


Keywords: channelopathy, epilepsy, intellectual disability, KCNQ5, M current
Abstract
We identified six novel de novo human KCNQ5 variants in children with motor/language delay, intellectual disability (ID), and/or epilepsy by whole exome sequencing. These variants, comprising two nonsense and four missense alterations, were functionally characterized by electrophysiology in HEK293/CHO cells, together with four previously reported KCNQ5 missense variants (Lehman A, Thouta S, Mancini GM, Naidu S, van Slegtenhorst M, McWalter K, Person R, Mwenifumbo J, Salvarinova R; CAUSES Study; EPGEN Study; Guella I, McKenzie MB, Datta A, Connolly MB, Kalkhoran SM, Poburko D, Friedman JM, Farrer MJ, Demos M, Desai S, Claydon T. Am J Hum Genet 101: 65–74, 2017). Surprisingly, all eight missense variants resulted in gain of function (GOF) due to hyperpolarized voltage dependence of activation or slowed deactivation kinetics, whereas the two nonsense variants were confirmed to be loss of function (LOF). One severe GOF allele (P369T) was tested and found to extend a dominant GOF effect to heteromeric KCNQ5/3 channels. Clinical presentations were associated with altered KCNQ5 channel gating: milder presentations with LOF or smaller GOF shifts in voltage dependence [change in voltage at half-maximal conduction (ΔV50) = ∼−15 mV] and severe presentations with larger GOF shifts in voltage dependence (ΔV50 = ∼−30 mV). To examine LOF pathogenicity, two Kcnq5 LOF mouse lines were created with CRISPR/Cas9. Both lines exhibited handling- and thermal-induced seizures and abnormal cortical EEGs consistent with epileptiform activity. Our study thus provides evidence for in vivo KCNQ5 LOF pathogenicity and strengthens the contribution of both LOF and GOF mutations to global pediatric neurological impairment, including ID/epilepsy.
NEW & NOTEWORTHY Six novel de novo human KCNQ5 variants were identified from children with neurodevelopmental delay, intellectual disability, and/or epilepsy. Expression of these variants along with four previously reported KCNQ5 variants from a similar cohort revealed GOF potassium channels, negatively shifted in V50 of activation and/or delayed deactivation kinetics. GOF is extended to KCNQ5/3 heteromeric channels, making these the predominant channels affected in heterozygous de novo patients. Kcnq5 LOF mice exhibited seizures, consistent with in vivo pathogenicity.
INTRODUCTION
Discovery of de novo variants in ion channel genes associated with human epilepsies and developmental disabilities has increased rapidly, although functional characterization and validations of causality have lagged (1–3). Among neuronal ion channels, potassium channels comprise the most functionally diverse class. Potassium channels are encoded by 79 α-subunit genes in the human genome subdivided into six major subclasses based on structural and functional similarities, modified by multiple families of subclass-specific accessory β-subunits (4, 5).
Among the major classes of potassium channels, KCNQ (Kv7) potassium channels have been prominently linked to multiple human channelopathies (6–8). Because KCNQ potassium channels underlie M currents (9–11), they are powerful regulators of basal excitability. KCNQ channels exhibit pronounced modulation by diverse cellular and intracellular signaling mechanisms including Gq-coupled G protein coupled receptors (GPCRs) via depletion of phosphatidylinositol 4,5-bisphosphate (PIP2) (9, 12, 13), Ca2+/calmodulin (14–16), reactive oxygen species (ROS) (17), arginine methylation (18), allosteric activation by GABA (19), and kinases preassembled by scaffolding proteins such as AKAP9 and AKAP150 (20) and yotiao (21). Thus, KCNQ channels serve as critical effectors linking numerous neuromodulatory transmitters and intracellular signaling pathways to sustained shifts in the electrical excitability of neurons and other excitable cells, or transport epithelia. Human KCNQ1-4 mutations are linked to multiple hereditary diseases underlying cardiac dysfunction (KCNQ1: LQT1) (22, 23), cardio-auditory disorder (KCNQ1: JLNS1) (24–26), childhood epilepsy (KCNQ2/KCNQ3: BFNS1, BFNS2) (27–29), and nonsyndromic early-onset hearing loss (KCNQ4: DFNA2A) (30).
In mammalian genomes, KCNQ potassium channel subunits are encoded by a conserved family of five genes (KCNQ1–5). Of these, KCNQ2–5 are expressed primarily in neurons. KCNQ subunits functionally assemble as tetramers in homomeric or heteromeric combinations, the latter most often with KCNQ3 (KCNQ2/3, KCNQ4/3, KCNQ5/3) as an obligatory subunit. Neuronal KCNQ2–5 channels are widely expressed and primarily composed of KCNQ2, KCNQ3, and KCNQ5 subunits, with KCNQ4 displaying a more restricted expression pattern, primarily associated with cochlear outer hair cells and central auditory nuclei (31, 32).
KCNQ5 was the last member of the KCNQ gene family to be cloned (33, 34). KCNQ5 expression is widespread in the brain, with enrichment in cortical layers 1–4 and CA3 hippocampus (35–37). Whereas KCNQ2/3 heteromeric channels localize at axon initial segments (AISs) via AnkyrinG binding (38, 39), homomeric KCNQ5 channels may specifically traffic to glutamatergic presynaptic terminals, exemplified by the calyx of Held (Refs. 40, 41, but see Ref. 42). KCNQ5 also expresses in vascular smooth muscle, where it provides a major component of resting potassium conductance and may contribute to neurovascular function by regulating vascular tone (43–46).
Recently, four de novo missense variants in KCNQ5 were reported in patients with intellectual disability (ID) and/or epilepsy (47), three reported to be loss of function (LOF) and one gain of function (GOF). Evidence for LOF was equivocal based on modest changes in gating properties but in the presence of substantial functional currents. Importantly, despite the association of altered KCNQ5 channel function with missense variations, demonstration of in vivo pathogenicity remained unproven.
Here we report six new de novo variants in KCNQ5 identified in individuals with ID and/or epilepsy, strengthening the association of human KCNQ5 variants with ID/epilepsy (47). From our analysis, our four reported missense variants along with all four previously reported missense variants (47) resulted in GOF channels due to hyperpolarizing shifts in voltage dependence of activation or slowed deactivation kinetics. We also report two nonsense variants, both associated with ID/epilepsy, and demonstrate that these variants result in LOF potassium channels. To examine possible in vivo KCNQ5 LOF pathology, two independent Kcnq5 LOF mouse lines were generated with CRISPR/Cas9. Both Kcnq5 LOF lines exhibited handling- and thermal-induced seizures and abnormal interictal cortical electroencephalograms (EEGs) consistent with epileptiform activity. Taken together, these results strengthen the association of both LOF and GOF KCNQ5 variants to human ID and epilepsy and support the in vivo pathophysiological potential for KCNQ5 LOF.
MATERIALS AND METHODS
Institutional Study Approval
Animal procedures used in this study were approved by the Institutional Animal Care and Use Committee at Seattle Children’s Research Institute. Use of human genetic data was conducted in accordance with the standards of the Ethics Committees and Institutional Review Boards at each institution. Written informed consent was obtained for all patients (or their guardians) in the study.
Clinical Diagnosis
Clinical diagnoses were performed as part of routine diagnostic evaluations and patient care at the following medical centers: patient LR16-507, Department of Human Genetics, Radboud University Medical Center, Nijmegen, The Netherlands; patient LR16-508, Department of Human Genetics, Erasmus Medical Center, Rotterdam, The Netherlands; patient LR16‐509, The Academic Center for Epileptology Kempenhaeghe and Maastricht University Medical Centre, The Netherlands; patient LR17-519, Medical Genetics, Cook Children’s Hospital, Fort Worth, TX; patient LR17-520, Department of Pediatrics, University of Texas Health Science Center, Houston, TX; patient LR17-518, Epilepsy Center, Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL.
Whole Exome Sequencing and Analysis
Trio-based whole exome sequencing (WES) was performed on genomic DNA from patients and parents on multiple academic and commercial next-generation sequencing platforms: in-house WES core services at Radboud University Medical Center and Maastricht University Medical Centre (patients LR16-507, LR16-508, and LR16-509); IDT xGen Exome Research Panel v1.0 at Ambry Genetics, Aliso Viejo, CA (patients LR17-519 and LR17-520); GeneDx WES at Gaithersburg, MD (patient LR17-518). Bioinformatics and data analysis were performed as previously described (48). Identified clinically relevant genetic variants were confirmed by Sanger sequencing.
Electrophysiology with HEK293 and CHO Cells
Human KCNQ5 (33) (kindly provided by K. Steinmeyer, Sanofi-Aventis, Paris, France) and catalytically inactive β-Secretase 1 [BACE1(D289N)] (49) (kindly provided by Tobias Huth and Christian Alzheimer, Friedrich-Alexander-Universitat Erlangen-Nurnberg, Erlangen, Germany) were cloned into pcDNA3 (Invitrogen), a cytomegalovirus (CMV)-driven expression plasmid modified to include either an IRES-GFP or a IRES-mCherry cassette downstream of the multicloning site. Site-directed mutagenesis of KCNQ5 was performed by overlap PCR or Gibson assembly strategies (HiFi DNA Assembly Kit, NEB, Beverly, MA) with oligonucleotide primers from Integrated DNA Technologies (Coralville, IA). An incomplete human KCNQ3 cDNA missing the first exon was purchased from Origene (RG234832; Rockville, MD) and modified by the addition of exon 1 generated by high-fidelity PCR from human genomic DNA, This full-length human KCNQ3 cDNA was cloned into pcDNA3 with IRES-GFP or IRES-mCherry. All constructs were sequenced to confirm the accuracy of introduced mutations and cloning manipulations with a commercial sequencing service (SimpleSeq, Eurofins Genomics, Louisville, KY).
For expression studies, plasmids were acutely transfected into HEK293 (CRL-1573, ATCC, Manassas, VA) or Chinese hamster ovary (CHO)-K1 (CCL-61, ATCC, Manassas, VA) cells with Lipofectamine 2000 (Invitrogen). CHO cells provided a lower background level of endogenous potassium currents compared with HEK293 cells (50, 51). Briefly, low-passage-number (<25 passages) cells were plated in 12-well plates (2.5 cm diameter/well) at a density to yield ∼80–95% confluence (∼1.5 × 106 cells/well). After cells were allowed to settle and attach, cells in each well were transfected with combinations of KCNQ5-IRES-GFP [wild type (WT) or mutant] (1.0 µg) and BACE1(D289N)-IRES-mCherry (0.3 µg) plasmids, with 3.75 µL of Lipofectamine 2000 per well, according to the manufacturer’s protocol. Combinations of KCNQ5-IRES-GFP (WT or P369T) (1.0 µg) and KCNQ3-IRES-mCherry (1.0 µg) were similarly cotransfected with Lipofectamine 2000. After overnight incubation, transfected cells were trypsinized (TrypLE; Thermo Fisher Scientific, Waltham, MA) and replated at low density (∼20 × 103 cells/well) on poly-lysine-coated 12-mm coverslips (Neuvitro Corporation, Vancouver, WA) in 24-well plates. HEK293 cells were maintained in DMEM (Gibco) supplemented with 10% fetal calf serum and 1% (vol/vol) penicillin-streptomycin (10,000 U/mL) in a 37°C incubator with 5% CO2. CHO cells were similarly cultured in DMEM-F-12 medium (Gibco) supplemented with 10% fetal calf serum and 1% (vol/vol) penicillin-streptomycin (10,000 U/mL). Cells were recorded after 2–5 days of incubation after transfection.
For patch-clamp recordings, cultured cells were transferred to the stage of a Zeiss Axio Examiner.A1 upright microscope equipped with a ×40 water-immersion objective for fluorescence. Data were acquired with an Axopatch-1D amplifier (Molecular Devices, Sunnyvale, CA) using pClamp v10.6 software (Molecular Devices, Sunnyvale, CA). Data were sampled at 10 kHz and filtered at 2.0 kHz. Whole cell recording mode was performed with patch pipettes with resistances of 3–5 MΩ filled with internal solution (in mM: 140 K-gluconate, 1.0 CaCl2, 2.0 MgSO4, 10 HEPES, 4.0 Na2ATP, 0.3 Na2GTP, 2.4 EGTA; pH to 7.4) This internal solution buffers free Ca2+ concentration ([Ca2+]) to 101 nM. Bath solution used (in mM) 4.0 KCl, 145 NaCl, 10 HEPES, 2.0 CaCl2, 2.0 MgCl2, and 10 glucose, pH to 7.2. Typical on-cell seal resistances were 2–4 GΩ before the patch was ruptured. Membrane resistances were typically >20 times the access resistances (<15 MΩ) in whole cell mode after the patch was ruptured. Voltage-clamp protocols utilized a family of 3.0-s voltage steps from −100 mV to 60 mV from a holding potential of −80 mV, followed by either a 0.4-s poststep to −60 mV or a 1.0-s voltage step to −100 mV, using an interstep interval of 10 s to allow channels to fully equilibrate to steady state at −80 mV. For current-clamp recordings, the amplifier was switched to current-clamp mode with zero current injection, to measure resting membrane potentials (Vms).
