

Abstract
HOXA5 is a transcription factor and tumor suppressor that promotes differentiation of breast epithelial cells and is frequently lost during malignant transformation. HOXA5 loss alone, however, does not confer tumorigenicity. To determine which molecular alterations combined with loss of HOXA5 expression can transform cells, we examined isogenic derivatives of a non-malignant breast epithelial cell line containing knock-in or knock-out mutations in key breast cancer genes. Knockdown (KD) of HOXA5 in cells harboring double knock-in (DKI) of mutated PIK3CA (E545K) and HER2 (V777L) induced epithelial-mesenchymal transition and migration and promoted invasive tumor outgrowth within mouse mammary ducts. The NF-κB pathway was significantly upregulated in DKI cells following HOXA5 KD. HOXA5 KD upregulated multiple NF-κB target genes, including IL6. IκB-α protein, but not RNA, expression was reduced in HOXA5-KD cells. HOXA5 bound and stabilized IκB-α, forming a nuclear HOXA5-IκB-α complex. ChIP-seq database queries revealed that HOXA5 and IκB-α are co-enriched at 528 genomic loci. In breast cancer patients, high co-expression of HOXA5 and IκB-α conferred a significantly better overall- and progression-free survival. Collectively, these data suggest that HOXA5 suppresses malignancy in breast epithelial cells by blunting NF-κB action via stabilization of its inhibitor IκB-α.
Keywords: HOXA5, IΚB-α, breast cancer, IL-6, NFKB
INTRODUCTION
Homeobox or ‘Hox’ transcription factors are encoded by a family of 39 genes, arranged in 4 contiguous groups on 4 chromosomes (1–3). They are characterized by the presence of the homeodomain, a highly conserved DNA binding region that binds a 5’-TAAT-3’ DNA motif (1–3). HOXA5 is a powerful differentiation factor and has been shown to be important during organ fate specification of the mammary gland (4), digestive tract (5), lung (6) and musculoskeletal system (7).
In the adult breast, HOXA5 maintains breast ductal epithelial cells in a differentiated state (8,9). HOXA5 expression is lost in around 60–70% of all breast cancers (9–12) . This often occurs at an early stage of disease due to promoter methylation of HOXA5 (12) or through the action of upstream regulators of HOXA5, such as RARβ (9,13).
HOXA5 loss plays a crucial role in breast cancer progression, since it acts as a transcriptional regulator of p53 (14) and the progesterone receptor (15). Expression of HOXA5 in breast cancer cells causes apoptosis, both through p53 (14) and activation of Caspase 6 and 8 (16). It also up regulates the expression of key genes E-Cadherin and CD24 that determine the epithelial cell fate of breast cells, the loss of these proteins in the absence of HOXA5 pushes cells toward a more stem-like, mesenchymal state (9).
While HOXA5’s action as a tumor suppressor in breast cancer is supported by strong experimental evidence, loss of HOXA5 alone is insufficient to initiate mammary tumorigenesis. Hoxa5 null mice display skeletal and other malformations, but do not develop mammary tumors (5,17). However, the complete loss of Hoxa5 on a p53+/− background enhances mammary tumor formation in mice (18). In the current study, we used genetically engineered MCF10A mammary epithelial cells possessing two mutations: a HER2 missense kinase domain mutation- V777L and a PIK3CA, E545K mutation generated by knocking-in the mutant allele at the endogenous genomic locus (19). The HER2 mutation alone activated the HER2-signaling pathway, but did not fully transform MCF10A cells (19). Even the presence of both HER2 and PIK3CA mutations, while promoting migratory properties of MCF10A cells, did not support the outgrowth of mammary carcinomas in nude mice (19).
NF-κB is a transcription factor that plays critical roles in inflammation, control of cell death pathways and cell proliferation. It consists of a family of five proteins: NF-κB1/p105, NF-κB2/p100, RelA/p65, RelB and c-Rel (20). Under unstimulated conditions, they exist as homo- or heterodimers bound by inhibitory κB (IκB) proteins in the cytoplasm. Upon stimulation by cytokines, IκB kinase activation leads to IκB phosphorylation and subsequent ubiquitin-dependent IκB degradation. The released NF-κB transcription factor with an unmasked nuclear localization signal then accumulates in the nucleus to regulate the expression of its target genes (21).
