

Significance Statement
Membranous nephropathy (MN) is the leading cause of adult nephrotic syndrome, in which the complement system is highly activated. This paper describes the discovery that the C3a/C3aR pathway is a crucial effector of complement-mediated podocyte injuries in MN. Levels of plasma C3a and glomerular C3aR are elevated in patients with MN compared with healthy controls and are associated with severity and prognosis. In vitro, C3a from MN patients’ plasma damages the physiologic function and cellular activity of podocytes, which C3aR antagonists block. In vivo, C3aR antagonists can also attenuate renal injuries of Heymann nephritis (a classic disease model of MN) rats. Collectively, C3aR blockade may be a potentially viable treatment for this disease.
Keywords: membrane nephropathy, complement, C3a receptor, podocyte, Heymann nephritis
Visual Abstract

Abstract
Background
The complement system is highly activated in primary membranous nephropathy (MN). Identifying the complement components that damage podocytes has important therapeutic implications. This study investigated the role of C3a and the C3a receptor (C3aR) in the pathogenesis of MN.
Methods
C3aR expression in kidneys and circulating levels of C3a of MN patients were examined. Human podocyte damage was assessed after exposure to MN plasma +/− C3aR blockade (SB290157, JR14a). C3aR antagonists were administered to rats with Heymann nephritis on day 0 or after proteinuria. Clinical and pathologic parameters, specific IgG and complement activation, and podocyte injuries were then assessed.
Results
In the glomeruli, C3aR staining merged well with podocin. Overexpression of C3aR correlated positively with proteinuria, serum creatinine, and no response to treatments. Human podocytes exposed to MN plasma showed increased expression of PLA2R, C3aR, and Wnt3/β-catenin, reduced expression of synaptopodin and migration function, downregulated Bcl-2, and decreased cell viability. C3aR antagonists could block these effects. In Heymann nephritis rats, C3aR blockade attenuated proteinuria, electron-dense deposition, foot process width, and glomerular basement membrane thickening in glomeruli. The increased plasma C3a levels and overexpression of C3aR were also alleviated. Specific, but not total, IgG levels decreased, with less deposition of rat IgG in glomeruli and subsequent reduction of C1q, factor B, and C5b-9.
Conclusion
C3a anaphylatoxin is a crucial effector of complement-mediated podocyte damage in MN. The C3aR antagonist may be a potentially viable treatment for this disease.
Membranous nephropathy (MN) is the leading cause of adult nephrotic syndrome and is characterized by immune complex deposits in the subepithelial area of glomerular capillary walls.1 Antigens of the immune complexes may be endogenous from glomeruli in primary MN, such as podocyte autoantigens, M-type phospholipase A2 receptor (PLA2R),2 and thrombospondin type 1 domain containing 7A (THSD7A),3 or exogenous from circulation in secondary MN, such as hepatitis B virus antigens.1 In primary MN, antibodies activate the complement cascade, mainly the lectin complement pathway, which results in the activation of the common terminal pathway and the formation of the membrane attack complex (MAC, C5b-9)4 on podocytes. C5b-9 leads to sublytic damage on podocytes5 and eventually results in proteinuria in patients with MN. However, inhibition of C5b-9 formation in complement- (C6-)deficient rats did not fully prevent proteinuria induced by passive Heymann nephritis, suggesting that additional mechanisms may play a role in causing podocyte injury.6
During the complement activation, C3 is cleaved by the C3 convertase, C4b2a or C3bBb, into two fragments between arginine 77 and serine 78. The smaller one (9 kDa), C3a, is an anaphylatoxin and binds to the C3a receptor (C3aR) on cell membrane. C3aR belongs to G-protein–coupled receptor family, and is widely expressed in organs and immune cells.7 In the kidney, the expression of C3aR is mainly on glomerular podocytes and tubular epithelial cells.8 The expressions of C3aR are increased on the podocytes of patients with lupus nephritis,9 focal segmental glomerulosclerosis (FSGS),10 IgA nephropathy,11 and diabetes nephropathy,12 and C3aR blockade attenuates kidney injuries effectively.
Cross-sectional studies including ours have shown that C3a levels are markedly elevated in the plasma and urine of patients with primary MN, especially those with positive anti-PLA2R antibodies.13,14 The deposition of anti-PLA2R IgG4 activates the complement cascade in glomeruli, by directly binding to the MBL/MASP-1/2 complex in a glycosylation-dependent manner, thereby inducing proteolysis of the podocyte proteins, synaptopodin and NEPH1, and resulting in perturbations of the podocyte cytoskeleton.15 However, only a few studies have investigated C3aR expression and its roles in the mechanism of primary MN. In this study in patients with primary MN, we measured plasma C3a levels and detected glomerular C3aR, examined C3aR-mediated podocyte damage using human immortalized podocyte assays, and assessed the therapeutic effects of C3aR antagonists on rats with Heymann nephritis. These investigations may provide some evidence for potential complement treatments in primary MN therapy that target C3a/C3aR.
Methods
C3a/C3aR in Patients with Primary MN
Patients and Clinical Data
Thirty-seven patients with primary MN were assessed. They all received kidney biopsy and showed IgG and C3 granular deposits along glomerular capillary walls, diffuse thickening of glomerular basement membrane (GBM), and electron-dense deposits under epithelial cells. Anti-PLA2R antibodies were detected by ELISA (Euroimmun, Lubeck, Germany). PLA2R in the kidneys was stained by immunofluorescence. Patients with secondary MN were excluded, including systemic lupus erythematosus (SLE), hepatitis B virus infection, malignancy, medication, and heavy metal poisoning.
This study included 10 patients with diabetic nephropathy, seven patients with primary FSGS and 10 patients with minimal change disease as disease controls. Thirteen healthy individuals were assessed as healthy controls.
All the blood samples were collected on the day of kidney biopsy, before the patients received any steroids or immunosuppressive agents. The samples were stored frozen at −80°C, avoiding repeated freezing and thawing. Clinical data were collected from medical records at biopsy and during follow-up.
Assessment of C3aR in Kidneys by Immunofluorescence or Immunohistochemistry
Acetone-fixed cryostat sections (5 μm) of kidneys were blocked with 3% BSA at room temperature for 1 hour and incubated with mouse anti-human/rat C3aR monoclonal antibodies (sc-133172; Santa Cruz Biotechnology, Dallas, TX) diluted 1:500 and rabbit anti-human podocin polyclonal antibodies (PA5-797;Invitrogen, Carlsbad, CA) diluted 1:100 at 4°C overnight. Next, the sections were incubated with goat anti-mouse IgG (H+L) (Alexa Fluor 555, A-21422; Invitrogen) diluted 1:500 and goat anti-rabbit IgG (H+L) (Alexa Fluor 488, A-11034; Invitrogen) diluted 1:500, at 37°C for 30 minutes, and examined under a fluorescence microscope (Nikon, Tokyo, Japan).
