

Abstract
Alzheimer's disease (AD) is the most commonly seen neurodegenerative brain disorder. The paracrine effects of mesenchymal stem cells (MSCs) signify to trigger immunomodulation and neural regeneration. However, the role and mechanism of bone marrow MSC- (BMSC-) derived CX3CL1 in AD remains elusive. In this study, Aβ1-42-intervened SH-SY5Y cells were used for AD cell model construction. pcDNA-ligated CX3CL1 overexpression plasmids were transfected into BMSCs. The levels of soluble and membrane-bound CX3CL1 were detected by ELISA and Western blotting (WB), respectively. The growth, apoptosis, and pathology of AD model cells were evaluated by CCK-8, flow cytometry, immunofluorescence, morphology observation, biochemical examination, and WB. It was found that Aβ1-42 significantly reduced CX3CL1 expression either in soluble or membrane-bound form, cell viability, relative protein expression of synaptic markers, SOD, CAT, and GSH-Px contents, as well as Trx protein expression; in addition, it enhanced the apoptosis rate, the relative expression of cleaved caspase-3, Aβ, tau, p-Tau, Iba1, MDA, TXNIP, and NLRP3 in SH-SY5Y cells; however, the above effects were prominently reversed by the coculture of BMSCs. Moreover, overexpression of CX3CL1 in BMSCs observably strengthened the corresponding tendency caused by BMSCs. In conclusion, through the TXNIP/NLRP3 pathway, CX3CL1 derived from BMSCs inhibited pathological damage in Aβ1-42-induced SH-SY5Y.
1. Introduction
As a progressive neurodegenerative brain disease, Alzheimer's disease (AD) is acknowledged to be the prime reason for dementia [1]. It is also the sixth main cause of death, with an estimate of about 75.6 million and 135.5 million sufferers worldwide as well as $2.54 trillion and $9.12 trillion costs by 2030 and 2050, respectively [2, 3]. AD is pathologically featured by the accumulation of aggregated β-amyloid (Aβ) plaques and neurofibrillary tangles (NFTs) that consist of hyperphosphorylated Tau proteins [4]. Thus, therapies targeting Aβ and antitau agents have attracted extensive attention [5]. However, the benefits and side effects of even the promising anti-Aβ drug, aducanumab, still need further investigations [6, 7]. Besides, antitau vaccine (AADvac1) and monoclonal antitau antibodies, including gosuranemab, tilavonemab, zagotenemab, and semorinemab, have only arrived phase II at present [5]. Even several clinical drugs, such as galantamine, memantine, donepezil, and rivastigmine, only show modest benefits to symptomatic patients with AD [8]. Therefore, potential mechanisms and therapeutic strategies for AD necessitate more attention.
As a type of pluripotent progenitor cells that can be separated from various tissues like bone marrow, fatty tissue, and umbilical cord blood, mesenchymal stem cells (MSCs) have the advantages of high proliferation and easy manipulation [9]. MSCs possess multifaceted functions, such as differentiating into glial and neuronal cells, secreting growth factor and anti-inflammatory cytokines, modulating immune cells, and stimulating endogenous repair mechanisms [10, 11], making them a novel and promising therapy for AD [12]. Bone marrow MSCs (BMSCs) are shown to alleviate cognitive decline of AD model rats [13]. Bae et al. [14] also presented that BMSCs decreased Aβ deposits and ameliorated synaptic transmission in AD mice models. Kim et al. [15] demonstrated the feasibility of AD treatment by human umbilical cord blood MSCs (hUC-MSCs) via stereotactic brain injection in a phase 1 clinical trial. Lee et al. [16] displayed that BMSCs induced the reduction of Aβ deposits and promoted microglia activation in AD mice models. Thus, MSCs are a potential and effective approach for the treatment of AD.
It has been demonstrated that the clinical efficacy of MSCs may be related to their paracrine effects on other effector cells [17]. It was found in vitro and in vivo that the suppression of BMSC-derived exosomal miR-1231 plays a role in restricting the characteristics of pancreatic cancer [18]. While supporting acute myeloid leukemia bioenergetics, BMSCs can also enhance antioxidant defense and escape from chemotherapy [19]. Meanwhile, MSCs can modulate the activation of retinal microglia by chemokine CX3CL1 signaling [20]. Moreover, CX3CL1-expressing MSCs played neuroprotective and immunomodulatory roles in the retinal degeneration rat model [21]. Also known as fractalkine, chemokine CX3CL1 is the one and only member of the CX3C family that binds to its just receptor, CX3CR1, which is expressed on the surface of microglia [22, 23]. CX3CL1 is mainly distributed in neurons within the central nervous system (CNS) [24]. Thus, there is a signaling network formed by the binding of CX3CL1 and CX3CR1 between neurons and microglia in the physiological CNS [25, 26]. There are two forms of CX3CL1 in CNS, namely, membrane-bound and soluble CX3CL1. Therein, membrane-bound CX3CL1 can be protease-cleaved to produce soluble CX3CL1 [27, 28]. Reportedly, CX3CL1 is neuroprotective via the reduced microglial generation of inflammatory factors [24]. However, the role of CX3CL1 derived from MSCs in the development and progression of AD remains elusive.
Accordingly, the motivation and novelty of this work is to clarify the role played by BMSC-derived CX3CL1 and the underlying mechanisms via Aβ1-42-induced SH-SY5Y cell model establishment. BMSC-derived CX3CL1 inhibited Aβ1-42-induced SH-SY5Y cell pathological damage via the TXNIP/NLRP3 axis.
