

Abstract
Rheumatoid arthritis (RA) is a common autoimmune disease that causes inflammation of the joints and damage to the cartilage and bone. The pathogenesis of RA is characterized in many patients by the presence of antibodies against citrullinated proteins. Proteoglycans are key structural elements of extracellular matrix in the joint articular cartilage and synovium and are secreted as lubricants in the synovial fluid. Alterations of proteoglycans contribute to RA pathogenesis. Proteoglycans such as aggrecan can be citrullinated and become potential targets of the rheumatoid autoimmune response. Proteoglycans are also upregulated in RA joints and/or undergo alterations of their regulatory functions over cytokines and chemokines, which promotes inflammation and bone damage. Recent studies have aimed to not only clarify these mechanisms but also develop novel proteoglycan-modulating therapeutics. These include agents altering the function and signaling of proteoglycans as well as tolerizing agents targeting citrullinated aggrecan. This mini-review summarizes the most recent findings regarding the dysregulation of proteoglycans that contributes to RA pathogenesis and the potential for proteoglycan-modulating agents to improve upon current RA therapy.
Keywords: aggrecan, citrullination, proteoglycans, rheumatoid arthritis, syndecan
INTRODUCTION
Rheumatoid arthritis (RA) is characterized by inflammation of the synovium, or joint lining tissue, which manifests with joint pain, swelling, stiffness, and progressive cartilage damage and bone erosions, which can lead to permanent deformity and functional limitation (1). RA is the most common systemic autoimmune disease and is estimated to affect ∼0.5% of the global world population. It can occur in all age groups, though the most prevalent incidence occurs in women aged 50–60 yr.
The etiology and pathogenesis of RA have been summarized in authoritative reviews and rely on a complex interaction between key genetic risk variants such as HLA-DRB1*04:01 (HLA-DR4) and a variety of environmental triggers such as smoking (2). In a subset of patients, these triggers can cause protein citrullination, which leads to “altered self” recognition by the immune system and the development of anticitrullinated protein antibodies (ACPAs). ACPAs have many pathogenic actions including activating T cells, which enhances the autoimmune response and triggers innate immune cell (e.g., macrophages and neutrophils)-driven joint inflammation. Adaptive and innate cells infiltrating the synovium release vasoactive agents, chemokines, and proinflammatory cytokines including tumor necrosis factor-α (TNF), interleukin (IL)-1, and IL-6 (Fig. 1), which together amplify immune infiltration. The inflamed RA synovium (also called “pannus”) displays an upregulation of vascular endothelial growth factor, promoting extensive neoangiogenesis. In addition, there is an activation and expansion of synovial fibroblasts (SF), local fibroblast-like cells that display abnormal migration and proliferation, loss of contact inhibition, and production of pathogenic extracellular matrix (ECM)-digesting enzymes. Rheumatoid SF also produce large amounts of receptor activator of nuclear factor-κB ligand (RANKL), a key stimulator of osteoclastogenesis, which ultimately promotes bone erosion (3).
Figure 1.
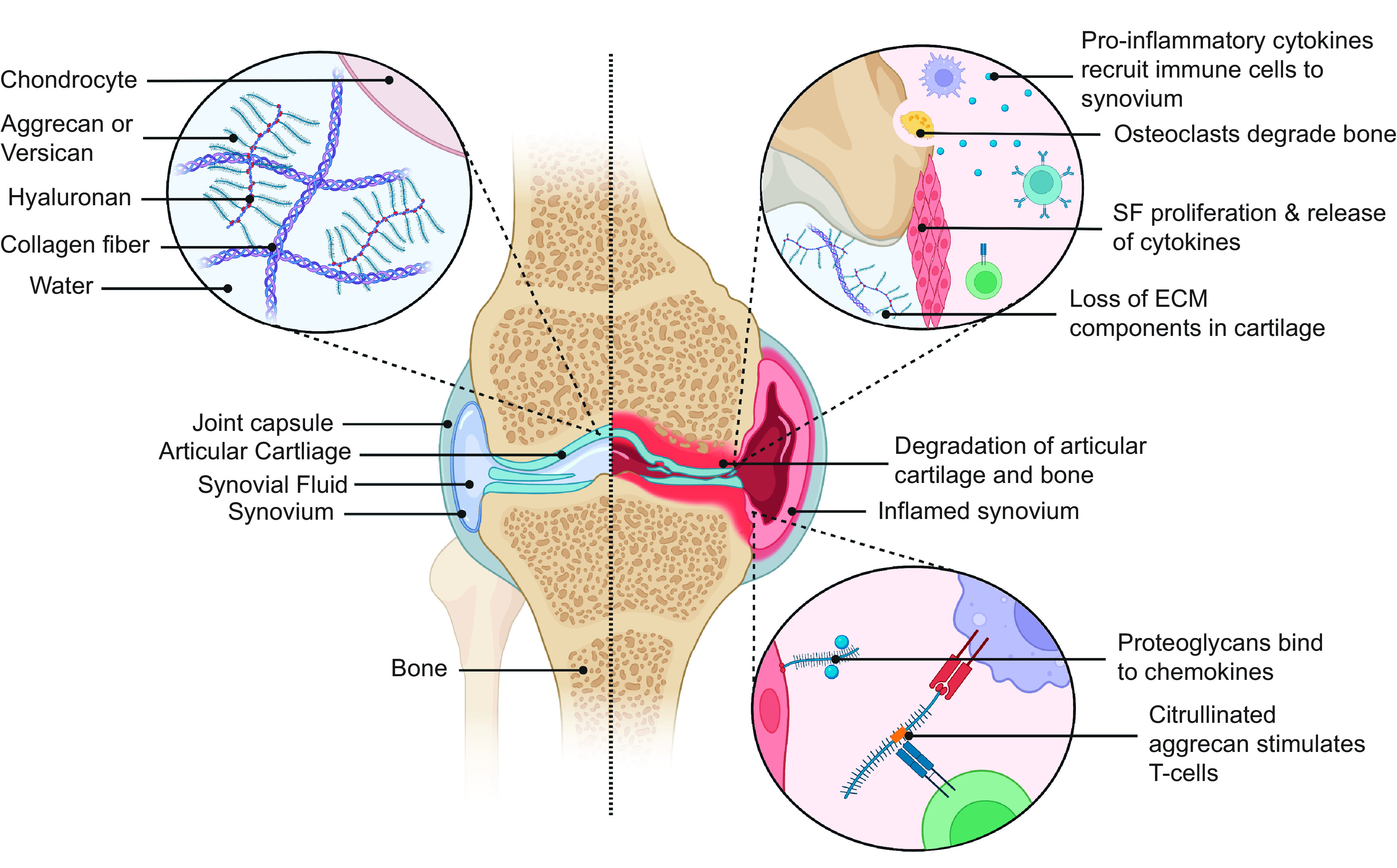
Proteoglycans contribute to adaptive and innate immune responses in RA. Proteoglycans are prevalent in normal articular cartilage where they interact with collagen fibers and hyaluronan creating a negatively charged aggregate that is highly hydrophilic and important for the biomechanical properties of the joint (left). In RA, aggrecan is degraded, proinflammatory cytokines bind proteoglycans and cause the trafficking of immune cells to the joint, and citrullinated aggrecan (citrullination shown in orange) is presented by antigen-presenting cells and activates T cells (right). (Created with Biorender.com). RA, rheumatoid arthritis; ECM, extracellular matrix; SF, synovial fibroblasts.