The NF-κB pathway is important for breast epithelial proliferation, architecture and branching during early post-natal development (22). While the pathway is not typically active in the adult breast, it plays a key role in the progression of breast cancer by aiding cell survival, inflammation, immunity and self-renewal of stem cells (23). Inhibition of NF-κB in the Polyoma Middle T transgenic mouse mammary tumor model impedes tumor progression and increases tumor latency (24). The NF-κB pathway is frequently activated in the HER2-positive and estrogen receptor (ER) and progesterone (PR)-negative subtypes of breast cancer (23), and can promote bone metastasis and osteolysis in patients with the ER/PR/HER2-negative subtype of breast cancer (25). While nuclear p65 is not detectable in the immortalized, MCF10A mammary epithelial cell line, it is frequently present in breast cancer cell lines and in carcinogen-induced rat primary mammary tumors (26).
A relationship between the NF-κB pathway and HOX proteins in developmental biology, cancer biology and inflammation has been previously documented (27). In liposarcoma cells, HOXA5 sequesters p65/RelA in the nucleus without affecting p65 transcriptional activity (28). Another member of the HOX family, HOXA1, acts as an oncogene in breast cancer, and was found to potentiate NF-κB activity via a physical interaction with the NF-κB upstream regulators, TRAF2 and RBCK1/HOIL-1 (29). In prostate cancer, HOXB13 was found to increase intracellular zinc levels, which enhanced NF-κB pathway activity (30).
The goal of this study was two-fold; to understand the importance of HOXA5 loss in the context of other common breast cancer-initiating mutations and to determine additional mechanisms underlying HOXA5-mediated tumor suppressive function. Here, we report that HOXA5 loss enhances tumorigenesis in a MCF10A cell line harboring knock-in PIK3CA and HER2 mutations. Using these MCF10A-knock-in cells and other breast cell lines, we delineate events underlying HOXA5 effects on NF-κB signaling. Our results suggest that HOXA5 mediates its tumor suppressor action through a protein-protein interaction-mediated stabilization of IκB-α, which inhibits NF-κB.
MATERIALS AND METHODS
Additional details are presented in Supplementary Methods.
Cell Culture
Isogenic MCF10A sublines with single allele mutations in oncogenes, KRAS (G12V), PIK3CA (E545 and H1047R), HER2 (V777L and L755S), and mutations in tumor suppressor genes, BRCA1 (185 del AG, R71G, C61G) and p53 (R248W), or p53 null, or double knock-in mutations in both HER2 (V777L) and PIK3CA (E545) were generated by Cre recombinase-mediated excision/insertion and generously provided by Dr. Ben Ho Park (Vanderbilt). Genomic DNA extraction using the Qiagen Blood DNA extraction kit (QIAamp DNA Blood Mini Kit, # 51104) was followed by PCR to amplify the correct genomic locus. Sanger sequencing was used to confirm genotype, as described previously (19). Cells were cultured in MCF10A media (DMEM /F12 Ham’s 1:1 mixture supplemented with 5% Horse Serum (Corning), EGF 20 ng/ml (Sigma), insulin 10 μg/ml (Sigma), hydrocortisone 0.5 mg/ml (Sigma), cholera toxin 100 ng/ml (Sigma), 100 units/ml penicillin and 100 μg/ml streptomycin. Cells with knock-in PIK3CA mutations were grown in MCF10A media lacking EGF. Cells transfected with lentiviral constructs were maintained in 5 μg/ml puromycin as a selection agent. MCF10A, SUM159, V777L DKI, p53R248W, HEK293, MCF10A-tet-sh528 and -sh529 (with doxycycline-inducible knock-down of HOXA5) (9), and MCF10A-Kras cells were maintained at 37°C with 5% CO2 in a humidified atmosphere. Cells were treated with the retinoid, ATAL (Sigma #R2500) (1 μM for 7 days). BAY 11–7085 was purchased from Selleckchem (#S7352). Cycloheximide was purchased from Cell Signaling Technology (#2112S). The retinoid, AM580, was purchased from Sigma (#A8843). All the cell lines (including derivatives of MCF10A) were authenticated by STR sequencing and confirmed to be Mycoplasma free with the Mycosensor qPCR Assay Kit (Agilent).