2. Materials and Methods
2.1. Cells and Cultivation
BMSCs and the human neuroblastoma SH-SY5Y cells, supplied by Cyagen Biosciences Inc. (Guangzhou, China) and Procell (CL-0208, Wuhan, China), respectively, were cultivated under the conditions of 5% carbon dioxide (CO2) at 37°C in a medium comprising Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA, USA) as well as 10% fetal bovine serum (FBS; Gibco, Rockville, MD, USA) and 1% streptomycin-penicillin (Solarbio, Beijing, China).
2.2. BMSC Transfection
BMSCs were cultivated (5% CO2, 37°C) after being seeded at 1 × 104 cells/cm2 into the wells of 6-well plates. When the cell confluency reached 80%, 50 ng pcDNA-ligated CX3CL1 overexpression plasmid (CX3CL1-ov) and its corresponding empty vector plasmid (NC-ov) supplied by RiboBio (Guangdong, China) were added for transfection following Lipofectamine® 3000 (Invitrogen; Thermo Fisher Scientific, Waltham, MA, USA) recommendations. The media were replaced with DMEM after 6 h of transfection, and BMSCs were then cultured for 72 h for further assays.
2.3. Experimental Groups and Treatment
SH-SY5Y cells were grouped as follows: control, Aβ, Aβ+MSC, Aβ+MSC-NC-ov, and Aβ+MSC-CX3CL1-ov. An AD cell model was built via treating SH-SY5Y with 20 μM Aβ1-42 [29], while the control group was administrated with phosphate buffer saline (PBS) with an equal amount. Moreover, SH-SY5Y cells in Aβ+MSC, Aβ+MSC-NC-ov, and Aβ+MSC-CX3CL1-ov groups were coincubated with BMSCs, BMSCs transfected with NC-ov, and BMSCs transfected with CX3CL1-ov, respectively. The observation of cell morphology and axon growth was performed with a microscope (Olympus, Tokyo, Japan). After 48 h of coculture, SH-SY5Y cells at the plate bottom were trypsinized and prepared for the following assays.
2.4. Enzyme-Linked Immunosorbent Assay (ELISA)
Soluble CX3CL1 concentration in cell culture supernatant, generated after 18 hours of incubation in the complete medium, was detected by commercial human fractalkine/CX3CL1 ELISA kit (ab100522, Abcam, Cambridge, UK) based on the operating instructions.
2.5. Western Blotting Analysis
SH-SY5Y cells after three PBS rinses were immersed in a RIPA lysis buffer (Boster, Wuhan, China) to obtain the total proteins. Following isolation by 10% SDS-PAGE, the protein samples were processed for electrical transfer onto PVDF membranes (EMD Millipore, Billerica, MA, USA) that were then sealed at an ambient temperature in bovine serum albumin (BSA; 3%) for 60 minutes and subsequently cultivated at 4°C with the following Ι antibodies all ordered from Abcam overnight: CX3CL1 (1 : 1000, ab25088), cleaved caspase-3 (1 : 500, ab2302), Aβ (1 : 1000, ab11132), tau (1 : 10000, ab76128), phosphorylated tau (p-Tau) (1 : 50000, ab109390), SNAP25 (1 : 1000, ab41455), Synapsin1 (1 : 1000, ab64581), PSD95 (1 : 1000, ab18258), TXNIP (1 : 2000, ab188865), Trx (1 : 1000, ab26320), and NLRP3 (1 : 1000, ab263899). At an ambient temperature, they were cultured with the corresponding II antibody for 60 minutes after triple PBS washes. Protein expressions were analyzed relative to β-actin. Band visualization was made with an ECL kit (EMD Millipore) based on the operating manual. Image-ProPlus (Media Cybernetics, Inc., Rockville, MD, USA) determined the gray value.
2.6. Cell Counting Kit-8 Assay
After each group of SH-SY5Y cells were planted into 96-well plates at 1 × 105/well for overnight cultivation under the conditions of 5% CO2 and 37°C, the cell proliferation was analyzed following the Cell Count Kit-8 (Dojindo Laboratories, Kumamoto, Japan) operating manual. A microplate reader, supplied by Thermo Fisher Scientific, Waltham, MA, USA, was adopted to read the absorbance450nm.
2.7. Flow Cytometry Assay
90% confluent cells were washed with PBS for 2 to 3 times. Then, cell digestion with trypsin-EDTA solution and resuspension in 500 μL binding buffer were performed. Subsequently, the cell resuspension was stained with a mixture comprising allophycocyanin (APC) and phycoerythrin (PE), both supplied by Sigma-Aldrich with an amount of 5 μL, for 15 min at an ambient temperature in a dark environment. Flow cytometry (BD FACSVerse, USA) determined the apoptosis rate.
2.8. Immunofluorescence Assay
Cells were maintained and adhered on chamber slides. After triple PBS rinses, they were treated with 4% paraformaldehyde fixation (10 min), 1% BSA blocking, and overnight cultivation (4°C) with the Iba1 antibody (1 : 1000, ab48004, Abcam). Subsequently, they were processed for triple PBS washes and 60 min of goat anti-rat IgG H&L (Alexa Fluor® 647) (1 : 500, ab150167, Abcam) incubation (37°C). This was followed by nuclear staining with DAPI (C1002, Beyotime, Shanghai, China), and the final image was captured with a confocal microscopy (LSM700; Zeiss, Oberkochen, Germany).
2.9. MDA, SOD, CAT, and GSH-Px Measurements
Malondialdehyde (MDA), superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GSH-Px) measurements used MDA (A003-1-1), total SOD (T-SOD) (A001-1-1), CAT (A007-1-1), and GSH-Px test kits (A005-1-2; all from Nanjing Jiancheng Bioengineering Institute, China), respectively, following the operating manual of the corresponding kit. The microplate reader read the absorbance of wells at 532, 560, 405, and 412 nm for MDA, SOD, CAT, and GSH, respectively.