Electrophysiological data were analyzed in Clampfit (Molecular Devices, Sunnyvale, CA) and Origin8 (OriginLab, Northampton, MA). Conductances (Gs) were measured as isochronic currents at the end of 3-s voltage steps, divided by driving force for K+, assuming a K+ reversal potential of −89.5 mV based on the recording solutions used and a Nernst equilibrium potential. Alternatively, conductance was measured from tail currents measured at −100 mV, following a family of activating voltage steps. For conductance/voltage plots, conductances were normalized to maximal conductance (G/Gmax). Each data set was fitted by a first-order Boltzmann function:
where n is the slope factor reflecting intrinsic voltage sensitivity; V50 is the midpoint of half-maximal conductance; k is the Boltzmann constant; and T is temperature (K). Tail current kinetics were derived by fits to either single- or double-exponential functions:
where τ1 and τ2 are the taus and A1 and A2 are the proportional fractions of the first and second exponential components, respectively, and C is the baseline offset.
Generation of Cas9/sgRNA-Targeted Kcnq5 Deletional LOF Mice
Kcnq5 was targeted for mutagenesis by cloning a short dsDNA adaptor encoding a single guide RNA (sgRNA) into the bicistronic plasmid pX330 for expressing Streptococcus pyogenes Cas9 (pX330-U6-Chimeric_BB-CBh-hSpCas9 was a gift from Feng Zhang, MIT; Addgene plasmid no. 42230) (52). This sgRNA targeted the sequence in Kcnq5 exon 5 encoding the NH2-terminal edge of the P-helix (5′- CGACATATGCGGATGCTCTC-3′).
The F0 ES cell line (C57BL/6J × Black Swiss) was derived and maintained essentially as described previously (53). One subconfluent 12-well dish of F0 cells was transfected with 1 µg of pX330 construct (containing Cas9 and sgRNA) and 1 µg of a puromycin-resistant plasmid with FuGene HD transfection reagent (Promega Corporation, Madison, WI) at the recommended concentrations. After 12 h, the cells were replated onto 2 × 100 mm dishes and 1.5 µg/mL puromycin was added to the media. After 24 h puromycin was removed, and the surviving colonies were picked on day 7–8.
Clonal ES cells (ESCs) recovered after transfection and selection were screened for targeted indels by T7 Endonuclease I assays (NEB, Beverley, MA) after hybridization to a 652-bp wild-type DNA template encompassing exon 4 and exon 5 generated by PCR with primers:
5′- CCTGATCATGTGTGACTTATGGCTGTCTCAACCTG-3′
5′- CCCGGCCGTTCAACGGAGACATTCCATAATAGCTAATAGAT-3′
A high fraction of clonal ESCs (70–80%) exhibited targeted indels. A selected pool of ESC clones was sequenced to confirm indels. One ESC clone was selected for blastocyst implantation. This ESC clone contained biallelic deletions of exon 5/intron 6 [removing 37 bp (Q5d1) and 33 bp (Q5d2)], both of which were predicted to result in prematurely truncated proteins.
Blastocyst microinjections were performed under an inverted microscope (Olympus IX71) equipped with Eppendorf micromanipulators. The procedure was carried out at room temperature in DME/High Glucose medium (HyClone Laboratories, Logan, UT). Approximately four to six ESCs were injected into four-cell or eight-cell embryos before compaction. After microinjection, embryos were immediately transferred into pseudopregnant CD-1 females (Charles River Laboratories, Hollister, CA), or cultured in BMOC medium (Thermo Fisher Scientific, Waltham, MA) at 37.5°C in 5% CO2 in air overnight and transferred the next day.
Four male chimeric founder mice (P0) were recovered and bred to C57BL/6J, of which three exhibited transmission to the next generation (F1) based on genotyping. Heterozygous F1 siblings were interbred to yield N1 homozygous lines carrying each deletion allele (Q5d1, Q1d2). N1 animals were further outcrossed to C57BL/6J to create N5 homozygous lines carrying each deletion allele. Genotyping was performed with the following primers:
5′-GAACCATATGTAGAGTTCATGTTTCCATTT CC-3′
5′- CCCGGCCGTTCAACGGAGACATTCCATAATAGCTAATAGAT-3′
which generate PCR products for WT (260 bp), Q5d1 (223 bp), and Q5d2 (228 bp).
Animals were housed and maintained in an AOAC/ALACC-approved vivarium at Seattle Children’s Research Institute under a 12:12-h (light-dark) cycle with lights on at 7:00 AM and off at 7:00 PM.
qRT-PCR
Total RNA was extracted from whole brains freshly dissected from adult mice [∼postnatal day (P)50–60] with RNAzol RT (Molecular Research Center, Cincinnati, OH) according to the manufacturer’s protocol. First-strand synthesis was performed with 1.0 µg of total RNA, random hexamers (6 µM), and M-MuLV Reverse Transcriptase (NEB, Beverley, MA). Individual qRT-PCR reactions were performed with 0.64% of each first-strand reaction with Power SYBR Green (Applied Biosystems, Foster City, CA) on an ABI 7300 Real Time PCR System (Applied Biosystems, Foster City, CA). Each qRT-PCR was replicated three or four times. Primer sequences used are presented in Supplemental Table S1 (all Supplemental Materials are available at https://doi.org/10.6084/m9.figshare.17077898).
Western Blots
Membrane protein lysates were prepared from freshly dissected brains of adult mice (∼P40). Brain tissues were manually homogenized at 0°C in a glass-Teflon pestle in 1–2 mL of sucrose buffer (0.32 M sucrose, 4.0 mM HEPES, pH 7.4) supplemented with 1× protease inhibitor cocktail (PIC) (Sigma-Aldrich, St. Louis, MO). Crude lysate was centrifuged at 1,000 g for 10 min (4°C) to pellet unlysed cellular debris. The supernatant was recovered and centrifuged at 15,000 g for 15 min (4°C) to pellet the membrane/synaptosomal fraction. This membrane fraction was dissolved in 1% NP-40 lysis buffer [20 mM Tris·HCl, 1% (vol/vol) NP-40, 1.0 mM NaF, 1× PIC] and recentrifuged at >15,000 g for 15 min (4°C) to pellet undissolved proteins. This last supernatant was kept and stored at −80°C as soluble membrane protein fractions for Western blots.
For Western blots, ∼34 µg of each soluble membrane protein sample was loaded per lane on 10% SDS-acrylamide gels and electrophoresed in 1× SDS-PAGE running buffer [25 mM Tris, 192 mM glycine, 0.1% (wt/vol) SDS, pH 8.3]. Proteins were transferred to PVDF membranes by electrophoresis at 0–4°C in Towbin buffer [25 mM Tris, 192 mM glycine, 0.1% (wt/vol) SDS, pH 8.3, 20% (vol/vol) methanol] and used immediately for immunodetection.
Blots were blocked in 5% (wt/vol) nonfat dry milk powder in 1× Tris-buffered saline-Tween (TBST) [20 mM Tris, 150 mM NaCl, pH 7.6, 0.1% (vol/vol) Tween 20] for 1 h at 24°C (RT). Blots were subsequently probed with a primary rabbit polyclonal antibody raised against full-length KCNQ5 peptide (ABN_1372: MilliporeSigma, Burlington, MA) at 1:2,000 for ∼8–12 h on a rolling shaker at 4°C. After washing in 1× TBST (3 times), blots were reprobed with a secondary donkey anti-rabbit IgG antibody conjugated to horseradish peroxidase (HRP) (16284; Abcam, Cambridge, MA) at 1:10,000 for 45 min at RT. After washing in 1× TBST (3 times), blots were visualized by chemiluminescence with SuperSignal West Femto (Thermo Fisher Scientific, Waltham, MA) substrate and a FluorChem R gel imager (Protein Simple, Wallingford, CT).
Video-Electroencephalography-Electromyography Recordings
Experiments were performed as previously described (54–56). Briefly, mice underwent survival surgery to implant fine (diameter: 130 µm bare, 180 µm coated) silver wire electroencephalography (EEG) and electromyography (EMG) electrodes under isoflurane anesthesia (3.5% induction, 2% maintenance). Two monopolar EEG electrodes were placed bilaterally through cranial burr holes over the sensorimotor cortices and fixed in position with cyanoacrylate glue and dental cement (Lang Dental Manufacturing Co., Inc., Wheeling, IL). Similarly, one reference electrode was placed above the cerebellum. A ground electrode was inserted subcutaneously over the back. EMG electrodes were placed in back muscles. Mice recovered after surgeries for 2–3 days before recordings. Electrode impedances were typically <10 KΩ. Simultaneous video-EEG-EMG recordings were recorded from awake mice during subjective night (mornings) with a PowerLab 8/35 data acquisition unit using LabChart 8.0 software (AD Instruments, Colorado Springs, CO). Recordings were acquired at 1.0 kHz and processed post hoc: EEGs through a 1– to 70-Hz band-pass filter and EMGs through a 3-Hz high-pass filter. Interictal spikes were identified as transients clearly distinguished from background activity, with sharp peaks followed by a slow wave and short durations (<5.0 ms). Myoclonic seizures were identified as sudden twitches of limb or back musculature on video, coincident with increases in EMG activity and a spike or polyspike wave complex on EEGs. Power spectral density analyses were performed with LabChart 8.0 (AD Instruments, Colorado Springs, CO).
Thermal Induction of Seizures
For each animal, core body temperature was continuously monitored by a rectal temperature probe controlled through a feedback circuit in line with a heat lamp. Body temperature was progressively increased in 0.5°C increments at 2-min intervals from 37°C until either a seizure was induced or a temperature of 43°C was reached. Animals were then cooled immediately and returned to home cages. Seizure activity was continuously monitored by simultaneous cortical EEGs, EMGs, and video recordings.
Molecular Modeling
Structural homology modeling of human KCNQ5 was performed with SwissModel, a web-based server (https://swissmodel.expasy.org), using the cryo-electron microscopy (cryo-EM) structure of Xenopus KCNQ1 (PDB: 5VMS) (57) as a template. Molecular models were visualized with DeepView/Swiss-PDBviewer and UCSF Chimera (https://www.cgl.ucsf.edu/chimera).
Additional Methods
Additional methods describing immunohistochemistry of HEK293 cells are found in Supplement SVI.
Statistics
All statistical data were analyzed in Prism 5.04 (GraphPad, La Jolla, CA) and expressed as mean ± SE or median and interquartile range (IQR), as noted. Statistical significances were determined by either pairwise unpaired t tests or nonparametric Mann–Whitney U test, as noted, with Prism 5.04. Sample sizes were determined for a power (1 − β) of 0.8 and a P value (α) of 0.05 with a Microsoft Excel add-in (Real Statistical Resource Pack: https://www.real-statistics.com, written by Dr. Charles Zaiontz).
Data and Research Material Availability
Human variants of KCNQ5 described in this study are deposited in the Leiden Open Variant Database (LOVD v.3.0). CRISPR-generated mouse Kcnq5 deletional alleles are deposited in the NIH-funded Mutant Mouse Resource and Research Centers (MMRRC) repository and will also be provided upon request from Seattle Children’s Research Institute. All plasmids generated for this study are made available through Addgene.
RESULTS
Neurological Impairment in Six Pediatric Patients Carrying de Novo KCNQ5 Variants
Six unrelated children with severe neurological impairment (age range 0.8–9 yr; 4 females and 2 males) were referred for whole exome sequencing from clinics in The Netherlands and the United States after standard metabolic and microarray assays proved uninformative. Trio analysis identified one heterozygous de novo variant in KCNQ5 in each child, together comprising four missense and two nonsense mutations (Table 1). Five of six variants were absent from the Genome Aggregation Database (gnomAD; gnomad.broadinstitute.org), whereas one variant carried by patient LR16-508 (c.1328G>A; p.R443Q) appeared once in gnomAD. All but one patient developed epilepsy during early childhood, with onsets of seizures ranging from 0.8 mo to 5 yr. As toddlers, all six children showed significant intellectual disability (ID) by neurological examination. Quantitative IQ assessment of two children with Wechsler Intelligence Scales provided full-scale IQ (FSIQ) scores at least 2 standard deviations (SDs) below the mean (FSIQ < 70) in both, indicative of low intellectual functioning. Each of the six children showed motor impairment, including delayed walking (5 of 6), hypotonia (2 of 6), and/or opisthotonus (1 of 6). All six children exhibited delayed language development. Of note, two children differed from the others by exhibiting mild loss of brain volume at infancy by magnetic resonance imaging (MRI), followed by severe regression of motor and language skills as toddlers (patients LR17-520 and LR16-509; Table 1, mutation group: GOF severe). In summary, each of the six de novo KCNQ5 variants was associated with clinical presentations of global neurological impairment, which included epilepsy and ID.
Table 1.
