

Abstract
Introducción.
La enfermedad de Pompe es una miopatía metabólica rara con espectro clínico heterogéneo, especialmente la de inicio tardío, cuya sintomatología es de progresión más lenta y representa un gran reto diagnóstico.
Objetivo.
Describir el genotipo y las características clínicas de pacientes mexicanos con Pompe de inicio tardío (LOPD).
Material y métodos.
Se incluyó a 19 pacientes mexicanos con LOPD confirmada mediante actividad enzimática y estudio molecular del gen GAA. Se evaluaron datos clínicos y se revisaron las mutaciones en bases de datos genómicas.
Resultados.
La mediana de edad de inicio de los síntomas fue de 19 años (rango: 2-43 años), y la edad de diagnóstico, de 36 años (rango: 9-52 años). Los síntomas más frecuentes fueron debilidad axial y proximal (n = 17; 89,5%), marcha basculante (n = 17; 89,5%) e hiperlordosis (n = 7; 36,8%). A 16 pacientes (84,2%) se les realizó electromiografía; 11 (57,8%) describieron patrón miopático y sólo en cinco pacientes (26%) se incluyó la valoración de los músculos paraespinales. Las variantes patogénicas más frecuentes en nuestra casuística fueron c.-32-13T>G, c.1799G>A y c.1082C>T.
Conclusiones.
Parecido a lo comunicado en publicaciones internacionales, la LOPD en México es clínicamente heterogénea; los pacientes pueden tardar años en llegar al diagnóstico. La debilidad muscular axial y proximal es el dato clínico más frecuente, por lo que la electromiografía debe incluir valoración de los músculos paraespinales. A excepción de una, las mutaciones encontradas en nuestra serie de casos se encuentran previamente descritas en las bases de datos de enfermedad de Pompe.
Palabras clave: Debilidad muscular, Electromiografía, Enfermedad de Pompe de inicio tardío, México, Miopatía, Músculos paraespinales
Abstract
Introduction.
Pompe disease (PD) is a rare metabolic myopathy with an ample and heterogeneous clinical spectrum, particularly late onset PD (LOPD), which is characterized by appearance at older age and slower disease progression, leading to diagnostic confirmation difficulty and delay.
Aim.
To describe the genotype and clinical characteristics of Mexican patients with LOPD.
Material and methods.
Clinical information from 19 Mexican patients with LOPD confirmed with enzyme activity and GAA gene analysis was reviewed. Genetic information of our population was crossed with international genetic databases.
Results.
Median age between onset of symptoms and diagnosis was 19 years (range 2-43) and diagnostic confirmation 36 years (range 9-52). Most frequently referred symptoms were proximal axial weakness (n = 17; 89.5%), waddling gait (n = 17; 89.5%) and hyperlordosis (n = 7; 36.8%). Sixteen patients (84.2%) were evaluated with electromyography; a myopathic pattern was reported in 11 (57.8%), but only in 5 patients (26%) paraspinal muscle evaluation was included. The most pathogenic mutations in our group were c.-32-13T>G, c.1799G>A and c.1082C>T.
Conclusions.
Similar to other international publications, LOPD in Mexico is clinically heterogeneous; patients may delay years before diagnosis is established. Axial and proximal weakness is the most frequent clinical feature; thus, electromyography with paraspinal muscle evaluation is essential. Except for one, the mutations found in our patients have been previously reported in PD genetic databases.
Key words: Electromyography, Late onset Pompe disease, Mexico, Myopathy, Paraspinal muscle, Weakness
Introducción
La enfermedad de Pompe (OMIM #232300) es una miopatía metabólica rara y progresiva causada por mutaciones en el gen GAA, que dan lugar a deficiencia o ausencia de alfa-glucosidasa ácida lisosómica, con el consecuente almacenamiento de glucógeno en los lisosomas de los músculos. La incidencia de la enfermedad varía según el origen étnico y la geografía, y se estima en alrededor de 1:40.000 habitantes [1].
El espectro clínico es heterogéneo, pero, en un intento de clasificar la enfermedad en función de la presentación clínica, se ha dividido en formas infantiles (clásicas y no clásicas) y formas de inicio tardío (juvenil y del adulto) [2].