Paraffin-embedded formalin-fixed sections (3 μm) were dehydrated and underwent antigen retrieval by EDTA (pH 9.0) under high temperature and high pressure for 5 minutes. Next, 3% H2O2 and 3% BSA were applied for blocking. The sections were incubated with mouse anti-human/rat C3aR antibodies (sc-133172; Santa Cruz Biotechnology) at 4°C overnight. Two-step kit (PV-9005; ZGSB-BIO, Beijing, China) and DAB (ZLI-9018; ZGSB-BIO) were used for color development.
Assessment of Circulating C3a/C3a des-Arg by ELISA
Commercial ELISA kits (ab133037; Abcam, Cambridge, United Kingdom) were used according to the manufacturer’s instructions. Plasma and standards samples, along with an alkaline phosphatase–conjugated C3a/C3a des-Arg antigen and polyclonal rabbit antibodies specific to C3a/C3a des-Arg, were added into 96-well plates precoated with goat anti-rabbit IgG antibodies. After incubation at 37°C for 2 hours, the excess reagents were washed away and pNpp substrate was added. OD was read by a microplate reader (Bio-Rad, Tokyo, Japan) at 405 nm.
C3a/C3aR Mediates Podocyte Injuries In Vitro
Human Podocytes Culture and Experimental Design
Conditionally immortalized human podocytes, kindly provided by Prof. Jochen Reiser (Rush University Medical Center, Chicago, IL), were cultivated and allowed to differentiate as described previously.16 The differentiated podocytes on six-well plates were cultured in RPMI 1640 medium for 6 hours to achieve synchronization, then stimulated in RPMI 1640 medium containing 10% plasma from patients with primary MN, 10% MN plasma + C3aR antagonist SB290157 (HY-101502A; MedChemExpress, Monmouth Junction, NJ), 10% MN plasma + C3aR antagonist JR14a (HY-138161; MedChemExpress), 10% MN plasma + recombinant human C3a (2 μg/ml, 3677-C3; R&D Systems, Minneapolis, MN), 10% plasma from healthy controls, C3a, or 5% FBS (10100147;Gibco, CA). All plasma samples were collected with sodium heparin as anticoagulator. All media had been filtered by 0.22-μm filters and were free of LPS.
Assessment of mRNA by Quantitative Real-Time PCR
Total RNA was extracted by an RNAprep Pure Cell/Bacteria Kit (DP430; Tiangen, Beijing, China), reversely transcribed to cDNA by a high-capacity cDNA reverse transcription kit (4368813; Applied Biosystems, Beverly, MA) and applied to real-time PCR with PowerUp SYBR Green Master Mix (A25741; Applied Biosystems). GAPDH was used as control gene. The primer sequences were designed (Supplemental Table 1) and synthesized (Tianyihuiyuan, Beijing, China). The optimal time point was selected by previous time course experiments (Supplemental Figure 1).
Assessment of Proteins by Western Blot
The podocytes were stimulated as described above, and the total proteins were extracted. Boiled or not-boiled, reduced or unreduced, samples and markers were separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride membranes. The membrane was incubated with primary antibodies (Supplemental Table 2) at 4°C overnight and secondary antibodies (Supplemental Table 2) at room temperature for 1 hour. The belts were observed under a chemiluminescence imaging system (GE Healthcare, Westborough, MA).
Migration Assay
One cross scratch was performed with a 200-μl sterile tip on each well of plates. The podocytes were stimulated as described above, and the scratches were observed under a microscope at 0 and 12 hours and at subsequent 12-hour intervals until 72 hours. Ten points were randomly selected on each scratch, and the podocyte migration distances were recorded at each time point. Average migration distance at x hours = ([scratch width of 0 hours at point 1 − scratch width of x hours at point 1] + … + [scratch width of 0 hours at point 10 − scratch width of x hours at point 10])/10.
Methyl Thiazolyl Tetrazolium Cell Viability Assay
The podocytes were stimulated as described above and collected at 24, 48, and 72 hours with 0.25% EDTA-free trypsin. The cells were counted and resuspended in plasma-free RPMI 1640 medium to 1 × 106 cells/ml. Methyl thiazolyl tetrazolium (MTT) reagent (ab211091; Abcam, Cambridge, United Kingdom) was added and incubated at 37°C for 3 hours. MTT solvent was added and the absorbance was read at 590 nm.
Effects of C3aR Antagonists on Rats of Heymann Nephritis
Experimental Design of Passive Heymann Nephritis
Sprague-Dawley rats (Supplemental Figure 2), male, 5 weeks old, received a single injection of sheep anti-rat Fx1A serum (0.8 ml/100 g, PTX-002S; Probetex, San Antonio, TX) via the tail vein. The rats in the negative control group were injected with normal saline.
SB290157 Group
The rats with Heymann nephritis were divided into three groups. The rats in the early-treatment group (n=6) were treated with a C3aR antagonist SB290157 (30 mg/kg, HY-101502A; MedChemExpress) from day 0 by daily intraperitoneal injection. The rats in the late-treatment group (n=8) were administered from day 21 when the rats presented with proteinuria. The rats in the disease control group (n=10) were treated with normal saline from day 0. All the rats were euthanized at week 7.
JR14a Group
The rats in the early-treatment group (n=8) were treated with a C3aR antagonist JR14a (10 mg/kg, HY-138161, MedChemExpress) from day 0 by daily intragastrical administration. The rats in the late-treatment group (n=6) were administered from day 21 when the rats presented with proteinuria. Other processes were the same as above.
Detection of Clinical Parameters and Circulating IgG, C3a, and C5a
Urine and blood samples were collected every week. Urinary protein, serum creatinine, BUN, and total cholesterol were analyzed by an automatic biochemical analyzer (UniCel DxC 600 Synchron; Beckman Coulter, Brea, CA).
The circulating levels of rat total IgG, IgA, IgM, C3a, and C5a were detected by commercial ELISA kits (IgG: JER-081, IgA: JER-082, IgM: JER-083; Joyeebio, Anhui, China; C3a: NBP2–70039, C5a: NBP2–82137; Novus Biologicals, Littleton, CO). The steps were carried out according to the manufacturers’ instructions.
The specific rat anti-sheep IgG in circulation was assessed by ELISA. Sheep anti-Fx1A serum (PTX-002S; Probetex) was coated on 96-well microplates with carbonate buffer (pH=9.6) at 1:300 and 4°C for overnight. After blocking with 3% BSA, rat plasma samples were added at 1:50 and incubated at 37°C for 1 hour. Then, alkaline phosphatase–conjugated rabbit anti-rat IgG was added and incubated at 37°C for 1 hour. The absorbance was read at 450 nm.
Pathologic Examination of the Kidneys
Kidney tissues were fixed in 4% paraformaldehyde and embedded in paraffin, or frozen in liquid nitrogen. The deposition of IgG (sheep and rat) and complement (C1q, MBL, factor B, C3d, C4d, C5b-9), the expression of C3aR and C5aR, and the inflammation cells (macrophage, CD4+ T cells, CD8+ T cells, regulatory T cells) were detected by immunohistochemistry or immunofluorescence appropriately (Supplemental Table 3). Image-Pro Plus (version 7.0, Media Cybernetics, Rockville, MD) was used to measure the staining intensity by integral OD.