Clinical features in 6 individuals with heterozygous KCNQ5 mutations
| Subject | LR17-518 | LR17-519 | LR16-507 | LR16-508 | LR17-520 | LR16-509 |
|---|---|---|---|---|---|---|
| Mutation group | LOF mild | LOF mild | GOF mild | GOF mild | GOF severe | GOF severe |
| Mutation (short form) | R178X | R443X | K289N | R443Q | P369T | H576R |
| KCNQ5 variants | ||||||
| NM_001160133.1 | c.532C>T | c.1327C>T | c.867G>T | c.1328G>A | c.11005C>A | c.1727A>G |
| Protein change | p.Arg178Ter | p.Arg443Ter | p.Lys289Asn | p.Arg443Gln | p.Pro369Thr | p.His576Arg |
| Demographics | ||||||
| Age of presentation | 5 yr | 1 yr | 9 yr | 4.5 yr | 0.8 yr | 1.8 yr |
| Age last evaluation | 6 yr | 6 yr | 21 yr | 14 yr | 1–11/12 yr | 16 yr (died) |
| Sex | F | F | F | M | M | F |
| Ethnicity | W Eur/Asian | AA | W Eur | W Eur | W Hisp | W Eur |
| Neurodevelopment | ||||||
| Early motor delay | Global mild | Mild | Mild | Mild to moderate | Global severe | Global severe |
| Walking | Delayed | ND | 1 yr 6 mo (delayed) | 1 yr 9 mo (delayed) | Delayed | 1 yr 8 mo (later lost) |
| First words | ND | ND | 2.5 yr | 3 yr | None at 2 yr | 2 yr |
| Language last evaluation | Delayed | Delayed | Delayed | Delayed | Delayed | Delayed |
| Intellectual disability | Mild ID | Mild ID | Mild (FSIQ 66) | Mod (FSIQ 50) | Severe ID | Severe ID |
| Autism | No | AUT | AUT | No | Unable to assess | Unable to assess |
| Mood dysfunction | Mood swings | No reports | No reports | No reports | Always irritable | Mood swings |
| Regression | No | No | No | No | Yes, severe | Yes, severe |
| Neurological | ||||||
| Head size (OFC) | 52.2 cm (84%) at 6 yr | ND | 55.5 cm (86%) at 21 yr | 53 cm (14%) at 14 yr | ∼10–20% | ∼10% |
| Tone | Hypotonia mild | Normal | Normal | Normal | Hypotonia severe | ND |
| Other features | CVI, opisthotonus | Head banging | ||||
| Brain imaging (MRI) | ND | Normal at 6 yr | ND | Normal | Mild diffuse volume loss at 6 mo | Mild diffuse volume loss |
| Epilepsy | ||||||
| Seizure age of onset | 5 yr | ∼1 yr | None | 4.5 yr | 10 mo | 1–10/12 yr |
| Seizure types | Atonic drop, atypical absence | Staring spells | None | Symmetric GTCS, often with fever | Frequent symmetric and asymmetric GTS; febrile SZ | Atonic SZ at onset, myoclonic SZ at 5 yr; GTCS with rotating eyes and laughter at 13 yr |
| EEG | Diffuse slowing, multifocal spikes | Centrotemporal spikes, BECTS-like | Normal (9 yr) | R > L centro-temporal spikes, frequent in sleep | Hypsarrhymia with GTS | Frontal 1- to 2-Hz high-voltage slow waves and 10- to 14-Hz sharps |
| Medical history | ||||||
| Feeding and other | Apnea, asthma, skin nevi | Dysmorphic face, skin nevi | Poor feeding, later GT, apnea | Poor feeding, later GT |
AA, African-American; AUT, autism; BECTS, benign epilepsy with centrotemporal spikes; CVI, cortical visual impairment; EUR, European; FSIQ, full-scale IQ; GT, gastrostomy tube; GTCS, generalized tonic-clonic seizures; GTS, generalized tonic seizures; Hisp, Hispanic; ID, intellectual disability; mod, moderate; MRI, magnetic resonance imaging; ND, no data; OFC, occipitofrontal circumference; SZ, seizures; W, white. Other notes: subject LR16-509 born at 35 wk of gestation.
Location of de Novo KCNQ5 Variants Identified in Children with Epilepsy and ID
The locations of the six patient-derived de novo KCNQ5 variants were mapped onto the topology of the KCNQ5 channel subunit modeled by homology to the cryo-EM structure of the homologous KCNQ1 channel (57), along with the four de novo KCNQ5 variants previously reported by Lehman et al. (47) from a separate but comparable patient cohort (Fig. 1, A and C). All 10 variants altered residues highly conserved between all five members of the KCNQ1–5 gene family, with the exception of S448I located in the weakly conserved linker between cytosolic Helix-A and Helix-B (Fig. 1B).
Figure 1.
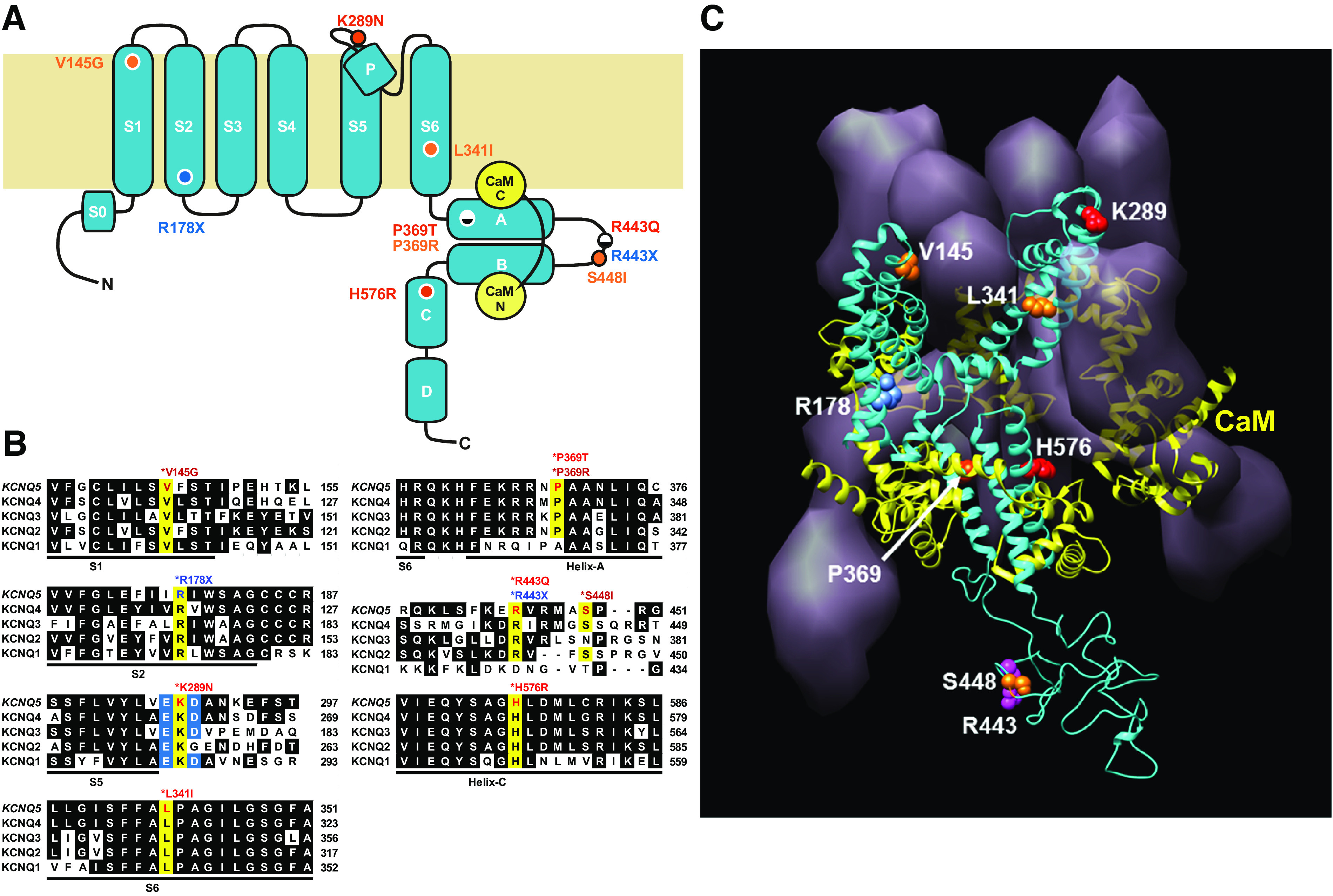
Human KCNQ5 de novo mutations identified from patients with intellectual disability (ID)/epilepsy alter highly conserved residues distributed throughout the channel subunit. A: location of de novo KCNQ5 mutations mapped to the structural domains of the KCNQ5 subunit defined by cryo-electron microscopy (cryo-EM) of the homologous Xenopus KCNQ1 subunit (57). R178X and R443X (blue) encode predicted truncated proteins, prematurely terminated at the base of S2 (R178X) and between Helix-A and Helix-B (R443X) in the cytosolic COOH-terminal tail. K289N, P369T, R443Q, and H576R (red) encode missense mutations distributed at the extracellular edge of S5 (K289N) and throughout the COOH-terminal tail (P369T, R443Q, H576R). Previously reported de novo KCNQ5 missense mutations (47) are depicted in orange: V145G in S1, L341I in S6, P369R in Helix-A, and S448I in the loop between Helix-A and Helix-B. B: alignment of amino acid residues for each KCNQ5 residue affected by de novo mutations (highlighted in yellow), with the sequences of all 5 human KCNQ subunits. KCNQ5 mutations alter residues highly conserved in all 5 KCNQ subunits. K289N neutralizes a positive charged residue located between 2 flanking negatively charged residues, E288 and D290 (highlighted in blue). C: structural homology model of a tetrameric KCNQ5 channel based on the structure of KCNQ1 (57), viewed obliquely from the extracellular side. The ribbon structure of a single subunit is shown (blue) with the surface volume representations of the other 3 subunits and a ring of 4 calmodulin accessory subunits (yellow). Locations of residues affected by de novo mutations are shown in the ribbon structure, using the same color scheme as in A.
The locations of the four KCNQ5 missense variants that we report were highly dispersed and heterogeneous throughout the channel subunit (Table 1, Fig. 1). Patient LR16-507 carried a variation (p.K289N, c.867G>T) that neutralized the positive charge of a highly conserved basic residue located within a local charge cluster with two adjacent acidic residues, at the extracellular edge of S5 immediately preceding the turret region leading to the pore α-helix. Patient LR16-508 carried a variant (p.R443Q, c.1328G>A) altering a conserved residue located in the unstructured linker segment connecting Helix-A to Helix-B in the cytosolic COOH-terminal tail domain (CTD), for which no structural data are available. The last two missense variants affected residues lodged within cytosolic α-helical segments in the CTD. The variant carried by patient LR17-520 was located in Helix-A, critical for channel preassembly with calmodulin- and Ca2+-dependent modulation of gating (p.P369T, c.1105C>A) (58), whereas the variant in patient LR16-509 was located in Helix-C, a site implicated in subunit-subunit interaction (p.H576R, c.1727A>G) (59, 60). Of note, the latter two missense variants in the CTD were carried by children who uniquely presented with intellectual and motor skill regression, accompanied by mild loss of brain volume (Table 1, mutation group: GOF severe).
The KCNQ5 nonsense variants carried by two patients (LR17-518, LR17-519; Table 1) were both predicted to result in LOF by prematurely truncating the conceptual channel protein either near the cytosol base of S2 (p.R178X, c.532C>T) or within the linker between Helix-A and Helix-B (p.R443X, c.1327C>T). The second truncation (R443X) occurred at the same residue altered as a missense variant in patient LR16-508 (R443Q). Unlike the first truncation (R178X), the second truncation (R443X) preserved all membrane-spanning segments.
De Novo Missense KCNQ5 Variants Produce Homomeric GOF Channels
To investigate the functional consequences of these variants in vitro, WT human KCNQ5 cDNA (33) and KCNQ5 cDNAs engineered with these de novo variants were transiently expressed in human embryonic kidney (HEK293) or Chinese hamster ovary (CHO) cells. Currents and membrane potentials were recorded by patch-clamp recordings in whole cell configuration under voltage clamp and current clamp. WT and de novo variants of KCNQ5 were cloned into a bicistronic IRES-GFP expression plasmid to ensure positive visual identification of transfected cells. KCNQ5 expression studies in HEK293 cells were performed by coexpression with the β-Secretase 1 putative KCNQ accessory subunit, using a BACE1(D289N)-IRES-mCherry expression plasmid (49). Control experiments revealed no significant changes in G/V parameters, deactivation kinetics, or current amplitudes by the coexpression of β-Secretase 1 (see Fig. 5, Fig. 6B, Supplemental Fig. S1).
Figure 5.

Coexpression of wild-type (WT) KCNQ5 with BACE1(D289N) and in combinations of KCNQ5(P369T) and WT KCNQ3: dominant gain-of-function (GOF) effect of P369T on heteromeric KCNQ3/KCNQ5(P369T) channels and lack of effect with BACE1(D289N). A, left: current traces from Chinese hamster ovary (CHO) cells transfected with WT KCNQ5 and WT KCNQ3 [denoted KCNQ5(+) and KCNQ3(+)] controls (black) compared with KCNQ5(+)+BACE1(D289N) (red) and KCNQ5(P369T) (blue). Right: similar current traces from KCNQ5(+)+KCNQ3(+) control (black) compared with KCNQ5(P369T)+KCNQ3(+) (red) and KCNQ5(P369T)+KCNQ5(+) (blue). For all recordings, currents evoked under voltage clamp from a holding potential of −80 mV stepped through a family of 3-s voltage steps from −110 mV to 60 mV and returned to a poststep potential of −100 mV. B: conductance-voltage (G/V) plots derived from tail currents for KCNQ5(+), KCNQ5(+) + BACE1(D289N), and KCNQ5(P369T). G/V plots for KCNQ5(+) and KCNQ5(+) + BACE1(D289N) are statistically indistinguishable. KCNQ5(P369T) is left-shifted relative to both KCNQ5(+) and KCNQ5(+) + BACE1(D289N). Single Boltzmann fitted values: KCNQ5(+) [voltage at half-maximal conductance (V50) = −10.1 mV, N = 10], KCNQ5(+) + BACE1(D289N) (V50= −18.4 mV, N = 5), KCNQ5(P369T) (V50 = −52.0 mV, N = 7). C: G/V plots for KCNQ5(+) + KCNQ3(+), KCNQ5(P369T) + KCNQ3(+), and KCNQ5(P369T) + KCNQ5(+). G/V plots for KCNQ5(P369T) + KCNQ3(+) and KCNQ5(P369T) + KCNQ5(+) are both significantly left-shifted from KCNQ5(+) + KCNQ3(+), demonstrating a dominant GOF effect by the mutant KCNQ5(P369T) subunit. Single Boltzmann fitted values: KCNQ5(+) + KCNQ3(+) (V50 = −8.9 mV, N = 8), KCNQ5(P369T) + KCNQ3(+) (V50 = −21.0 mV, N = 7), KCNQ5(P369T) + KCNQ5(+) (V50 = −20.8 mV, N = 12). G/Gmax, conductance normalized to maximal conductance; Vm, membrane potential.