2.10. Statistical Processing
All data collected were present in the form of the mean ± standard deviation, and the statistical significance threshold was p < 0.05. Each assay was replicated 3 times. One-way analysis of variance and Duncan's test were employed to identify inter- and multigroup differences, respectively. SPSS 20.0 (SPSS Inc., Chicago, IL, USA) was used for analysis.
3. Results
3.1. BMSC-Derived CX3CL1 Reversed the Aβ1-42-Reduced Level of CX3CL1 in SH-SY5Y
To explore the role played by CX3CL1 derived from BMSCs in a Aβ1-42-induced SH-SY5Y cell model, pcDNA-ligated CX3CL1 overexpression plasmid was first transfected into BMSCs to upregulate CX3CL1 expression in BMSCs. ELISA (Figure 1(a)) and western blotting (Figures 1(b) and 1(c)) results showed that overexpression of CX3CL1 in BMSCs observably increased the levels of both soluble and membrane-bound CX3CL1, while there were no differences between the NC-ov and control groups. Besides, statistically decreased soluble and membrane-bound CX3CL1 levels were observed in SH-SY5Y under Aβ1-42 inducement, which were significantly rescued by the coculture of BMSCs (Figures 1(d)–1(f)). Moreover, overexpression of CX3CL1 in BMSCs further statistically enhanced CX3CL1 levels of both the soluble and membrane-bound forms, whereas transfection of NC-ov showed no statistical effect (Figures 1(d)–1(f)). These results indicated that CX3CL1 derived from BMSCs notably reversed the Aβ1-42-reduced CX3CL1 expression in SH-SY5Y cells.
Figure 1.

CX3CL1 derived from BMSCs significantly inverted Aβ1-42-decreased CX3CL1 in SH-SY5Y. (a) Soluble CX3CL1 expression in BMSCs was detected by ELISA. (b and c) Western blotting determined CX3CL1 expression relative to β-actin in BMSCs. (d) ELISA measured soluble CX3CL1 levels in SH-SY5Y under different interventions. (e and f) Western blotting examined CX3CL1 expression relative to β-actin in SH-SY5Y under different interventions. ns: nonsignificance; ∗p < 0.05 versus control group; #p < 0.05 versus Aβ group; &p < 0.05 versus Aβ+MSC group.
3.2. CX3CL1 Derived from BMSCs Increased Aβ1-42-Induced SH-SY5Y Viability but Suppressed Apoptosis
Then, we investigated the role played by CX3CL1 derived from BMSCs in SH-SY5Y cell growth under Aβ1-42 inducement. Statistically declined Aβ1-42-intervened SH-SY5Y cell viability was found compared to the control group, which was markedly inverted by the coculture of BMSCs (Figure 2(a)). Also, transfection of CX3CL1 overexpression plasmid (not empty plasmid) into BMSCs further markedly elevated SH-SY5Y cell viability compared to SH-SY5Y with Aβ1-42 and BMSC coculture (Figure 2(a)). However, the opposite tendency was discovered in SH-SY5Y cell apoptosis, as indicated by that cocultured with BMSCs significantly reduced the Aβ1-42-increased apoptosis rate (Figures 2(b) and 2(c)) and the relative protein expression of cleaved caspase-3 (Figures 2(d) and 2(e)) of SH-SY5Y cells, which was prominently antagonized with the overexpression of CX3CL1 in BMSCs. These data suggested that CX3CL1 derived from BMSCs significantly enhanced Aβ1-42-induced SH-SY5Y cell growth but inhibited apoptosis.
Figure 2.

CX3CL1 derived from BMSCs statistically augmented Aβ1-42-induced SH-SY5Y viability but repressed apoptosis. (a) CCK-8 assay assessed SH-SY5Y viability. (b and c) Flow cytometry evaluated SH-SY5Y apoptosis. (d and e) Western blotting determined cleaved caspase-3 expression relative to β-actin. ∗p < 0.05 versus control group; #p < 0.05 versus Aβ group; &p < 0.05 versus Aβ+MSC group.
3.3. CX3CL1 Derived from BMSCs Ameliorated the Pathology-Induced Aβ1-42 in SH-SY5Y
Next, the role played by CX3CL1 derived from BMSCs in the pathology of Aβ1-42-induced SH-SY5Y was investigated. Aβ, tau, and p-Tau were found to be markedly increased with the induction of Aβ1-42 in SH-SY5Y, which was statistically improved with the coculture of BMSCs (Figures 3(a)–3(d)). Moreover, transfection of CX3CL1 overexpression plasmid (not empty plasmid) into BMSCs further notably diminished the relative protein expression of Aβ, tau, and p-Tau (Figures 3(a)–3(d)). Also, similar changes were found in the relative integrated density of Iba1 (Figures 3(e) and 3(f)). Besides, coculture of BMSCs obviously improved the cell growth and axon length of Aβ1-42-induced SH-SY5Y, which was further ameliorated with the overexpression of CX3CL1 in BMSCs (Figure 3(g)). Furthermore, the relative protein expressions of synaptic markers, including SNAP25, Synapsin1, and PSD95, were statistically declined in SH-SY5Y with Aβ1-42 inducement, which were notably reversed by the coculture of BMSCs (Figures 3(h)–3(k)). Consistently, upregulation of CX3CL1 in BMSCs further statistically elevated SNAP25, Synapsin1, and PSD95 protein levels (Figures 3(h)–3(k)). Taken together, these results demonstrated that CX3CL1 derived from BMSCs obviously improved the pathology-induced Aβ1-42 in SH-SY5Y.
Figure 3.