The cortical tissues (2 mm × 2 mm) were fixed at 4°C, and specimens were produced and scanned under a transmission electron microscope. Two glomeruli were randomly selected from each kidney sample and 20 representative nonoverlapping digital micrographs were taken from each glomerulus under ×10,000 magnification. The number of subepithelial electron-dense deposits was counted in 13.5 × 18.0 μm electron microscope pictures. The thickness of the GBM was calculated as ΣGBM area/ΣGBM length. The width of the podocyte foot process was calculated as (π/4) × (ΣGBM length/Σnumber of foot processes). The data were analyzed by ImageJ (version 1.48, National Institutes of Health, Bethesda, MD).
Rat Podocyte Injuries Mediated by Plasma from Rats with Heymann Nephritis
The primary rat podocytes (NBCC, Henan, China) were used in vitro and exposed to rat plasma samples obtained from disease control groups, early-treatment groups, late-treatment groups, and negative control groups at week 7 with sodium heparin as anticoagulator. The real-time PCR, migration assay, and MTT assay were performed as described above (Supplemental Table 1).
Statistical Analyses
Statistical analysis was performed using SPSS 23.0 (IBM, Armonk, NY). Shapiro–Wilk test was used to determine the distribution of data. For data of normal distribution, the results were expressed as mean±SD. For data of nonnormal distribution, the results were expressed as median (interquartile range). t tests and Mann–Whitney U tests were used to assess the differences of quantitative and semiquantitative data. According to distributions, one-way ANOVA or Kruskal–Wallis test was used to compare differences among groups for continuous variables, and chi-squared test or Fisher exact test was used to compare differences among groups for categorical variables. The probabilities were two-sided, and P<0.05 was considered statistically significant.
Ethical Statement
This study was performed in compliance with the Declaration of Helsinki and approved through the Ethics Committee and Experimental Animal Ethics Committee of Peking University First Hospital. Written informed consent was obtained from all participants for the collection of clinical data, blood, urine, and kidney samples.
Results
Circulating C3a Levels and Expression of C3aR in the Kidneys of Patients with Primary MN
Clinical and Pathologic Data of Patients
The clinical features of the 37 patients with kidney biopsy-proven primary MN are shown in Supplemental Table 4. At diagnosis, the median (interquartile range) proteinuria was 6.1 (3.3, 10.1) g/24 h, serum albumin was 26.0 (23.0, 31.0) g/L, and eGFR was 92.2 (59.6, 100.3) ml/min per 1.73 m2. Twenty-five (67.6%) patients had positive anti-PLA2R antibodies (>20 U/ml), with a median level of 150.0 (22.0, 437.5) U/ml. Granular staining of PLA2R was observed in 33 (89.2%) patients. The median plasma level of C3a/C3a des-Arg was 1817.7 (1142.9, 6721.4) ng/ml, markedly higher than that observed in healthy controls (111.7 [99.2, 174.8] ng/ml, P=0.001).
A total of 25 (67.6%) patients received steroids and/or immunosuppressive treatments, and the remaining 12 received angiotensin-converting enzyme inhibitors and/or angiotensin receptor blockers only. During the follow-up period of 29.0 (27.0, 31.0) months, 33 (89.2%) patients achieved clinical remission: 13 (35.1%) achieved complete remission and 20 (54.1%) partial remission.
Expression of C3aR on Podocytes
Intense granular staining of C3aR was observed along the glomerular capillary walls of the patients with primary MN (integral OD 0.61 [0.59, 0.65]), but was weaker in the patients with diabetic nephropathy (0.38 [0.31, 0.46], P<0.001) or FSGS (0.52 [0.46, 0.60], P<0.001), and negative in the patients with minimal change disease (0.29 [0.26, 0.35], P<0.001) and healthy controls (0.24 [0.21, 0.25], P<0.001) (Figure 1, A and B).
Figure 1.

The expression of C3aR in the kidneys of patients with primary MN and its correlations with clinical features. The granular staining of C3aR along glomerular capillary walls was strongly shown in patients with primary MN, weaker in patients with diabetes nephropathy (DN) and FSGS, and was negative in patients with minimal change disease (MCD) and healthy controls (A, B). The staining intensity was measured by integral OD (IOD). The expression of C3aR merged well with podocin on the podocytes (red: C3aR; green: podocin) (C). In patients with primary MN, the level of C3aR expression was positively correlated with proteinuria (D) and serum creatinine (E). It was significantly lower in the patients who achieved clinical remission after treatments compared with that in the patients of no remission (F).
Staining of C3aR merged well with podocin, indicating that C3aR is expressed on the cell membrane of podocytes (Figure 1C).
Correlations between C3aR Expression and Clinical Features
At the time of kidney biopsy, overexpression of C3aR on podocytes correlated positively with the level of proteinuria (r=0.569, P<0.001) and serum creatinine (r=0.642, P<0.001) (Figure 1, D and E). However, the overexpression was not related to the circulating levels of anti-PLA2R antibodies (P=0.735) or C3a (P=0.806), or staining in the kidneys of PLA2R (P=0.499), IgG (P=0.508), or C3 (P=0.055).
The expression of C3aR was lower in the patients who achieved clinical remission after treatment compared with that in patients who did not (0.60 [0.59, 0.63] versus 0.67 [0.60, 0.68], P=0.021, Figure 1F). Expression was also lower in patients in complete remission than those with partial remission (0.60 [0.58, 0.62] versus 0.61 [0.59, 0.65], P=0.045).
C3a/C3aR Pathway Mediates Podocyte Injury In Vitro
Human podocytes were exposed to medium containing 10% MN plasma (with C3a 1966.7±259.0 ng/ml, Supplemental Table 5), 10% MN plasma + C3aR antagonist SB290157, 10% MN plasma + C3aR antagonist JR14a, 10% MN plasma + human C3a (2 μg/ml), 10% healthy plasma (with C3a 144.5±52.7 ng/ml, Supplemental Table 5), C3a, or blank control.
Expression of PLA2R and C3aR on Podocytes
PLA2R was expressed weakly on cultured human podocytes (Figure 2A). Expression was increased significantly after exposure to MN plasma (mRNA: 17.4 [17.1, 25.0] versus 0.6 [0.6, 0.7], P=0.011; protein: 2.0 [2.0, 2.0] versus 0.0 [0.0, 0.0], P<0.001) compared with podocytes exposed to healthy plasma. This upregulation of PLA2R by MN plasma was blocked by both C3aR antagonists, SB290157 (mRNA: 1.3 [0.5, 8.6], P=0.049; protein: 1.5 [1.1, 1.9], P=0.047) and JR14a (mRNA: 1.2 [0.4, 17.0], P<0.001; protein: 0.8 [0.2, 1.0], P=0.001), in a dose-dependent manner (Supplemental Figure 3A).
Figure 2.