La enfermedad de Pompe de inicio tardío (LOPD) se caracteriza por debilidad muscular lentamente progresiva de predominio axial y proximal. La afectación del diafragma y los músculos accesorios conlleva una neumopatía restrictiva y problemas respiratorios del sueño. Pocos pacientes presentan alteraciones cardíacas, pero en algunos pueden presentarse arritmias. La principal causa de muerte es el fallo respiratorio [3].
El diagnóstico clínico se confirma por la ausencia o la marcada reducción de la actividad de la enzima alfa-glucosidasa ácida e identificando variantes patogénicas de la enfermedad de Pompe en el análisis molecular del gen GAA [4].
En este trabajo describimos el genotipo y las características clínicas de 19 pacientes mexicanos con LOPD.
Material y métodos
Se realizó una revisión retrolectiva de expedientes clínicos de pacientes con LOPD, de médicos diagnosticadores y/o tratantes de estos pacientes en México que aceptaron participar enviando información de forma anónima y confidencial. Todos los pacientes contaban con consentimiento informado para el tratamiento de datos, de acuerdo con los lineamientos de cada institución de salud y con las buenas prácticas clínicas internacionales. La enfermedad se confirmó mediante determinación de actividad enzimática de alfa-glucosidasa ácida en gota de sangre seca y estudio molecular del gen GAA.
Las mutaciones documentadas en nuestra casuística se cotejaron con las bases de datos de variantes patogénicas para enfermedad de Pompe documentadas en la bibliografía; la clasificación de las variantes se realizó de acuerdo con las directrices del Colegio Americano de Genética Médica y Genómica, y se clasificaron como patogénica, probablemente patogénica, de significado incierto, probablemente benigna y benigna. Se analizaron los datos clínicos con estadística descriptiva.
Resultados
Se incluyó a 19 pacientes mexicanos con LOPD que fueron diagnosticados en Sinaloa (cinco), Jalisco (cuatro), Nuevo León (tres), Veracruz (tres), Ciudad de México (dos), Tlaxcala (uno) y Sonora (uno) (Figura). Dieciséis (84%) eran del sexo femenino. Algunos pacientes pertenecen a una misma familia y se identifican en la tabla con número y letra.
Figura.
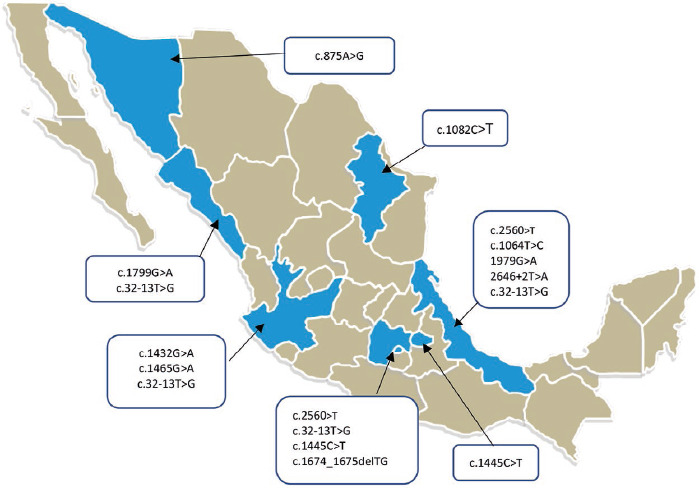
Mapa de México con la distribución de las variantes patogénicas del gen GAA encontradas en esta serie de casos. De particular relevancia son c.2560C>T (frecuente en afrodescendientes y documentada en Veracruz, donde existe una importante población afromexicana) y c.1674_1675delTG en el centro del país, y que a nuestro conocimiento no se ha descrito previamente en bases de datos de enfermedad de Pompe. La variante c.-32-13T>G se encontró en heterocigosis en pacientes de varios estados, que, al combinarse con otra variante patogénica, parece predecir una enfermedad menos grave.
