

Abstract

A highly enantioselective one-pot synthesis of functionalized triflones, bearing a quaternary stereocenter, has been developed, exploiting the Michael reaction of α-(trifluoromethylsulfonyl) aryl acetic acid esters with N-acryloyl-1H-pyrazole catalyzed by commercially available Takemoto’s catalyst, followed by nucleophilic acyl substitution with alcohols. Preliminary investigations highlighted the attractive potential of the triflinate anion as the leaving group for stereocontrolled postfunctionalizations.
Chiral nonracemic sulfones are a class of compounds of great importance in different areas, from organic synthesis, medicinal chemistry to material science. In particular, those bearing the sulfone group directly connected to the stereogenic center are endowed with different biological activities, such as antifungal agents (Agelasidine A),1 β-lactamase inhibitors (tazobactan),2 and γ-secretase inhibitor.3 The sulfonyl group is an accredited bioisoster of the carbonyl group and a strong H-bonding acceptor able to increase the interactions with the biological targets.4 Moreover, sulfones are highly useful synthetic building blocks amenable of different transformations.5
The asymmetric synthesis of sulfones, having this group directly attached to the stereogenic center is a challenging task,6 which has been mainly accomplished via metal-catalyzed substitution,7 hydrosulfonylation,8 hydrogenation,9 and conjugate addition.10 However, most of the protocols so far developed are focused on the generation of optically enriched secondary sulfones. In comparison, the stereoselective preparation of aryl and alkyl sulfones featuring a quaternary stereocenter is largely underdeveloped.6,7,10c In this context, scant examples have been reported on the stereoselective preparation of either secondary and tertiary triflones (Scheme 1). Nakamura and Toru illustrated an interesting asymmetric reaction of nBuLi generated α-carbanion of benzyl trifluoromethylsulfone with aldehydes in the presence of 30 mol % of bis(oxazoline) ligands (Scheme 1a).11 The products were obtained in good to high diastereo- and enantioselectivity.
Scheme 1. Approaches to Optically Enriched Triflones.

Raabe and Gais, developed a five-step sequence from optically enriched secondary alcohols as the reagent to obtain secondary triflones, mantaining the level of enantioselectivity.12
The latter were then alkylated, under controlled conditions, to provide triflones with an all-carbon quaternary stereocenter in comparable ee values (Scheme 1b). We recently developed a one-pot α-trifluoromethylthiolation of readily available N-acyl pyrazoles, followed by oxidation to access α-trifluoromethansulfonyl aryl acetic acid esters.13a The process has been also improved under continuous flow conditions, starting from carboxylic acids.14 The triflyl group is the strongest neutral electron-withdrawing group,15 showing mild lipophilicity. This prompted its introduction onto molecular scaffolds, as it affects the activity of fluorinated drugs16 and more in general the properties of the materials.17 As illustrated in Scheme 1, the asymmetric synthesis of tertiary triflones remains an elusive goal, where catalytic approaches still have to be developed.18 Having in hand a viable route to trifluoromethansulfonyl aryl acetic acid esters, we envisaged that they might serve as suitable pronucleophiles15c,15d to employ in Michael reactions under mild organocatalytic conditions. Herein, we report a first catalytic and highly enantioselective preparation of triflones, featuring a quaternary stereocenter. Michael reaction of trifluoromethansulfonyl aryl acetic acid esters with N-acryloyl-1H-pyrazole has been mediated by Takemoto’s catalyst, followed by nucleophilic acyl substitution with alcohols in one pot. The final bis-ester triflones also demonstrated to be useful compounds for interesting postfunctionalizations.