Given the autoimmune etiology of RA, immune-targeting interventions have been the cornerstone of RA therapy for decades. The emergence in the past 20 years of novel disease-modifying antirheumatic agents (DMARDs) such as biologics targeting TNF has dramatically improved the prognosis of RA. However, a large unmet need remains in the field since >30% of patients do not achieve remission on current regimens (4). Importantly, all the current DMARDs target immune cells and mechanisms that are ubiquitous, and many DMARDs cause immunosuppression to some degree with a heightened risk of serious infections. Thus, there is a renewed appreciation in the field of the importance of understanding and targeting joint-localized determinants of disease, especially the joint ECM and factors that influence the interaction of immune cells with the joint ECM.
The joint ECM is proteoglycan (PG)-rich. Chondrocytes make up only 2% of the volume of joint cartilage while the majority of the volume is occupied by water trapped by a hydrophilic web of PGs, collagens, and hyaluronan (Fig. 1) (5). This water-dense structure of cartilage ECM provides terrific resilience and enables friction-less operation of the joint. PGs can also be found on the surface of most cells in the synovium and in the synovial fluid, where they are important joint lubricant factors. Since PGs are such important components of the joint ECM, several studies have focused on the potential dysregulation of PGs homeostasis in RA and/or on PG-regulating enzymes as pathogenic players and therapeutic targets for RA. Several members of the key-ECM regulating matrix metalloproteinase (MMP) (6) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) (7) enzyme families are involved in the turnover of PGs. Despite the early disappointment with MMP inhibitors in early clinical trials, there has been a recent revival of interest in MMPs and other PG-regulating enzymes for RA therapy. The research on the expression and role of MMPs, ADAMTS, and other proteases in RA has been excellently summarized elsewhere (for example, see Ref. 8). Thus, the present review will focus specifically on recent reports documenting the roles PGs play in RA and the potential for novel RA therapeutics that directly target PGs.
All PGs share a core structure consisting of a long peptide with at least one glycosaminoglycan (GAG) chain. The structural and functional diversity of PGs has been reviewed elsewhere in this issue and will not be detailed beyond the scheme in Fig. 2 showing the PGs reviewed here.
Figure 2.
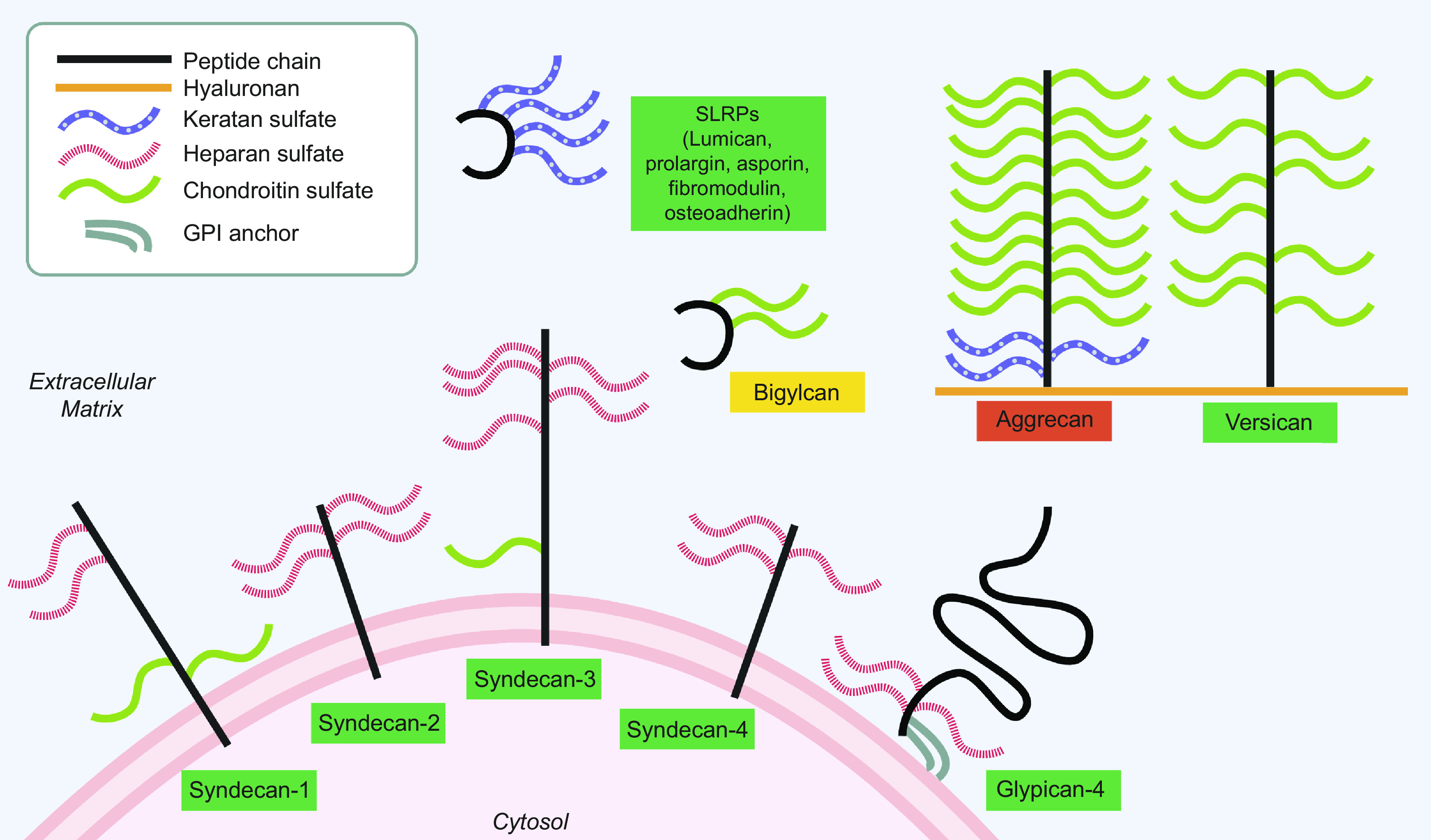
Structure of proteoglycans implicated in RA and their subcellular location. The legend is located in the upper left-hand box. Upregulated in RA (green), downregulated (red), unknown, or no change (yellow). PGs differ by type and number of glycosaminoglycan chains. Aggrecan and versican are usually found bound to a hyaluronan backbone. SLRPs share a similar structure with 2-7 keratan sulfate chains. Membrane-bound PGs can be shed and consequently also found in the ECM. (Created with Biorender.com). ECM, extracellular matrix; PG, proteoglycan; RA, rheumatoid arthritis; SLRP, small leucine-rich proteoglycan; GPI, glycophosphatidylinositol.
AGGRECAN
The chondroitin sulfate PG aggrecan (Agg) is the most prevalent PG in cartilage. It has three globular domains (G1, G2, and G3) and a GAG-enriched region between G2 and G3. The G1 domain mediates noncovalent interactions with free GAGs, hyaluronic acid, and the glycoprotein link protein, which lead to the formation of large aggregates that in turn organize in a stiff network with collagen (Fig. 1) (9). In RA, Agg found in the articular cartilage is degraded by MMPs and aggrecanases leaving degradation-derived neoantigens in RA cartilage (10). A decrease in Agg paired with an increase in aggrecanase-generated fragments is also found in the serum of patients with RA (11). A recent study of acetazolamide as a potential therapeutic in rats with adjuvant-induced arthritis (AA), a model where arthritis is induced by exogenously injected inactivated mycobacteria, found increased Agg expression in cartilage, which was used as a readout correlating to reduced arthritis severity (12). An increase in Agg staining of cartilage paired with a reduction of arthritis clinical score in AA rat’s knees also pointed to the therapeutic potential of a nanofiber sheet implanted into the knee containing mesenchymal stem cells (MSCs) pretreated with soluble IL-6R (13). Another study employed Agg fragment neoepitope immunostaining of mouse knee joint cartilage to assess the potential of mesenchymal stem cell (MSC) media as an RA therapeutic. In the study, MSC media administered via intra-articular injections reduced joint aggrecan degradation in mice with antigen-induced arthritis (AIA), a model where arthritis is induced by immunization with Bordetella pertussis toxin (14).