La mediana de edad de inicio de los síntomas fue de 19 años (rango: 2-43 años), y la edad de diagnóstico, de 36 años (rango: 9-52 años). Dos mujeres se referían asintomáticas, pero se estudiaron por ser familiares de pacientes previamente diagnosticados; en ambas se confirmó LOPD. La mediana de tiempo transcurrido desde el inicio de los síntomas y el diagnóstico fue de 16 años (rango: 2-30 años). A excepción de las dos pacientes asintomáticas, todos los demás tenían debilidad axial y proximal con marcha basculante y siete (36,8%) presentaban hiperlordosis. A cuatro pacientes (21%) se les hizo biopsia de músculo que describió necrosis, distrofia de cinturas, distrofia de Becker y muestra insuficiente. Los niveles de creatinfosfocinasa oscilaron entre 221 y 3.000 U/L, con una mediana de 462,5 U/L. Se realizó electromiografía a 16 (84,2%) pacientes: 11 (57,8%) tenían patrón miopático; sin embargo, solo en cinco casos (26%) se incluyó la valoración de los músculos paraespinales. Once pacientes (57,8%) presentaron disnea durante su evolución, y ocho (42%), neumonías. Quince pacientes (78,9%) recibían o habían recibido terapia de reemplazo enzimático con alfa-glucosidasa (Tabla).
Tabla.
Características sociodemográficas, clínicas, resultados de estudios y genotipo de 19 pacientes con enfermedad de Pompe de inicio tardío.
| Pacientea | Procedencia | Sexo | Edad de inicio | Edad de Dx | Debilidad muscular proximal/axial | Marcha basculante | Hiperlordosis | Biopsia muscular | CPK basal (UI/L) | EMG | TRE | Variante patogénica 1 | Variante patogénica 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Veracruz | F | 33 | 47 | Sí | Sí | Sí | Sí | 1.011 | Sí | Sí | Exón 18 c.2560>T | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 2 | Tlaxcala | M | 13 | 21 | Sí | Sí | No | No | 3.000 | Sí | Sí | Exón 10 c.1445C>T | Exón 10 c.1445C>T |
|
| |||||||||||||
| 3 | CdMx | F | 25 | 52 | Sí | Sí | Sí | No | 340 | Sí | Sí | Exón 18 c.2560C>T | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 4a | Nuevo León | F | 13 | 29 | Sí | Sí | Sí | No | 358 | Sí | Sí | Exón 7 c.1082C>T | Exón 7 c.1082C>T |
|
| |||||||||||||
| 4b | Nuevo León | F | 15 | 40 | Sí | Sí | Sí | No | 402 | Sí | Sí | Exón 7 c.1082C>T | Exón 7 c.1082C>T |
|
| |||||||||||||
| 4c | Nuevo León | F | 14 | 30 | Sí | Sí | Sí | No | 432 | Sí | Sí | Exón 7 c.1082C>T | Exón 7 c.1082C>T |
|
| |||||||||||||
| 5 | Jalisco | F | 13 | 29 | Sí | Sí | Sí | No | 439 | Sí | No | Exón 9 c.1432G>A | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 6 | CdMx | F | 2 | 9 | Sí | Sí | No | No | 926 | No | No | Exón 10 c.1445C>T | Exón 12 c.1674_1675delTG |
|
| |||||||||||||
| 7a | Sinaloa | M | 39 | 51 | Sí | Sí | Sí | Sí | 216 | Sí | Sí | Exón 13 c.1799G>A | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 7b | Sinaloa | F | 19 | 49 | Sí | Sí | No | No | 496 | Sí | Sí | Exón 13 c.1799G>A | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 7c | Sinaloa | M | 36 | 48 | Sí | Sí | No | No | 454 | Sí | Sí | Exón 13 c.1799G>A | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 7d | Sinaloa | F | 31 | 36 | Sí | Sí | No | No | 495 | Sí | Sí | Exón 13 c.1799G>A | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 7e | Sinaloa | F | 25 | 33 | Sí | Sí | No | No | 659 | Sí | Sí | Exón 13 c.1799G>A | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 8 | Veracruz | F | 10 | 13 | Sí | Sí | No | No | 926 | Sí | Sí | Exón 6 c.1064T>C | Exón 14 1979G>A |
|
| |||||||||||||
| 9 | Sonora | F | 43 | 49 | Sí | Sí | No | No | 221 | Sí | Sí | Exón 5 c.875A>G | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 10 | Veracruz | F | 38 | 40 | Sí | Sí | No | Sí | 498 | Sí | Sí | Intrón 18 2646+2T>A | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 11a | Jalisco | F | 14 | 37 | Sí | Sí | No | Sí | 307 | Sí | Sí | Exón 10 c.1465G>A | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 11b | Jalisco | F | Asintomático | 40 | No | No | No | No | 471 | Sí | No | Exón 10 c.1465G>A | Intrón 1 c.-32-13T>G |
|
| |||||||||||||
| 11c | Jalisco | F | Asintomático | 31 | No | No | No | No | NN | Sí | No | Exón 10 c.1465G>A | Intrón 1 c.-32-13T>G |
CdMx: Ciudad de México; CPK: creatinfosfocinasa; Dx: diagnóstico; EMG: electromiografía; F: femenino; M: masculino; NN: no notificado; TRE: terapia de reemplazo enzimático. a Los números corresponden a pacientes y las letras a los miembros de la misma familia.