WT KCNQ5 currents in HEK293 or CHO cells closely resembled those obtained by expression in Xenopus oocytes (33, 34). From a holding potential of −80 mV, slowly activating outward currents were elicited by 3-s step depolarizations, typical of KCNQ currents. WT KCNQ5 currents exhibited isochronic voltage dependence of activation fitted by a single Boltzmann function, with a half-maximal conduction-voltage relationship (V50) equal to −25.0 mV (Fig. 2, black).
Figure 2.

Human KCNQ5 channels carrying de novo missense mutations from patients with intellectual disability (ID)/epilepsy result in gain of function (GOF) expressed in HEK293 cells. Left: current traces evoked from HEK293 cells transfected with wild-type (WT) KCNQ5 (black) and KCNQ5 missense mutants (K289N, P369T, R443Q, H576R) (red), under voltage clamp from a holding potential of −80 mV, stepped through a family of 3-s voltage steps from −100 mV to 60 mV and returned to a poststep potential of −60 mV. Right: conductance/voltage (G/V) plots are shown for each construct, calculated from isochronic current measurements at the end of 3-s voltage steps and the Nernst potassium reversal potential based on the recording solutions. All KCNQ5 missense mutations (K289N, P369T, R443Q, H576R) resulted in channels negatively shifted in voltage dependence of activation toward hyperpolarizing potentials relative to WT (+). All transfections were performed in a background of cotransfected catalytically inactive BACE1(D289N) to assist channel expression levels (49) (but see text). Plots displayed as means ± SE, with fits to single Boltzmann functions and values for fitted voltage at half-maximal conduction (V50) and N (in brackets). G/Gmax, conductance normalized to maximal conductance; Vm, membrane potential.
All four KCNQ5 missense variants identified in this study produced homomeric channels that conducted outward currents dramatically shifted in voltage dependence of activation toward negative potentials, consistent with GOF. Fitted V50 values for these variants were K289N (−40.4 mV), R443Q (−41.6 mV), H567R (−50.0 mV), and P369T (−58.0 mV), corresponding to hyperpolarizing shifts of K289N (−15.4 mV), R443Q (−16.6 mV), H567R (−24.0 mV), and P369T (−33.0 mV) relative to WT KCNQ5 (Fig. 2, red). All missense variants, affecting residues dispersed throughout the transmembrane domains and CTD, produced GOF channels due to varying degrees of hyperpolarizing shifts in voltage dependence. Under current clamp, all GOF KCNQ5 variants exhibited significantly hyperpolarized membrane potentials ranging from −45.5 ± 3.1 mV (R443Q) to −59.5 ± 2.5 mV (P369T), relative to untransfected controls (−14.8 ± 4.1 mV) (see Fig. 7C, red).
Figure 7.

De novo KCNQ5 nonsense mutations R178X and R443X are loss-of-function (LOF) alleles. No functional currents were recorded from HEK293 cells transfected with R178X or R443X. A: examples of current traces from untransfected (HEK293) and GFP-, R178X-, and R443X-transfected cells. R178X- and R443X-transfected cells produced currents indistinguishable from untransfected and GFP-transfected controls. Currents evoked from a holding potential of −80 mV stepped through a family of 3-s voltage steps from −100 mV to 60 mV and a poststep potential of −100 mV. B: R178X and R443X current densities (blue) are indistinguishable from GFP-transfected controls (green). By contrast, wild-type (WT, gray) and all de novo KCNQ5 mutations (red and orange) expressed 10- to 20-fold higher current densities (I/C) compared with either GFP controls or these missense mutations. C: R178X and R443X exhibited membrane potentials (Vm) indistinguishable from untransfected and BACE1(D289N)- or GFP-transfected controls (∼−20 mV). By contrast, WT and all de novo KCNQ5 mutations produced cells with hyperpolarized Vm (−50 mV to −70 mV). Plots displayed as median and interquartile range (IQR). Pairwise Mann–Whitney tests for statistical significance: ***P < 0.0001, **P < 0.008, *P < 0.020; ns, nonsignificant.
Using similar expression and electrophysiological methods, four additional KCNQ5 missense mutations previously reported (47) to be associated with ID/epilepsy (V145G, L341I, P369R, S448I) were reexamined in HEK293 cells (Fig. 3). Voltage dependence of activation was examined under voltage clamp by conductance/voltage (G/V) plots derived from tail currents recorded at −100 mV, following voltage steps from a holding potential of −80 mV. WT KCNQ5 G/V plots were well fitted by a single Boltzmann function with a V50 equal to −20.3 mV (Fig. 3, black). Strong hyperpolarizing shifts in half-maximal conductance were observed from three variants, V145G (−55.3 mV), P369R (−61.4 mV), and S448I (−48.9 mV), and a slightly depolarized shift for L341I (−14.1 mV) located in S6 (Fig. 3, red). However, L341I, along with V145G and P369R were found to exhibit substantial constitutive fractions of maximal conductance (∼25%) at the holding potential of −80 mV, suggestive of an energetic perturbation between open and closed states favoring the open state, caused by these variants. Consistent with this interpretation, deactivation kinetics for V145G, L341I, and P369R variants were two- to fourfold slower than WT, whereas S448I was statistically indistinguishable from WT (Fig. 4).
Figure 3.

Previously reported de novo KCNQ5 mutations associated with intellectual disability (ID)/epilepsy [Lehman et al. (47)] result in channels with gain of function (GOF) expressed in HEK293 cells. Left: current traces evoked from HEK293 cells transfected with wild-type (WT) KCNQ5 (black) and KCNQ5 missense mutations (V145G, L341I, P369R, S448I) previously reported by Lehman et al. (47). Currents evoked under voltage clamp from a holding potential of −80 mV stepped through a family of 3-s voltage steps from −100 mV to 60 mV and returned to a poststep potential of −100 mV. Right: conductance/voltage (G/V) plots are shown for each construct, measured from tail currents at −100 mV. All mutations (V145G, P369R, S448I) resulted in channels shifted in voltage dependence of activation toward hyperpolarized potentials relative to WT (+), except for L341I, which exhibited a voltage at half-maximal conduction (V50) (−14.1 mV) slightly depolarized to WT (−20.3 mV). However, L341I, along with V145G and P369R, all exhibited significant constitutive conductance at the holding potential of −80 mV, evident from their G/V plots and instantaneous currents at the beginning of voltage steps. Both properties of hyperpolarized voltage dependence of activation (V145G, P369R, S448I) and constitutive conductance at the holding potential (V145G, L341I, P369R) contributed to GOF for all these mutations. Plots displayed as means ± SE, with fits to single Boltzmann functions and values for fitted V50 and N (in brackets). G/Gmax, conductance normalized to maximal conductance; Vm, membrane potential.
Figure 4.

Delayed deactivation kinetics contribute to gain of function (GOF) for de novo KCNQ5 mutations V145G, L341I, and P369R. A: examples of tail current traces recorded at −100 mV, following a 3-s activation voltage step to 0 mV for wild-type (WT), V145G, L341I, P369R, and S448I. Delayed deactivation results in GOF due to accumulation of open channels with repetitive depolarizations. Tail currents fitted with either single- or double-exponential functions (blue): Itail(t) = A1exp(−t/τ1) + A2exp(−t/τ2) + C (baseline offset). B: examples of exponential fits of tail currents (top), superimposed upon recorded tail current traces (gray) normalized to maximal current and baseline current at −100 mV (bottom). C: fitted taus of deactivation (τ1, τ2). V145G, L341I, and P369R fitted by the sum of 2 exponentials; WT and S448I fitted by single exponentials. The primary components of deactivation (τ1) for V145G, L341I, and P369R were 2- to 4-fold slower compared to WT, with no significant difference between WT and S448I. Plots displayed as medians and interquartile ranges (IQRs), with values for the median and N (in brackets) for each sample set. Pairwise Mann–Whitney tests for significance: ***P < 0.0001; ns, nonsignificant. D: the secondary fast component of deactivation (Α2, τ2) contributes to 10–20% of total tail currents for V145G, L341I, and P369R.
KCNQ5(P369T) Subunits Exert GOF in Heteromeric KCNQ5/3 Channels
KCNQ5/3 heteromeric channels are likely the predominant molecular species of KCNQ5-derived channels in the central nervous system (33, 34). Therefore, for heterozygous de novo KCNQ5 mutant alleles of clinical relevance, it is important to investigate the functional properties of homomeric and heteromeric channels assembled by combinations of mutant KCNQ5 subunits with WT KCNQ5 or WT KCNQ3 subunits [hereafter denoted as KCNQ5(+) and KCNQ3(+)]. For these studies, we focused on P369T identified in patient LR17-520. This variant produced an extremely negative shift in voltage dependence of activation as a homomeric channel and was associated with one of the most severely impaired neurological presentations in our patient cohort (Table 1, Fig. 2).
KCNQ5(P369T) was coexpressed with either KCNQ5(+) or KCNQ3(+) by acute transfections in CHO cells, followed by patch-clamp recordings. Homomeric KCNQ5(P369T) (Fig. 5, A and B, left, blue) displayed a robust hyperpolarized G/V plot relative to KCNQ5(+) (Fig. 5, A and B, left, black) [KCNQ5(P369T) V50 = −52 mV; KCNQ5(+) V50 = −10 mV], similar to currents observed in HEK293 cells. By comparison, currents expressed by 1:1 heteromeric combinations of KCNQ5(P369T) and KCNQ3(+) (Fig. 5, A and B, right, red) [KCNQ5(P369T)+KCNQ3(+); V50 = −20.9 mV] or KCNQ5(+) (Fig. 5, A and B, right, blue) [KCNQ5(P369T)+KCNQ5(+); V50 = −20.8 mV] exhibited smaller hyperpolarizing shifts in G/V relative to KCNQ5(+) + KCNQ3(+) (V50 = −8.9 mV) or KCNQ5(+) (V50 = −10 mV) controls (Fig. 5, A and C, black). However, only the negative G/V shift by KCNQ5(P369T) + KCNQ3(+) (Fig. 5, A and B, right, red) compared with KCNQ5(+)+KCNQ3(+) (Fig. 5, A and B, right, black) heteromeric currents rose to statistical significance (Fig. 5C; Supplemental Tables S5 and S6).
Expression of KCNQ3(+) alone failed to produce sufficient current that could be analyzed in CHO cells, consistent with previous reports (33, 34) (Supplemental Fig. S1). In Xenopus oocytes, previous studies observed that coexpression of KCNQ3(+) with KCNQ5(+) resulted in a four- to fivefold increase in current amplitude compared with KCNQ5(+) alone (33, 34). Surprisingly, in CHO cells cotransfection of KCNQ3(+) with KCNQ5(+) failed to increase current amplitudes (Supplemental Fig. S1). However, deactivation kinetics were dramatically accelerated threefold in KCNQ5/3 heteromeric currents (τ1 = 58 ms) compared with KCNQ5(+) currents (τ1 = 178 ms). Accordingly, tail currents from heteromeric KCNQ5(P369T) + KCNQ3(+) (τ1 = 336 ms) and homomeric KCNQ5(P369T) + KCNQ5(+) (τ1 = 219 ms) channels were slowed, relative to WT controls [KCNQ5/3 and KCNQ5(+), respectively] (Fig. 6).
Figure 6.

KCNQ5(P369T) subunits slow deactivation when coexpressed in homomeric KCNQ5 or heteromeric KCNQ5/3 channels. A: tail currents recorded from Chinese hamster ovary (CHO) cells transfected with KCNQ3(+) + KCNQ5(+) and KCNQ5(+) controls (black, left) compared with KCNQ3(+) + KCNQ5(P369T) and KCNQ5(+) + KCNQ(P369T) (red, right). Coexpression of KCNQ5(P369T) significantly slowed deactivation kinetics only when coexpressed with KCNQ3(+), compared with controls. Coexpression of KCNQ5(P369T) + KCNQ5(+) exhibited a trend toward slower deactivation relative to KCNQ5(+), but this trend did not reach statistical significance. Tail currents recorded at −100 mV under voltage clamp, following a family of 3-s voltage steps from −110 mV to 60 mV, from a holding potential of −80 mV. B: taus of deactivation (τ1, τ2) for tail currents from KCNQ5(+) (Q5), KCNQ5(+) + BACE1(D289N) (Q5+BACE1), KCNQ5(P369T) (P369T), KCNQ5(P369T) + KCNQ5(+) (P369T+Q5), KCNQ5(+) + KCNQ3(+) (Q5+Q3), and KCNQ5(P369T) + KCNQ3(+) (P369T+Q3) fitted to either single- or double-exponential functions: Itail(t)= A1exp(−t/τ1) + A2exp(−t/τ2) + C (baseline offset). Mean τ1 and τ2 values with SE shown for each condition, with N in brackets. Deactivation rates (τ1) between KCNQ5(+) and KCNQ5(+) + BACE1(D287N) were statistically indistinguishable. Wild-type (WT) KCNQ5/3 heteromeric channels [Q5(+) + Q3(+)] exhibited 3-fold accelerated deactivation (τ1 = 58 ms) relative to KCNQ5(+) homomeric channels (τ1 = 178 ms). Coexpression of KCNQ5(P369T) with KCNQ3(+) [P369T + Q3(+); τ1 = 336 ms] significantly slowed deactivation compared with WT KCNQ5/3 currents (τ1 = 58 ms). Coexpression of KCNQ5(P369T) with KCNQ5 [P369T + Q5(+); τ1 = 216 ms] produced tail currents with deactivation kinetics that were insignificantly different from KCNQ5(+) (τ1 = 178 ms). C: the secondary fast component of deactivation (A2, τ2) accounted for ∼3–20% of total tail currents, although most tail currents were well fitted by single exponentials. Pairwise Mann–Whitney test for significance: ***P < 0.001, **P < 0.002, *P < 0.05; ns, nonsignificant. WT KCNQ5 and WT KCNQ3 denoted by KCNQ5(+) and KCNQ3(+), respectively.