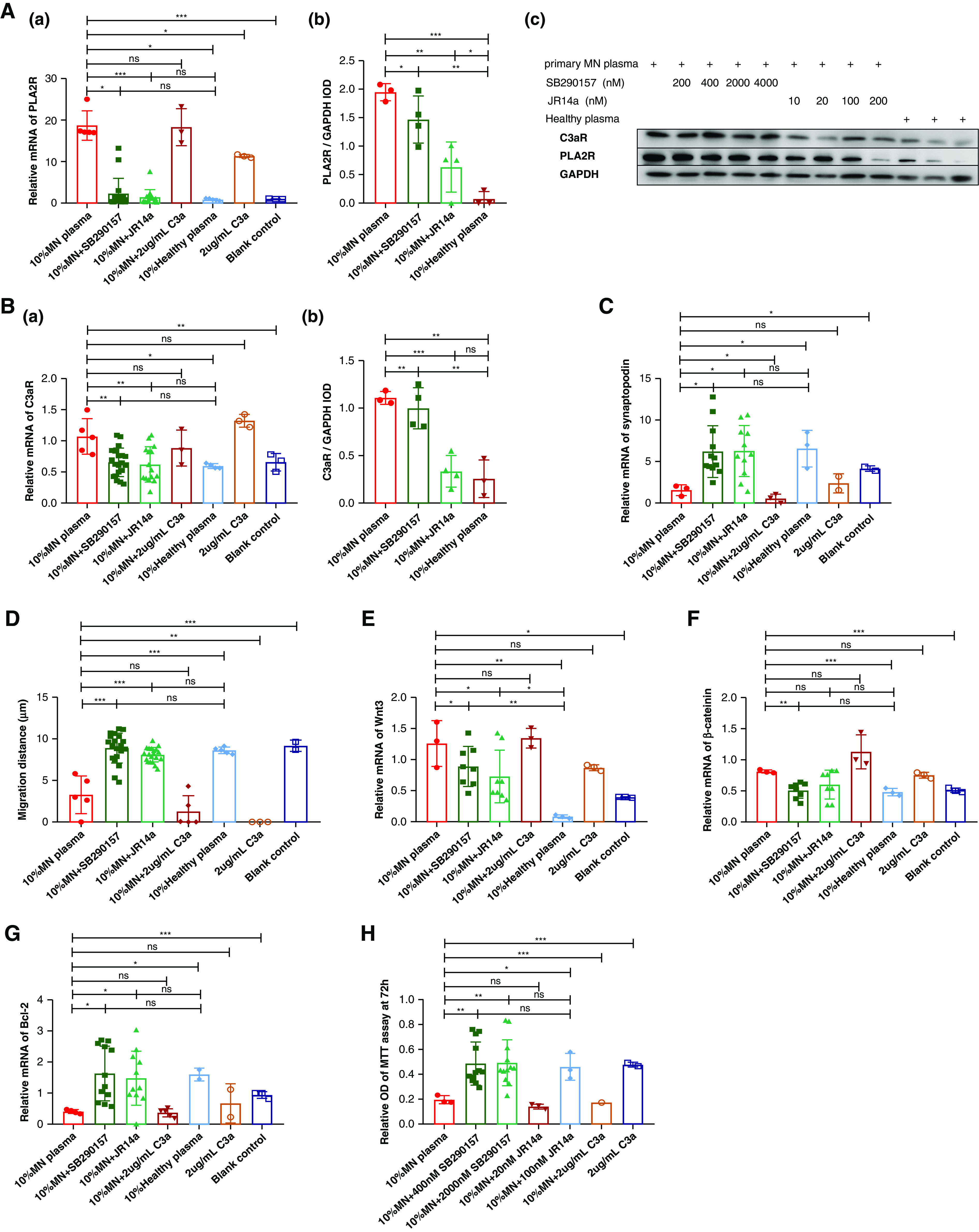
The effects on podocytes after exposure to the plasma from MN patients and C3aR antagonists. Human podocytes were exposed to medium containing 10% MN plasma (with C3a 1966.7±259.0 ng/ml), 10% MN plasma + C3aR antagonist SB290157, 10% MN plasma + C3aR antagonist JR14a, 10% MN plasma + human C3a (2 μg/ml), 10% healthy plasma, C3a, or blank control. The expression of PLA2R (A) and C3aR (B) was significantly elevated after exposure to MN plasma, and blocked by C3aR antagonists. Synaptopodin (C) was downregulated in podocytes exposed to MN plasma and recovered by the addition of C3aR antagonists. The migration distance of podocytes (D) was shortened after exposure to MN plasma. C3aR antagonists could attenuate it. The expression of Wnt3 (E) and β-catenin (F) in podocytes was upregulated by exposure of MN plasma and blocked by C3aR antagonists. The apoptosis inhibition gene Bcl-2 (G) was downregulated and the viability of podocytes (H) was decreased in MN plasma, which was attenuated by C3aR antagonists.
The expression of C3aR (Figure 2B) on podocytes was increased after stimulation by MN plasma (mRNA: 1.1 [0.8, 1.3] versus 0.6 [0.6, 0.7], P=0.011; protein: 1.2 [1.2, 1.2] versus 0.2 [0.1, 0.5], P=0.001) and attenuated by the C3aR antagonists, SB290157 (mRNA: 0.6 [0.5, 0.8], P=0.002; protein: 1.0 [0.8, 1.2], P=0.006) and JR14a (mRNA: 0.5 [0.4, 0.9], P=0.007; protein: 0.3 [0.2, 0.5], P<0.001).
Expression of Synaptopodin and Migration Function of Podocytes
Expression of synaptopodin (Figure 2C) was downregulated in podocytes exposed to MN plasma (mRNA: 1.8 [0.8, 2.1] versus 7.0 [4.2, 8.5], P=0.020). The addition of C3aR antagonists recovered this expression.
The migration distance of podocytes (Figure 2D and Supplemental Figure 4) was shortened after exposure to MN plasma (3.0 [1.2, 5.4] versus 8.7 [8.2, 9.0] μm, P<0.001). C3aR antagonists attenuated this change, SB290157 (9.4 [7.6, 10.3] μm, P<0.001) and JR14a (8.5 [7.5, 8.9] μm, P<0.001).
Wnt/β-Catenin Pathway in Podocytes
The expression of Wnt3 (Figure 2E) and β-catenin (Figure 2F) in podocytes was upregulated significantly by exposure to MN plasma (Wnt3: 1.2 [0.9, 1.6] versus 0.1 [0.1, 0.1], P=0.002; β-catenin: 0.8 [0.8, 0.8] versus 0.5 [0.4, 0.5], P<0.001) and blocked by the C3aR antagonists, SB290157 (Wnt3: 0.7 [0.5, 1.0], P=0.040; β-catenin: 0.5 [0.4, 0.6], P=0.001) and JR14a (Wnt3: 0.5 [0.4, 0.7], P=0.015; β-catenin: 0.6 [0.4, 0.8], P=0.100).
The apoptosis inhibition gene17 Bcl-2 (Figure 2G) was downregulated in MN plasma (0.4 [0.3, 0.5] versus 1.6 [1.5, 1.7], P=0.018), which was arrested by the C3aR antagonists, SB290157 (1.4 [0.8, 2.6], P=0.017) and JR14a (1.2 [0.9, 2.2], P=0.030).