At the ouset of the study, methyl vinyl ketone was reacted with compound 1a, using readily available bifunctional organocatalysts at 20 mol % loading, in toluene at room temperature (Table 1). Pleasingly, quinidine (QD) catalyzed the conjugate addition, providing product 3a in 82% yield and 20% ee (entry 1). This result prompted us to use Cinchona alkaloids-derived thiourea eQNT and eQDT, which unfortunately were much less effective promoters (entries 2 and 3). Sterically hindered amine-thiourea 4 gave a small improvement in the enantioselectivity up to 37% ee (entry 4). Takemoto’s catalyst 5 proved to be more active, leading to 3a in 75% yield and 55% ee, after a short reaction time (entry 5). Next, phenyl vinyl ketone was treated with compound 1a using catalyst 5, observing the formation of the adduct 3b with an increased level of enantioselectivity (entry 6). Readily available amine-thiourea 6 was then checked in the process, giving disappointing results (entry 7), as well as the commercially available squaramide 7, which afforded racemic 3b in only moderate yield (entry 8). When more sterically hindered isopropyl ester 1a′ (R1 = 4-BrC6H4) was reacted, a decreased level of enantioselectivity was observed (entry 9). For the purpose of improving the enantiocontrol, 1-naphthyl vinyl ketone was employed with 1a in the presence of catalyst 5 (entry 10). However, the adduct 3d was isolated in 85% yield and 44% ee. Activated acrylic acid derivatives were then checked, such as the 1,1,1,3,3,3-hexafluoroisopropyl acrylate, but it proved to be poorly reactive (entry 11). Given the utility displayed over recent years by α,β-unsaturated N-acyl pyrazoles in asymmetric catalysis,19 the corresponding N-acryloyl-1H-3-phenyl pyrazole was reacted under the optimized conditions (entry 12). Pleasingly, it was smoothly converted into the corresponding adduct 3e,20 which was isolated in 42% yield and 75% ee. The same reaction when conducted at −20 °C afforded product 3e with improved 86% ee (entry 13). Finally, when using N-acryloyl-1H-pyrazole as the acceptor, adduct 3f was recovered in 50% yield20 and 89% ee (entry 14). Reduction of the catalyst loading to 10 mol % as well as the temperature as low as −20 °C enabled the product to be satisfactorily obtained in high yield21 and 94% ee (entries 15 and 16).
Table 1. Reaction Optimizationa.

| entry | cat. | R2 | t (h) | 3 yield (%)b | 3 eec |
|---|---|---|---|---|---|
| 1 | QD | Me 2a | 7 | 82 (3a) | 20 |
| 2 | eQNT | Me | 23 | 10 (3a) | 5 |
| 3 | eQDT | Me | 23 | 57 (3a) | rac |
| 4 | 4 | Me | 23 | 41 (3a) | 37 |
| 5 | 5 | Me | 6 | 75 (3a) | 55(−) |
| 6 | 5 | Ph 2b | 16 | 79 (3b) | 67 |
| 7 | 6 | Ph | 16 | 44 (3b) | 37 |
| 8 | 7 | Ph | 16 | 23 (3b) | rac |
| 9d | 5 | Ph | 17 | 76 (3c) | 63 |
| 10 | 5 | 1-naphthyl 2c | 17 | 85 (3d) | 44 |
| 11 | 5 | OCH(CF3)22d | 25 | <10 | n.d. |
| 12 | 5 | 3-Phpyrazole 2e | 17 | 42 (3e) | 75 |
| 13e | 5 | 3-Phpyrazole 2e | 40 | 61 (3e) | 86 |
| 14 | 5 | pyrazole 2f | 17 | 50 (3f) | 89 |
| 15f | 5 | pyrazole 2f | 24 | 90 (3f) | 93 |
| 16g | 5 | pyrazole 2f | 24 | 95 (3f) | 94 |
Reactions performed at 0.1 mmol scale of 1a (C 0.2 M) using 2 (1.2 equiv).
Isolated yield after chromatography.
Determined by chiral HPLC analysis; n.d. = not determined. Negative sign indicates enantiomeric excess for the opposite enantiomer.
The isopropyl ester of compound 1a was used.
Run at −20 °C.
10 mol % of 5 was used at 0 °C.
10 mol % of 5 was used at −20 °C.
N-Acyl pyrazoles behave as useful carboxylic acid ester surrogates,19 due to the good leaving group ability of the pyrazole group. Hence, we thought to develop a simple one-pot methodology to directly obtain the bis-ester triflones 8, treating compounds 3 with an alcohol at room temperature, after the end of the conjugate addition step. Under the optimized reaction conditions reported in Scheme 1, entry 16, the scope of the one-pot process was next investigated (Scheme 2). As illustrated in Scheme 2, triflones 1, bearing halogens at para- and ortho-position of the phenyl ring, were converted into the corresponding methyl esters 8a–d in excellent yields and high enantioselectivity (82–95% ee).