We conclude that KCNQ5(P369T) mutant subunits exert a dominant GOF effect when assembled in heteromeric KCNQ5/3 channels, shifting voltage dependence toward negative potentials and slowing deactivation by five- to sixfold. As experimentally examined, currents produced by KCNQ5(P369T) in combination with KCNQ5(+) subunits were not detectably different from those produced by homomeric KCNQ5(+) channels. Taken together, we conclude that KCNQ5(P369T) extends a dominant GOF effect in heteromeric channel combinations with KCNQ3(+) subunits, under assembly conditions mimicking those in heterozygous de novo KCNQ5 patients.
Nonsense KCNQ5 de Novo Variants Yield LOF
HEK293 cells expressing one of two truncation variants (R178X, R443X) did not express currents that were distinguishable from untransfected controls or cells expressing either GFP and or BACE1(D287N) alone (Fig. 7A). Baseline transfections with WT KCNQ5 and all missense variants of KCNQ5 examined exhibited ∼10- to 20-fold higher current density compared with GFP-transfected control cells (WT: 12.98 ± 1.48 vs. GFP: 1.48 ± 0.39 pA/pF) (Mann–Whitney, P = 0.0004) (Fig. 7B). By contrast, HEK293 cells expressing R178X failed to produce current densities significantly different from GFP controls (R178X: 0.80 ± 0.57 vs. GFP: 1.48 ± 0.39 pA/pF) (Mann–Whitney, P = 0.7619). Similarly, R443X-transfected cells also failed to produce current densities different from GFP controls (R443X: 0.81 ± 0.20 vs. GFP: 1.48 ± 0.39 pA/pF) (Mann–Whitney, P = 0.1807). Accordingly, both R178X- and R443X-transfected cells displayed significantly lower current densities compared with WT KCNQ5 cells [Mann–Whitney: P = 0.0028 (R178X); P < 0.0002 (R443X)] and were indistinguishable from endogenous HEK293 currents observed in untransfected and GFP controls.
LOF was further confirmed by recording resting membrane potential under current-clamp recording conditions. Under current clamp, expression of WT KCNQ5 significantly hyperpolarized resting membrane potentials compared with untransfected and BACE1(D289N)- or GFP-transfected controls (Fig. 7C). Untransfected HEK293 cells and GFP controls displayed resting membrane potentials near −20 mV, whereas BACE1(D289N)-transfected cells were slightly depolarized (0.6 ± 2.5 mV) compared with untransfected cells (−14.8 ± 4.1 mV) (Mann–Whitney, P = 0.0159). By contrast, WT KCNQ5 membrane potentials (−51.8 ± 2.9 mV) and all missense variants of KCNQ5 examined (−58 to −70 mV) were significantly hyperpolarized compared with GFP controls (−18.8 ± 7.2 mV) (Mann–Whitney, P < 0.005). As expected, neither R178X nor R443X cells were hyperpolarized (R178X: −17.83 ± 6.45 mV; R443X: −9.5 ± 2.7 mV) relative to controls, displaying membrane potentials indistinguishable from GFP controls [Mann–Whitney, P = 0.9358 (R178X); P = 0.1596 (R443X)]. Notably for R443X, despite a conceptual truncation that preserved all membrane-spanning segments, no evidence was observed for the expression of functional channels.
Severity of Human Pathologies Associates with Mutant KCNQ5 Channel Phenotypes
All six pediatric patients reported in this study had significant developmental and neurological deficits, but with clinical severities separable into two groups. One group presented with comparatively milder dysfunctions (Table 1, mutation groups: LOF mild and GOF mild). In this group of four patients (LR17-518, LR17-519, LR16-507, and LR16-508), two carried LOF (R178X, R443X; mutation group: LOF mild) and two GOF (K289N, R443Q; mutation group: GOF mild) mutations, the latter with currents mildly shifted in voltage dependence toward negative potentials (ΔV50 = ∼−15 mV relative to WT). All patients displayed mild developmental delays, walking at 1.5–2 yr and speaking first words at 2–3 yr, with moderate intellectual disability (IQ 66 and 50 in the 2 individuals with available data). Other clinical features included epilepsy (3 of 4) with onset at 1, 4, and 5 yr, autism (2 of 4), and mood swings in one child. EEGs showed centrotemporal or diffuse multifocal spikes (3 of 4).
The second group of two patients (Table 1, mutation group: GOF severe) exhibited severe dysfunctional presentations. Both patients (LR17-520, LR16-509; mutation group: GOF severe) carried strong GOF mutations (P369T, H576R), as defined by currents markedly shifted in voltage dependence toward negative potentials (ΔV50 = ∼−30 mV relative to WT). Both patients had severe global developmental delays, with limited language development in the oldest child, severe intellectual disability (too severe to evaluate for autism), early onsets of epilepsy at 10 and 22 mo that proved difficult to treat with medications, and striking mood swings or irritability. Both patients had severe feeding problems requiring placement of gastrostomy tubes, and one had opisthotonus with cortical visual impairment. Both patients also had marked regression of skills following the onset of epilepsy and brain MRI scans that showed mild diffuse volume loss. One of these patients died at 16 yr.
Generation and Confirmation of Kcnq5 LOF Mice by CRISPR
To investigate the pathogenic potential of KCNQ5 LOF, murine Kcnq5 was successfully targeted for deletional mutagenesis by CRISPR/Cas9, generating two independent LOF alleles (Fig. 8).
Figure 8.

Murine Kcnq5 loss-of-function (LOF) deletion alleles created by targeted Cas9/single guide RNA (sgRNA). A: site of Cas9/sgRNA targeted deletions in a diagram of the Kcnq5 subunit. A sgRNA was selected to target Cas9 to exon 5 encoding the NH2-terminal edge of the pore α-helix (P). Two targeted deletion mouse lines were recovered (Q5d1, Q5d2). B: genomic DNA chromatograms of wild-type (WT) and homozygous Kcnq5 deletion alleles (Q5d1, Q5d2) sequenced through the Cas9-targeted site. Targeted deletion of 37 base pairs (Q5d1) and 33 base pairs (Q5d2) observed, deleting the native 3′ splice site of exon 5 and the 5′ end of the following intron 5. C: DNA sequences and predicted open reading frames (ORFs) of Kcnq5 exon 5 for WT (Q5 WT, green ORF) and deletion alleles (Q5d1, blue ORF; Q5d2, yellow ORF), with the sgRNA shown in red. Both deletion alleles ablate the native donor splice site of exon 5 and generate predicted ORFs, which truncate the channel protein prematurely. These deletion alleles likely result in LOF from in vivo nonsense-mediated decay (see Fig. 9) (61).
To confirm Kcnq5 LOF in these targeted deletional mouse strains, qRT-PCR and Western blot analyses were performed. qRT-PCR assays with whole brain RNA using primers directed across the Kcnq5 exon 5/6 splice site revealed a selective 105- to 106-fold loss of Kcnq5 transcripts in both Q5d1 and Q5d2 deletion strains, without an appreciable effect on the abundance of Kcnq1–4 transcripts (Fig. 9A). Western blots with whole brain synaptosomal lysates detected a band with the predicted size of a single KCNQ5 subunit (∼103 kDa) in WT C57BL/6J brains with a commercial polyclonal antibody raised against the entire KCNQ5 polypeptide. By contrast, no detectable band of that size was observed in either Kcnq5 deletional strain (Q5d1, Q5d2) (Fig. 9B). Taken together, these results verified selective LOF of Kcnq5 in both Q5d1 and Q5d2 deletional strains, without appreciable compensatory regulation by Kcnq1–4.
Figure 9.

Kcnq5 Cas9-generated alleles are loss of function (LOF) by selective loss of mRNA and protein. Kcnq5 LOF mice exhibit late-onset seizures. A: quantitative RT-PCR (qRT-PCR) assessment of transcript abundance for Kcnq1–5 relative to Gapdh, with whole brain RNA from wild-type (WT) (BL6) and homozygous Kcnq5 LOF alleles (Q5d1, Q5d2). Both Kcnq5 LOF alleles selectively reduce Kcnq5 transcript abundance by ∼10,000-fold relative to WT, without appreciably altering the expression levels of Kcnq1-4. Each measurement plotted as mean ± SE, for N = 3 or 4. dCt = (sample Ct − GAPDH Ct). B: Western blot of membrane lysates from cerebral cortex from WT (BL6) and homozygous Kcnq5 LOF alleles (Q5d1, Q5d2) probed with a commercial anti-KCNQ5 antibody raised against full-length KCNQ5 peptide. KCNQ5 protein detected as an ∼103-kDa band in WT lysate, which is absent in lysates from both Kcnq5 LOF alleles. N = 3, with similar results. (See Supplemental Fig. S4 for full molecular mass range.) C: example of handling-induced tonic-clonic seizure observed in a Kcnq5 LOF adult. Neck and tail flexion observed at 2.20 (s), loss of standing posture at 3.73 (s). (See Supplemental Fig. S3, Supplemental Video.) D: summary of age of onset of handling-induced seizures observed for homozygous Kcnq5 LOF alleles outcrossed to C57BL/6J for 1–2 (N1/2) and 5 generations (N5). Onset of seizures occurred in mature adults of both LOF alleles at ∼25 postnatal wk with a penetrance of ∼20–24%, in N1/2 animals. Onset of seizures was significantly increased to ∼40 postnatal wk in N5 animals, without altering penetrance (23–37%). Plots displayed as median and interquartile range (IQR). Statistical significance determined by pairwise Mann–Whitney tests: *P = 0.0135, ***P < 0.0001.
Kcnq5 LOF Mice Exhibit Handling-Induced Seizures in Adulthood
Seizures were observed in both Kcnq5 LOF lines, homozygous for Q5d1 or Q5d2, during routine handling such as when cages were moved from housing racks to an observation hood and uncovered (Fig. 9C; Supplemental Video). Seizures were observed within 5 min of uncovering cages, presumably triggered by multisensory stimulation associated with handling. Both absence-type and myoclonic seizures were observed, with seizure episodes often consisting of combinations of both types. Absence-type seizures were characterized by a frozen posture and elevated breathing rate, during which time the animal became unresponsive to gentle prodding or manual repositioning. To varying degrees of severity, myoclonic seizures were observed, consisting of repetitive twitches of facial or neck muscles, forelimb clonus and face wiping, tail flexion, and whole body tonic-clonic convulsions. By video quantitation, seizure durations ranged from 30 to 197 s, with a mean of 70 ± 23 s (N = 7). These seizures were observed in adult animals, with an age of onset at ∼25 postnatal wk in homozygous LOF animals outcrossed to C57BL/6J 1–2 generations [Q5d1(N1/2): 27.7 ± 2.1 postnatal wk; Q5d2(N1/2): 22.9 ± 2.2 postnatal wk], with a penetrance of ∼20–24% (Fig. 9D). Seizures were retained in both LOF alleles outcrossed 5 generations to C57BL/6J, but with the age of seizure onset further delayed by ∼10–20 wk [Q5d1(N1/2) vs. Q5d1(N5): 37.1 ± 2.3 postnatal wk, Mann–Whitney, P = 0.0048; Q5d2(N1/2) vs. Q5d2(N5): 47.1 ± 3.6 postnatal wk, Mann–Whitney, P = 0.0003], without significantly altering penetrance (∼23–37%) (Fig. 9D). Handling-induced seizures were never observed under similar conditions in a control population of C57BL/6J animals (39 males and 35 females) monitored until 26–73 postnatal wk.
Kcnq5 LOF Mice Exhibit Abnormal Interictal Cortical EEGs and Thermal-Induced Seizures
To examine interictal cortical activity, cortical electroencephalograms (EEGs) were recorded from the dorsal neocortical surface of WT and Kcnq5 LOF animals (3 Q5d1 and 4 Q5d2; ages P60 to P230; 6 males and 1 female), during the restful awake state. Cortical EEGs from Kcnq5 LOF animals revealed significantly elevated persistent electrical activity in the high alpha and beta bands (8–20 Hz) compared with WT C57BL/6J control animals (Fig. 10, A and B). Examination of Kcnq5 LOF EEGs at higher time resolution revealed numerous epileptiform events including generalized sharp wave spikes, polyspike discharges, and generalized synchronous high-amplitude oscillations across left and right cortices during interictal periods, not present in WT control animals (Fig. 10C). In addition, mild elevation of core body temperature (39–41°C) triggered seizures in Kcnq5 LOF mice, suggesting susceptibility to thermal-induced seizures reminiscent of Dravet syndrome mice carrying a hemizygous loss of SCN1A voltage-gated sodium channels (Fig. 10, D and E) (54).