The viability of podocytes (Figure 2H) was decreased after exposure to MN plasma (relative OD in the MTT assay: 0.2 [0.2, 0.2] versus 0.4 [0.4, 0.6], P=0.015) compared with that measured in healthy plasma. After the addition of C3aR antagonists, the viability of the podocytes was recovered.
We also assessed the expression of NF-κB, IL-1, IL-6, IL-10, TNF-α, vinculin, α-actinin 4, PI3K, GSK3β, and AKT1, and showed no significant change in any factor (P>0.05, Supplemental Figures 5 and 6).
C3aR Antagonist Treatments Arrested and Attenuated Heymann Nephritis in Rats
Clinical and Pathologic Parameters of Kidney Injuries
Passive Heymann nephritis was established in Sprague-Dawley rats by a single intravenous injection of sheep anti-Fx1A serum (Figure 3 and Supplemental Figure 2). At week 2, the rats developed proteinuria that gradually worsened. At week 7, the rats presented with severe proteinuria (P<0.001), although the serum creatinine, BUN, and cholesterol levels remained in the normal range (P>0.05). Subepithelial electron-dense deposits (P<0.001), a diffusive podocyte effusion process (P<0.001), and thickening of the GBM (P<0.010) were shown in the kidneys using electron microscopy.
Figure 3.

The treatments of C3aR antagonist reduced proteinuria of Heymann nephritis. Sprague-Dawley rats were injected with anti-Fx1A antibody serum 0.8 ml/100 g via the tail vein. The rats developed proteinuria and aggravated gradually (A). At week 7, the rats presented with severe proteinuria (B) and the kidneys (C–E) showed electron-dense deposit in subepithelial area, effusive podocyte foot process fusion, and basement membrane thickening. In the early-treatment groups, the C3aR antagonist, SB290157 (30 mg/kg intraperitoneally, n=6) or JR14a (10 mg/kg intragastrically, n=8), was administrated daily from day 0. In the late-treatment groups, both C3aR antagonists (SB290157, n=8; JR14a, n=6) were administrated from week 3 after the proteinuria was detected. Compared with the disease controls treated with normal saline, the level of proteinuria was significantly reduced in both the early-treatment and late-treatment groups of two kinds of C3aR antagonists (A, B). It was even lower than that at the time point when the treatments were initiated. The amount of electron-dense deposits, the foot process width, and the thickening of GBM were alleviated (C–E). *P<0.05, **P<0.01, ***P<0.001.
In the early-treatment groups (Figure 3), C3aR antagonists were administrated daily from day 0, SB290157 (30 mg/kg intraperitoneally) and JR14a (10 mg/kg intragastrically). At week 7, the levels of urinary protein were markedly reduced compared with those in the disease controls treated with normal saline (SB290157: 120.6 [76.0, 161.0] versus 299.1 [237.2, 367.0] mg/d, P<0.001; JR14a: 282.6 [247.3, 306.0] versus 484.9 [460.7, 512.6] mg/d, P<0.001). In the kidneys, the amount of electron-dense deposits under epithelial cells was reduced (SB290157: P<0.001; JR14a: P<0.001), the foot process width was decreased (SB290157: P=0.012; JR14a: P<0.001), and the thickening of the basement membrane was alleviated (SB290157: P=0.160; JR14a: P<0.001).
In the late-treatment groups (Figure 3), the C3aR antagonists were administrated from week 3 after the rats presented with proteinuria. The rats were divided randomly into disease control and treatment groups, with no difference between the groups for the presence of proteinuria (P>0.5). After treatments, proteinuria was reduced significantly (SB290157: 196.3 [119.1, 289.8] versus 299.1 [37.2, 367.0] mg/d, P=0.039; JR14a: 255.3 [308.8, 345.0] versus 484.9 [460.7, 512.6] mg/d, P<0.001). The level of proteinuria was even lower than that measured at the time point when the treatments were initiated. In the kidneys, the subepithelial electron-dense deposits (SB290157: P<0.001; JR14a: P<0.001), podocytes effusion process (SB290157: P=0.002; JR14a: P=0.001), and GBM thickening (SB290157: P=0.998; JR14a: P=0.002) were also alleviated.
Effects of C3aR Antagonists on Specific IgG and Complement Activation
In the rats with Heymann nephritis, the plasma level of C3a (P<0.001) and glomerular expression of C3aR (SB290157: P<0.001; JR14a: P=0.021) were increased significantly (Figure 4). After C3aR antagonist treatment, the plasma C3a level was reduced in both the early-treatment groups (SB290157: P=0.001; JR14a: P<0.001) and late-treatment groups (SB290157: P<0.001; JR14a: P=0.001). Staining of C3aR in glomeruli (Figure 4) was decreased in both the early-treatment groups (SB290157: P<0.001; JR14a: P=0.032) and late-treatment groups (SB290157: P<0.001; JR14a: P=0.221). The expression of C5aR1 was not influenced by the C3aR antagonists (Supplemental Figure 7, P>0.05).
Figure 4.

The treatments of C3aR antagonist alleviated plasma level of C3a and glomerular expression of C3aR on Heymann nephritis rats. Sprague-Dawley rats were injected with anti-Fx1A antibody serum via the tail vein. The plasma C3a was significantly elevated in disease controls (A, C). After C3aR antagonist treatments, it was decreased remarkably, both in the early-treatment groups and in the late-treatment groups: SB290157 (B), JR14a (D). In the kidneys, the staining of C3aR was strong in the rats with Heymann nephritis (E), and was attenuated in both treatment groups (F, G). *P<0.05, **P<0.01, ***P<0.001.
The rats with Heymann nephritis produced large amounts of specific anti-sheep IgG due to the passive transfer of sheep anti-Fx1A serum (P<0.001) (Figure 5). Both sheep IgG and rat IgG were deposited in a granular pattern along the capillary walls of the kidneys (Figure 5). The complement cascade was activated (C3d, C4d, and C5b-9 deposits) through the classic pathway (C1q deposit) and the alternative pathway (factor B deposit), and MBL staining was negative (Figure 5, Supplemental Figure 7).
Figure 5.

The treatments of C3aR antagonist inhibited specific IgG and complement activation on Heymann nephritis rats. Sprague-Dawley rats were injected with anti-Fx1A antibody serum via the tail vein. In the early-treatment groups, C3aR antagonists (SB290157 or JR14a) was administrated daily from day 0. In the late-treatment groups, both C3aR antagonists were administrated from week 3 after proteinuria was detected. Compared with the disease controls treated with normal saline, the plasma level of specific anti-sheep IgG was decreased in treatment groups of both C3aR antagonists, SB290157 (A) and JR14a (B), whereas the level of total IgG was comparable among the groups. In the glomeruli, the deposit of sheep IgG (C) was comparable among the disease controls and treatment groups. However, the deposit of rat IgG (D) was alleviated in both treatment groups. Along with that, the staining of C1q (E), factor B (F), and C5b-9 (G) was significantly decreased in the treatment groups. *P<0.05, **P<0.01, ***P<0.001.
