

Inherited Metabolic Disorders (IMD) can present with different movement disorders, from infancy to adulthood. Movements can range from subtle to be the main feature of the disease. Fumaric aciduria (FA) is a rare IMD resulting from a deficiency of fumarate hydratase, an enzyme of the Krebs tricarboxylic acid cycle. 1 We present a child with FA and progressive dystonia and parkinsonism, previously unexplored features of this disease.
A 4‐year‐old girl of Caucasian origin born to nonconsanguineous parents presented with speech and gait abnormalities. Pregnancy was uneventful, and delivery was at 39 weeks by cesarean. She had no family history. She had developed independent language and walking before difficulties started at the age of 3. Her first motor symptoms were lower limb abnormal postures, with hopping gait leading to falls. It was particularly noticed while running, and she was described as a “clumsy” child. Neurological examination was compatible with asymmetric lower limb dystonia, with more prominent right‐side involvement (Video 1, first sequence). There were no abnormalities of the cranial somatometry or dysmorphic facial features. Within the following years, dystonia became generalized with knee flexion and involvement of face and upper limbs, axial hypotonia developed and broad‐base ataxic gait (Video 1, second sequence). She also had versive seizures with secondary generalization. Severe behavioral and cognitive regression was measured by the Wechsler Intelligence Scale for Children (median 51, very low level) and the Vineland Adaptive Behavior Scale (global level from 5 years and 1 month to 3 years and 5 months).
Video 1.
Neurological examination showing bilateral lower limb dystonia at 6‐years‐old producing gait impairment which became progressively generalized and, at the age of 12 and 15, included elements of parkinsonism, hypotonia and myoclonus.
At the time of her last visit, aged 15, the examination revealed scarce spontaneous speech, spasmodic dysphonia, neck hypotonia with head drop, limb dystonia with overflow and striatal left hallux. She also had parkinsonism with akinesia and amimia and lower limb pyramidal signs, including sustained ankle clonus and brisk patellar stretch reflexes. There was mild distal myoclonus. The stance was unstable and marked difficulties with walking and impaired balance prevented independent gait, leaving the patient wheelchair bounded (Video 1, third sequence).
Brain MRI showed thin corpus callosum and mild cortico‐subcortical generalized atrophy but no signal changes in the basal ganglia, cerebellum, or pyramidal tracts (Fig. 1A). Interictal electroencephalography at the age of 8 revealed unspecific bilateral paroxysmal activity and mild slowing (frequency of 6–7 Hz). Urine organic acid analysis revealed excessive urinary excretion of fumaric acid (55.0 μmol/mmol creatinine; lab reference values 1.4–9.9) and fumarase activity in cultured skin fibroblasts was 6.71% of normal values (lab reference values 42.8–156.0 nmol/min/mg). Blood levels of lactate and pyruvate were within the normal range and a biochemical analysis of the mitochondrial respiratory chain was normal. Only lactate in the cerebrospinal fluid was elevated (1.406 mmol/L, lab reference values 0.77–1.29 mmol/L) with normal pyruvate levels and normal lactate‐to‐pyruvate ratio. Other organic acidurias and neurotransmitter disorders were excluded. DNA analysis (expanded NGS panel—6110 genes—including genes involved in dystonia and parkinsonism) found two heterozygous pathogenic variants of the FH gene [NM_000143.3:c.267 +1_267 + 10del, p.(?), and NM_000143.3:c.1431_1433dup, p.(Lys477dup)], that were confirmed by Sanger sequencing. Segregation analysis was performed in the parents and confirmed the trans configuration (Fig. 1B, C). Muscle pathology had no changes.
FIG. 1.
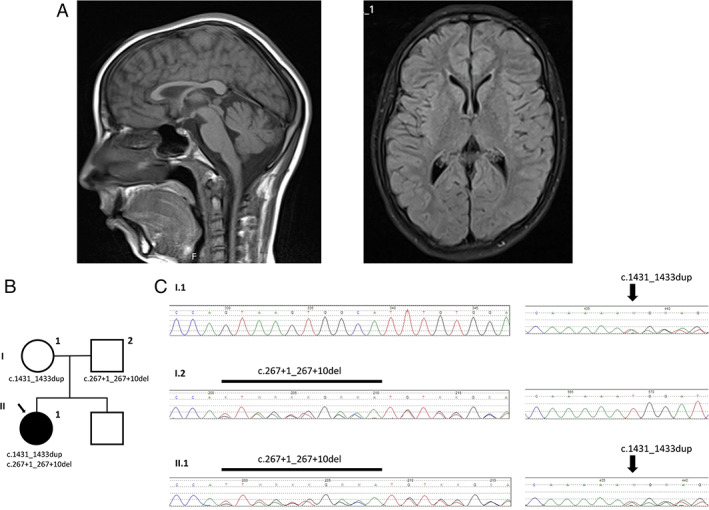
(A) Brain MRI at age 12, with thin corpus callosum and mild cortico‐subcortical generalized atrophy. No lesions involved the basal ganglia, cerebellum, or pyramidal tracts. (B) Pedigree depicting the index case (arrow) and her unaffected parents. (C) Chromatograms of the c.267 + 1_267 + 10del and c.1431_1433dup FH mutations.