Figure 10.

Kcnq5 loss of function (LOF) mice exhibit abnormal interictal electroencephalographs (EEGs) and thermal-induced seizures. A: power spectral density plots of cortical EEGs from C57BL/6J [wild type (WT)] and Kcnq5 LOF adult mice, recorded during the resting awake state for 2 h. Kcnq5 LOF mice exhibited persistent higher power densities at high frequency bands (>8 Hz) compared with WT. f, frequency. B: summary of EEG power density as a function of frequency (f) for WT and Kcnq5 LOF mice. Significant enhancement of power densities for frequencies >8 Hz for Kcnq5 LOF mice compared with WT, consistent with increased epileptiform activity. Plots displayed as mean ± SE (envelopes); N = 7. C: example of EEGs from a Kcnq5 LOF mouse exhibiting epileptiform spikes (asterisks), polyspikes, and generalized high-amplitude oscillations and polyspikes. D: EEGs recorded at baseline (24°C) and during thermal-induced seizure (40°C) from a Kcnq5 LOF mouse. E: summary of thermal-induced seizure susceptibility for WT and Kcnq5 LOF mice. Seizure susceptibility plotted as fraction of animals that seized as a function of core body temperature. Approximately 50% of Kcnq5 LOF animals exhibited a thermal-induced seizure at 40.5°C compared with 0% of WT animals. N = 7 (combined data from 4 Q5d1 and 3 Q5d2 animals).
Taken together, these in vitro molecular characterizations and in vivo genetic results provide independent lines of corroborative evidence for a strong genetic association between clinical KCNQ5 variants and human KCNQ5 channelopathies.
DISCUSSION
Neurodevelopmental Severity of KCNQ5 Variant Phenotypes Associates with the Degree of Altered GOF Channel Gating
We report six new de novo variants in KCNQ5 identified in individuals with intellectual disability (ID) and epilepsy, four missense and two nonsense variants. The two nonsense variants (R178X, R443X) were nonfunctional, as assayed by heterologous expression in HEK293 cells. Since both nonsense mutations occur in exons 5′ to the last KCNQ5 exon, the entire transcripts from both of these nonsense alleles are likely to be effectively degraded by the cellular quality control mechanism of nonsense-mediated decay (NMD) (61), resulting in hemizygous loss of function (LOF). However, failure of normal trafficking to the plasma membrane may also contribute to LOF of KCNQ5(R443X), for rare transcripts that escape NMD (Supplement SII). Notably, this result contrasts with the three missense variants (V145G, L341I, S448I) reported by Lehman et al. (47) as LOF, but with subtle alterations in gating properties and considerable currents, as reported by heterologous expression in Xenopus oocytes. Our reexamination of these missense variants in HEK293 cells revealed that all four missense variants (V145G, L341I, S448I, P369R) reported by Lehman et al. (47) generated functional channels with gain-of-function (GOF) properties due to negatively shifted voltage dependence of activation (V145G, P369R, S448I) and slowed deactivation kinetics (V145G, L341I, P369R). Moreover, all four missense mutations that we report here (K289N, R443Q, P369T, H576R) resulted in GOF channels with hyperpolarizing shifts in voltage dependence of activation. Together all eight de novo missense variants produced GOF channels. This discovery is surprising, as the majority of human pathological missense mutations in the related KCNQ1–4 genes result from LOF (Refs. 8, 62, but see Refs. 63–65).
Combining our data with two prior reports (47, 66), 11 individuals with variants of KCNQ5 have now been reported associated with ID and epilepsy, with phenotypes separable into two groups based on phenotypic severity. The first group of seven affected individuals (64%) had mild developmental delay, late-onset moderate ID, and normal brain imaging studies (R178X, V133X, R443X, V145G, L341I, K289N, R443Q). Seizures were documented in four of seven individuals (57%) with onsets at 1–5 yr, which were well controlled with medications (R178X, V133X, R443X, R443Q). Functional expression studies classified these KCNQ5 variants as LOF [R178X, R443X, V133X (66) from exon 2–11 skipping] or mild GOF due to little to modest negative shifts in voltage dependence (L341I, K289N, R443Q). The sole outlier to this functional characterization scheme is V145G, which we observe to exhibit a strong GOF negative shift in voltage dependence (ΔV50= −35 mV) relative to WT in HEK293 cells, whereas Lehman et al. (47) report a positive shift in voltage-dependent (ΔV50 = 7.4 mV) relative to WT in Xenopus oocytes.
The second group of four affected individuals (36%) [S448I, P369R (47); P369T, H576R] all had severe developmental encephalopathies associated with global developmental delay, severe ID with obvious neurological and behavioral regression in childhood, mood disorders, poor feeding leading to tube feeding in childhood, and progressive diffuse volume loss upon brain MRI scans. All four individuals had seizures, including infantile spasms in two individuals with onsets at 5–24 mo. All progressed to present with multiple seizure types with poor responsiveness to medications, and one died at 16 yr. Functional expression studies of these four KCNQ5 mutations [S448I, P369R (47); P369T, H576R] demonstrated large negative shifts in voltage dependence (ΔV50 = −25 to −41 mV relative to WT, measured in HEK293 cells), supporting severe GOF changes in channel gating function. These observations support an association between the magnitude of negative shifts in voltage dependence resulting in channel GOF and the severity of clinical phenotypes due to KCNQ5 variants. Functional in vitro characterization of KCNQ5 channel variants may thus provide a potential prognostic guide to the severity and progression of associated neurodevelopmental phenotypes. However, more case studies will be required to confirm the validity of this generalization.
Possible molecular mechanisms of KCNQ5 GOF channels.
Although all eight missense variants in KCNQ5 (V145G, K289N, L341I, P369T, P369R, R443Q, S448I, H576R) produced GOF channels resulting from hyperpolarizing shifts in voltage dependence of activation (V50) or slowed deactivation, these variants were distributed widely throughout the channel subunit. Because of the highly allosteric nature of channel gating (67), shifts in V50 are likely caused by multiple molecular mechanisms, as discussed below for the variants described in this study.
KCNQ5(V145G) is located at the extracellular edge of S1, at the homologous site of two human variants of KCNQ1 (S140G and V141M) associated with hereditary atrial fibrillations, in combination with the KCNE1 β-subunit (68–71). This portion of the KCNQ1 subunit directly interacts with gating charges in the S4 voltage sensor domain either with or without KCNE1 (70), to slow the relaxation of S4 gating currents upon repolarization and the deactivation kinetics of ionic tail currents. Voltage dependence of activation is negatively shifted by these KCNQ1 variants in the presence of KCNE1 (71). An analogous interaction may occur in KCNQ5 between V145G and homologous S4 gating charges, underlying the negative shifts in voltage dependence and slowed deactivation kinetics we observed.
KCNQ5(K289N) is located at the extracellular edge of S5. This mutation neutralizes a highly conserved basic residue, flanked by two conserved acidic residues (E288, D290), in the “turret” domain immediately preceding the pore α-helix. By analogy with site-directed cysteine mutagenesis analysis in Shaker potassium channels, this region has been shown to physically interact with residues at the extracellular edge of S4 in the voltage-sensing domain (VSD) during voltage-dependent activation, allowing for state-dependent disulfide cross-linkage (72). In addition, Larsson and colleagues (73) have shown that charged residues in this turret region electrostatically tune the pH dependence of the gating effect of KCNE1 in KCNQ1/KCNE1 heteromeric channels during outward S4 movements. These studies suggest that a similar interaction may underlie KCNQ5(K289N) GOF, perhaps through a direct electrostatic interaction with S4 gating residues or an indirect effect with lipid head groups in the outer membrane bilayer.
KCNQ5(L341I) is located immediately adjacent to the conserved “PAG” motif of S6, which undergoes a critical expansion upon the gating of KCNQ channels. Cryo-EM studies comparing the structures of KCNQ1/CaM/KCNE3 in the closed conformation and KCNQ1/CaM/KCNE3/PIP2 in the open conformation revealed a remarkable extension and rotation of the S6 α-helix upon transition from the closed to open state, resulting in a near 180° rotation of the Helix-A/Helix-B/CaM complex, and the lateral dilation of the intracellular gate located at the base of S6 (74–76). This gating scheme is also seen in a recent cryo-EM study of KCNQ4 complexed with PIP2 and the KCNQ channel activator ML213 (76) and is consistent with additional intermediate structures of KCNQ4 and KCNQ2 in preopen late closed state conformations (77, 78). This dramatic conformational transition pivots at P342, immediately next to L341I, and is thought to occur after outward voltage-dependent movements of S4. Conceivably, steric interactions between L341 and neighboring residues may be influenced by the L341I variant, increasing the energy barrier between open-to-closed state transitions, resulting in slowing of deactivation without shifting voltage dependence of activation.
KCNQ5(P369T) as well as the previously reported KCNQ5(P369R) (47) are located within the A-helix of the COOH-terminal tail. Crystallographic and cryo-EM structures reveal that the A-helix and B-helix in each subunit together form an antiparallel coil/coil structure that provides a flexible docking site for Ca2+/calmodulin (58). Disruption of A-helix residues that might be critical for A/B-helical interaction or binding by Ca2+/calmodulin may alter voltage-dependent gating, perhaps by altering steric interactions between calmodulin and the overlying VSD (57). In addition, P369 is located immediately adjacent to a conserved cluster of positively charged lysine/arginine residues that forms one of several phosphatidylinositol 4,5-bisphosphate (PIP2) binding sites located at the protein/inner leaflet bilayer interface, critical for channel function (76). PIP2 binding at this site is essential for coupling either voltage-dependent movements of the VSD or binding of the opener retigabine in a conserved pocket in S5 (W270 in KCNQ5) to conformational changes that gate the channel open (79, 80). Mutations at P369 are thus well positioned to influence PIP2 binding and voltage-dependent gating.
KCNQ5(R443Q) and KCNQ5(S448I) are located within the linker sequence connecting the A-helix to the B-helix, but not within a putative PIP2-binding region (81). This region shows relatively low sequence conservation, variable lengths between different KCNQ paralogs, and alternative splicing that yields several KCNQ5 splice variants with different lengths of A/B-helix linkers (33, 34). No structural information is available, because of the presumed high conformational flexibility of this region precluding structural resolution. The Helix-A/B linker is nonessential, since deletion of this linker still yields functional channels (58). However, mutagenesis experiments have identified a conserved poly-cationic site within the linkers of KCNQ2 and KCNQ3, responsible for high PIP2 sensitivity (81). How this site may alter V50 remains unclear. However, this region may act synergistically with the PIP2 binding sites described above by “funneling” PIP2 from distal surrounding membrane to more centrally located PIP2 binding sites (74, 82) at the cytosolic channel protein-lipid interface defined by residues within the S2-S3 and S4-S5 linkers and post-S6 region, which have been demonstrated to couple VSD activation to channel gating (79, 80).
KCNQ5(H576R) is mechanistically the most intriguing. This mutation is predicted to disrupt the C-helix, which together with the D-helix are thought to hold the COOH-terminal tails of KCNQ1–5 subunits together via homomeric (C- and D-helixes) and compatible heteromeric (D-helix) parallel α-helical tetrameric structures, based on crystallographic studies (57, 60). The D-helix is the major determinant of normal subunit/subunit assembly based on domain swapping experiments (83), but the functional role of the C-helix is poorly understood (59). The dramatic GOF observed with H576R suggests that stability of the C-helix α-helical structure may be a significant and unexpected determinant of gating, perhaps by regulating the overall conformational flexibility of the assembled COOH-terminal tails. Lateral contacts between C-helical side residues and the N-lobe of preassembled calmodulin may provide a physical substrate for allosteric coupling to the gating mechanism (57, 74, 76).
Based on coexpression experiments with KCNQ5(P369T) and WT KCNQ5 or KCNQ3, we observed a significant dominant GOF effect only for heteromeric channels assembled by combinations of KCNQ5(P369T) and KCNQ3 subunits. This dominant GOF effect was observed as both a hyperpolarizing shift in G/V plots and a five- to sixfold slowing of deactivation relative to WT KCNQ5/3. Coexpression of KCNQ5(P369T) with WT KCNQ5 appeared to mitigate the severity of GOF observed from homotetrameric KCNQ5(P369T) channels using a 1:1 ratio of cotransfected plasmids, although other cotransfection ratios were not examined. However, KCNQ5/3 heteromeric channels are thought to be the dominant molecular species of KCNQ5-derived channels in the central nervous system (31, 33, 34). Based on our results and assuming random assortment and assembly of subunits expressed from one de novo GOF copy of KCNQ5, one WT copy of KCNQ5, and two WT copies of KCNQ3, our data best fit a model in which at least 25% of all channels derived from KCNQ5 and KCNQ3 subunits are conferred GOF by the incorporation of one or more GOF KCNQ5 subunits (Supplemental Fig. S5). Therefore, for patients carrying a heterozygous de novo KCNQ5(P369T) mutation, we would expect the consequences of GOF to be manifested primarily by heteromeric KCNQ5/3 channels and not homomeric KCNQ5 channels, assembled with one or more mutant KCNQ5 subunits. We speculate that the other KCNQ5 GOF missense mutations characterized in this study may behave similarly, although this remains to be tested experimentally.