Injections of Agg or recombinant Agg G1 domain are able to induce arthritis in Balb/c mice in the PG-induced arthritis model (PGIA) (15) and early studies showed that Agg could be an autoantigen in RA (16). In 2010, it was documented that patients with RA uniquely carry proinflammatory T cells that responded to a specific citrullinated Agg (cit-Agg) peptide that encompassed aa84-103 and was citrullinated at position 93 (17). More recently, Rims et al. (18) generated Agg peptides citrullinated at other positions using a scanning algorithm to identify likely HLA-DR4-binding motifs. Ultimately, six cit-Agg peptides were identified that could bind to recombinant biotinylated HLA-DR4 tetramers and induce T cell expansion in an ex vivo assay. A higher percentage of peripheral blood CD4+ T cells from patients with RA than from healthy subjects were stained by tetramers specific for two of the cit-Agg peptides, and the frequency of cit-Agg tetramer-positive CD4+ T cells correlated with serum levels of antibodies against the Agg G1 domain in these patients (18). Becart et al. (19) used peptides from several known citrullinated RA autoantigens, including the aforementioned Agg aa84-103 peptide, to elucidate the relationship between HLA-DR affinity and immunogenicity of citrullinated peptides. However, in a collagen-competition-binding assay, no differences were found between the arg- and cit-Agg peptides for binding to HLA-DRB1*01:01 (HLA-DR1), and neither peptide could outcompete collagen for binding to HLA-DR4. Humanized DR1 or DR4-bearing mice were subsequently immunized with arg- or cit-Agg and after 10 days, T cells from draining lymph nodes were challenged with arg- or cit-Agg. Intriguingly, in both mouse models, T cell proliferation was the highest in the cit-Agg immunized group and cit-Agg responsive cells cross-recognized arg-Agg, albeit with reduced proliferation. When cytokine levels were assessed from supernatants from the T cell proliferation assays, both arg- and cit-Agg induced the production of inflammatory cytokines including interferon (IFN)γ and IL-6 in cit-Agg immunized mice. These results suggest that while there is no clear relationship between HLA-binding affinity and immunogenicity of cit-Agg, cit-Agg can induce a cross-reactive response against endogenous Agg, which might help perpetuate inflammation in RA. Extending this type of analysis in humanized mice to additional predicted or observed citrullination sites of Agg would be desirable and potentially useful to design personalized protocols to induce tolerance to autoantigens for nonimmunosuppressive RA treatments. Another team scanned candidate citrullinated peptides from the G1 domain of Agg for their immunogenicity in Balb/c mice and ex vivo using cells from patients with RA. They found a peptide encompassing aa294-308 citrullinated at position 305 that caused a greater release of proinflammatory cytokines such as IFNγ, IL-6, and IL-17 than its arg counterpart in Balb/c mice. When RA and healthy human peripheral blood mononuclear cell (PBMC) cultures were treated with the citrullinated peptide, a greater release of the same proinflammatory cytokines was observed from RA PBMC (20). A tolerizing vaccine was then designed using an approach called Ligand Epitope Antigen Presenting System (LEAPS) where the citrullinated peptide was conjugated with a peptide derived from the β-chain of human major histocompatibility complex II that acts as an immune cell-binding ligand. Vaccinated PGIA mice displayed a promising amelioration of disease correlating with a higher ratio of anti-inflammatory to proinflammatory cytokines produced by spleen cells (21).