En la tabla se muestran las variantes patogénicas del gen GAA encontradas (en los dos alelos) y en la figura se muestran por estados. La más frecuente fue c.-32-13T>G (n = 13, todos en heterocigosis); la siguiente más frecuente fue c.1445C>T. Otras variantes frecuentes fueron c.1465G>A y c.2560C>T. Cuatro pacientes (21%) eran homocigotos, y 15 (79%), heterocigotos.
Discusión
Nuestra casuística involucra a pacientes de distintos estados, y Veracruz y Ciudad de México son los que concentran el mayor número de familias en las cuales se documentó la enfermedad. En Sinaloa, Jalisco y Nuevo León se diagnosticó a pacientes pertenecientes a una misma familia en cada estado, lo cual refuerza la importancia de interrogar sobre los antecedentes familiares para diagnosticar a otros miembros de la familia que tienen síntomas incipientes o incluso asintomáticos. En Jalisco se detectaron dos pacientes asintomáticas a partir de un miembro familiar con síntomas.
La LOPD incluye a pacientes cuya sintomatología comienza en la niñez, en la juventud y en la adultez, aun la avanzada. Los datos pivote para el diagnóstico son la debilidad de los músculos proximales y axiales, incluyendo el diafragma, con la consecuente afección respiratoria. La marcha basculante y la hiperlordosis compensatoria son frecuentes [5]. Nuestro grupo de pacientes fue concordante en cuanto al cuadro clínico muscular con lo comunicado en la bibliografía internacional.
Aproximadamente la mitad de los pacientes presentaron disnea y neumonías durante su evolución. Esto es similar a lo hallado en otras publicaciones, que describen que hasta el 30% de los pacientes comienzan con datos a nivel pulmonar, y el fallo respiratorio es la principal causa de muerte [5].
Una encuesta realizada en varios países de Europa a pacientes con enfermedad de Pompe comunicó una mediana de tiempo transcurrido desde el inicio de los síntomas hasta el diagnóstico de 12 años. El retraso en el diagnóstico se atribuyó a que no se enviaron directamente a centros especializados o de referencia para pacientes con enfermedad de Pompe, debido al desconocimiento sobre a quién y a dónde deben referirse estos pacientes una vez que se sospecha la enfermedad [6]. En otro estudio se incluyó a 647 pacientes del Registro de Enfermedad de Pompe, dividiéndolos en cuatro grupos según la clasificación clínica, y se comunicó que los pacientes con enfermedad de Pompe de inicio juvenil fueron los que más tardaron en ser diagnosticados, con un promedio de 12,6 años [7]. En nuestra serie de casos, ocho pacientes (42%) iniciaron sintomatología en la edad pediátrica (Pompe juvenil); sin embargo, si tenemos en cuenta a todos los pacientes de nuestro estudio, la mediana entre el inicio de los síntomas y el diagnóstico es de 16 años, mayor de lo descrito en la bibliografía, y consideramos que este retraso en el diagnóstico se debe al desconocimiento y a la baja sospecha de esta enfermedad en el ámbito médico.