Apparent Discrepancies with Previous KCNQ5 Mouse Model
In contrast to the proepileptic phenotype that we observe in both of our Kcnq5 LOF lines, seizures were not reported in the Kcnq5 dominant-negative mouse line created by engineering a nonconducting mutation into the native Kcnq5(+) locus by homologous recombination (36, 42). Despite a stronger mutant phenotype expected from suppression of both homomeric KCNQ5 and heteromeric (KCNQ5/3, KCNQ5/4) channels, overt seizures were not reported in this line.
The seizure phenotype we observed was characterized by late onset and modest penetrance compared with other mouse models of KCNQ2 and KCNQ3 LOF (84–87) and thus may been overlooked in previous studies. In addition, our Kcnq5 LOF seizure phenotype showed evidence for strain dependence, a feature commonly observed in other mouse genetic models for epilepsy. For example, mice carrying epileptogenic knockin alleles of Kcnq2 or Kcnq3 exhibit more severe seizure phenotypes in an FVB genetic background compared with C57BL/6J (85, 88). Likewise, mouse models for Dravet syndrome, due to hemizygous loss of Scn1a, also show a strong strain dependence for seizures, with more severe phenotypes in a C57BL/6J compared to 129/SvJ background (88). We suggest that background strain differences between mice generated in this study (Black Swiss and C57BL/6J), and that previously reported by Tzingounis et al. (36) (129/SvJ) may also contribute to why these authors did not report an epileptic phenotype.
Future follow-up studies will be important to fully characterize the phenotypes of heterozygous Kcnq5 LOF mice, which would more closely model the genetic background of patients carrying heterozygous de novo KCNQ5 LOF mutations. It is reasonable to expect that the epileptiform phenotypes that we see in homozygous Kcnq5 LOF mice would be mitigated in heterozygous LOF mice. Additional generation and phenotypic characterization of Kcnq5 GOF mice will provide essential complementary data for interpreting murine models of KCNQ5 channelopathies.
Potential Basis of KCNQ5 Neurodevelopmental Disorders
Neurodevelopmental disorders have been reported with mutations in multiple potassium channel genes, notably KCNJ2 (Kir2.1), KCNJ10 (Kir4.1), KCNT1 (Slo2.2/SLACK), and KCNH1 (EAG1) (3, 89–92), suggesting that defects in electrical excitability can lead to abnormal brain development as well as epilepsy. Indeed, direct in vivo manipulation of membrane potential in neuronal progenitors by exogenous expression of GOF potassium channels demonstrates pronounced effects of silencing electrical activity on the incorporation and differentiation of neuronal progenitors into mature cortical circuits (93). Similar cell-autonomous manipulations of intrinsic excitability have been shown to alter the survival of postmitotic interneurons in postnatal cortical circuits by altering the normal extent of activity-dependent developmental culling through apoptosis (94). We hypothesize that KCNQ5 variants, particularly strong GOF mutations, may exert a similar neurodevelopmental influence on developing brains, independent of seizure-related maladaptive effects. Neurodevelopmental KCNQ5 variants may thus alter normal electrical activity in neuronal progenitors and shift their developmental trajectories, resulting in sensorimotor or cognitive defects in the mature brain. Collectively, the demonstration of both LOF and GOF KCNQ5 channelopathies suggests that the clinical phenotypes of these two patient groups are likely due to complex underlying etiologies and may thus necessitate tailoring different therapeutic interventions, informed by further circuit-based and neurodevelopmental investigations.
DATA AVAILABILITY
All patient genetic information into the Leiden Open Variant Database (LOVD): https://databases.lovd.nl/shared/variants/KCNQ5/unique.
SUPPLEMENTAL DATA
Supplemental Materials: https://www.doi.org/10.6084/m9.figshare.17077898.
GRANTS
This study was funded by the National Institutes of Health under National Institute of Neurological Disorders and Stroke and National Heart, Lung, and Blood Institute Grants R01NS092772 (W.B.D.), R01HL126523 (J.-M.R.), R01NS102796 (F.K.K.), and R01NS099027 (K.J.M.) and a grant from Citizens United for Research in Epilepsy (CURE) (F.K.K.).
DISCLOSURES
D.N.S. and K.L.H were employed by Ambry Genetics during this study. J.-M. R.is an editor of the Journal of Neurophysiology and was not involved and did not have access to information regarding the peer review process or final disposition of this article. An alternate editor oversaw the peer review and decision-making process for this article. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
Jan-Marino Ramirez is an editor of Journal of Neurophysiology and was not involved and did not have access to information regarding the peer-review process or final disposition of this article. An alternate editor oversaw the peer-review and decision-making process for this article.
AUTHOR CONTRIBUTIONS
A.D.W., P.W., T.A.Z., W.B.D., and F.K.K. and conceived and designed research; A.D.W., P.W., T.A.Z., A.M.B., N.S., M.H.W., H.J.S., A.P.A.S., J.S.V. S.A.d., M.W.W., T.K., D.N.S., K.L.H., A.B., V.F.W., D.R.-B., E.B., J.J.M., and F.K.K. performed experiments; A.D.W., W.B.D., and F.K.K. analyzed data; A.D.W. and F.K.K. interpreted results of experiments; A.D.W. and F.K.K. prepared figures; A.D.W. drafted manuscript; A.D.W. and J.-M.R. edited and revised manuscript; A.D.W., K.J.M., J.-M.R., and F.K.K. approved final version of manuscript.
ENDNOTE
At the request of the authors, readers are herein alerted to the fact that additional materials related to this manuscript may be found at https://databases.lovd.nl/shared/variants/KCNQ5/unique. These materials are not a part of this manuscript and have not undergone peer review by the American Physiological Society (APS). APS and the journal editors take no responsibility for these materials, for the website address, or for any links to or from it.
ACKNOWLEDGMENTS
We thank the patients’ families for important contributions to our work on this disorder. We gratefully acknowledge Dr. Steve Smith for advice with Western blots, Dr. Manuel Covarrubias for advice with transfections of cultured cell lines, and Dr. Eric Turner for use of laboratory equipment. Drs. John Welsh and Lawrence Salkoff provided critical comments on the manuscript. BioRender (www.biorender.com) and VectorStock (www.VectorStock.com) provided stock images for the graphical abstract, which are published with permission.
REFERENCES
- 1.D’Adamo MC, Catacuzzeno L, Di Giovanni G, Franciolini F, Pessia M. K+ channelepsy: progress in the neurobiology of potassium channels and epilepsy. Front Cell Neurosci 7: 134, 2013. [Erratum in Front Cell Neurosci 8: 9, 2014]. doi: 10.3389/fncel.2013.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet 101: 664–685, 2017. doi: 10.1016/j.ajhg.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kessi M, Chen B, Peng J, Tang Y, Olatoutou E, He F, Yang L, Yin F. Intellectual disability and potassium channelopathies: a systematic review. Front Genet 11: 614, 2020. doi: 10.3389/fgene.2020.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexander SP, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA; CGTP Collaborators. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: voltage-gated ion channels. Br J Pharmacol 174, Suppl 1: S160–S194, 2017. doi: 10.1111/bph.13884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei A, Jegla T, Salkoff L. Eight potassium channel families revealed by the C. elegans genome project. Neuropharmacology 35: 805–829, 1996. doi: 10.1016/0028-3908(96)00126-8. [DOI] [PubMed] [Google Scholar]
- 6.Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol 156: 1185–1195, 2009. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 1: 21–30, 2000. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- 8.Soldovieri MV, Miceli F, Taglialatela M. Driving with no brakes: molecular pathophysiology of Kv7 potassium channels. Physiology (Bethesda) 26: 365–376, 2011. doi: 10.1152/physiol.00009.2011. [DOI] [PubMed] [Google Scholar]
- 9.Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283: 673–676, 1980. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- 10.Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282: 1890–1893, 1998. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 11.Yang WP, Levesque PC, Little WA, Conder ML, Ramakrishnan P, Neubauer MG, Blanar MA. Functional expression of two KvLQT1-related potassium channels responsible for an inherited idiopathic epilepsy. J Biol Chem 273: 19419–19423, 1998. doi: 10.1074/jbc.273.31.19419. [DOI] [PubMed] [Google Scholar]
- 12.Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 314: 1454–1457, 2006. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang H, Craciun LC, Mirshahi T, Rohács T, Lopes CM, Jin T, Logothetis DE. PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37: 963–975, 2003. doi: 10.1016/S0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]
- 14.Alaimo A, Villarroel A. Calmodulin: a multitasking protein in Kv7.2 potassium channel functions. Biomolecules 8: 57, 2018. doi: 10.3390/biom8030057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wen H, Levitan IB. Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J Neurosci 22: 7991–8001, 2002. doi: 10.1523/JNEUROSCI.22-18-07991.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yus-Najera E, Santana-Castro I, Villarroel A. The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J Biol Chem 277: 28545–28553, 2002. doi: 10.1074/jbc.M204130200. [DOI] [PubMed] [Google Scholar]
- 17.Gamper N, Zaika O, Li Y, Martin P, Hernandez CC, Perez MR, Wang AY, Jaffe DB, Shapiro MS. Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. EMBO J 25: 4996–5004, 2006. doi: 10.1038/sj.emboj.7601374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HJ, Jeong MH, Kim KR, Jung CY, Lee SY, Kim H, Koh J, Vuong TA, Jung S, Yang H, Park SK, Choi D, Kim SH, Kang K, Sohn JW, Park JM, Jeon D, Koo SH, Ho WK, Kang JS, Kim ST, Cho H. Protein arginine methylation facilitates KCNQ channel-PIP2 interaction leading to seizure suppression. eLife 5: e17159, 2016. doi: 10.7554/eLife.17159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manville RW, Papanikolaou M, Abbott GW. Direct neurotransmitter activation of voltage-gated potassium channels. Nat Commun 9: 1847, 2018. doi: 10.1038/s41467-018-04266-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H. AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci 6: 564–571, 2003. doi: 10.1038/nn1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295: 496–499, 2002. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 22.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 384: 80–83, 1996. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 23.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 12: 17–23, 1996. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 24.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KVLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature 384: 78–80, 1996. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 25.Chouabe C, Neyroud N, Richard P, Denjoy I, Hainque B, Romey G, Drici MD, Guicheney P, Barhanin J. Novel mutations in KvLQT1 that affect Iks activation through interactions with Isk. Cardiovasc Res 45: 971–980, 2000. doi: 10.1016/S0008-6363(99)00411-3. [DOI] [PubMed] [Google Scholar]
- 26.Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Fauré S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 15: 186–189, 1997. doi: 10.1038/ng0297-186. [DOI] [PubMed] [Google Scholar]
- 27.Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science 279: 403–406, 1998. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- 28.Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet 18: 53–55, 1998. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- 29.Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales TO, Peiffer A, Anderson VE, Leppert M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet 18: 25–29, 1998. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 30.Kubisch C, Schroeder BC, Friedrich T, Lütjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96: 437–446, 1999. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- 31.Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, Brown DA. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci 23: 5012–5019, 2003. doi: 10.1523/JNEUROSCI.23-12-05012.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El-Amraoui A, Petit C, Jentsch TJ. KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci USA 97: 4333–4338, 2000. doi: 10.1073/pnas.97.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem 275: 22395–22400, 2000. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- 34.Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem 275: 24089–24095, 2000. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- 35.Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 563: 72–78, 2018. doi: 10.1038/s41586-018-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tzingounis AV, Heidenreich M, Kharkovets T, Spitzmaul G, Jensen HS, Nicoll RA, Jentsch TJ. The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc Natl Acad Sci USA 107: 10232–10237, 2010. doi: 10.1073/pnas.1004644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yus-Nájera E, Muñoz A, Salvador N, Jensen BS, Rasmussen HB, Defelipe J, Villarroel A. Localization of KCNQ5 in the normal and epileptic human temporal neocortex and hippocampal formation. Neuroscience 120: 353–364, 2003. doi: 10.1016/s0306-4522(03)00321-x. [DOI] [PubMed] [Google Scholar]
- 38.Cooper EC. Made for “anchorin”: Kv7.2/7.3 (KCNQ2/KCNQ3) channels and the modulation of neuronal excitability in vertebrate axons. Semin Cell Dev Biol 22: 185–192, 2011. doi: 10.1016/j.semcdb.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci 26: 2599–2613, 2006. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caminos E, Garcia-Pino E, Martinez-Galan JR, Juiz JM. The potassium channel KCNQ5/Kv7.5 is localized in synaptic endings of auditory brainstem nuclei of the rat. J Comp Neurol 505: 363–378, 2007. doi: 10.1002/cne.21497. [DOI] [PubMed] [Google Scholar]