SYNDECANS
The four mammalian syndecans: syndecan-1, syndecan-2, syndecan-3, and syndecan-4 are transmembrane PGs that interact with a variety of ligands and are major players in inflammation in part due to their ability to bind chemokines that regulate leukocyte extravasation (22). This multitude of interactions implicates syndecans in a variety of different cell processes including migration, proliferation, differentiation, adhesion, and angiogenesis. Although syndecan-1 and syndecan-3 have both heparan sulfate and chondroitin sulfate chains, the syndecans predominantly function as heparan sulfate PGs (Fig. 2) (23). In addition to being transmembrane, the ectodomain of syndecans can be shed as a result of cleavage by zinc-dependent endopeptidases and MMPs, and thus syndecans may exist as soluble proteoglycans in the ECM (23).
Over-expression of syndecan-1 was observed in mononuclear immune cells within human RA synovial tissue (24). Syndecan-1 promotes the binding of IL-34 to macrophage colony-stimulating factor receptor (M-CSFR) on myeloid cells (25). IL-34 is elevated in RA serum and was shown to induce and promote progressive inflammation in mice in a syndecan-1-dependent manner (26). An experiment using syndecan-1 knockout (KO) mice showed that syndecan-1 is necessary not only for IL-34-induced joint inflammation but also for osteoclast differentiation. IL-34-induced osteoclast differentiation occurs via the RANK pathway and RANK expression is partially reduced in bone marrow-derived macrophages from syndecan-1 KO mice through a mechanism that is not completely understood (26). Soluble syndecan-1, which is shed by MMP-9, is also found in the serum of patients with RA and following 6 wk treatment with either methotrexate or TNF inhibitors, the methotrexate group had a significant reduction in syndecan-1 levels (27). Thus, syndecan-1 could play a pathogenic role in RA inflammation and bone damage and its expression/shedding correlate with response to DMARD treatment.
Syndecan-2 can be found over-expressed on endothelial cells, pericytes, and smooth muscle cells in RA synovium blood vessels (24). In 2014, a research group linked syndecan-2 shed by mesenchymal stem cells to angiogenesis and found shed syndecan-2 to inhibit endothelial cell migration and inflammation (28). Since angiogenesis is a key feature of RA, there are possible links between syndecan-2 and RA that have yet to be explored.
Endothelial syndecan-3 is hypothesized to bind chemokines such as chemokine C-X-C ligand-1 and ligand-8 (CXCL1 and CXCL8) contributing to their presentation to leukocyte receptors, which promotes leukocyte recruitment and cartilage damage in RA (29). Using a comparison of WT and syndecan-3 KO mice either intra-articularly or intradermally injected with CXCL1, syndecan-3 was found to have a proinflammatory role in the joint and an anti-inflammatory role in the skin of mice (29). Recent studies have shown that the addition of soluble syndecan-3 reduced chemokine-induced leukocyte migration in vitro. Administration of soluble syndecan-3 in vivo reduced RA disease severity in mice with AIA and collagen-induced arthritis (CIA, a model where arthritis is induced by immunization with collagen), by binding to chemokines and preventing them from triggering their receptors on leukocytes. Patients with RA were also found to have greater amounts of shed syndecan-3 in their sera, indicating a possible feedback attempt to downregulate excessive inflammation (30). In a related study, McNaughton et al. (31) synthesized peptides encompassing the GAG-binding regions of chemokines CCL5, CXCL8, and CXCL12γ, hypothesizing they could competitively bind to syndecan-3 and inhibit chemokine interaction. After verifying the ability of a lead peptide, pCXCL8-1, to bind to syndecan-3, and compete with CXCL8 for binding to immortalized human bone marrow endothelial cells, they showed that treatment of AIA mice with pCXCL8-1 decreased the concentration of serum TNF and recruitment of neutrophils to the inflamed synovium (31). Another study linking syndecan-3 to RA built off of a 2014 finding where intra-articular injection of MSCs was shown to improve disease in AIA (32). Since syndecan-3 is expressed on MSCs and involved in establishing the stromal “niche” required for maintaining and generating hemopoietic cells (33), the researchers sought to understand whether MSCs syndecan-3 influences the ability of MSCs to ameliorate inflammatory arthritis. They found that syndecan-3 KO MSCs had more marked anti-inflammatory effects than WT MSCs. Mechanistically, syndecan-3 KO MSCs showed increased MSC adhesion to collagen, which promoted MSC cell proliferation and survival, likely via regulation of the AKT signaling pathway (34). Put together, these findings make syndecan-3 a target worth more investigation for potential novel RA therapies.