After C3aR antagonist treatments, the circulating levels of rat IgG against sheep serum were decreased significantly in both the early-treatment groups (SB290157: P=0.015; JR14a: P=0.009) and late-treatment groups (SB290157: P<0.001; JR14a: P=0.067) (Figure 5). In the kidneys, the deposition of rat IgG along the capillary walls was alleviated in both treatment groups (early-treatment: SB290157 P=0.038, JR14a P=0.049; late-treatment: SB290157 P=0.305, JR14a P=0.053), whereas the deposition of sheep IgG was comparable between the disease group and the treatment groups (P>0.05). Subsequently, the activation of complement cascade was attenuated with less deposition of C1q (early-treatment: SB290157 P=0.037, JR14a P=0.023; late-treatment: SB290157 P=0.190, JR14a P=0.011), factor B (early-treatment: SB290157 P=0.007, JR14a P<0.001; late-treatment: SB290157 P=0.011, JR14a P=0.007), and C5b-9 (early-treatment: SB290157 P=0.013, JR14a P=0.049; late-treatment: SB290157 P=0.004, JR14a P=0.045) (Figure 5). The deposition of MBL, C3d, and C4d were not influenced by C3aR antagonist treatments (Supplemental Figure 7).
We also assessed the inflammatory cells infiltration in the kidneys, including CD4+ T cells, CD8+ T cells, regulatory T cells (foxp3+), and macrophages (CD68+), and there was no significant difference after C3aR antagonist treatments (P>0.05, Supplemental Figure 8).
Plasma from Rats with C3aR Antagonists Attenuated Rat Podocyte Injuries In Vitro
The plasma from rats in the disease control, treatment groups, and negative control were collected to stimulate primary rat podocytes (Figure 6). Exposure to the plasma of rats with Heymann nephritis induced upregulation of C3aR (P=0.010) and β-catenin (P=0.013) on rat podocytes, and inhibited synaptopodin (P=0.001) and Bcl-2 (P=0.004), compared with those exposed to the plasma from negative control rats. In the treatment groups, the overexpression of C3aR was blocked (early-treatment: SB290157 P=0.001, JR14a P=0.005; late-treatment: SB290157 P=0.001, JR14a P=0.009), the downregulation of synaptopodin was recovered (early-treatment: SB290157 P=0.007, JR14a P=0.002; late-treatment: SB290157 P=0.094, JR14a P<0.001), the upregulation of β-catenin was inhibited (early-treatment: SB290157 P=0.017, JR14a P=0.005; late-treatment: SB290157 P=0.015, JR14a P=0.006), and Bcl-2 was also recovered.
Figure 6.

The rat podocyte injuries after exposure to the plasma from rats with Heymann nephritis and those with C3aR antagonists. On primary rat podocytes, the expression of C3aR (A) and β-catenin (B) was upregulated, and the expression of synaptopodin (C) and Bcl-2 (D) was downregulated, after exposure to the plasma from disease rats. These effects were attenuated in the treatment groups of both C3aR antagonists. The migration distance (E) and cell viability (F) were reduced after the podocytes were exposed to the disease plasma, which was recovered in treatment groups.
The migration function of podocytes was damaged after exposure to the plasma from disease rats (P<0.001, Figure 6, Supplemental Figure 9). Migration function was recovered significantly in the plasma from rats under C3aR antagonist treatments (early-treatment: SB290157 P=0.007, JR14a P<0.001; late-treatment: SB290157 P=0.021, JR14a P=0.014).
The cell viability of podocytes was decreased in the plasma from disease rats (P<0.001), but recovered in the plasma from rats under C3aR antagonist treatments (early-treatment: SB290157 P<0.001, JR14a P<0.001; late-treatment: SB290157 P<0.001, JR14a P=0.013).
Discussion
Accumulating evidence supports the essential role of complement activation in the pathogenesis of primary MN.18 This has led to the identity of complement effector mechanisms of important therapeutic implications. This study showed that C3a was markedly increased in the circulation of patients with primary MN. C3aR was overexpressed on their podocytes and was related with clinical severity and treatment responses. The plasma from MN patients containing high levels of C3a promoted the upregulation of PLA2R and overexpression of C3aR on human podocytes in vitro, and impeded the expression of synaptopodin and the migration function and cell viability of podocytes, possibly through the Wnt/β-catenin pathway. These effects could be blocked by the C3aR antagonists, SB290157 and JR14a. Based on these results, we firstly tried C3aR antagonist therapy in vivo in Heymann nephritis rats, and showed that the proteinuria was reduced in both early- and late-treatment groups. Immune complex deposition, foot process effacement, and GBM thickening were also alleviated in the kidneys. Specific rat IgG against sheep serum was reduced in circulation, whereas the total IgG level was not influenced. In addition, the deposition of rat IgG and subsequent complement activation in the kidneys was attenuated. The plasma from diseased rats induced injuries on primary rat podocytes similar to those observed with the plasma from MN patients. C3aR antagonists also blocked these effects. Taken together, these results demonstrate that the C3a/C3aR pathway plays a crucial role in the pathogenesis of MN and that C3aR antagonists may have potential clinical value for the treatment of MN patients.
Complement-mediated podocyte injuries in MN have been attributed mainly to C5b-9,4,18,19 although inhibition of C5b-9 formation in C6-deficient rats did not fully prevent proteinuria associated with Heymann nephritis.6 Our previous study13 and this study showed that the level of C3a des-Arg (a biomarker for elevated C3a) was markedly elevated (>10-fold increase) in the plasma and urine of MN patients, and was even higher than that observed in other glomerular diseases characteristic of podocyte injuries, such as FSGS and minimal change disease. In this study we found that C3aR was overexpressed on the podocytes of MN patients. This elevated expression of C3aR was related to severe proteinuria, kidney dysfunction, and no response to treatments. These findings from patients imply that C3a/C3aR may be a crucial effector of complement-mediated podocyte damage in MN.
We exposed cultured human podocytes to the plasma from MN patients containing high levels of C3a, to assess the in vitro effects on podocytes. PLA2R is expressed weakly in this cell line, similar to that observed on podocytes in the kidneys of healthy individuals.2 After exposure to MN plasma, the expression of PLA2R was markedly upregulated, and could also be induced by C3a and blocked by C3aR antagonists. The expression of PLA2R on podocytes is essential for antibody binding and complement activation within the mechanism of MN.2,15,20–22 Our results provide one possibility that overexpression of PLA2R in MN patients might be mediated by elevated levels of C3a in any inflammatory condition, before disease onset, and then be sustained during the course of disease.
We also showed that C3aR expression on podocytes was upregulated, and synaptopodin was reduced after exposure to MN plasma. These results are consistent with those from Haddad et al.15 Their study showed overexpression of C3aR on podocytes after anti-PLA2R IgG4 deposition in the presence of complement, and further revealed a reduction in synaptopodin levels as a result of the proteolytic pathway mediated by cysteine proteinase. The destruction and loss of cytoskeleton protein, in turn, leads to the functional incapacitation and the migratory dysfunction of podocytes observed in this study. Increased C3aR expression has also been reported in rodent models of diabetic nephropathy,23 suggesting that the overexpression of C3aR might be induced by C3a activation, but not directly by anti-PLA2R antibodies.