CX3CL1 derived from BMSCs alleviated the pathology-induced Aβ1-42 in SH-SY5Y. (a–d) Western blotting examined Aβ, tau, and p-Tau protein expressions relative to β-actin. (e and f) The relative integrated density of Iba1 was evaluated by IF. (g) The axons of SH-SY5Y cells were observed using inverted microscope. (h–k) Western blotting examined SNAP25, Synapsin1, and PSD95 protein levels relative to β-actin. ∗p < 0.05 versus control group; #p < 0.05 versus Aβ group; &p < 0.05 versus Aβ+MSC group.
3.4. The Influence of BMSC-Derived CX3CL1 on Aβ1-42-Induced SH-SY5Y Was Associated with TXNIP/NLRP3 Axis
Furthermore, the potential mechanisms associated with the role of CX3CL1 derived from BMSCs in SH-SY5Y with Aβ1-42 inducement were studied. Coculture of BMSCs markedly declined the Aβ1-42-enhanced MDA concentration, which was further statistically decreased by overexpression of CX3CL1 in BMSCs (Figure 4(a)). On the contrary, coculture of BMSCs markedly elevated the Aβ1-42-reduced concentrations of SOD, CAT, and GSH-Px, which was further significantly increased by overexpression of CX3CL1 in BMSCs (Figures 4(b)–4(d)). Mechanically, coculture of BMSCs prominently diminished the Aβ1-42-enhanced TXNIP and NLRP3 protein levels while aggrandized the Aβ1-42-attenuated Trx protein expression (Figures 4(e)–4(h)). Moreover, upregulation of CX3CL1 in BMSCs further statistically strengthened the corresponding tendency (Figures 4(e)–4(h)). Together, these data clarified that the effect of CX3CL1 derived from BMSCs on Aβ1-42-induced SH-SY5Y involved the TXNIP/NLRP3 signaling axis.
Figure 4.
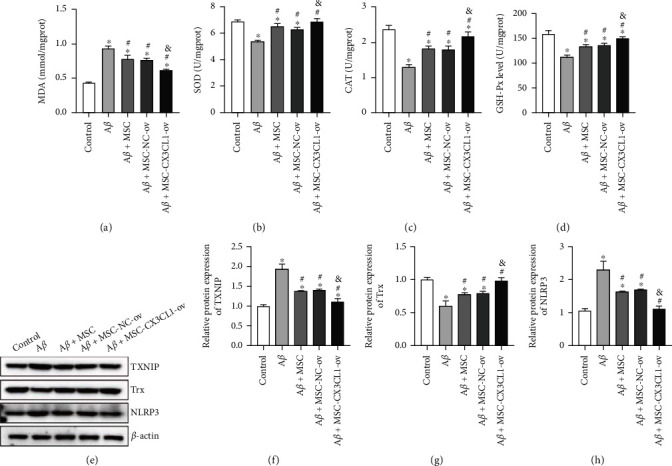
The role played by CX3CL1 derived from BMSCs in Aβ1-42-induced SH-SY5Y was related to TXNIP/NLRP3 axis. (a–d) MDA, SOD, CAT, and GSH-Px concentrations measured by commercial kits. (e–h) Western blotting tested TXNIP, NLRP3, and Trx protein levels relative to β-actin. ∗p < 0.05 versus control group; #p < 0.05 versus Aβ group; &p < 0.05 versus Aβ+MSC group.
4. Discussion
AD is the most prevalent neurodegenerative brain disorder that imposes a severe burden on patients and their families, even leading to social and economic losses [30]. It is demonstrated that MSC therapy is a novel and promising treatment for AD, whose paracrine action is meant to trigger immunomodulation and neural regeneration [17]. CX3CL1 is essential in the regulation of microglia activation, neuroprotection, and immunomodulation by MSCs [20, 21]. In this paper, Aβ1-42 treatment markedly reduced both soluble and membrane-bound CX3CL1 levels, cell viability, protein levels of synaptic markers (SNAP25, Synapsin1, and PSD95), SOD, CAT, and GSH-Px concentrations, and Trx protein expression, while it enhanced apoptosis, cleaved caspase-3, Aβ, tau, and p-Tau protein levels, relative integrated density of Iba1, MDA concentration, and TXNIP and NLRP3 protein levels in SH-SY5Y; however, the above effects were prominently reversed by the coculture of BMSCs. Moreover, we overexpressed CX3CL1 in BMSCs and found that overexpression of CX3CL1 in BMSCs observably strengthened the corresponding tendency caused by the coculture of BMSCs.
The membrane-bound CX3CL1 and soluble CX3CL1 play diverse roles under physiological conditions. Membrane-bound CX3CL1 functions is involved in infiltrating leukocyte recruitment and adhesion [31], while soluble CX3CL1 serves as a chemoattractant associated with the cellular migration, as well as a neuroprotective signaling molecule to contribute to the sustainability of the resting state of microglia [32, 33]. Besides, soluble CX3CL1 modulates the anti-inflammatory effect in the brain, and the receptors of membrane-bound CX3CL1 are located on microglial cell surfaces [32, 34]. During fibrosis amelioration, MSCs can participate in Ly-6C-high (inflammatory) to Ly-6C low (anti-inflammatory) macrophage polarization by increasing CX3CL1 levels, exerting significant effects on ameliorating liver disease [35]. The cerebrospinal fluid CX3CL1 level is reported to be notably diminished in AD patients [36]. Also, our results revealed statistically reduced soluble and membrane-bound CX3CL1 levels in SH-SY5Y cells under Aβ1-42 inducement. However, coculture of BMSCs markedly rescued decreased CX3CL1 by Aβ1-42 in SH-SY5Y, implying that BMSCs might express and secrete CX3CL1 to compensate for the loss of CX3CL1 [37]. Overexpression of CX3CL1 in BMSCs further confirmed this conclusion. Besides, we discovered that CX3CL1 derived from BMSCs significantly enhanced Aβ1-42-induced SH-SY5Y growth but declined apoptosis rate and cleaved caspase-3 at the protein level. Cleaved caspase-3 is the activation form of caspase-3 that can regulate different phases of the apoptotic pathway [38]. Therefore, these findings elucidated that Aβ1-42 treatment resulted in low level of CX3CL1, which was positively associated with the growth and negatively related to the apoptosis of SH-SY5Y cells. Upregulation of CX3CL1 in BMSCs obviously increased the reduced CX3CL1 and proliferation of SH-SY5Y cells but inhibited increased apoptosis by Aβ1-42. Taken together, CX3CL1 derived from BMSCs reversed the Aβ1-42-reduced CX3CL1, which increased the viability but suppressed the apoptosis of SH-SY5Y under Aβ1-42 inducement.