Scheme 2. Substrate Scope of the One-Pot Process,,

First step: 0.1 mmol scale of 1 (C 0.2 M) using 2f (1.5 equiv). Second step: addition of R2OH (50 equiv), in case of morpholine (3 equiv).
Isolated yield after chromatography.
Ee determined by chiral HPLC analysis.
Pleasingly, more sterically encumbered ortho-fluoro derivative 8d was isolated with excellent ee value (95%). Electron-donating or -withdrawing substitution at the para-, meta-, and ortho- positions, including the phenyl and 2-naphthyl moieties, were well tolerated, as the products 8e–j were recovered in good to high yields and 91–96% ee values. Only the sterically demanding ortho-methyl derivative 8g was isolated in 40% yield, although a 96% ee value was observed. Then, we surveyed the suitability of other alcohols as nucleophiles in the second step on differently substituted trifluoromethansulfonyl phenyl acetic acid esters 1. Ethanol, n-butanol to more sterically hindered isopropanol and allylic alcohol could be employed, performing the esterification at 50 °C. The corresponding triflones 8k–o, bearing single or double substitution at the phenyl ring, have been obtained in fairly good to high yields and ee values (83–94%).
Finally, the pyrazole displacement with morpholine performed at room temperature led to the corresponding product 8p, bearing a tertiary amide group, in 92% yield and 93% ee. The practicality of the process was investigated scaling-up reagent 1a to 1.0 mmol. Triflone 8a was isolated maintaining a high yield and enantioselectivity. Further experiments, carried out during the preparation of tertiary amide 8p, allowed us to disclose a synthetically appealing derivatization, involving triflyl group displacement (Scheme 3).
Scheme 3. One-Pot Derivatizations of Compounds 8 Involving Triflyl Group Displacement.

When the second step was performed using morpholine in the presence of water (2 equiv) at 50 °C, for a prolonged reaction time, the α-hydroxyl ester 9 was efficiently formed in 95% yield and 92% ee. This remarkable result would be rationalized invoking SN2 displacement of the triflinate anion, which is an excellent leaving group,22 by an in situ generated hydroxyl anion.23 The transformation is noteworthy, being a formal enantioselective hydroxylation at a congested α-position of an ester. The absolute configuration of compound 9 was determined to be S by X-ray crystallographic analysis (CCDC No.: 2165089).
Consequently, compounds 8 were assigned as R-configured, which was found to be consistent with DFT calculations (Figure 1).
Figure 1.
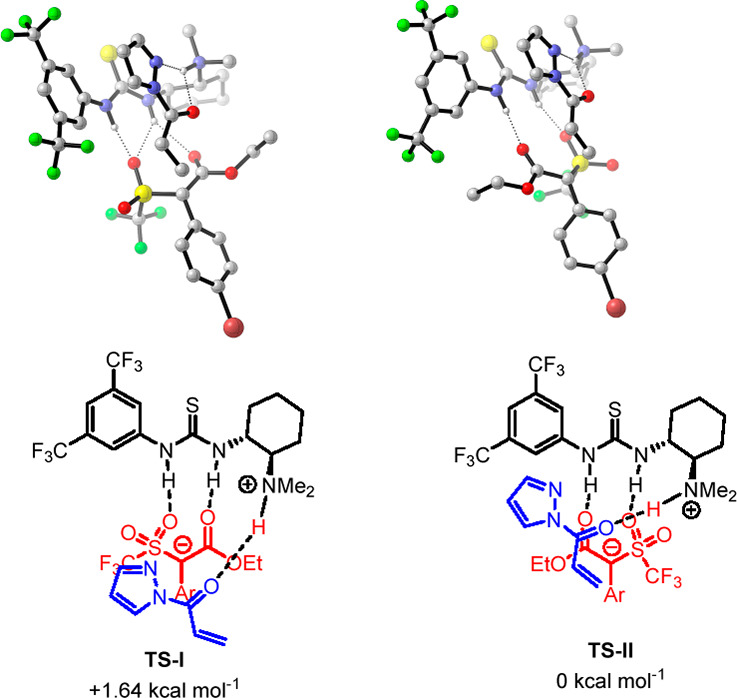
Proposed model of stereoselection. Geometries and ΔΔG0 (kcal mol–1) of transition states related to the synthesis of enriched triflone 8a were calculated at the M062X/6-31G(d,p)/PCM (toluene) level of theory. Hydrogens are omitted for clarity.
