develop. 68, 49–56. 10.1016/j.gde.2021.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo C. S., Varela M. C., de Castro C. I. E., Otto P. A., Perez A. B. A., Lourenço C. M., et al. (2018). Chromosomal Microarray Analysis in the Genetic Evaluation of 279 Patients with Syndromic Obesity. Mol. Cytogenet. 11, 14. 10.1186/s13039-018-0363-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvignes J.-P., Bartoli M., Delague V., Krahn M., Miltgen M., Béroud C., et al. (2018). VarAFT: a Variant Annotation and Filtration System for Human Next Generation Sequencing Data. Nucleic Acids Res. 46 (W1), W545–w553. 10.1093/nar/gky471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginete C., Serrasqueiro B., Silva-Nunes J., Veiga L., Brito M. (2021). Identification of Genetic Variants in 65 Obesity Related Genes in a Cohort of Portuguese Obese Individuals. Genes 12 (4), 603. 10.3390/genes12040603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi M. O., Lehman D. M., Taylor K. D., Guo X., Cui J., Quiñones M. J., et al. (2007). SORCS1: a Novel Human Type 2 Diabetes Susceptibility Gene Suggested by the Mouse. Diabetes 56 (7), 1922–1929. 10.2337/db06-1677 [DOI] [PubMed] [Google Scholar]
- Gu W., Zhang F., Lupski J. R. (2008). Mechanisms for Human Genomic Rearrangements. Pathogenetics 1 (1), 4. 10.1186/1755-8417-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N., Lee I., Marcotte E. M., Hurles M. E. (2010). Characterising and Predicting Haploinsufficiency in the Human Genome. Plos Genet. 6 (10), e1001154. 10.1371/journal.pgen.1001154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis N. M., Rothstein J. H., Pejaver V., Middha S., McDonnell S. K., Baheti S., et al. (2016). REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 99 (4), 877–885. 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M., Girirajan S. (2019). An Interaction-Based Model for Neuropsychiatric Features of Copy-Number Variants. Plos Genet. 15 (1), e1007879. 10.1371/journal.pgen.1007879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konyukh M., Delorme R., Chaste P., Leblond C., Lemière N., Nygren G., et al. (2011). Variations of the Candidate SEZ6L2 Gene on Chromosome 16p11.2 in Patients with Autism Spectrum Disorders and in Human Populations. PLoS One 6 (3), e17289. 10.1371/journal.pone.0017289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopanos C., Tsiolkas V., Kouris A., Chapple C. E., Albarca Aguilera M., Meyer R., et al. (2019). VarSome: the Human Genomic Variant Search Engine. Bioinformatics 35 (11), 1978–1980. 10.1093/bioinformatics/bty897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P., Henikoff S., Ng P. C. (2009a). Predicting the Effects of Coding Non-synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 4 (7), 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Kumar R. A., Marshall C. R., Badner J. A., Babatz T. D., Mukamel Z., Aldinger K. A., et al. (2009b). Association and Mutation Analyses of 16p11.2 Autism Candidate Genes. PLoS One 4 (2), e4582. 10.1371/journal.pone.0004582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., et al. (2016). Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 536 (7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loid P., Mustila T., Mäkitie R. E., Viljakainen H., Kämpe A., Tossavainen P., et al. (2020). Rare Variants in Genes Linked to Appetite Control and Hypothalamic Development in Early-Onset Severe Obesity. Front. Endocrinol. 11, 81. 10.3389/fendo.2020.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo S. F., Stanghellini I., Predieri B., Ciancia S., Leo F., Bruzzi P., et al. (2021). Copy Number Variation Analysis Increases the Number of Candidate Loci Associated with Pediatric Obesity. Horm. Res. Paediatr. 94, 251–262. 10.1159/000519299 [DOI] [PubMed] [Google Scholar]
- Morson S., Yang Y., Price D. J., Pratt T. (2021). Expression of Genes in the 16p11.2 Locus during Development of the Human Fetal Cerebral Cortex. Cereb. Cortex 31 (9), 4038–4052. 10.1093/cercor/bhab067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowska B. (2017). Clinical Interpretation of Copy Number Variants in the Human Genome. J. Appl. Genet. 58 (4), 449–457. 10.1007/s13353-017-0407-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson M., Viljakainen H., Loid P., Mustila T., Pekkinen M., Armenio M., et al. (2017). Copy Number Variants Are Enriched in Individuals with Early-Onset Obesity and Highlight Novel Pathogenic Pathways. J. Clin. Endocrinol. Metab. 102 (8), 3029–3039. 10.1210/jc.2017-00565 [DOI] [PubMed] [Google Scholar]