Aβ accumulation and hyperphosphorylated tau are the dominating pathological hallmarks of AD [39, 40], which may explain prominently high Aβ, Tau, and p-Tau protein levels in SH-SY5Y with Aβ1-42 inducement in this research. Recently, Qin et al. [41] have reported that BMSC transplantation mitigated neuropathology and ameliorated cognitive deficits in AD animal models. Consistently, we found that coculture of BMSCs statistically reduced the Aβ1-42-increased Aβ, Tau, and p-Tau protein levels, which were further notably decreased by the upregulation of CX3CL1 in BMSCs. Tau and Aβ are the prime suspects driving AD pathology and, as such, have become the focus of therapeutic development. There is mounting evidence that these proteins may perform many crucial physiological functions during AD pathological development that can be disrupted by Aβ- or tau-lowering therapeutics [42]. In the past decades, a large number of research has demonstrated that neuroinflammation is a central risk factor of AD; thus, strategies of decreasing neuroinflammation have been regarded as the promising therapeutics for AD [43, 44]. Neuroinflammation in the brain is mediated by microglial cells, so microglial activation is a characteristic of neuroinflammation [45, 46]. Here, we observed that coculture of BMSCs significantly declined the Aβ1-42-enhanced relative integrated density of Iba1, a marker of microglial cells, which was further obviously diminished by the overexpression of CX3CL1 in BMSCs, indicating that CX3CL1 derived from BMSCs inhibited neuroinflammation in SH-SY5Y with Aβ1-42 inducement. Moreover, it confirmed the fact that soluble CX3CL1 plays an anti-inflammatory role in the brain [32, 34]. In addition, it has been demonstrated that Aβ and Tau oligomers, as well as neuroinflammation, can conduce to synaptic loss, which results in synaptic dysfunction [46–48]. Our findings revealed that coculture of BMSCs obviously improved Aβ1-42-induced SH-SY5Y cell growth and axon length, which was further notably ameliorated by the overexpression of CX3CL1 in BMSCs. Also, coculture of BMSCs statistically elevated the Aβ1-42-attenuated relative protein levels of SNAP25, Synapsin1, and PSD95, as well as the presynaptic and postsynaptic markers, which was further markedly promoted by the overexpression of CX3CL1 in BMSCs. Based on the above, we can draw the conclusion that CX3CL1 derived from BMSCs notably suppressed pathological damage in Aβ1-42-induced SH-SY5Y cells.
Furthermore, neuroinflammation can induce oxidative stress in the brain [48]. In the present study, Aβ1-42 treatment remarkably reduced SOD, CAT, and GSH-Px concentrations in SH-SY5Y while enhancing MDA levels, which were prominently reversed by the coculture of BMSCs. Overexpression of CX3CL1 in BMSCs observably strengthened the corresponding tendency caused by the coculture of BMSCs, which suggested statistically inhibited oxidative stress in SH-SY5Y with Aβ1-42 inducement by BMSC-derived CX3CL1. TrX is an intracellular antioxidative protein via thiol reduction and the scavenging of reactive oxygen species (ROS) [49]. This research identified that TrX expression was predominately decreased in SH-SY5Y with Aβ1-42 inducement, which was in line with the reduction in AD brain regions [50]. TrX can be bound by TXNIP to dampen TrX activation and facilitate oxidative stress [51]. Oakley et al. [52] exhibited that the level of TXNIP was upregulated in the hippocampus of mice in a 5 × AD mouse model. Consistently, SH-SY5Y with Aβ1-42 inducement presented overexpressed TXNIP in our study. Moreover, TXNIP is a key link between inflammasome activation and oxidative stress [53]. Under oxidative stress, dissociated TXNIP from TrX binds to NLRP3 that conduces to inflammasome formation and activation. Consequently, a series of proinflammatory mediators are secreted to enhance neuroinflammation, which promotes synaptic dysfunction and the subsequent AD progression. Furthermore, Wang et al. [54] have reported an enhancement of binding between TXNIP and NLRP3 in the APP/PS1 mouse brain, and NLRP3 inflammasome and Nrf2/TXNIP/TrX axis are involved in the pathological changes of AD. Here, we discovered that Aβ1-42 treatment notably decreased Trx protein expression and elevated TXNIP and NLRP3 protein levels in SH-SY5Y that were prominently reversed by the coculture of BMSCs. Upregulation of CX3CL1 in BMSCs significantly intensified the corresponding tendency caused by the coculture of BMSCs. Therefore, these results elaborated that the role of CX3CL1 derived from BMSCs in Aβ1-42-iintervened SH-SY5Y was linked to the TXNIP/NLRP3 signaling axis.