The transition states leading to the formation of both (R)-8a and (S)-8a for the Michael reaction, promoted by (R,R)-catalyst 5, were fully optimized by DFT calculation at the M062X-631g(d,p)/PCM (toluene) level of theory using the M062X functional.24 Different models of substrates-catalyst coordination were investigated.25 The energetically most affordable calculated transition states would involve the activation model previously proposed by Papai,26 where triflone 1a is coordinated to the thiourea group of the catalyst and acyl pyrazole 2f to the tertiary amine group. According to this model, TS-II leading to product (R)-8a was found to be more stable by 1.64 kcal mol–1 than TS-I leading to product (S)-8a, which features weaker hydrogen bonding of thiourea with the SO2CF3 group, and, possibly, a destabilizing interaction between SO2CF3 and one of the CF3 residues in the catalyst. Noteworthy, remarkably good agreement between calculated (91% ee) and experimental (93% ee in entry 15, Table 1) ee values has been achieved.
We further applied the displacement to develop an asymmetric one-pot Michael/SN2 displacement/esterification to γ-butyrolactone, bearing a γ-quaternary stereocenter (Scheme 3). The in situ generated adduct 8p was treated with Et3N, water at 50 °C, affording the expected lactone 10 in 44% yield and 95% ee. Although the process needs to be optimized, it represents an interesting and useful application of optically active triflones 8 as intermediates toward difficult to prepare γ-butyrolactones 10.27
Additional postfunctionalizations on representative compound 8a have been performed under reductive conditions (Scheme 4). Unexpectedly, treatment with DIBALH under controlled conditions afforded alcohol 11 in 76% yield, without erosion of the ee value. Reduction of the less activated methyl ester might be likely ascribed to the congested nature of the ethyl ester portion. Having ascertained that selective reduction of the ester to aldehyde occurred in a shorter reaction time, a one-pot process from reagent 1a, involving asymmetric Michael reaction/reduction to aldehyde/Horner–Emmons olefination, has been developed. The diversely functionalized product 12 was recovered in satisfactory 50% overall yield and 93% ee.
Scheme 4. Additional Derivatizations of Compounds 8.

In summary, we successfully develop a first enantioselective catalytic route to triflones, featuring a quaternary stereocenter. The asymmetric Michael reaction between α-(trifluoromethylsulfonyl) aryl acetic acid esters with N-acryloyl-1H-pyrazole was efficiently catalyzed by commercial Takemoto’s catalyst, followed by nucleophilic acyl substitution with alcohols. The bis-ester triflones were obtained in good to excellent yields and high enantioselectivity in one-pot. Moreover, this work provides useful knowledge on the application of tertiary triflones in stereoselective organic synthesis. The utility of the products has been demonstrated via triflone displacement and under reductive conditions to conveniently access a variety of attractive enantioenriched scaffolds.
Acknowledgments
F.F. thanks MUR for a PON RI 2014-2020 PhD fellowship. S.M. thanks MUR and European Union for AIM – international attraction and mobility call for researchers funded by PON RI 2014-2020. University of Salerno is acknowledged for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.2c01589.