Transfection
For generating cell lines with stable overexpression of HOXA5 or HOXA5 shRNAs, the following plasmids were used: pLKO.1 vector (containing Scr or -sh528, or -sh529 for HOXA5), packaging and envelop plasmids: pVSV-G and pCMV-dR8.91 or pLHCX vector (containing HOXA5) that was not susceptible to degradation by -sh528 and -sh529 (7 μg) and packaging plasmid pCL-Ampho (7 μg). For lentiviral transfection, 2 × 106 HEK293T cells were plated in 10 cm plates, and transfection using calcium phosphate precipitate method with 20 μg transfer vector, 15 μg packaging plasmid and 6 μg envelope plasmid was performed the next day. For retroviral transfection, Lipofectamine 2000 (Life technologies, Carlsbad, CA, USA) was used per manufacturer’s directions. The supernatant was collected and filtered after 48 hours (lentiviral transfection) or 72 hours (retroviral transfection). For infection of target cells, 1 ml of supernatant was added to each well of cells in a 6-well plate. Puromycin (5μg/ml, Sigma- # P7255) or Hygromycin (100 μM, Roche- #10843555001) was used to select for positive clones. For the generation of CRISPR/Cas9 mediated knockout (KO) of HOXA5, the LentiCRISPRv2 plasmid (a gift from Feng Zhang, Addgene, 52961) was used. The LentiCRISPRv2 was digested and purified, after which it was incubated with phosphorylated, annealed oligos targeting HOXA5 in a ligation reaction, and transformed into Stbl3 bacteria (ThermoFisher Scientific, C7373-03). The ligated plasmid was subsequently used to transfect HEK293T and infect target cells as described above.
RNA isolation, real time PCR analysis
RNA was isolated and purified using the QIAGEN RNeasy mini kit (Qiagen, Valenica, CA, U.S.A.) and converted to cDNA using iScript cDNA synthesis kit (Biorad). PCR primer sequences are presented in Table S1. Real time PCR was performed using Sybr green (Biorad), in the Step-one Real Time PCR System (Thermo Fisher).
Luciferase assay
For luciferase assays, 5 × 104 SUM159 or 2 × 105 MCF10A-tet-sh528 were seeded per well in 24-well plates. MCF10A-tet-sh528 was treated with doxycycline (100 nM) the following day to initiate sh-mediated shut down of HOXA5 expression. Luciferase readings were taken at the indicated time points, using the Dual-Glo luciferase assay kit (Promega, Madison, WI, USA) per manufacturers’ directions. A microplate reader (BMG Labtech) was used to measure chemiluminescence.
Western blot analysis
For western blot analysis, cells were scraped after a 10-minute incubation (4° C) in RIPA buffer (Sigma) supplemented with protease inhibitor and PMSF. After a freeze-thaw cycle, lysates were and passed through a 215/8-gauge needle and protein was quantified using the bicinchonic Acid (BCA) assay. A 10% SDS-PAGE gel was used to resolve proteins in 20–60 μg of whole cell lysate. The proteins were transferred onto a nitrocellulose or polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA) and probed with primary antibody overnight at 4°C. The antibodies used were: HOXA5 (Sigma H9790, Santa Cruz sc-365784), E-cadherin (Cell Signaling, 3195) Vimentin (Cell Signaling, 5741), N-cadherin (Cell Signaling, 13116) Slug (Santa Cruz, sc-166476), Progesterone receptor (Santa Cruz, sc-166169), COX-2 (BD Biosciences,610203), p65 (Cell Signaling, 824), Lamin B (Santa Cruz, sc-6216) GAPDH, (Cell Signaling, 97166), IKK-α (Cell Signaling, 11930), IκB-α (Cell Signaling, 4814), p-IKK-α (Cell Signaling, 2697), P53 (Santa Cruz sc-126), Myc tag (Cell Signaling 2276).