Syndecan-4 KO is disease protective in mouse CIA through a mechanism partially mediated by the promotion of B cell migration and formation of germinal centers (35). However, syndecan-4 also promotes arthritis pathogenesis by acting on SF. Cai et al. (36) found that syndecan-4 silencing in human RA SF inhibited a wide range of pathogenic SF phenotypes including levels of reactive oxygen species, and nitric oxide (both known to correlate with the proinflammatory effect of SF in RA), the expression of proinflammatory cytokines by SF, and SF apoptosis. In SF, syndecan-4 dimerization was also found to be important for caveolin vesicle-mediated trafficking of the interleukin-1 receptor type 1 (IL1R1) receptor, which determines the sensitivity of fibroblasts to IL-1, a key proarthritogenic cytokine. Consistent with these findings, an antibody that prevented the dimerization of syndecan-4 was found to reduce arthritis severity in the human TNF transgenic mouse model of RA (37). Our team has focused on syndecan-4-mediated regulation of protein tyrosine phosphatase receptor type S (PTPRS). PTPRS is inhibited by heparan sulfate PGs and activated by chondroitin sulfate PGs, through a mechanism termed the “PG switch” (38). In mouse and RA SF, syndecan-4 inhibits PTPRS activity through oligomerization of the phosphatase which allows proinflammatory platelet-derived growth factor (PDGF) signaling to proceed unabated. However, treatment with a decoy peptide that occupies the PTPRS-binding site of syndecan-4 flips the PG switch, allowing PTPRS to remain active and inhibit SF migration. The syndecan-4 binding decoy peptide effectively reduced SF migration in vitro and its injection in mice ameliorated serum transfer-induced arthritis (STIA, a model induced by injection of serum from a mouse that expresses a transgenic T cell receptor (TCR) that recognizes the cartilage antigen glucose-6-phosphate isomerase) (39). More recently, we also showed that this treatment could synergize with a TNF inhibitor in significantly ameliorating mouse CIA (40).
ASPORIN
Asporin is a small leucine-rich PG (SLRP), which is lowly expressed in adult cartilage and might play a role in osteoarthritis (OA) where it is hypothesized to inhibit transforming growth factor-β signaling and bind to collagen causing collagen mineralization (41, 42). A D-repeat genetic polymorphism of asporin has been studied in both OA and RA and in RA it displays an association with disease variability rather than incidence (43). One recent study in rat CIA focused on the mechanism of the medicinal effects of paeoniflorin, the active ingredient of white peony root (Radix Paeoniae Alba), an anti-inflammatory herb used in Chinese traditional medicine to treat RA. Quantitative proteomics performed on synovial tissue of animals treated with 1 mg/kg paeoniflorin showed that asporin expression was significantly reduced, correlating with a reduction of arthritis score and serum levels of TNF and IL-1 (44).
PROLARGIN AND BIGLYCAN
Another SLRP, prolargin, has also been examined for a potential role in RA. Prolargin is involved in the anchoring of basement membranes to connective tissues (45). Protein gels paired with mass spectrometry were used to identify differentially expressed proteins in RA and OA human articular cartilage, and prolargin levels were found to be higher in RA extracts than in OA extracts (46). Due to the known role of prolargin in controlling RANK-dependent osteoclastogenesis, the authors speculate that prolargin might play a role in the pathogenesis of bone erosions in RA. However, the absence of a healthy control in this study makes it difficult to understand if prolargin is upregulated in RA, downregulated in OA, or both. The levels of another SLRP, biglycan, were also assessed in this study, but there was no significant difference between RA and OA samples (46). However, previous reports identified the presence of anti-biglycan antibodies in RA that have been reviewed elsewhere (47).
OTHER PGs
Other PGs for which data suggest a potential role in RA but have not been published recently include osteoadherin, fibromodulin, glypicans, versican, and lumican. Osteoadherin and fibromodulin are able to activate the complement pathway via C1q binding in human sera, which could potentially promote joint inflammation given the critical importance of complement in RA pathogenesis (48). Glypicans are a group of six PGs anchored to the cell membrane by glycosylphosphatidylinositol (GPI) anchors that are widely expressed in tissues and play a role in growth and regeneration. Glypican-4 expression is higher in the synovial lining layer of RA versus healthy subjects and is also expressed in the rheumatoid endothelium (24). Versican is another PG involved in many different cellular processes such as wound healing and blood vessel formation (49) whose mRNA is over-expressed in proliferating RA SF (50) and also in peripheral blood mononuclear cells of CIA rats (51). Lumican, a PG well-known for promoting recruitment of macrophages in corneal wound healing, is also upregulated in human RA synovial fluid and can also be citrullinated thus constituting a potential target for ACPAs (52–54).
CONCLUSIONS AND FUTURE DIRECTIONS
PGs high prevalence in the joint and their complex, multifaceted roles render them important and interesting players in RA. This review included a summary of what is currently known regarding how dysregulation of PGs can be causal or serve as a marker for RA and recent studies of potential PG modulating therapeutics (summarized in Fig. 3). Recent studies have shown that syndecan-3 not only has proinflammatory versus anti-inflammatory effects in joints versus skin but also could play a pro- or anti-inflammatory role in its transmembrane versus shed forms (29, 30). It remains unknown whether other PGs display similar functional differences depending on tissue or subcellular locations. Other studies have linked PGs to signaling pathways that are important for inflammation or tissue damage in RA, e.g., syndecan-1 plays a role in IL-34-induced osteoclasts differentiation (26), but more work is warranted to clarify PG ligands and intracellular pathways that are relevant for these functions. Deeper knowledge about the role of PGs in RA would be desirable and could guide the development of new PG-based therapeutics including PG-targeted peptide mimetics or antibodies and tolerizing agents based on citrullinated-proteoglycan epitopes.
Figure 3.

Current therapeutic approaches to modulate PG activity. (Created with Biorender.com). PG, proteoglycan. GAG, glycosaminoglycan; PTPRS, protein tyrosine phosphatase receptor type S; cit-Agg, citrullinated aggrcan; IL1R1, interleukin-1 receptor type 1.
GRANTS
B.H. was supported in part by the University of California-San Diego (UCSD) Graduate Training Program in Cellular and Molecular Pharmacology through an institutional training grant from the National Institute of General Medical Sciences, T32GM007752. N.B. was supported in part by National Institute of Arthritis and Musculoskeletal and Skin Diseases Grants R01AR066053 and P30AR073761.
DISCLOSURES
N.B. has a relationship with Knoubis Bio, Inc., which consists of being a scientific founder with equity and interim officer of the company. The terms of this arrangement have been reviewed and approved by the University of California-San Diego in accordance with its conflict of interest policies.
This article is part of the special collection “Deciphering the Role of Proteoglycans and Glycosaminoglycans in Health and Disease.” Liliana Schaefer, MD, served as Guest Editor of this collection.
AUTHOR CONTRIBUTIONS
N.B. conceived and designed research; B.H. prepared figures; B.H. drafted manuscript; B.H. and N.B. edited and revised manuscript; and B.H. and N.B. approved final version of manuscript.