El diagnóstico clínico de la enfermedad de Pompe se confirma por la ausencia o la reducción de la actividad de la enzima alfa-glucosidasa ácida. Todos nuestros pacientes tenían actividad enzimática baja, y posteriormente se hizo el análisis molecular para confirmar el diagnóstico. A cuatro pacientes (21%) se les realizó biopsia de músculo, pero con resultados diversos. Lo anterior revela que se requiere experiencia en su obtención, manejo e interpretación; también es congruente con otros informes que consideran que hasta el 80% de los pacientes con enfermedad de Pompe tuvieron un diagnóstico erróneo de otra miopatía [8]. Por este motivo, consideramos que el estudio más sencillo para el diagnóstico de certeza es mediante gotas de sangre seca en papel filtro, el cual es fácil de realizar y de guardar durante varios días sin que la muestra sufra degradación. En él se determina la actividad enzimática de la alfa-glucosidasa ácida, y, en caso de estar disminuida, se procede a realizar un estudio molecular con la misma muestra [9].
Algunos estudios muestran que los pacientes con LOPD pueden tener niveles elevados (raramente por encima de 2.000 U/L) o normales de creatinfosfocinasa [10]. En nuestra serie de casos, los niveles de creatinfosfocinasa fueron similares a lo notificado en otros estudios.
La electromiografía en la enfermedad de Pompe revela un patrón miopático con descargas miotónicas; sin embargo, es importante incluir los músculos proximales de las cinturas y axiales, específicamente paraespinales, músculos en los cuales se ha documentado mayor asertividad para el diagnóstico de la enfermedad de Pompe. Kassardjian et al informan de los hallazgos electromiográficos en 37 pacientes, de los cuales 28 (76%) tenían descargas miotónicas por lo menos en un músculo, más comúnmente en los músculos paraespinales y proximales de las piernas; incluso tres pacientes también las presentaron en el diafragma [11]. A la mayoría de nuestros pacientes se les realizó electromiografía; sin embargo, sólo en una cuarta parte de ellos se incluyeron los músculos paraespinales. En todo paciente con miopatía proximal es importante solicitar/realizar la valoración de músculos paraespinales en busca de descargas miotónicas que orienten al diagnóstico.
La terapia de reemplazo enzimático con alfa-glucosidasa es el tratamiento específico actual para la enfermedad de Pompe [12]. Quince de nuestros pacientes (78.9%) recibían o habían recibido terapia de reemplazo enzimático.
El gen GAA se localiza en el cromosoma 17q25 y contiene 20 exones. La base de datos de variantes de GAA de la enfermedad de Pompe (http://www.pompevariantdatabase.nl) es una base de datos que enumera y clasifica todas las variantes descritas del gen GAA [13].
Con respecto a las variantes patogénicas encontradas en esta serie de casos, la variante c.-32-13T>G fue la más frecuente, concordante con otros estudios de poblaciones diversas donde se ha encontrado con una frecuencia alélica del 40-70% en pacientes con LOPD, la mayoría en heterocigosis (sólo el 1% de los casos corresponde a pacientes homocigotos) [14]. La variante c.1082C>T se presentó en homocigosis en tres pacientes de una misma familia. Esta se ha descrito en pacientes que inician sintomatología desde la infancia, como sucedió en estas pacientes, y la progresión de la enfermedad es lenta y potencialmente menos grave [15].
La combinación en heterocigosis de las variantes c.1799G>A y c.-32-13T>G, hallada en cinco pacientes de nuestra serie, se ha descrito previamente, asociándose a una evolución clínica considerablemente heterogénea, aun dentro de una misma familia. Lo mismo sucede con la combinación de variantes c.1465G>A y c.-32-13T>G, descrita en población italiana y en otras partes del mundo, que igualmente predice una enfermedad que puede empezar en edades muy variables de la vida. Tres de nuestras pacientes tenían estas variantes: una había iniciado con síntomas a los 14 años y las otras dos estaban asintomáticas a los 30 y los 40 años [16,17].