Animals and tumorigenicity assays
All animals were maintained in accordance with the IACUC policy at Johns Hopkins Medical Institutions. For the intraductal injection of tumor cells, ex-breeder, female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (NSG mice, The Jackson Laboratory), approximately 8 months of age were used since the larger teats enabled easier intraductal (i.duc) injection of cells though the teat (31,32). Cells were trypsinized and 2 × 106 cells were suspended in a 100 ul mixture of serum-free medium and Matrigel (Corning); 80% Matrigel and 20% medium. Cells were injected i.duc into both right and left fourth mammary gland teats of each mouse under anesthesia. For mammary fat pad injection, 2 × 106 cells were suspended in a 200 μl mixture of 80% Matrigel and 20% PBS. Both primary tumors and liver and lungs were fixed in 10% formalin for 48 hours. Paraffin embedded blocks were sectioned at the Reference Histology core facility at Johns Hopkins Hospital and stained with hematoxylin/eosin for histopathological examination by the study veterinary pathologist (KLG).
Statistical analysis
For the dataset analysis of expression, the R2 platform was used (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi). After choosing the dataset, the KEGG PathwayFinder by Gene correlation tool was used to assess HOXA5 gene correlation with KEGG pathways. A p value of less than or equal to 0.05 was chosen and data was log2 transformed. One-way Anova test was used to determine significance. The R2 algorithm Hugoonce was used to choose a single probe-set to represent a gene.
All RT-qPCR data was analyzed using two tailed T test with unequal variance.
Data availability statement
All pertinent data is presented the main paper and in the Supplementary Data files of this paper. Raw data for this study were generated at SKCCC Microarray Core. Derived data supporting the findings of this study are available from the corresponding author upon request.
RESULTS
HOXA5 protein and transcript levels are altered in MCF10A cell line derivatives
Studies have shown that while HOXA5 loss confers a less differentiated and more aggressive phenotype to epithelial cells (9,33,34), but its loss alone in mammary epithelial cells is unable to initiate neoplastic transformation (9,18). We hypothesized that loss of HOXA5 might be a key step to tumorigenic transformation of mammary epithelial cells harboring certain key mutations. Twelve isogenic derivatives of MCF10A harboring mutations in a single oncogene or tumor suppressor gene that were generated by Cre recombinase-mediated excision/insertion were used. These were MCF10A cell lines with single point mutations in oncogenes -KRAS (G12V) (35) PIK3CA (E545, H1047R) (36), and HER2 (V777L, L755S) (19), and in tumor suppressor genes [BRCA1 (185delAG, R71G, C61G) (37), and p53 (R248W) (38), or p53 null cells (39). In addition, two double knock-in cell lines harboring PIK3CA-E545 + HER2-V777L (V777L DKI), or PIK3CA E545K + HER2-L755S (L755S DKI) (19) were used (Table S2). Sanger sequencing confirmed the genotype of each cell line (Fig. S1A). RT- qPCR showed that levels of HOXA5 mRNA were significantly low in seven of the 12 MCF10A derivatives, relative to MCF10A (Fig. S1B). The V777L DKI cell line retained wild-type level of expression of HOXA5 (Fig. S1B, C, D). We selected two cell lines for further functional analysis, V777L-DKI, with high HOXA5 (Fig. S1B, C, D), and P53-R248W with low HOXA5 mRNA and protein expression (Fig. S1 E, F).