REFERENCES
- 1.Sparks JA. Rheumatoid arthritis. Ann Intern Med 170: ITC1–ITC16, 2019. doi: 10.7326/AITC201901010. [DOI] [PubMed] [Google Scholar]
- 2.Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res 6: 15, 2018. doi: 10.1038/s41413-018-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim K-W, Cho M-L, Lee S-H, Oh H-J, Kang C-M, Ju JH, Min S-Y, Cho Y-G, Park S-H, Kim H-Y. Human rheumatoid synovial fibroblasts promote osteoclastogenic activity by activating RANKL via TLR-2 and TLR-4 activation. Immunol Lett 110: 54–64, 2007. doi: 10.1016/j.imlet.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Feldmann M, Maini RN. Perspectives from masters in rheumatology and autoimmunity: can we get closer to a cure for rheumatoid arthritis? Arthritis Rheumatol 67: 2283–2291, 2015. doi: 10.1002/art.39269. [DOI] [PubMed] [Google Scholar]
- 5.Lohmander S. Proteoglycans of joint cartilage. Structure, function, turnover and role as markers of joint disease. Baillière's Clin Rheumatol 2: 37–62, 1988. doi: 10.1016/S0950-3579(88)80004-9. [DOI] [PubMed] [Google Scholar]
- 6.Murphy G, Knäuper V, Atkinson S, Butler G, English W, Hutton M, Stracke J, Clark I. Matrix metalloproteinases in arthritic disease. Arthritis Res 4, Suppl 3: S39–S49, 2002. doi: 10.1186/ar572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelwick R, Desanlis I, Wheeler GN, Edwards DR. The ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol 16: 113, 2015. doi: 10.1186/s13059-015-0676-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itoh Y. Metalloproteinases in rheumatoid arthritis: potential therapeutic targets to improve current therapies. Prog Mol Biol Transl Sci 148: 327–338, 2017. doi: 10.1016/bs.pmbts.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Kiani C, Chen L, Wu YJ, Yee AJ, Yang BB. Structure and function of aggrecan. Cell Res 12: 19–32, 2002. doi: 10.1038/sj.cr.7290106. [DOI] [PubMed] [Google Scholar]
- 10.Lark MW, Bayne EK, Flanagan J, Harper CF, Hoerrner LA, Hutchinson NI, Singer II, Donatelli SA, Weidner JR, Williams HR, Mumford RA, Lohmander LS. Aggrecan degradation in human cartilage. Evidence for both matrix metalloproteinase and aggrecanase activity in normal, osteoarthritic, and rheumatoid joints. J Clin Invest 100: 93–106, 1997. doi: 10.1172/JCI119526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rousseau JC, Sumer EU, Hein G, Sondergaard BC, Madsen SH, Pedersen C, Neumann T, Mueller A, Qvist P, Delmas P, Karsdal MA. Patients with rheumatoid arthritis have an altered circulatory aggrecan profile. BMC Musculoskelet Disord 9: 74, 2008. doi: 10.1186/1471-2474-9-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai L, Chen W-N, Li R, Hu C-M, Lei C, Li C-M. Therapeutic effect of acetazolamide, an aquaporin 1 inhibitor, on adjuvant-induced arthritis in rats by inhibiting NF-κB signal pathway. Immunopharmacol Immunotoxicol 40: 117–125, 2018. doi: 10.1080/08923973.2017.1417998. [DOI] [PubMed] [Google Scholar]
- 13.Yamagata K, Nakayamada S, Zhang T, Zhang X, Tanaka Y. Soluble IL-6R promotes chondrogenic differentiation of mesenchymal stem cells to enhance the repair of articular cartilage defects using a rat model for rheumatoid arthritis. Clin Exp Rheumatol 38: 670–679, 2020. [PubMed] [Google Scholar]
- 14.Kay AG, Long G, Tyler G, Stefan A, Broadfoot SJ, Piccinini AM, Middleton J, Kehoe O. Mesenchymal stem cell-conditioned medium reduces disease severity and immune responses in inflammatory arthritis. Sci Rep 7: 18019, 2017. doi: 10.1038/s41598-017-18144-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glant TT, Radacs M, Nagyeri G, Olasz K, Laszlo A, Boldizsar F, Hegyi A, Finnegan A, Mikecz K. Proteoglycan-induced arthritis and recombinant human proteoglycan aggrecan G1 domain-induced arthritis in BALB/c mice resembling two subtypes of rheumatoid arthritis. Arthritis Rheum 63: 1312–1321, 2011. doi: 10.1002/art.30261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li NL, Zhang DQ, Zhou KY, Cartman A, Leroux JY, Poole AR, Zhang YP. Isolation and characteristics of autoreactive T cells specific to aggrecan G1 domain from rheumatoid arthritis patients. Cell Res 10: 39–49, 2000. doi: 10.1038/sj.cr.7290034. [DOI] [PubMed] [Google Scholar]
- 17.von Delwig A, Locke J, Robinson JH, Ng W-F. Response of Th17 cells to a citrullinated arthritogenic aggrecan peptide in patients with rheumatoid arthritis. Arthritis Rheum 62: 143–149, 2010. doi: 10.1002/art.25064. [DOI] [PubMed] [Google Scholar]
- 18.Rims C, Uchtenhagen H, Kaplan MJ, Carmona-Rivera C, Carlucci P, Mikecz K, Markovics A, Carlin J, Buckner JH, James EA. Citrullinated aggrecan epitopes as targets of autoreactive CD4+ T cells in patients with rheumatoid arthritis. Arthritis Rheumatol 71: 518–528, 2019. [Erratum in Arthritis Rheumatol 71: 907, 2019]. doi: 10.1002/art.40768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becart S, Whittington KB, Prislovsky A, Rao NL, Rosloniec EF. The role of posttranslational modifications in generating neo-epitopes that bind to rheumatoid arthritis-associated HLA-DR alleles and promote autoimmune T cell responses. PLoS One 16: e0245541, 2021. doi: 10.1371/journal.pone.0245541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Markovics A, Ocskó T, Katz RS, Buzás EI, Glant TT, Mikecz K. immune recognition of citrullinated proteoglycan aggrecan epitopes in mice with proteoglycan-induced arthritis and in patients with rheumatoid arthritis. PLoS One 11: e0160284, 2016. doi: 10.1371/journal.pone.0160284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zimmerman DH, Mikecz K, Markovics A, Carambula RE, Ciemielewski JC, Toth DM, Glant TT, Rosenthal KS. Vaccination by two DerG LEAPS conjugates incorporating distinct proteoglycan (PG, aggrecan) epitopes provides therapy by different immune mechanisms in a mouse model of rheumatoid arthritis. Vaccines (Basel) 9: 448, 2021. doi: 10.3390/vaccines9050448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gopal S. Syndecans in inflammation at a glance. Front Immunol 11: 227, 2020. doi: 10.3389/fimmu.2020.