5. Conclusion
In conclusion, our results revealed that through the TXNIP/NLRP3 signaling pathway, CX3CL1 derived from BMSCs alleviates Aβ1-42-induced SH-SY5Y cell injury. However, there are still some limitations in our study. First, Lee et al. [55] have reported that membrane-bound CX3CL1 but not soluble CX3CL1 mediated the Aβ pathology. Our results showed that CX3CL1 derived from BMSCs reversed the Aβ1-42-reduced soluble and membrane-bound CX3CL1 levels; hence, whether the membrane-bound CX3CL1 consistently improved the Aβ pathology or not needs further exploration in our subsequent studies. Besides, multiple research has demonstrated that the CX3CL1 is closely involved in inflammation [56–58]. However, in our study, we failed to further reveal the role of CX3CL1 derived from BMSCs in neuroinflammation. Also, the findings are not verified in vivo. Thus, more experiments are expected to be conducted in further studies to explore the underlying mechanism. In brief, it is hoped that our results can lay a foundation for AD treatment and even the management of other neurodegenerative disorders.
Acknowledgments
The present work was supported by the Severe phenotypic transformation of microglia mediated by TLR3/TRIF signaling pathway and its regulation and mechanism on neurons after intracerebral hemorrhage, Sichuan Provincial Health Commission (No. 21PJ113); Study on the mechanism of icariin in the treatment of anti-neuro-inflammation and anti-oxidation after spinal cord injury based on Nrf2 signaling pathway, Sichuan Administration of Traditional Chinese Medicine (No. 2020JC0025); and Research on the polarization and directional induction regulation of microglia by icariin after spinal cord injury, the First Affiliated Hospital of Chengdu Medical College High-level Talents Research Start-up Fund (No. CYFY-GQ25).
Data Availability
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors report that there are no competing interests to declare.
Authors' Contributions
CG, QL, and XX designed the experiments; CG, QL, and JX carried out the experiments; XZ, MT, and LX analyzed the experimental results and interpreted the data; CG, QL, and XX wrote and revised the manuscript. All authors have approved the final manuscript. Chuan Guo and Qinxuan Li contributed equally to this work and are co-first authors.
References
- 1.Wiley J. Alzheimer’s disease facts and figures. Alzheimers Dementia . 2021;17 doi: 10.1002/alz.12328. [DOI] [PubMed] [Google Scholar]
- 2.Jia J., Wei C., Chen S., et al. The cost of Alzheimer's disease in China and re-estimation of costs worldwide. Alzheimer's & Dementia . 2018;14(4):483–491. doi: 10.1016/j.jalz.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Zhang D.-F., Xu M., Bi R., Yao Y.-G. Genetic analyses of Alzheimer’s disease in China: achievements and perspectives. ACS Chemical Neuroscience . 2019;10(2):890–901. doi: 10.1021/acschemneuro.8b00435. [DOI] [PubMed] [Google Scholar]
- 4.d‘Errico P., Meyer-Luehmann M. Mechanisms of pathogenic tau and aβ protein spreading in Alzheimer’s disease. Frontiers in Aging Neuroscience . 2020;12:p. 265. doi: 10.3389/fnagi.2020.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaz M., Silvestre S. Alzheimer's disease: recent treatment strategies. European Journal of Pharmacology . 2020;887, article 173554 doi: 10.1016/j.ejphar.2020.173554. [DOI] [PubMed] [Google Scholar]
- 6.Long J. M., Holtzman D. M. Alzheimer disease: an update on pathobiology and treatment strategies. Cell . 2019;179(2):312–339. doi: 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sevigny J., Chiao P., Bussière T., et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature . 2016;537(7618):50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 8.Breijyeh Z., Karaman R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules . 2020;25(24):p. 5789. doi: 10.3390/molecules25245789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding D.-C., Shyu W.-C., Lin S.-Z. Mesenchymal stem cells. Cell Transplantation . 2011;20(1):5–14. doi: 10.3727/096368910X. [DOI] [PubMed] [Google Scholar]
- 10.Chen X., Wang S., Cao W. Mesenchymal stem cell-mediated immunomodulation in cell therapy of neurodegenerative diseases. Cellular Immunology . 2018;326:8–14. doi: 10.1016/j.cellimm.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Vasic V., Barth K., Schmidt M. H. Neurodegeneration and neuro-regeneration—Alzheimer’s disease and stem cell therapy. International Journal of Molecular Sciences . 2019;20(17):p. 4272. doi: 10.3390/ijms20174272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alipour M., Nabavi S. M., Arab L., et al. Stem cell therapy in Alzheimer’s disease: possible benefits and limiting drawbacks. Molecular Biology Reports . 2019;46(1):1425–1446. doi: 10.1007/s11033-018-4499-7. [DOI] [PubMed] [Google Scholar]
- 13.Babaei P., Soltani Tehrani B., Alizadeh A. Transplanted bone marrow mesenchymal stem cells improve memory in rat models of Alzheimer's disease. Stem Cells International . 2012;2012:8. doi: 10.1155/2012/369417.369417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bae J. S., Jin H. K., Lee J. K., Richardson J. C., Carter J. E. Bone marrow-derived mesenchymal stem cells contribute to the reduction of amyloid-β deposits and the improvement of synaptic transmission in a mouse model of pre-dementia Alzheimer's disease. Current Alzheimer Research . 2013;10(5):524–531. doi: 10.2174/15672050113109990027. [DOI] [PubMed] [Google Scholar]