La variante c.2560C>T, asociada a Pompe infantil y frecuente en afrodescendientes [18], es una mutación que por lo general predice una enfermedad grave; sin embargo, en combinación con la variante c.-32-13T>G, parece asociarse a un fenotipo de LOPD, como se describe en dos de nuestras pacientes, una procedente de Veracruz, estado con una importante población afromexicana.
La variante c.1445C>T (p.Pro482Leu) es una mutación comunicada en pacientes españoles con enfermedad de Pompe [19]. Dos de nuestros pacientes presentaron esta variante, uno en homocigosis y el otro en heterocigosis. La variante c.875A>G se ha descrito en la enfermedad de Pompe infantil, pero también en pacientes que inician a los 2 y 7 años, lo que va en relación con la otra variante patogénica con la que se combina, ya que pueden comenzar desde la infancia o hasta los 60 años, cuando se combina con la variante c.-32-13T>G [19]. Una paciente de Sonora presentaba esta última combinación y había empezado con síntomas hasta los 43 años. La variante c.1674_1675delTG no se encontró en bases de datos de enfermedad de Pompe; sin embargo, los modelos en sílice predicen que la variante es patogénica.
En nuestro conocimiento, hasta la fecha ésta es la casuística más grande de pacientes con LOPD descrita en México. Nuestros hallazgos concuerdan con lo descrito en la bibliografía internacional en cuanto a genética, hallazgos clínicos y paraclínicos. El dato más alarmante, tanto en casuísticas internacionales como en la nuestra, es la demora en la confirmación diagnóstica. Ante ello, el mayor reto es aumentar el conocimiento de la enfermedad en el país para acortar la travesía diagnóstica y ofrecer un tratamiento oportuno.
Conclusiones
La LOPD es una enfermedad heterogénea que puede iniciar con sintomatología desde la infancia o hasta la edad adulta. Los datos clínicos pivote son la debilidad muscular axial y proximal, y problemas respiratorios. La enfermedad debe sospecharse en todo paciente con debilidad progresiva y/o fallo respiratorio de etiología indeterminada. Junto con la clínica, la creatinfosfocinasa y la electromiografía son herramientas auxiliares para la evaluación de estos pacientes. Una vez que se sospecha la enfermedad, la confirmación se realiza mediante un estudio sencillo en papel filtro con gotas de sangre seca. Llegar al diagnóstico puede llevar años, lo que hace imperativo diseminar el conocimiento de esta enfermedad para poder acortar la travesía diagnóstica de los pacientes.
Agradecimientos:
A los pacientes y sus familias por no rendirse durante el largo camino para llegar al diagnóstico. A todos los médicos comprometidos con los pacientes con enfermedades raras.
Bibliografía
- 1.Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, et al. Pompe disease diagnosis and management guideline. Genet Med 2006;8:267-88. [DOI] [PMC free article] [PubMed]; Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8:267–88. doi: 10.1097/01.gim.0000218152.87434.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dubroysky A, Fulgenzi A, Amartino H, Carlés D, Corderi J, De Vito E, et al. Consenso argentino para el diagnóstico, seguimiento y tratamiento de la enfermedad de Pompe. Neurología Argentina 2014;6:96-113.; Dubroysky A, Fulgenzi A, Amartino H, Carlés D, Corderi J, De Vito E, et al. Consenso argentino para el diagnóstico, seguimiento y tratamiento de la enfermedad de Pompe. Neurología Argentina. 2014;6:96–113. [Google Scholar]
- 3.Barba-Romero MA, Barrot E, Bautista-Lorite J, Gutierrez-Rivas E, Illa I, Jimenez LM, et al. Clinical guidelines for late-onset Pompe disease. Rev Neurol 2012;54:497-507. [PubMed]; Barba-Romero MA, Barrot E, Bautista-Lorite J, Gutierrez-Rivas E, Illa I, Jimenez LM, et al. Clinical guidelines for late-onset Pompe disease. Rev Neurol. 2012;54:497–507. [PubMed] [Google Scholar]