HOXA5 loss promotes epithelial to mesenchymal transition
Knock down of HOXA5 in V777L DKI using -sh528 and HOXA5 KO using CRISPR resulted in reduced E-cadherin mRNA and other epithelial markers such as Occludin, DSG3, Claudin1 as well as gain in mesenchymal markers such as N-cadherin, Slug and Vimentin (Fig. 1A–D). No change was observed in the level of HOXA5 mRNA in the CRISPR-KO cells, suggesting that the transcript was resistant to nonsense-mediated decay. Western blot confirmed changes in E-cadherin, Vimentin, N-cadherin and Slug (Fig. 1C, D). Immunofluorescence (IF) showed loss of E-cadherin and gain of Vimentin expression upon loss of HOXA5 (Fig. S1G). Three-dimensional cultures in Matrigel showed significantly higher number of stellate colonies in the MCF10A-sh528 and a second cell line, MCF10A-HOXA5-sh529 cells, relative to the -Scr control (Fig. 1E, F). KD of HOXA5 also led to enhanced colony formation (Fig. 1G, H), invasiveness and wound healing (Fig. S1H, I). In contrast, ectopic expression of HOXA5 in the MCF10A-p53R248W cell line (Fig. 1I, J) resulted in decreased colony formation (Fig. 1 K, L) and wound healing (Fig. S1J). Transcripts of HOXA5-induced genes, Occludin, CDH1, CLDN1, CD24 and ALDH1A3 were also increased (Fig. 1M) in these cells. Inducing endogenous HOXA5 in MCF10A-p53R248W using two retinoid derivatives, All-trans-Retinol (ATAL) and AM580, increased HOXA5 mRNA (Fig. 1N), and protein levels (Fig. 1O) and significantly suppressed proliferation (Fig. S1K).
Figure 1: Ablation of HOXA5 results in epithelial to mesenchymal transition (EMT) and restoration of HOXA5 reverses EMT.

(A) Quantitative PCR of cDNA from V777L DKI-sh528 cells (HOXA5 shRNA) and (B) V777L DKI-A5-KO (HOXA5 CRISPR knock-out) cell lines showed reduced RNA levels of E-cadherin (CDH1), Occludin, DSG3, Zo-1, Claudin-1 (CLDN1) and increased levels of Vimentin and N-cadherin (CDH1), while western blotting of protein from both (C) shRNA and (D) CRISPR cells showed that ablation of HOXA5 resulted in reduced E-cadherin and increased Vimentin, N-cadherin and Slug (E). Phase contrast images of V777L DKI-Scr and -sh529 knock-down HOXA5 cells grown in Matrigel: Collagen I matrix (6 days), with quantitative analysis (F) of branched or stellate structures formed (n=4) with both -sh528 and -529 showed significantly increased number of stellate structures in the HOXA5 knock-down cells, * p value < 0.05. (G) Crystal violet staining of colonies formed by V777L DKI-SCR, -sh528 and -sh529, with representative images, showing significantly increased colony formation (H) in the knock-down cells. Staining intensity was measured spectrophotometrically at 590 nm, * p value <0.05. HOXA5 was overexpressed in the MCF10A p53R248W cell line possessing mutant p53. (I) qPCR and (J) western blotting were used to confirm over-expression, * p value <0.05 (K, L). Colony formation assay showed reduced colony formation in MCF10A p53R248W HOXA5 cells relative to vector control, p value < 0.05. Experiment performed in triplicate, repeated thrice. (M) qPCR shows increased levels of epithelial transcripts in cells overexpressing HOXA5, * p value <0.05 (N) Treatment with retinoids ATAL and AM580 (1 μM) for 1 week showed increased expression of HOXA5 and its target gene- CD24 but not E-cadherin (CDH1) using qPCR and, * p value <0.05 (O) western blot, which showed increased HOXA5 and Progesterone receptor (PR) expression.
HOXA5 loss enhances tumorigenicity
Based on enhanced invasiveness and migratory properties bestowed by HOXA5 KD in vitro, we investigated if HOXA5 loss in V777L DKI cells promoted tumorigenesis in this previously reported nontumorigenic cell line (19). We used two mouse models, mammary fat pad (MFP) and mammary intraductal (MIND) cell implantation in NSG mice (Fig. 2A). Four weeks following implantation of V777L DKI-Scr cells in three MFPs of each NSG mouse, mammary ductal structures with a single layer epithelium (Fig. 2B) were observed, resembling those previously observed with parental MCF10A cells (40). V777L DKI -sh528 cells, on the other hand, formed pre-neoplastic outgrowths resembling atypical hyperplasia in all nine injected mammary glands, but there was no evidence of frankly malignant growth. RT-qPCR of xenograft RNA showed that HOXA5 loss was retained, as was the reduced level of its transcriptional targets, CD24 and E-cadherin (Fig. 2C).