- 15.Kim H. J., Seo S. W., Chang J. W., et al. Stereotactic brain injection of human umbilical cord blood mesenchymal stem cells in patients with Alzheimer's disease dementia: a phase 1 clinical trial. Alzheimer's & Dementia: Translational Research & Clinical Interventions . 2015;1(2):95–102. doi: 10.1016/j.trci.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J. K., Jin H. K., Bae J. S. Bone marrow-derived mesenchymal stem cells reduce brain amyloid-β deposition and accelerate the activation of microglia in an acutely induced Alzheimer's disease mouse model. Neuroscience Letters . 2009;450(2):136–141. doi: 10.1016/j.neulet.2008.11.059. [DOI] [PubMed] [Google Scholar]
- 17.Phinney D. G., Pittenger M. F. Concise review: MSC-derived exosomes for cell-free therapy. Stem Cells . 2017;35(4):851–858. doi: 10.1002/stem.2575. [DOI] [PubMed] [Google Scholar]
- 18.Shang S., Wang J., Chen S., et al. Exosomal mirna-1231 derived from bone marrow mesenchymal stem cells inhibits the activity of pancreatic cancer. Cancer Medicine . 2019;8(18):7728–7740. doi: 10.1002/cam4.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forte D., García-Fernández M., Sánchez-Aguilera A., et al. Bone marrow mesenchymal stem cells support acute myeloid leukemia bioenergetics and enhance antioxidant defense and escape from chemotherapy. Cell metabolism . 2020;32(5):p. 829. doi: 10.1016/j.cmet.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang L., Xu G., Guo J., Xie M., Chen L., Xu W. Mesenchymal stem cells modulate light-induced activation of retinal microglia through CX3CL1/CX3CR1 signaling. Ocular Immunology and Inflammation . 2016;24(6):684–692. doi: 10.3109/09273948.2015.1071405. [DOI] [PubMed] [Google Scholar]
- 21.Huang L., Xu W., Xu G. Transplantation of CX3CL1-expressing mesenchymal stem cells provides neuroprotective and immunomodulatory effects in a rat model of retinal degeneration. Ocular Immunology and Inflammation . 2013;21(4):276–285. doi: 10.3109/09273948.2013.791925. [DOI] [PubMed] [Google Scholar]
- 22.Limatola C., Ransohoff R. M. Modulating neurotoxicity through CX3CL1/CX3CR1 signaling. Frontiers in Cellular Neuroscience . 2014;8:p. 229. doi: 10.3389/fncel.2014.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo P., Chu S. F., Zhang Z., Xia C. Y., Chen N. H. Fractalkine/CX3CR1 is involved in the cross-talk between neuron and glia in neurological diseases. Brain Research Bulletin . 2019;146:12–21. doi: 10.1016/j.brainresbull.2018.11.017. [DOI] [PubMed] [Google Scholar]
- 24.Hatori K., Nagai A., Heisel R., Ryu J. K., Kim S. U. Fractalkine and fractalkine receptors in human neurons and glial cells. Journal of Neuroscience Research . 2002;69(3):418–426. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- 25.Finneran D. J., Nash K. R. Neuroinflammation and fractalkine signaling in Alzheimer’s disease. Journal of Neuroinflammation . 2019;16(1):1–8. doi: 10.1186/s12974-019-1412-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandur E., Tamási K., Pap R., Varga E., Miseta A., Sipos K. Fractalkine induces hepcidin expression of BV-2 microglia and causes iron accumulation in SH-SY5Y cells. Cellular and Molecular Neurobiology . 2019;39(7):985–1001. doi: 10.1007/s10571-019-00694-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garton K. J., Gough P. J., Blobel C. P., et al. Tumor necrosis factor-α-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) Journal of Biological Chemistry . 2001;276(41):37993–38001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- 28.Hundhausen C., Misztela D., Berkhout T. A., et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood . 2003;102(4):1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- 29.Xu M., Huang H., Mo X., et al. Quercetin-3-O-glucuronide alleviates cognitive deficit and toxicity in Aβ1-42-induced ad-like mice and SH-SY5Y cells. Molecular Nutrition & Food Research . 2021;65(6, article e2000660) doi: 10.1002/mnfr.202000660. [DOI] [PubMed] [Google Scholar]
- 30.Hugo J., Ganguli M. Dementia and cognitive impairment: epidemiology, diagnosis, and treatment. Clinics in Geriatric Medicine . 2014;30(3):421–442. doi: 10.1016/j.cger.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imai T., Hieshima K., Haskell C., et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell . 1997;91(4):521–530. doi: 10.1016/S0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 32.Morganti J. M., Nash K. R., Grimmig B. A., et al. The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson's disease. Journal of Neuroscience . 2012;32(42):14592–14601. doi: 10.1523/JNEUROSCI.0539-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ostuni M. A., Guellec J., Hermand P., et al. CX3CL1, a chemokine finely tuned to adhesion: critical roles of the stalk glycosylation and the membrane domain. Biology open . 2014;3(12):1173–1182. doi: 10.1242/bio.20149845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nash K. R., Moran P., Finneran D. J., et al. Fractalkine over expression suppresses α-synuclein-mediated neurodegeneration. Molecular Therapy . 2015;23(1):17–23. doi: 10.1038/mt.2014.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baghaei K., Varjavand P., Malmir A., et al. Mesenchymal stem cells foster Ly-6C low macrophages polarization through the CX3CL1 pathway to ameliorate liver fibrosis. Cytotherapy . 2020;22(5):p. S67. doi: 10.1016/j.jcyt.2020.03.103. [DOI] [Google Scholar]
- 36.Perea J. R., Lleó A., Alcolea D., Fortea J., Ávila J., Bolós M. Decreased CX3CL1 levels in the cerebrospinal fluid of patients with Alzheimer’s disease. Frontiers in Neuroscience . 2018;12:p. 609. doi: 10.3389/fnins.2018.00609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Honczarenko M., Le Y., Swierkowski M., Ghiran I., Glodek A. M., Silberstein L. E. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells . 2006;24(4):1030–1041. doi: 10.1634/stemcells.2005-0319. [DOI] [PubMed] [Google Scholar]