Full experimental procedures, characterization data, NMR spectra, HPLC traces, computational details and crystallographic data for 9 are available. (PDF)
Accession Codes
CCDC 21065089 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Stout E. P.; Yu L. C.; Molinski T. F. Antifungal Diterpene Alkaloids from the Caribbean Sponge Agelas citrina: Unified Configurational Assignments of Agelasidines and Agelasines. Eur. J. Org. Chem. 2012, 2012, 5131–5135. 10.1002/ejoc.201200572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan S.; Kerry P. S.; Bance N.; Niikura M.; Pinto B. M. Serendipitous Discovery of a Potent Influenza Virus A Neuraminidase Inhibitor. Angew. Chem., Int. Ed. 2014, 53, 1076–1080. 10.1002/anie.201308142. [DOI] [PubMed] [Google Scholar]
- Scott J. P.; Oliver S. F.; Brands K. M. J.; Brewer S. E.; Davies A. J.; Gibb A. D.; Hands D.; Keen S. P.; Sheen F. J.; Reamer R. A.; Wilson R. D.; Dolling U.-H. Practical Asymmetric Synthesis of a γ-Secretase Inhibitor Exploiting Substrate-Controlled Intramolecular Nitrile Oxide Olefin Cycloaddition. J. Org. Chem. 2006, 71, 3086–3092. 10.1021/jo060033i. [DOI] [PubMed] [Google Scholar]
- Velázquez F.; Sannigrahi M.; Bennett F.; Lovey R. G.; Arasappan A.; Bogen S.; Nair L.; Venkatraman S.; Blackman M.; Hendrata S.; Huang Y.; Huelgas R.; Pinto P.; Cheng K.-C.; Tong X.; McPhail A. T.; Njoroge F. G. Cyclic Sulfones as Novel P3-Caps for Hepatitis C Virus NS3/4A (HCV NS3/4A) Protease Inhibitors: Synthesis and Evaluation of Inhibitors with Improved Potency and Pharmacokinetic Profiles. J. Med. Chem. 2010, 53, 3075–3085. 10.1021/jm9016027. [DOI] [PubMed] [Google Scholar]
- a Trost B. M. Chemical Chameleons. Organosulfones as Synthetic Building Blocks. Bull. Chem. Soc. Jpn. 1988, 61, 107–124. 10.1246/bcsj.61.107. [DOI] [Google Scholar]; b El-Awa A.; Noshi M. N.; du Jourdin X. M.; Fuchs P. L. Evolving Organic Synthesis Fostered by the Pluripotent Phenylsulfone. Moiety. Chem. Rev. 2009, 109, 2315–2349. 10.1021/cr800309r. [DOI] [PubMed] [Google Scholar]; c Xu X. H.; Matsuzaki K.; Shibata N. Synthetic Methods for Compounds Having CF3-S Units on Carbon by Trifluoromethylation, Trifluoromethylthiolation, Triflylation, and Related Reactions. Chem. Rev. 2015, 115, 731–764. 10.1021/cr500193b. [DOI] [PubMed] [Google Scholar]
- For recent reviews, see:; a Zhu C.; Cai Y.; Jiang H. Recent Advances for the Synthesis of Chiral Sulfones with the Sulfone Moiety Directly Connected to the Chiral Center. Org. Chem. Front. 2021, 8, 5574–5589. 10.1039/D1QO00663K. [DOI] [Google Scholar]; b Huang Y.; Li J.; Chen H.; He Z.; Zeng Q. Recent Progress on the Synthesis of Chiral Sulfones. Chem. Rec. 2021, 21, 1216–1239. 10.1002/tcr.202100023. [DOI] [PubMed] [Google Scholar]; c Liang X.; Shen Y. Advances in Synthesis of Enantioenriched Chiral Sulfones by Enantioselective Conjugate Addition Reactions. Asian J. Org. Chem. 2022, 11, e202100598 10.1002/ajoc.202100598. [DOI] [Google Scholar]