- 41.Huang H, Trussell LO. KCNQ5 channels control resting properties and release probability of a synapse. Nat Neurosci 14: 840–847, 2011. doi: 10.1038/nn.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fidzinski P, Korotkova T, Heidenreich M, Maier N, Schuetze S, Kobler O, Zuschratter W, Schmitz D, Ponomarenko A, Jentsch TJ. KCNQ5 K+ channels control hippocampal synaptic inhibition and fast network oscillations. Nat Commun 6: 6254, 2015. doi: 10.1038/ncomms7254. [DOI] [PubMed] [Google Scholar]
- 43.Greenwood IA, Ohya S. New tricks for old dogs: KCNQ expression and role in smooth muscle. Br J Pharmacol 156: 1196–1203, 2009. doi: 10.1111/j.1476-5381.2009.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jepps TA, Olesen SP, Greenwood IA. One man’s side effect is another man's therapeutic opportunity: targeting Kv7 channels in smooth muscle disorders. Br J Pharmacol 168: 19–27, 2013. doi: 10.1111/j.1476-5381.2012.02133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mani BK, Robakowski C, Brueggemann LI, Cribbs LL, Tripathi A, Majetschak M, Byron KL. Kv7.5 potassium channel subunits are the primary targets for PKA-dependent enhancement of vascular smooth muscle Kv7 currents. Mol Pharmacol 89: 323–334, 2016. doi: 10.1124/mol.115.101758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manville RW, van der Horst J, Redford KE, Katz BB, Jepps TA, Abbott GW. KCNQ5 activation is a unifying molecular mechanism shared by genetically and culturally diverse botanical hypotensive folk medicines. Proc Natl Acad Sci USA 116: 21236–21245, 2019. doi: 10.1073/pnas.1907511116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lehman A, Thouta S, Mancini GM, Naidu S, van Slegtenhorst M, McWalter K, Person R, Mwenifumbo J, Salvarinova R; CAUSES Study; EPGEN Study; Guella I, McKenzie MB, Datta A, Connolly MB, Kalkhoran SM, Poburko D, Friedman JM, Farrer MJ, Demos M, Desai S, Claydon T. Loss-of-function and gain-of-function mutations in KCNQ5 cause intellectual disability or epileptic encephalopathy. Am J Hum Genet 101: 65–74, 2017. doi: 10.1016/j.ajhg.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neveling K, Feenstra I, Gilissen C, Hoefsloot LH, Kamsteeg EJ, Mensenkamp AR, et al. A post-hoc comparison of the utility of Sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat 34: 1721–1726, 2013. doi: 10.1002/humu.22450. [DOI] [PubMed] [Google Scholar]
- 49.Hessler S, Zheng F, Hartmann S, Rittger A, Lehnert S, Völkel M, Nissen M, Edelmann E, Saftig P, Schwake M, Huth T, Alzheimer C. b-Secretase BACE1 regulates hippocampal and reconstituted M-currents in a beta-subunit-like fashion. J Neurosci 35: 3298–3311, 2015. doi: 10.1523/JNEUROSCI.3127-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ponce A, Castillo A, Hinojosa L, Martinez-Rendon J, Cereijido M. The expression of endogenous voltage-gated potassium channels in HEK293 cells is affected by culture conditions. Physiol Rep 6: e13663, 2018. doi: 10.14814/phy2.13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu SP, Kerchner GA. Endogenous voltage-gated potassium channels in human embryonic kidney (HEK293) cells. J Neurosci Res 52: 612–617, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 52.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823, 2013. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nichols J, Ying QL. Derivation and propagation of embryonic stem cells in serum- and feeder-free culture. Methods Mol Biol 329: 91–98, 2006. doi: 10.1385/1-59745-037-5:91. [DOI] [PubMed] [Google Scholar]
- 54.Catterall WA. Dravet syndrome: a sodium channel interneuronopathy. Curr Opin Physiol 2: 42–50, 2018. doi: 10.1016/j.cophys.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalume F, Oakley JC, Westenbroek RE, Gile J, de la Iglesia HO, Scheuer T, Catterall WA. Sleep impairment and reduced interneuron excitability in a mouse model of Dravet syndrome. Neurobiol Dis 77: 141–154, 2015. doi: 10.1016/j.nbd.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, Catterall WA. Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain 138: 2219–2233, 2015. doi: 10.1093/brain/awv142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun J, MacKinnon R. Cryo-EM structure of a KCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell 169: 1042–1050.e9, 2017. doi: 10.1016/j.cell.2017.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang A, Abderemane-Ali F, Hura GL, Rossen ND, Gate RE, Minor DL Jr.. A calmodulin C-lobe Ca2+-dependent switch governs Kv7 channel function. Neuron 97: 836–852.e6, 2018. doi: 10.1016/j.neuron.2018.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haitin Y, Wiener R, Shaham D, Peretz A, Cohen EB, Shamgar L, Pongs O, Hirsch JA, Attali B. Intracellular domains interactions and gated motions of IKS potassium channel subunits. EMBO J 28: 1994–2005, 2009. doi: 10.1038/emboj.2009.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Howard RJ, Clark KA, Holton JM, Minor DL Jr.. Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron 53: 663–675, 2007. doi: 10.1016/j.neuron.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Popp MW, Maquat LE. The dharma of nonsense-mediated mRNA decay in mammalian cells. Mol Cells 37: 1–8, 2014. doi: 10.14348/molcells.2014.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miceli F, Soldovieri MV, Iannotti FA, Barrese V, Ambrosino P, Martire M, Cilio MR, Taglialatela M. The voltage-sensing domain of Kv7.2 channels as a molecular target for epilepsy-causing mutations and anticonvulsants. Front Pharmacol 2: 2, 2011. doi: 10.3389/fphar.2011.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miceli F, Soldovieri MV, Ambrosino P, De Maria M, Migliore M, Migliore R, Taglialatela M. Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J Neurosci 35: 3782–3793, 2015. doi: 10.1523/JNEUROSCI.4423-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Millichap JJ, Miceli F, De Maria M, Keator C, Joshi N, Tran B, Soldovieri MV, Ambrosino P, Shashi V, Mikati MA, Cooper EC, Taglialatela M. Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant. Epilepsia 58: e10–e15, 2017. doi: 10.1111/epi.13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mulkey SB, Ben-Zeev B, Nicolai J, Carroll JL, Grönborg S, Jiang YH, Joshi N, Kelly M, Koolen DA, Mikati MA, Park K, Pearl PL, Scheffer IE, Spillmann RC, Taglialatela M, Vieker S, Weckhuysen S, Cooper EC, Cilio MR. Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ2 gain-of-function variants R201C and R201H. Epilepsia 58: 436–445, 2017. doi: 10.1111/epi.13676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosti G, Tassano E, Bossi S, Divizia MT, Ronchetto P, Servetti M, Lerone M, Pisciotta L, Mancardi MM, Veneselli E, Puliti A. Intragenic duplication of KCNQ5 gene results in aberrant splicing leading to a premature termination codon in a patient with intellectual disability. Eur J Med Genet 62: 103555, 2019. doi: 10.1016/j.ejmg.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 67.Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol 120: 267–305, 2002. [Erratum in J Gen Physiol 120: 599, 2002]. doi: 10.1085/jgp.20028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y, Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J, Huang W. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 299: 251–254, 2003. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 69.Lee HC, Rudy Y, Liang H, Chen CC, Luo CH, Sheu SH, Cui J. Pro-arrhythmogenic effects of the V141M KCNQ1 mutation in short QT syndrome and its potential therapeutic targets: Insights from modeling. J Med Biol Eng 37: 780–789, 2017. doi: 10.1007/s40846-017-0257-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peng G, Barro-Soria R, Sampson KJ, Larsson HP, Kass RS. Gating mechanisms underlying deactivation slowing by two KCNQ1 atrial fibrillation mutations. Sci Rep 7: 45911, 2017. doi: 10.1038/srep45911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Restier L, Cheng L, Sanguinetti MC. Mechanisms by which atrial fibrillation-associated mutations in the S1 domain of KCNQ1 slow deactivation of IKs channels. J Physiol 586: 4179–4191, 2008. doi: 10.1113/jphysiol.2008.157511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gandhi CS, Clark E, Loots E, Pralle A, Isacoff EY. The orientation and molecular movement of a K+ channel voltage-sensing domain. Neuron 40: 515–525, 2003. doi: 10.1016/S0896-6273(03)00646-9. [DOI] [PubMed] [Google Scholar]
- 73.Larsson JE, Larsson HP, Liin SI. KCNE1 tunes the sensitivity of KV7.1 to polyunsaturated fatty acids by moving turret residues close to the binding site. eLife 7: e37257, 2018. doi: 10.7554/eLife.37257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cooper EC, Abreo T, Tran B. KCNQ channel PIP2 modulation: two loose links, three rings, and a twist. Neuron 110: 178–180, 2022. doi: 10.1016/j.neuron.2021.12.026. [DOI] [PubMed] [Google Scholar]
- 75.Sun J, MacKinnon R. Structural basis of human KCNQ1 modulation and gating. Cell 180: 340–347.e9, 2020. doi: 10.1016/j.cell.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zheng Y, Liu H, Chen Y, Dong S, Wang F, Wang S, Li GL, Shu Y, Xu F. Structural insights into the lipid and ligand regulation of a human neuronal KCNQ channel. Neuron 110: 237–247.e4, 2022. doi: 10.1016/j.neuron.2021.10.029. [DOI] [PubMed] [Google Scholar]
- 77.Li T, Wu K, Yue Z, Wang Y, Zhang F, Shen H. Structural basis for the modulation of human KCNQ4 by small-molecule drugs. Mol Cell 81: 25–37.e4 2021. doi: 10.1016/j.molcel.2020.10.037. [DOI] [PubMed] [Google Scholar]
- 78.Li X, Zhang Q, Guo P, Fu J, Mei L, Lv D, Wang J, Lai D, Ye S, Yang H, Guo J. Molecular basis for ligand activation of the human KCNQ2 channel. Cell Res 31: 52–61, 2021. doi: 10.1038/s41422-020-00410-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim RY, Pless SA, Kurata HT. PIP2 mediates functional coupling and pharmacology of neuronal KCNQ channels. Proc Natl Acad Sci USA 114: E9702–E9711, 2017. doi: 10.1073/pnas.1705802114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zaydman MA, Silva JR, Delaloye K, Li Y, Liang H, Larsson HP, Shi J, Cui J. Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc Natl Acad Sci USA 110: 13180–13185, 2013. doi: 10.1073/pnas.1305167110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hernandez CC, Zaika O, Shapiro MS. A carboxy-terminal inter-helix linker as the site of phosphatidylinositol 4,5-bisphosphate action on Kv7 (M-type) K+ channels. J Gen Physiol 132: 361–381, 2008. doi: 10.1085/jgp.200810007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pant S, Zhang J, Kim EC, Lam K, Chung HJ, Tajkhorshid E. PIP2-dependent coupling of voltage sensor and pore domains in Kv7.2 channel. Commun Biol 4: 1189, 2021. doi: 10.1038/s42003-021-02729-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwake M, Athanasiadu D, Beimgraben C, Blanz J, Beck C, Jentsch TJ, Saftig P, Friedrich T. Structural determinants of M-type KCNQ (Kv7) K+ channel assembly. J Neurosci 26: 3757–3766, 2006. doi: 10.1523/JNEUROSCI.5017-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci 8: 51–60, 2005. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- 85.Singh NA, Otto JF, Dahle EJ, Pappas C, Leslie JD, Vilaythong A, Noebels JL, White HS, Wilcox KS, Leppert MF. Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. J Physiol 586: 3405–3423, 2008. doi: 10.1113/jphysiol.2008.154971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soh H, Pant R, LoTurco JJ, Tzingounis AV. Conditional deletions of epilepsy-associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. J Neurosci 34: 5311–5321, 2014. doi: 10.1523/JNEUROSCI.3919-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Watanabe H, Nagata E, Kosakai A, Nakamura M, Yokoyama M, Tanaka K, Sasai H. Disruption of the epilepsy KCNQ2 gene results in neural hyperexcitability. J Neurochem 75: 28–33, 2000. doi: 10.1046/j.1471-4159.2000.0750028.x. [DOI] [PubMed] [Google Scholar]
- 88.Rubinstein M, Westenbroek RE, Yu FH, Jones CJ, Scheuer T, Catterall WA. Genetic background modulates impaired excitability of inhibitory neurons in a mouse model of Dravet syndrome. Neurobiol Dis 73: 106–117, 2015. doi: 10.1016/j.nbd.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, Chen H, Kronengold J, Abhyankar A, Cilio R, Nitschke P, Kaminska A, Boddaert N, Casanova JL, Desguerre I, Munnich A, Dulac O, Kaczmarek LK, Colleaux L, Nabbout R. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 44: 1255–1259, 2012. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Plaster NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptacek LJ. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 105: 511–519, 2001. doi: 10.1016/S0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 91.Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA 106: 5842–5847, 2009. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Simons C, Rash LD, Crawford J, Ma L, Cristofori-Armstrong B, Miller D, Ru K, Baillie GJ, Alanay Y, Jacquinet A, Debray FG, Verloes A, Shen J, Yesil G, Guler S, Yuksel A, Cleary JG, Grimmond SM, McGaughran J, King GF, Gabbett MT, Taft RJ. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet 47: 73–77, 2015. [Erratum in Nat Genet 47: 304, 2015]. doi: 10.1038/ng.3153. [DOI] [PubMed] [Google Scholar]
- 93.De Marco García NV, Karayannis T, Fishell G. Neuronal activity is required for the development of specific cortical interneuron subtypes. Nature 472: 351–355, 2011. doi: 10.1038/nature09865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wong FK, Bercsenyi K, Sreenivasan V, Portalés A, Fernández-Otero M, Marín O. Pyramidal cell regulation of interneuron survival sculpts cortical networks. Nature 557: 668–673, 2018. doi: 10.1038/s41586-018-0139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials: https://www.doi.org/10.6084/m9.figshare.17077898.
Data Availability Statement
All patient genetic information into the Leiden Open Variant Database (LOVD): https://databases.lovd.nl/shared/variants/KCNQ5/unique.