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leonova EI, Galzitskaya OV. Structure and functions of syndecans in vertebrates. Biochemistry (Mosc) 78: 1071–1085, 2013. doi: 10.1134/S0006297913100015. [DOI] [PubMed] [Google Scholar]
- 24.Patterson AM, Cartwright A, David G, Fitzgerald O, Bresnihan B, Ashton BA, Middleton J. Differential expression of syndecans and glypicans in chronically inflamed synovium. Ann Rheum Dis 67: 592–601, 2008. doi: 10.1136/ard.2006.063875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Segaliny AI, Brion R, Mortier E, Maillasson M, Cherel M, Jacques Y, Le Goff B, Heymann D. Syndecan-1 regulates the biological activities of interleukin-34. Biochim Biophys Acta 1853: 1010–1021, 2015. doi: 10.1016/j.bbamcr.2015.01.023. [DOI] [PubMed] [Google Scholar]
- 26.Van Raemdonck K, Umar S, Palasiewicz K, Volin MV, Elshabrawy HA, Romay B, Tetali C, Ahmed A, Amin MA, Zomorrodi RK, Sweiss N, Shahrara S. Interleukin-34 reprograms glycolytic and osteoclastic rheumatoid arthritis macrophages via syndecan 1 and macrophage colony-stimulating factor receptor. Arthritis Rheumatol 73: 2003–2014, 2021. doi: 10.1002/art.41792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deyab G, Reine TM, Vuong TT, Jenssen T, Hjeltnes G, Agewall S, Mikkelsen K, Førre Ø, Fagerland MW, Kolset SO, Hollan I. Antirheumatic treatment is associated with reduced serum syndecan-1 in rheumatoid arthritis. PLoS One 16: e0253247, 2021. doi: 10.1371/journal.pone.0253247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Rossi G, Evans AR, Kay E, Woodfin A, McKay TR, Nourshargh S, Whiteford JR. Shed syndecan-2 inhibits angiogenesis. J Cell Sci 127: 4788–4799, 2014. doi: 10.1242/jcs.153015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kehoe O, Kalia N, King S, Eustace A, Boyes C, Reizes O, Williams A, Patterson A, Middleton J. Syndecan-3 is selectively pro-inflammatory in the joint and contributes to antigen-induced arthritis in mice. Arthritis Res Ther 16: R148, 2014. doi: 10.1186/ar4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eustace AD, McNaughton EF, King S, Kehoe O, Kungl A, Mattey D, Nobbs AH, Williams N, Middleton J. Soluble syndecan-3 binds chemokines, reduces leukocyte migration in vitro and ameliorates disease severity in models of rheumatoid arthritis. Arthritis Res Ther 21: 172, 2019. doi: 10.1186/s13075-019-1939-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McNaughton EF, Eustace AD, King S, Sessions RB, Kay A, Farris M, Broadbridge R, Kehoe O, Kungl AJ, Middleton J. Novel anti-inflammatory peptides based on chemokine–glycosaminoglycan interactions reduce leukocyte migration and disease severity in a model of rheumatoid arthritis. J Immunol. 200: 3201–3217, 2018. doi: 10.4049/jimmunol.1701187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kehoe O, Cartwright A, Askari A, El Haj AJ, Middleton J. Intra-articular injection of mesenchymal stem cells leads to reduced inflammation and cartilage damage in murine antigen-induced arthritis. J Transl Med 12: 157, 2014. doi: 10.1186/1479-5876-12-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schofield KP, Gallagher JT, David G. Expression of proteoglycan core proteins in human bone marrow stroma. Biochem J 343: 663–668, 1999. doi: 10.1042/bj3430663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones FK, Stefan A, Kay AG, Hyland M, Morgan R, Forsyth NR, Pisconti A, Kehoe O. Syndecan-3 regulates MSC adhesion, ERK and AKT signalling in vitro and its deletion enhances MSC efficacy in a model of inflammatory arthritis in vivo. Sci Rep 10: 20487, 2020. doi: 10.1038/s41598-020-77514-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Endo T, Ito K, Morimoto J, Kanayama M, Ota D, Ikesue M, Kon S, Takahashi D, Onodera T, Iwasaki N, Uede T. Syndecan 4 regulation of the development of autoimmune arthritis in mice by modulating B cell migration and germinal center formation. Arthritis Rheumatol 67: 2512–2522, 2015. doi: 10.1002/art.39193. [DOI] [PubMed] [Google Scholar]
- 36.Cai P, Lu Z, Jiang T, Wang Z, Yang Y, Zheng L, Zhao J. Syndecan-4 involves in the pathogenesis of rheumatoid arthritis by regulating the inflammatory response and apoptosis of fibroblast-like synoviocytes. J Cell Physiol 235: 1746–1758, 2020. doi: 10.1002/jcp.29093. [DOI] [PubMed] [Google Scholar]
- 37.Godmann L, Bollmann M, Korb-Pap A, König U, Sherwood J, Beckmann D, Mühlenberg K, Echtermeyer F, Whiteford J, De Rossi G, Pap T, Bertrand J. Antibody-mediated inhibition of syndecan-4 dimerisation reduces interleukin (IL)-1 receptor trafficking and signalling. Ann Rheum Dis 79: 481–489, 2020. doi: 10.1136/annrheumdis-2019-216847. [DOI] [PubMed] [Google Scholar]