- For selected examples, see:; a Trost B. M.; Organ M. G.; O’Doherty G. A. Asymmetric Synthesis of Allylic Sulfones Useful as Asymmetric Building Blocks. J. Am. Chem. Soc. 1995, 117, 9662–9670. 10.1021/ja00143a007. [DOI] [Google Scholar]; b Ueda M.; Hartwig J. F. Iridium-Catalyzed, Regio- and Enantioselective Allylic Substitution with Aromatic and Aliphatic Sulfinates. Org. Lett. 2010, 12, 92–94. 10.1021/ol9023248. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Choi J.; Martín-Gago P.; Fu G. C. Stereoconvergent Arylations and Alkenylations of Unactivated Alkyl Electrophiles: Catalytic Enantioselective Synthesis of Secondary Sulfonamides and Sulfones. J. Am. Chem. Soc. 2014, 136, 12161–12165. 10.1021/ja506885s. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gómez J. E.; Cristòfol À.; Kleij A. W. Copper-Catalyzed Enantioselective Construction of Tertiary Propargylic Sulfones. Angew. Chem., Int. Ed. 2019, 58, 3903–3907. 10.1002/anie.201814242. [DOI] [PubMed] [Google Scholar]
- For selected examples, see:; a Jia S.; Chen Z.; Zhang N.; Tan Y.; Liu Y.; Deng J.; Yan H. Organocatalytic Enantioselective Construction of Axially Chiral Sulfone-Containing Styrenes. J. Am. Chem. Soc. 2018, 140, 7056–7060. 10.1021/jacs.8b03211. [DOI] [PubMed] [Google Scholar]; b Zhang Q.; Dong D.; Zi W. Palladium-Catalyzed Regio and Enantioselective Hydrosulfonylation of 1,3-Dienes with Sulfinic Acids: Scope, Mechanism, and Origin of Selectivity. J. Am. Chem. Soc. 2020, 142, 15860–15869. 10.1021/jacs.0c05976. [DOI] [PubMed] [Google Scholar]; c Li M. M.; Cheng L.; Xiao L. J.; Xie J. H.; Zhou Q. L. Palladium-Catalyzed Asymmetric Hydrosulfonylation of 1,3-Dienes with Sulfonyl Hydrazides. Angew. Chem., Int. Ed. 2021, 60, 2948–2951. 10.1002/anie.202012485. [DOI] [PubMed] [Google Scholar]
- a Shi L.; Wei B.; Yin X.; Xue P.; Lv H.; Zhang X. Rh-Catalyzed Asymmetric Hydrogenation of α-Substituted Vinyl Sulfones: An Efficient Approach to Chiral Sulfones. Org. Lett. 2017, 19, 1024–1027. 10.1021/acs.orglett.6b03845. [DOI] [PubMed] [Google Scholar]; b Vyas V. K.; Clarkson G. J.; Wills M. Sulfone Group as a Versatile and Removable Directing Group for Asymmetric Transfer Hydrogenation of Ketones. Angew. Chem., Int. Ed. 2020, 59, 14265–14269. 10.1002/anie.202004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples, see:; a Mauleon P.; Carretero J. C. Rhodium-Catalyzed Enantioselective Conjugate Addition of Organoboronic Acids to α,β-Unsaturated Sulfones. Org. Lett. 2004, 6, 3195–3198. 10.1021/ol048690p. [DOI] [PubMed] [Google Scholar]; b Landa A.; Maestro M.; Masdeu C.; Puente Á.; Vera S.; Oiarbide M.; Palomo C. Highly Enantioselective Conjugate Additions of Aldehydes to Vinyl Sulfones.. Chem.- Eur. J. 2009, 15, 1562–1565. 10.1002/chem.200802441. [DOI] [PubMed] [Google Scholar]; c Prakash G. K. S.; Wang F.; Stewart T.; Mathew T.; Olah G. A. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 4090–4094. 10.1073/pnas.0900179106. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Bera K.; Namboothiri I. N. N. Chem. Commun. 2013, 49, 10632–10634. 10.1039/c3cc45985c. [DOI] [PubMed] [Google Scholar]; h Deng R.; Wu S.; Mou C.; Liu J.; Zheng P.-c.; Zhang X.; Chi Y. R. Carbene-Catalyzed Enantioselective Sulfonylation of Enone Aryl Aldehydes: A New Mode of Breslow Intermediate Oxidation. J. Am. Chem. Soc. 2022, 144, 5441–5449. 10.1021/jacs.1c13384. [DOI] [PubMed] [Google Scholar]
- Nakamura S.; Hirata N.; Kita T.; Yamada R.; Nakane D.; Shibata N.; Toru T. Highly Enantioselective Reactions of α-Sulfonyl Carbanions of Trifluoromethyl Sulfone. Angew. Chem., Int. Ed. 2007, 46, 7648–7650. 10.1002/anie.200702185. [DOI] [PubMed] [Google Scholar]