Human C3aR is a 55-kDa seven-transmembrane domain receptor that belongs to G-protein coupled receptor family.7 Signal transduction after C3aR activation has been fully investigated in podocytes. This study showed that the Wnt/β-catenin pathway was promoted by MN plasma, associated with a downregulation of Bcl-2, and influenced the cell viability of podocytes. The Wnt/β-catenin pathway triggers a wide variety of damage to podocytes.24–27 The accumulated β-catenin translocates into cell nuclei and forms a transcription factor complex, the β-catenin/T cell factor (TCF)/lymphoid enhancer factor (LEF). This factor, in turn, promotes the transcription factor, Snail, that inhibits expression of cytoskeleton protein and suppresses podocytes’ viability by downregulation of Bcl-2. C3aR can also stimulate phosphatidylinositol 3-kinase (PI3K) to inhibit glycogen synthase kinase (GSK)-3β, thereby inducing downregulation of cytoskeleton proteins28,29 such as nephrin, Snail, and α-actinin 4, and focal adhesion proteins such as vinculin. However, we did not observe these changes in our experiments.
All the above podocyte injuries mediated by MN plasma could be blocked in vitro by C3aR antagonists. This provides a rationale for assessing the therapeutic implications of C3aR blockade. To the best of our knowledge, this is the first study to evaluate treatment with C3aR antagonists in passive Heymann nephritis rats, with these investigations achieving good responses. Proteinuria was obviously reduced, and the deposition of immune complexes, effacement of foot processes, and thickening of the GBM were alleviated in the kidneys. These effects were noted, not only in the early-treatment groups, but also in the late-treatment groups in which C3aR antagonists were administrated after the rats developed proteinuria.
We showed that plasma C3a levels and expression of glomerular C3aR were elevated in diseased rats, which was consistent with the findings in MN patients. This indicates that this model is appropriate for the assessment of C3aR antagonists in vivo. After C3aR antagonist treatment, the increase in plasma C3a levels and overexpression of C3aR were significantly reduced, whereas C5aR expression remained unchanged. In addition to the injuries on primary rat podocytes that were mediated by plasma from diseased rats and blocked by C3aR antagonists, consistent with similar findings on human podocytes, we also noticed the effects of C3aR antagonists on the adaptive immune system. In rats receiving treatment, the levels of specific rat IgG against passively transferred sheep serum were significantly reduced, and subsequent deposition of rat IgG and complement activation on glomeruli was alleviated. In contrast, total IgG levels did not change. Recently, similar findings have been reported in ovalbumin (OVA) immunization of C3ar1−/−C5ar1−/− mice, in which OVA-specific IgG production was eliminated. However, identical OVA immunization of decay acceleration factor (DAF)−/− mice, in which C3aR/C5aR signaling is potentiated, caused heightened anti-OVA total IgG production than that observed in wild-type mice.30 During the production of antigen-specific antibodies, blockade of C3aR inhibited activation of dendritic cells by downregulating the expression of major histocompatibility complex II (MCH II), B7.1, B7.2, and CD40.31,32 There is evidence that simultaneous blockade of C3aR and C5aR upregulates Foxp3 and CTLA4 in regulatory T cells, and suppresses conventional T cells.33 Blockade of C3aR has also been shown to suppress the proliferation, differentiation, and affinity maturation of germinal center B cells.34 Through these mechanisms, C3aR blockade attenuates the production of specific IgG in the adaptive immune response.35 This may be one mechanism by which C3aR antagonists arrested and attenuated passive Heymann nephritis. However, whether this mechanism contributes to human MN, especially PLA2R-associated MN, requires further investigations.
In summary, our results show that the C3a/C3aR pathway is a crucial effector of the complement-mediated podocyte damage in primary MN. C3aR antagonists may therefore be a potentially viable treatment for this disease. These findings could support studies in human MN patients targeting this therapeutic strategy.
Disclosures
M. Zhao reports consultancy with AstraZeneca, GlaxoSmithKline, InflaRx, Novartis, and Roche; honoraria from Chinese Medical Association and Chinese Society of Nephrology; and an advisory or leadership role with Chinese Society of Nephrology (vice president) and Asian Pacific Society of Nephrology (executive member). All remaining authors have nothing to disclose.
Funding
This work is supported by grants from the Natural Science Foundation of China (81870486, 82070732, 82090021), the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2019-I2M-5-046), and the X-Young Scholars Project of Peking University (PKU2021LCXQ013).
Supplementary Material
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Complement is Complimentary in Membranous Nephropathy,” on pages 1631–1633.
Author Contributions
Z. Cui and M.-H. Zhao conceptualized the study and were responsible for funding acquisition, methodology, project administration, resources, software, and supervision; S. Gao was responsible for data curation, formal analysis, investigation, methodology, project administration, validation, and visualization and wrote the original draft; M.-H. Zhao was responsible for formal analysis; Z. Cui was responsible for investigation; and all authors reviewed and edited the manuscript.
Data Sharing Statement
All data used in this study are available on request to the authors.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2021101384/-/DCSupplemental.
Supplemental Table 1. Primers for the target genes.
Supplemental Table 2. Primary antibodies and secondary antibodies used in Western blot assays.
Supplemental Table 3. Primary antibodies used on the kidney pathologic examination of rats with Heymann nephritis.
Supplemental Table 4. Clinical features of the patients with kidney biopsy-proven primary MN.
Supplemental Table 5. Plasma level of C3a from five patients with primary MN and healthy controls in podocytes experiments in vitro.
Supplemental Figure 1. Optimal time point for mRNA detection was selected by time course experiments.
Supplemental Figure 2. Flowchart of C3aR antagonist interventions on passive Heymann nephritis.
Supplemental Figure 3. Dose-dependent manner of C3aR blockade to the podocyte injuries mediated by plasma from MN patients.
Supplemental Figure 4. Migration assay of human podocytes at different time points to different exposure.
Supplemental Figure 5. Expression of NF-κB, IL-1, IL-6, IL-10, and TNF-α on human podocytes after exposure to MN plasma and C3aR antagonists.
Supplemental Figure 6. Expression of vinculin, α-actinin 4, PI3K, GSK3β, and AKT1 on human podocytes after exposure to MN plasma and C3aR antagonists.
Supplemental Figure 7. Staining of C5aR1, MBL, C3d, and C4d in the glomeruli of rats with Heymann nephritis.
Supplemental Figure 8. Staining of inflammatory cell infiltration in the kidneys of rats with Heymann nephritis.
Supplemental Figure 9. Migration assay of primary rat podocytes exposed to the plasma from rats with Heymann nephritis, C3aR antagonist treatment groups, and negative controls.