- 4.Kallwass H, Carr C, Gerrein J, Titlow M, Pomponio R, Bali D, et al. Rapid diagnosis of late-onset Pompe disease by fluorometric assay of alpha-glucosidase activities in dried blood spots. Mol Genet Metab 2007;90:449-52. [DOI] [PubMed]; Kallwass H, Carr C, Gerrein J, Titlow M, Pomponio R, Bali D, et al. Rapid diagnosis of late-onset Pompe disease by fluorometric assay of alpha-glucosidase activities in dried blood spots. Mol Genet Metab. 2007;90:449–52. doi: 10.1016/j.ymgme.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Mellies U, Lofaso F. Pompe disease:a neuromuscular disease with respiratory muscule involvement. Respiratory Med 2009;103:477-84. [DOI] [PubMed]; Mellies U, Lofaso F. Pompe disease:a neuromuscular disease with respiratory muscule involvement. Respiratory Med. 2009;103:477–84. doi: 10.1016/j.rmed.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 6.Lagler FB, Moder A, Rohrbach M, Hennermann J, Mengel E, Gökce S, Hundsberger T, Rösler KM, Karabul N, Huemer M, et al. Extent, impact, and predictors of diagnostic delay in Pompe disease:a combined survey approach to unveil the diagnostic odyssey. JIMD Rep 2019;49:89-95. [DOI] [PMC free article] [PubMed]; Lagler FB, Moder A, Rohrbach M, Hennermann J, Mengel E, Gökce S, Hundsberger T, Rösler KM, Karabul N, Huemer M, et al. Extent, impact, and predictors of diagnostic delay in Pompe disease:a combined survey approach to unveil the diagnostic odyssey. JIMD Rep. 2019;49:89–95. doi: 10.1002/jmd2.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kishnani PS, Amartino HM, Lindberg C, Miller TM, Wilson A, Keutzer J;Pompe Registry Boards of Advisors. Timing of diagnosis of patients with Pompe disease:data from the Pompe registry. Am J Med Genet A 2013;161A:2431-43. [DOI] [PubMed]; Kishnani PS, Amartino HM, Lindberg C, Miller TM, Wilson A, Keutzer J. Pompe Registry Boards of Advisors Timing of diagnosis of patients with Pompe disease:data from the Pompe registry. Am J Med Genet A. 2013;161A::2431–43. doi: 10.1002/ajmg.a.36110. [DOI] [PubMed] [Google Scholar]
- 8.Vissing J, Lukacs Z, Straub V. Diagnosis of Pompe disease:muscle biopsy vs blood-based assays. JAMA Neurol 2013;70:923-7. [DOI] [PubMed]; Vissing J, Lukacs Z, Straub V. Diagnosis of Pompe disease:muscle biopsy vs blood-based assays. JAMA Neurol. 2013;70:923–7. doi: 10.1001/2013.jamaneurol.486. [DOI] [PubMed] [Google Scholar]
- 9.Musumeci O, Toscano A. Diagnostic tools in late onset Pompe disease (LOPD). Ann Transl Med 2019;7:286. [DOI] [PMC free article] [PubMed]; Musumeci O, Toscano A. Diagnostic tools in late onset Pompe disease (LOPD) Ann Transl Med. 2019;7:286. doi: 10.21037/atm.2019.06.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Jing-Jing H, Zhi-Qiang W, Ning-Wang Z. Value of muscle enzyme measurement in evaluating different neuromuscular diseases. Clin Chim Acta 2012;413:520-4. [DOI] [PubMed]; Zhang Y, Jing-Jing H, Zhi-Qiang W, Ning-Wang Z. Value of muscle enzyme measurement in evaluating different neuromuscular diseases. Clin Chim Acta. 2012;413:520–4. doi: 10.1016/j.cca.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 11.Kassardjian CD, Engel AG, Sorenson EJ. Electromyographic findings in 37 patients with adult-onset acid maltase deficiency. Muscle Nerve 2015;51:759-61. [DOI] [PubMed]; Kassardjian CD, Engel AG, Sorenson EJ. Electromyographic findings in 37 patients with adult-onset acid maltase deficiency. Muscle Nerve. 2015;51:759–61. doi: 10.1002/mus.24620. [DOI] [PubMed] [Google Scholar]