- Hellmann G.; Hack A.; Thiemermann E.; Luche O.; Raabe G.; Gais H.-J. Chiral Fluorinated α-Sulfonyl Carbanions: Enantioselective Synthesis and Electrophilic Capture, Racemization Dynamics, and Structure. Chem.—Eur. J. 2013, 19, 3869–3897. 10.1002/chem.201204014. [DOI] [PubMed] [Google Scholar]
- a Franco F.; Meninno S.; Benaglia M.; Lattanzi A. Formal α-Trifluoromethylthiolation of Carboxylic Acid Derivatives via N-Acyl Pyrazoles. Chem. Commun. 2020, 56, 3073–3076. 10.1039/D0CC00116C. [DOI] [PubMed] [Google Scholar]; For an alternative route, see:; b Goumont R.; Faucher N.; Moutiers G.; Tordeux M.; Wakselman C. A Novel Synthesis of Deactivated Benzylic Triflones. Synthesis 1997, 1997, 691–695. 10.1055/s-1997-1390. [DOI] [Google Scholar]
- Franco F.; Meninno S.; Lattanzi A.; Puglisi A.; Benaglia M. J. Org. Chem. 2021, 86, 14207–142012. 10.1021/acs.joc.1c01270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sheppard W. A. The Effect of Fluorine Substitution on the Electronic Properties of Alkoxy, Alkylthio and Alkylsulfonyl Groups. J. Am. Chem. Soc. 1963, 85, 1314–1318. 10.1021/ja00892a021. [DOI] [Google Scholar]; b Terrier F.; Kizilian E.; Goumont R.; Faucher N.; Wakselman C. α-Sulfonyl Carbanions: Combined Kinetic, Thermodynamic, and NMR Approaches for the Study of the Ionization of Benzyltriflones in Me2SO and H2O-Me2SO Mixtures. J. Am. Chem. Soc. 1998, 120, 9496–9503. 10.1021/ja9806074. [DOI] [Google Scholar]; c Goumont R.; Kizilian E.; Buncel E.; Terrier F. Super Acidifiers: the Origin of the Exceptional Electron Transmission Capability of the SO2CF3 Group in Carbanion Stabilization. Org. Biomol. Chem. 2003, 1, 1741–1748. 10.1039/B302029K. [DOI] [PubMed] [Google Scholar]; d Berger S. T. A.; Ofial A. R.; Mayr H. Inverse Solvent Effects in Carbocation Carbanion Combination Reactions: The Unique Behavior of Trifluoromethylsulfonyl Stabilized Carbanions. J. Am. Chem. Soc. 2007, 129, 9753–9761. 10.1021/ja072135b. [DOI] [PubMed] [Google Scholar]
- For selected examples, see:; a Park C.-M.; Bruncko M.; Adickes J.; Bauch J.; Ding H.; Kunzer A.; Marsh K. C.; Nimmer P.; Shoemaker A. R.; Song X.; Tahir S. K.; Tse C.; Wang X.; Wendt M. D.; Yang X.; Zhang H.; Fesik S. W.; Rosenberg S. H.; Elmore S. W. Discovery of an Orally Bioavailable Small Molecule Inhibitor of Prosurvival B-Cell Lymphoma 2 Proteins. J. Med. Chem. 2008, 51, 6902–6915. 10.1021/jm800669s. [DOI] [PubMed] [Google Scholar]; c Sleebs B. E.; Czabotar P. E.; Fairbrother W. J.; Fairlie W. D.; Flygare J. A.; Huang D. C. S.; Kersten W. J. A.; Koehler M. F. T.; Lessene G.; Lowes K.; Parisot J. P.; Smith B. J.; Smith M. L.; Souers A. J.; Street I. P.; Yang H.; Baell J. B. Quinazoline Sulfonamides as Dual Binders of the Proteins B-Cell Lymphoma 2 and B-Cell Lymphoma Extra Long with Potent Proapoptotic Cell-Based Activity. J. Med. Chem. 2011, 54, 1914–1926. 10.1021/jm101596e. [DOI] [PubMed] [Google Scholar]
- a Mongin O.; Porres L.; Charlot M.; Katan C.; Blanchard-Desce M. Synthesis, Fluorescence, and Two-Photon Absorption of a Series of Elongated Rodlike and Banana-Shaped Quadrupolar Fluorophores: A Comprehensive Study of Structure-Property Relationships. Chem.—Eur. J. 2007, 13, 1481–1498. 10.1002/chem.200600689. [DOI] [PubMed] [Google Scholar]; b Rouxel C.; Le Droumaguet C. L.; Macé Y.; Clift S.; Mongin O.; Magnier E.; Blanchard-Desce M. Octupolar Derivatives Functionalized with Superacceptor Peripheral Groups: Synthesis and Evaluation of the Electron-Withdrawing Ability of Potent Unusual Groups. Chem.—Eur. J. 2012, 18, 12487–12497. 10.1002/chem.201103460. [DOI] [PubMed] [Google Scholar]