- Rice A. M., McLysaght A. (2017). Dosage Sensitivity Is a Major Determinant of Human Copy Number Variant Pathogenicity. Nat. Commun. 8, 14366. 10.1038/ncomms14366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M., Murtaza N., Scharrenberg R., White S. H., Johanns O., Walker S., et al. (2019). Altered TAOK2 Activity Causes Autism-Related Neurodevelopmental and Cognitive Abnormalities through RhoA Signaling. Mol. Psychiatry 24 (9), 1329–1350. 10.1038/s41380-018-0025-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumans N. J. T., Vink R. G., Bouwman F. G., Fazelzadeh P., van Baak M. A., Mariman E. C. M. (2017). Weight Loss-Induced Cellular Stress in Subcutaneous Adipose Tissue and the Risk for Weight Regain in Overweight and Obese Adults. Int. J. Obes. 41 (6), 894–901. 10.1038/ijo.2016.221 [DOI] [PubMed] [Google Scholar]
- Saari A., Sankilampi U., Hannila M.-L., Kiviniemi V., Kesseli K., Dunkel L. (2011). New Finnish Growth References for Children and Adolescents Aged 0 to 20 years: Length/height-For-Age, Weight-For-Length/height, and Body Mass index-for-age. Ann. Med. 43 (3), 235–248. 10.3109/07853890.2010.515603 [DOI] [PubMed] [Google Scholar]
- Saeed S., Arslan M., Manzoor J., Din S. M., Janjua Q. M., Ayesha H., et al. (2020). Genetic Causes of Severe Childhood Obesity: A Remarkably High Prevalence in an Inbred Population of Pakistan. Diabetes 69 (7), 1424–1438. 10.2337/db19-1238 [DOI] [PubMed] [Google Scholar]
- Schwarz J. M., Cooper D. N., Schuelke M., Seelow D. (2014). MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 11 (4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Selvanayagam T., Walker S., Gazzellone M. J., Kellam B., Cytrynbaum C., Stavropoulos D. J., et al. (2018). Genome-wide Copy Number Variation Analysis Identifies Novel Candidate Loci Associated with Pediatric Obesity. Eur. J. Hum. Genet. 26 (11), 1588–1596. 10.1038/s41431-018-0189-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subkhangulova A., Malik A. R., Hermey G., Popp O., Dittmar G., Rathjen T., et al. (2018). SORCS 1 and SORCS 3 Control Energy Balance and Orexigenic Peptide Production. EMBO Rep. 19 (4), e44810. 10.15252/embr.201744810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka S., Yanagiya A., Mohamed H. M. A., Higa R., Abe T., Inoue K. I., et al. (2021). Neuronal XRN1 is Required for Maintenance of Whole-Body Metabolic Homeostasis. iScience 24 (10), 103151. 10.1016/j.isci.2021.103151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillaume M.-L., Naudion S., Banneau G., Diene G., Cartault A., Cailley D., et al. (2014). New Candidate Loci Identified by Array-CGH in a Cohort of 100 Children Presenting with Syndromic Obesity. Am. J. Med. Genet. 164 (8), 1965–1975. 10.1002/ajmg.a.36587 [DOI] [PubMed] [Google Scholar]
- Vysotskiy M., Zhong X., Miller-Fleming T. W., Zhou D., Cox N. J., Weiss L. A. (2021). Integration of Genetic, Transcriptomic, and Clinical Data Provides Insight into 16p11.2 and 22q11.2 CNV Genes. Genome Med. 13 (1), 172. 10.1186/s13073-021-00972-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters R. G., Jacquemont S., Valsesia A., de Smith A. J., Martinet D., Andersson J., et al. (2010). A New Highly Penetrant Form of Obesity Due to Deletions on Chromosome 16p11.2. Nature 463 (7281), 671–675. 10.1038/nature08727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Li W.-D., Glessner J. T., Grant S. F. A., Hakonarson H., Price R. A. (2010). Large Copy-Number Variations Are Enriched in Cases with Moderate to Extreme Obesity. Diabetes 59 (10), 2690–2694. 10.2337/db10-0192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler E., Huang N., Bochukova E. G., Keogh J. M., Lindsay S., Garg S., et al. (2013). Genome-wide SNP and CNV Analysis Identifies Common and Low-Frequency Variants Associated with Severe Early-Onset Obesity. Nat. Genet. 45 (5), 513–517. 10.1038/ng.2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrei M., MacDonald J. R., Merico D., Scherer S. W. (2015). A Copy Number Variation Map of the Human Genome. Nat. Rev. Genet. 16 (3), 172–183. 10.1038/nrg3871 [DOI] [PubMed] [Google Scholar]
- Zufferey F., Sherr E. H., Beckmann N. D., Hanson E., Maillard A. M., Hippolyte L., et al. (2012). A 600 Kb Deletion Syndrome at 16p11.2 Leads to Energy Imbalance and Neuropsychiatric Disorders. J. Med. Genet. 49 (10), 660–668. 10.1136/jmedgenet-2012-101203 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in this article are not readily available because Data cannot be shared publicly because the data consists sensitive patient data. More specifically the data consists of individual clinical data and individual genotypes for young children. Data are available from the Helsinki University Hospital’s Institutional Data Access/Ethics Committee for researchers who meet the criteria for access to confidential data. Requests to access the datasets should be directed to Outi Mäkitie MD, Ph.D., outi.makitie@helsinki.fi.