- 12.Cupler EJ, Berger KI, Leshner RT, Wolfe GI, Han JJ, Barohn RJ, et al. Consensus treatment recommendations for late-onset Pompe disease. Muscle Nerve 2012;45:319-33. [DOI] [PMC free article] [PubMed]; Cupler EJ, Berger KI, Leshner RT, Wolfe GI, Han JJ, Barohn RJ, et al. Consensus treatment recommendations for late-onset Pompe disease. Muscle Nerve. 2012;45:319–33. doi: 10.1002/mus.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Faria DOS, 't Groen SLMI, Hoogeveen-Westerveld M, Nino MY, van der Ploeg AT, Bergsma AJ, et al. Update of the Pompe variant database for the prediction of clinical phenotypes:novel disease-associated variants, common sequence variants, and results from newborn screening. Hum Mutat 2021;42:119-34. [DOI] [PMC free article] [PubMed]; de Faria DOS, 't Groen SLMI, Hoogeveen-Westerveld M, Nino MY, van der Ploeg AT, Bergsma AJ, et al. Update of the Pompe variant database for the prediction of clinical phenotypes:novel disease-associated variants, common sequence variants, and results from newborn screening. Hum Mutat. 2021;42:119–34. doi: 10.1002/humu.24148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park KS. Carrier frequency and predicted genetic prevalence of Pompe disease based on a general population database. Mol Genet Metab Rep 2021;27:100734. [DOI] [PMC free article] [PubMed]; Park KS. Carrier frequency and predicted genetic prevalence of Pompe disease based on a general population database. Mol Genet Metab Rep. 2021;27:100734. doi: 10.1016/j.ymgmr.2021.100734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu YP, Sheng B, Lau KK, Chan HF, Kam GY, Lee HH, et al. Clinical manifestation of late onset Pompe disease patients in Hong Kong. Neuromuscul Disord 2016;26:873-9. [DOI] [PubMed]; Chu YP, Sheng B, Lau KK, Chan HF, Kam GY, Lee HH, et al. Clinical manifestation of late onset Pompe disease patients in Hong Kong. Neuromuscul Disord. 2016;26:873–9. doi: 10.1016/j.nmd.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Wens SC, van Gelder CM, Kruijshaar ME, de Vries JM, van der Beek NA, Reuser AJ, et al. Phenotypical variation within 22 families with Pompe disease. Orphanet J Rare Dis 2013;8:182. [DOI] [PMC free article] [PubMed]; Wens SC, van Gelder CM, Kruijshaar ME, de Vries JM, van der Beek NA, Reuser AJ, et al. Phenotypical variation within 22 families with Pompe disease. Orphanet J Rare Dis. 2013;8:182. doi: 10.1186/1750-1172-8-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pittis MG, Filocamo M. Molecular genetics of late onset glycogen storage disease II in Italy. Acta Myol 2007;26:67-71. [PMC free article] [PubMed]; Pittis MG, Filocamo M. Molecular genetics of late onset glycogen storage disease II in Italy. Acta Myol. 2007;26:67–71. [PMC free article] [PubMed] [Google Scholar]
- 18.Becker JA, Vlach J, Raben N, Nagaraju K, Adams EM, Hermans MM, et al. The African origin of the common mutation in African American patients with glycogen-storage disease type II. Am J Hum Genet 1998;62:991-4. [DOI] [PMC free article] [PubMed]; Becker JA, Vlach J, Raben N, Nagaraju K, Adams EM, Hermans MM, et al. The African origin of the common mutation in African American patients with glycogen-storage disease type II. Am J Hum Genet. 1998;62:991–4. doi: 10.1086/301788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gort L, Coll MJ, Chabás A. Glycogen storage disease type II in Spanish patients:high frequency of c.1076-1G>C mutation. Mol Genet Metab 2007;92:1837. [DOI] [PubMed]; Gort L, Coll MJ, Chabás A. Glycogen storage disease type II in Spanish patients:high frequency of c.1076-1G>C mutation. Mol Genet Metab. 2007;92:1837. doi: 10.1016/j.ymgme.2007.05.011. [DOI] [PubMed] [Google Scholar]