- For other synthetic examples, see:; a Huang Z.; Wang C.; Tokunaga E.; Sumii Y.; Shibata N. Stereoselective Synthesis of β-Lactam-Triflones under Catalyst-Free Conditions. Org. Lett. 2015, 17, 5610–5613. 10.1021/acs.orglett.5b02827. [DOI] [PubMed] [Google Scholar]; b Huang Z.; Jia S.; Wang C.; Tokunaga E.; Sumii Y.; Shibata J. Fluorine Chem. 2017, 198, 61–66. 10.1016/j.jfluchem.2016.12.012. [DOI] [Google Scholar]; c Luo J.; Cao Q.; Cao X.; Zhao X. Selenide-Catalyzed Enantioselective Synthesis of Trifluoromethylthiolated Tetrahydronaphthalenes by Merging Desymmetrization and Trifluoromethylthiolation. Nat. Commun. 2018, 9, 527–536. 10.1038/s41467-018-02955-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a recent review, see:; Meninno S.; Franco F.; Benaglia M.; Lattanzi A. Pyrazoleamides in Catalytic Asymmetric Reactions: Recent Advances. Adv. Synth. Catal. 2021, 363, 3380–3410. 10.1002/adsc.202100006. [DOI] [Google Scholar]
- 1H-NMR analysis of the crude reaction mixture showed high conversion to the adduct, which proved to be unstable over silica gel during the purification.
- The adduct 3f could be recovered without significant degradation when using flash silica gel pretreated in an oven overnight at 120 °C.
- a Hendrickson J. B.; Giga A.; Wareing J. Triflones (CF3SO2C). A Survey of Reactivity and Synthetic Utility. J. Am. Chem. Soc. 1974, 96, 2275–2276. 10.1021/ja00814a061. [DOI] [Google Scholar]; b Creary X. Nucleofugality of the Sulfinate Group in Carbocation-Forming Processes. J. Org. Chem. 1985, 50, 5080–5084. 10.1021/jo00225a017. [DOI] [Google Scholar]; c Nambo M.; Yim J. C.; Freitas L. B. O.; Tahara Y.; Ariki Z. T.; Maekawa Y.; Yokogawa D.; Crudden C. M. Modular Synthesis of α-Fluorinated Arylmethanes via Desulfonylative Cross-Coupling. Nat. Commun. 2019, 10, 4528. 10.1038/s41467-019-11758-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An intramolecular displacement of the triflyl group by the amide moiety, followed by hydrolysis, cannot be excluded.
- Zhao Y.; Truhlar D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- a Okino T.; Hoashi Y.; Takemoto Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. 10.1021/ja036972z. [DOI] [PubMed] [Google Scholar]; b Guo J.; Wong M. W. Cinchona Alkaloid-Squaramide Catalyzed Sulfa-Michael Addition Reaction: Mode of Bifunctional Activation and Origin of Stereoinduction. J. Org. Chem. 2017, 82, 4362–4368. 10.1021/acs.joc.7b00388. [DOI] [PubMed] [Google Scholar]; c Izzo J. A.; Myshchuk Y.; Hirschi J. S.; Vetticatt M. J. Transition State Analysis of an Enantioselective Michael Addition by a Bifunctional Thiourea Organocatalyst. Org. Biomol. Chem. 2019, 17, 3934–3939. 10.1039/C9OB00072K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza A.; Schubert G.; Soós T.; Pápai I. Theoretical Studies on the Bifunctionality of Chiral Thiourea-Based Organocatalysts: Competing Routes to C-C Bond Formation. J. Am. Chem. Soc. 2006, 128, 13151–13160. 10.1021/ja063201x. [DOI] [PubMed] [Google Scholar]
- For a review, see:; Murauski K. J. R.; Jaworski A. A.; Scheidt K. A. A Continuing Challenge: N-Heterocyclic Carbene-Catalyzed Syntheses of γ-Butyrolactones. Chem. Soc. Rev. 2018, 47, 1773–1782. 10.1039/C7CS00386B. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.