Figure 2: Loss of HOXA5 enhances in vivo tumorigenicity.
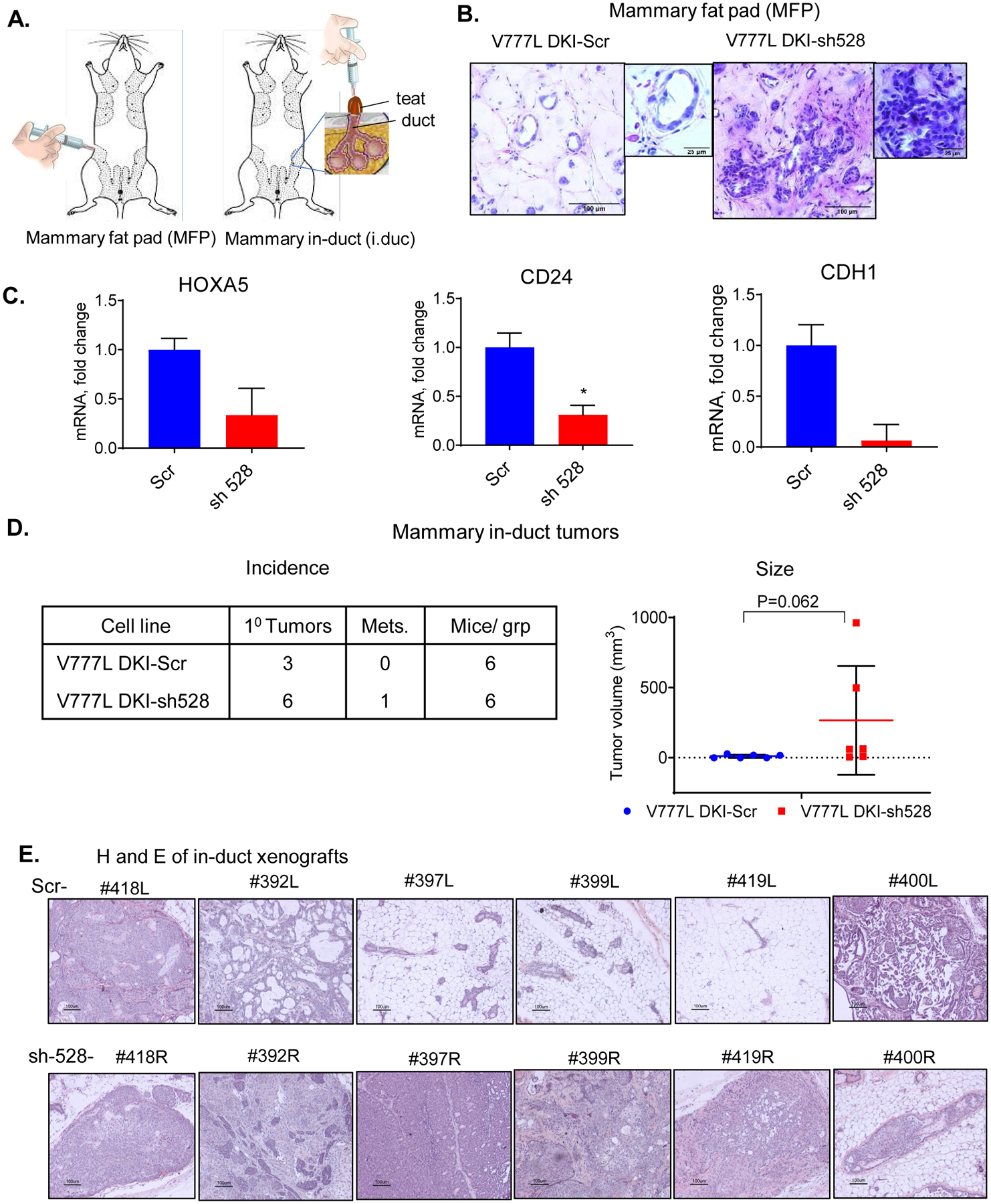
(A) Pictorial representation of mammary fat pad injection (left) and intraductal implantation (right). (B) Mammary fat pad injection in 6 teats (3 per cell line) of an NSG mouse with 2 × 106 cells suspended in a mixture of 80% Matrigel and 20% PBS was performed, teats implanted with V777L DKI-sh528 cells showed areas with dysplasia while those with -scr control cells had normal ducts, 6 weeks post-implantation. (C) RNA was extracted from the xenografts and qPCR using human specific primers was performed to confirm KD of HOXA5 and expression of target genes E-cadherin (CDH1) and CD24, * p value <0.05 where indicated. (D) Intraductal implantation of V777L DKI-Scr in the left fourth teat and V777L DKI-sh528 in the fourth right teat of the same NSG mouse showed enhanced tumor formation in the -sh528 cohort. Table shows number of tumors formed and mice per group. Th right panel shows that mean tumor volume of V777L DKI-sh528 tumors was higher than -scr tumors, p=0.062, n=6. Mice were sacrificed after 6 months or when moribund. (F) Hematoxylin and Eosin staining of tumor sections from all 6 mammary glands in both groups.