A trial with levodopa (maximum daily dose 400 mg), introduced only after the development of generalized dystonia and parkinsonism, did not modify motor symptoms. Ketogenic diet was tried and had limited success in seizure stabilization and no observable changes in dystonia or myoclonus. The patient is currently being treated with levetiracetam 700 mg daily which improved seizures and myoclonus.We described a long follow‐up of a child with FA, in whom dystonia was the first feature of the disease. Extrapyramidal signs developed later and did not respond to a levodopa trial.
Tonus abnormalities, namely axial hypotonia, appear to occur in most patients with severe forms of FA since birth or within the first months of life. 2 Anecdotal reports describe lower limb hypertonia in FA children, sometimes combined with axial hypotonia, 3 and Supplementary file. Hypotonia in our patient was revealed as head drop after the first decade. Otherwise, lower limb dystonia started early and was the major feature in this patient during the first decade. In addition to the previously recognized speech and cognitive delay, chronic progressive brain dysfunction with a slow development of multiple neurological abnormalities appears to be a milder phenotype of FA. 4 Intriguingly, our patient has the most common genetic variant of the disease, [NM_000143.3:c.1431_1433dup, p.(Lys477dup)], which has been linked to the most severe phenotypes, including severe neurodevelopmental impairment and early death. 5 The other FH gene mutation [NM_000143.3:c.267 + 1_267 + 10del, p.(?)] has otherwise been associated with cutaneous and uterine leiomyomas and renal cell carcinoma, which should prompt cancer screening. 6 Epilepsy in FA has been described to develop at the end of the first decade.
The diagnosis of FA can be made by biochemical screening in urine and blood and confirmed by molecular genetic testing. No clinical feature correlates with any levels of residual enzyme activity or fumaric acid excretion. 7 Previously described non‐specific MRI changes in FA include polymicrogyria and agenesis of corpus callosum. 2 At present, treatment of FA is only symptomatic. Also, cancer risk should not be overlooked.
Fumaric aciduria is a rare IMD with an easy diagnosis that should be considered in children with progressive generalized movement abnormalities and neurodevelopment delay or regression.
Author Roles
Research Project: A. Conception, B. Organization, C. Execution.
Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
D.R.C.: 1A, 1B, 1C, 2A, 2B.
C.R.: 1A, 1B, 1C, 2B.
M.V.: 1C, 2B.
M.L.: 1C, 2B.
L.D.: 1A, 1B, 1C, 2B.
Disclosures
Ethical Compliance Statement: The authors have received the consent form from the patient's family and have it on file. The obtention of a written informed consent in case reports implies that the study does not need Ethics Committee approval. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: No funding was received for this work. The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months: The authors declare that there are no additional disclosures to report.
Supporting information
Supplementary Table S1. Clinical and genetic information of previously reported cases of Fumaric Aciduria with movement disorders.
References
- 1. Zinn AB, Kerr DS, Hoppel CL. Fumarase deficiency: A new cause of mitochondrial encephalomyopathy. N Engl J Med 1986;315(8):469–475. 10.1056/NEJM198608213150801. [DOI] [PubMed] [Google Scholar]
- 2. Kerrigan JF, Aleck KA, Tarby TJ, Bird CR, Heidenreich RA. Fumaric aciduria: Clinical and imaging features. Ann Neurol 2000;47:583–588. [PubMed] [Google Scholar]
- 3. Bourgeron T, Chretien D, Poggi‐Bach J, et al. Mutation of the fumarase gene in two siblings with progressive encephalopathy and fumarase deficiency. J Clin Invest 1994;93(6):2514–2518. 10.1172/JCI117261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Whelan DT, Hill RE, McClorry S. Fumaric aciduria: A new organic aciduria, associated with mental retardation and speech impairment. Clin Chim Acta 1983;132:301–308. [DOI] [PubMed] [Google Scholar]
- 5. Peetsold M, Goorden S, Breuning M, et al. Fumarase deficiency: A case with a new pathogenic mutation and a review of the literature. J Child Neurol 2021;36(4):310–323. 10.1177/0883073820962931. [DOI] [PubMed] [Google Scholar]
- 6. Alam NA, Olpin S, Leigh IM. Fumarate hydratase mutations and predisposition to cutaneous leiomyomas, uterine leiomyomas and renal cancer. Br J Dermatol 2005;153(1):11–17. 10.1111/j.1365-2133.2005.06678.x. [DOI] [PubMed] [Google Scholar]
- 7. Bonioli E, Di Stefano A, Peri V, et al. Fumarate hydratase deficiency. J Inherit Metab Dis 1998;21(4):435–436. 10.1023/a:1005379330187 PMID: 9700607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Clinical and genetic information of previously reported cases of Fumaric Aciduria with movement disorders.