- 38.Coles CH, Shen Y, Tenney AP, Siebold C, Sutton GC, Lu W, Gallagher JT, Jones EY, Flanagan JG, Aricescu AR. Proteoglycan-specific molecular switch for RPTPsigma clustering and neuronal extension. Science 332: 484–488, 2011. doi: 10.1126/science.1200840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doody KM, Stanford SM, Sacchetti C, D. Svensson MN, Coles CH, Mitakidis N, Kiosses WB, Bartok B, Fos C, Cory E, Sah RL, Liu-Bryan R, Boyle DL, Arnett HA, Mustelin T, Corr M, Esko JD, Tremblay ML, Firestein GS, Aricescu AR, Bottini N. Targeting phosphatase-dependent proteoglycan switch for rheumatoid arthritis therapy. Sci Transl Med 7: 288ra76, 2015. doi: 10.1126/scitranslmed.aaa4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svensson MND, Zoccheddu M, Yang S, Nygaard G, Secchi C, Doody KM, Slowikowski K, Mizoguchi F, Humby F, Hands R, Santelli E, Sacchetti C, Wakabayashi K, Wu DJ, Barback C, Ai R, Wang W, Sims GP, Mydel P, Kasama T, Boyle DL, Galimi F, Vera D, Tremblay ML, Raychaudhuri S, Brenner MB, Firestein GS, Pitzalis C, Ekwall A-KH, Stanford SM. Synoviocyte-targeted therapy synergizes with TNF inhibition in arthritis reversal. Sci Adv 6: eaba4353, 2020. doi: 10.1126/sciadv.aba4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ikegawa S. Expression, regulation and function of asporin, a susceptibility gene in common bone and joint diseases. Curr Med Chem 15: 724–728, 2008. doi: 10.2174/092986708783885237. [DOI] [PubMed] [Google Scholar]
- 42.Xu L, Li Z, Liu SY, Xu SY, Ni GX. Asporin and osteoarthritis. Osteoarthritis Cartilage 23: 933–939, 2015. doi: 10.1016/j.joca.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 43.Torres B, Orozco G, García-Lozano JR, Oliver J, Fernández O, González-Gay MA, Balsa A, García A, Pascual-Salcedo D, López-Nevot MA, Núñez-Roldán A, Martín J, González-Escribano MF. Asporin repeat polymorphism in rheumatoid arthritis. Ann Rheum Dis 66: 118–120, 2007. doi: 10.1136/ard.2006.055426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang S, Xing Z, Liu T, Zhou J, Liang Q, Tang T, Cui H, Peng W, Xiong X, Wang Y. Synovial tissue quantitative proteomics analysis reveals paeoniflorin decreases LIFR and ASPN proteins in experimental rheumatoid arthritis. Drug Des Devel Ther 12: 463–473, 2018. doi: 10.2147/DDDT.S153927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bengtsson E, Mörgelin M, Sasaki T, Timpl R, Heinegård D, Aspberg A. The leucine-rich repeat protein PRELP binds perlecan and collagens and may function as a basement membrane anchor. J Biol Chem 277: 15061–15068, 2002. doi: 10.1074/jbc.M108285200. [DOI] [PubMed] [Google Scholar]
- 46.Pillai VS, Kundargi RR, Edathadathil F, Nair S, Thilak J, Mathew RA, Xavier T, Shenoy P, Menon KN. Identification of prolargin expression in articular cartilage and its significance in rheumatoid arthritis pathology. Int J Biol Macromol 110: 558–566, 2018. doi: 10.1016/j.ijbiomac.2018.01.141. [DOI] [PubMed] [Google Scholar]
- 47.Nastase MV, Young MF, Schaefer L. Biglycan: a multivalent proteoglycan providing structure and signals. J Histochem Cytochem 60: 963–975, 2012. doi: 10.1369/0022155412456380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sjöberg AP, Manderson GA, Mörgelin M, Day AJ, Heinegård D, Blom AM. Short leucine-rich glycoproteins of the extracellular matrix display diverse patterns of complement interaction and activation. Mol Immunol 46: 830–839, 2009. doi: 10.1016/j.molimm.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sotoodehnejadnematalahi F, Burke B. Structure, function and regulation of versican: the most abundant type of proteoglycan in the extracellular matrix. Acta Med Iran 51: 740–750, 2013. [PubMed] [Google Scholar]
- 50.Masuda K, Masuda R, Neidhart M, Simmen BR, Michel BA, Müller-Ladner U, Gay RE, Gay S. Molecular profile of synovial fibroblasts in rheumatoid arthritis depends on the stage of proliferation. Arthritis Res 4: R8, 2002. doi: 10.1186/ar427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shou J, Bull CM, Li L, Qian H-R, Wei T, Luo S, Perkins D, Solenberg PJ, Tan S-L, Chen X-YC, Roehm NW, Wolos JA, Onyia JE. Identification of blood biomarkers of rheumatoid arthritis by transcript profiling of peripheral blood mononuclear cells from the rat collagen-induced arthritis model. Arthritis Res Ther 8: R28, 2006. doi: 10.1186/ar1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karamanou K, Perrot G, Maquart F-X, Brézillon S. Lumican as a multivalent effector in wound healing. Adv Drug Deliv Rev 129: 344–351, 2018. doi: 10.1016/j.addr.2018.02.011. [DOI] [PubMed] [Google Scholar]
- 53.Bhattacharjee M, Sharma R, Goel R, Balakrishnan L, Renuse S, Advani J, Gupta ST, Verma R, Pinto SM, Sekhar NR, Nair B, Prasad TSK, Harsha HC, Jois R, Shankar S, Pandey A. A multilectin affinity approach for comparative glycoprotein profiling of rheumatoid arthritis and spondyloarthropathy. Clin Proteomics 10: 11, 2013. doi: 10.1186/1559-0275-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang X, Zhao Y, Wang Y, Chen Y, Yan X. Screening citrullinated proteins in synovial tissues of rheumatoid arthritis using 2-dimensional western blotting. J Rheumatol 40: 219–227, 2013. doi: 10.3899/jrheum.120751. [DOI] [PubMed] [Google Scholar]