References
- 1.Ronco P, Beck L, Debiec H, Fervenza FC, Hou FF, Jha V, et al. : Membranous nephropathy. Nat Rev Dis Primers 7: 69, 2021 [DOI] [PubMed] [Google Scholar]
- 2.Beck LH Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. : M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 361: 11–21, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomas NM, Beck LH Jr, Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, et al. : Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med 371: 2277–2287, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salant DJ, Belok S, Madaio MP, Couser WG: A new role for complement in experimental membranous nephropathy in rats. J Clin Invest 66: 1339–1350, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Couser WG: Pathogenesis of glomerular damage in glomerulonephritis. Nephrol Dial Transplant 13[Suppl 1]: 10–15, 1998 [DOI] [PubMed] [Google Scholar]
- 6.Spicer ST, Tran GT, Killingsworth MC, Carter N, Power DA, Paizis K, et al. : Induction of passive Heymann nephritis in complement component 6-deficient PVG rats. J Immunol 179: 172–178, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Gao S, Cui Z, Zhao MH: The complement C3a and C3a receptor pathway in kidney diseases. Front Immunol 11: 1875, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun MC, Reins RY, Li TB, Hollmann TJ, Dutta R, Rick WA, et al. : Renal expression of the C3a receptor and functional responses of primary human proximal tubular epithelial cells. J Immunol 173: 4190–4196, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Song D, Guo WY, Wang FM, Li YZ, Song Y, Yu F, et al. : Complement alternative pathway’s activation in patients with lupus nephritis. Am J Med Sci 353: 247–257, 2017 [DOI] [PubMed] [Google Scholar]
- 10.Han R, Hu S, Qin W, Shi J, Hou Q, Wang X, et al. : C3a and suPAR drive versican V1 expression in tubular cells of focal segmental glomerulosclerosis. JCI Insight 4: 122912, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu L, Zhang Y, Duan X, Peng Q, Liu Q, Zhou Y, et al. : C3a, C5a renal expression and their receptors are correlated to severity of IgA nephropathy. J Clin Immunol 34: 224–232, 2014 [DOI] [PubMed] [Google Scholar]
- 12.Li XQ, Chang DY, Chen M, Zhao MH: Complement activation in patients with diabetic nephropathy. Diabetes Metab 45: 248–253, 2019 [DOI] [PubMed] [Google Scholar]
- 13.Zhang MF, Huang J, Zhang YM, Qu Z, Wang X, Wang F, et al. : Complement activation products in the circulation and urine of primary membranous nephropathy. BMC Nephrol 20: 313, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y, Wang C, Jin L, He F, Li C, Gao Q, et al. : IgG4 anti-phospholipase A2 receptor might activate lectin and alternative complement pathway meanwhile in idiopathic membranous nephropathy: An inspiration from a cross-sectional study. Immunol Res 64: 919–930, 2016 [DOI] [PubMed] [Google Scholar]
- 15.Haddad G, Lorenzen JM, Ma H, de Haan N, Seeger H, Zaghrini C, et al. : Altered glycosylation of IgG4 promotes lectin complement pathway activation in anti-PLA2R1-associated membranous nephropathy. J Clin Invest 131: e140453, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, Liu G, Zhang YM, Cui Z, Wang F, Liu XJ, et al. : Urinary soluble urokinase receptor levels are elevated and pathogenic in patients with primary focal segmental glomerulosclerosis. BMC Med 12: 81, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swanton E, Savory P, Cosulich S, Clarke P, Woodman P: Bcl-2 regulates a caspase-3/caspase-2 apoptotic cascade in cytosolic extracts. Oncogene 18: 1781–1787, 1999 [DOI] [PubMed] [Google Scholar]
- 18.Ma H, Sandor DG, Beck LH Jr: The role of complement in membranous nephropathy. Semin Nephrol 33: 531–542, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cybulsky AV, Quigg RJ, Salant DJ: Experimental membranous nephropathy redux. Am J Physiol Renal Physiol 289: F660–F671, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer-Schwesinger C, Tomas NM, Dehde S, Seifert L, Hermans-Borgmeyer I, Wiech T, et al. : A novel mouse model of phospholipase A2 receptor 1-associated membranous nephropathy mimics podocyte injury in patients. Kidney Int 97: 913–919, 2020 [DOI] [PubMed] [Google Scholar]
- 21.Ronco P, Debiec H: Pathophysiological advances in membranous nephropathy: Time for a shift in patient’s care. Lancet 385: 1983–1992, 2015 [DOI] [PubMed] [Google Scholar]
- 22.Hoxha E, Kneißler U, Stege G, Zahner G, Thiele I, Panzer U, et al. : Enhanced expression of the M-type phospholipase A2 receptor in glomeruli correlates with serum receptor antibodies in primary membranous nephropathy. Kidney Int 82: 797–804, 2012 [DOI] [PubMed] [Google Scholar]
- 23.Morigi M, Perico L, Corna D, Locatelli M, Cassis P, Carminati CE, et al. : C3a receptor blockade protects podocytes from injury in diabetic nephropathy. JCI Insight 5: e131849, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou L, Liu Y: Wnt/β-catenin signalling and podocyte dysfunction in proteinuric kidney disease. Nat Rev Nephrol 11: 535–545, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai C, Stolz DB, Kiss LP, Monga SP, Holzman LB, Liu Y: Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J Am Soc Nephrol 20: 1997–2008, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou L, Chen X, Lu M, Wu Q, Yuan Q, Hu C, et al. : Wnt/β-catenin links oxidative stress to podocyte injury and proteinuria. Kidney Int 95: 830–845, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang K, Li M, Huang H, Li L, Yang J, Feng L, et al. : Dishevelled1-3 contribute to multidrug resistance in colorectal cancer via activating Wnt/β-catenin signaling. Oncotarget 8: 115803–115816, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Angeletti A, Cantarelli C, Petrosyan A, Andrighetto S, Budge K, D’Agati VD, et al. : Loss of decay-accelerating factor triggers podocyte injury and glomerulosclerosis. J Exp Med 217: e20191699, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Locatelli M, Buelli S, Pezzotta A, Corna D, Perico L, Tomasoni S, et al. : Shiga toxin promotes podocyte injury in experimental hemolytic uremic syndrome via activation of the alternative pathway of complement. J Am Soc Nephrol 25: 1786–1798, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paiano J, Harland M, Strainic MG, Nedrud J, Hussain W, Medof ME: Follicular B2 cell activation and class switch recombination depend on autocrine C3ar1/C5ar1 signaling in B2 cells. J Immunol 203: 379–388, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng Q, Li K, Anderson K, Farrar CA, Lu B, Smith RA, et al. : Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a-C3aR interaction. Blood 111: 2452–2461, 2008 [DOI] [PubMed] [Google Scholar]
- 32.Li K, Fazekasova H, Wang N, Peng Q, Sacks SH, Lombardi G, et al. : Functional modulation of human monocytes derived DCs by anaphylatoxins C3a and C5a. Immunobiology 217: 65–73, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwan WH, van der Touw W, Paz-Artal E, Li MO, Heeger PS: Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med 210: 257–268, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cumpelik A, Heja D, Hu Y, Varano G, Ordikhani F, Roberto MP, et al. : Dynamic regulation of B cell complement signaling is integral to germinal center responses. Nat Immunol 22: 757–768, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacPherson G, Kushnir N, Wykes M: Dendritic cells, B cells and the regulation of antibody synthesis. Immunol Rev 172: 325–334, 1999 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.