- 38.Asadi M., Taghizadeh S., Kaviani E., et al. Caspase-3: structure, function, and biotechnological aspects. Biotechnology and Applied Biochemistry . 2021 doi: 10.1002/bab.2233. [DOI] [PubMed] [Google Scholar]
- 39.Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer's disease. Annals of the New York Academy of Sciences . 1996;777(1):121–131. doi: 10.1111/j.1749-6632.1996.tb34410.x. [DOI] [PubMed] [Google Scholar]
- 40.Holtzman D. M., Mandelkow E., Selkoe D. J. Alzheimer disease in 2020. Cold Spring Harbor Perspectives in Medicine . 2012;2(11, article a011585) doi: 10.1101/cshperspect.a011585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin C., Lu Y., Wang K., et al. Transplantation of bone marrow mesenchymal stem cells improves cognitive deficits and alleviates neuropathology in animal models of Alzheimer’s disease: a meta-analytic review on potential mechanisms. Translational Neurodegeneration . 2020;9(1):1–20. doi: 10.1186/s40035-020-00199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kent S. A., Spires-Jones T. L., Durrant C. S. The physiological roles of tau and aβ: implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathologica . 2020;140(4):417–447. doi: 10.1007/s00401-020-02196-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ardura-Fabregat A., Boddeke E., Boza-Serrano A., et al. Targeting neuroinflammation to treat Alzheimer’s disease. CNS Drugs . 2017;31(12):1057–1082. doi: 10.1007/s40263-017-0483-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sánchez-Sarasúa S., Fernández-Pérez I., Espinosa-Fernández V., Sánchez-Pérez A. M., Ledesma J. C. Can we treat neuroinflammation in Alzheimer’s disease? International Journal of Molecular Sciences . 2020;21(22):p. 8751. doi: 10.3390/ijms21228751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamelin L., Lagarde J., Dorothée G., et al. Distinct dynamic profiles of microglial activation are associated with progression of Alzheimer's disease. Brain . 2018;141(6):1855–1870. doi: 10.1093/brain/awy079. [DOI] [PubMed] [Google Scholar]
- 46.Regen F., Hellmann-Regen J., Costantini E., Reale M. Neuroinflammation and Alzheimer's disease: implications for microglial activation. Current Alzheimer Research . 2017;14(11):1140–1148. doi: 10.2174/1567205014666170203141717. [DOI] [PubMed] [Google Scholar]
- 47.Compta Y., Revesz T. Neuropathological and biomarker findings in Parkinson’s disease and Alzheimer’s disease: from protein aggregates to synaptic dysfunction. Journal of Parkinson's Disease . 2021;11(1):107–121. doi: 10.3233/JPD-202323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao J. S., Kellom M., Kim H.-W., Rapoport S. I., Reese E. A. Neuroinflammation and synaptic loss. Neurochemical Research . 2012;37(5):903–910. doi: 10.1007/s11064-012-0708-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patenaude A., Murthy M., Mirault M.-E. Emerging roles of thioredoxin cycle enzymes in the central nervous system. Cellular and Molecular Life Sciences CMLS . 2005;62(10):1063–1080. doi: 10.1007/s00018-005-4541-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lovell M. A., Xie C., Gabbita S. P., Markesbery W. R. Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer's disease brain. Free Radical Biology and Medicine . 2000;28(3):418–427. doi: 10.1016/S0891-5849(99)00258-0. [DOI] [PubMed] [Google Scholar]
- 51.Junn E., Han S. H., Im J. Y., et al. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. The Journal of Immunology . 2000;164(12):6287–6295. doi: 10.4049/jimmunol.164.12.6287. [DOI] [PubMed] [Google Scholar]
- 52.Oakley H., Cole S. L., Logan S., et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. Journal of Neuroscience . 2006;26(40):10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou R., Tardivel A., Thorens B., Choi I., Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nature Immunology . 2010;11(2):136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 54.Wang C.-Y., Xu Y., Wang X., Guo C., Wang T., Wang Z.-Y. Dl-3-n-butylphthalide inhibits nlrp3 inflammasome and mitigates Alzheimer's-like pathology via Nrf2-TXNIP-TrX axis. Antioxidants & Redox Signaling . 2019;30(11):1411–1431. doi: 10.1089/ars.2017.7440. [DOI] [PubMed] [Google Scholar]
- 55.Lee S., Xu G., Jay T. R., et al. Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. Journal of Neuroscience . 2014;34(37):12538–12546. doi: 10.1523/JNEUROSCI.0853-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li A., Zhao J., Fan C., et al. Delivery of exogenous proteins by mesenchymal stem cells attenuates early memory deficits in a murine model of Alzheimer's disease. Neurobiology of Aging . 2020;86:81–91. doi: 10.1016/j.neurobiolaging.2019.10.012. [DOI] [PubMed] [Google Scholar]
- 57.Strobel S., Grünblatt E., Riederer P., et al. Changes in the expression of genes related to neuroinflammation over the course of sporadic Alzheimer’s disease progression: CX3CL1, TREM2, and PPARγ. Journal of Neural Transmission . 2015;122(7):1069–1076. doi: 10.1007/s00702-015-1369-5. [DOI] [PubMed] [Google Scholar]
- 58.Subbarayan M. S., Joly-Amado A., Bickford P. C., Nash K. R. CX3CL1/CX3CR1 signaling targets for the treatment of neurodegenerative diseases. Pharmacology & Therapeutics . 2022;231(article 107989) doi: 10.1016/j.pharmthera.2021.107989. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request.