Breast cancer cells show more robust growth when injected into the mammary duct- the site of origin of mammary tumors (31). By intraductal injection, 6 mice received the V777L DKI-Scr cells through the teat of the left 4th mammary gland, and the V777L DKI-sh528 cells in the right 4th mammary gland. Mice were monitored for a period of 6 months. We found that 6/6 teats implanted with V777L DKI-sh528 cells developed visible tumors (Fig. 2D, left panel, Fig. S2A) and liver metastasis was observed in 1/6 mice (Fig. S2B). Smaller, slower growing, more differentiated tumors developed in 3/6 of the mice implanted with V777L DKI-Scr, with a trend to significance compared to the HOXA5 KD cells; p value= 0.062 (Fig. 2D, right panel, S2C). Histopathologically, they were undifferentiated carcinomas that were locally invasive (Fig. 2E) Staining the intraductal mouse xenografted tumors with a human-specific Ku80 antibody ruled out the possibility that the tumors in both V777L DKI-scr- and -sh528-injected mice were spontaneous mouse mammary tumors (Fig. S2C) (41). Based on these data we conclude that HOXA5 loss was responsible for conferring the properties of rapid tumor-formation, invasion and metastasis to V777L DKI human mammary epithelial cells.
HOXA5 perturbs NF-κB/TNF signaling
Next, to gain an understanding of the pathways and functions deployed by HOXA5 to mediate tumor suppression in this model, we interrogated an Agilent Human v2, 4x 44K, 2-color expression array using RNA extracted from V777L-Scr and -sh528 cell lines. Ingenuity pathway analysis showed that IL-22 signaling was one of the most dysregulated pathways and TNF was an important upstream regulator (Table S3A, B). The top up-regulated and down-regulated genes, DCN, PTX-3, EPCAM and TMEM in the array (Table S3C), were validated by RT-qPCR (Fig. S3 A–D). Presented in the heatmap, among the 82 top-most differentially altered transcripts (filtered by p value < 0.05 and fold change > 1.5) were NF-κB target genes PTX3 (42) and BIRC3 (43) (Fig. 3A). Both these targets of canonical NF-κB signaling showed higher expression upon KD of HOXA5. RT-qPCR was used to confirm the suppressive effect of HOXA5 expression on these transcripts in V777L DKI and MCF-10A with HOXA5 KD, and SUM159 cells with overexpression of HOXA5 (Fig. 3B, C). Gene set enrichment analysis (GSEA) analysis showed that HOXA5 KD cells were enriched for RelA (p65) and inflammatory gene signatures (Fig. 3D, E). Since TNF was implicated as an upstream regulator, four publicly available breast cancer datasets were analyzed to determine the correlation between mRNA expression of HOXA5 and members of the NF-κB pathway. In all four clinical breast cancer RNA sequencing datasets examined, we found a significant inverse correlation between expression of HOXA5 and KEGG NF-κB pathway genes (Table 1).
Figure 3: HOXA5 loss perturbs inflammatory signaling.
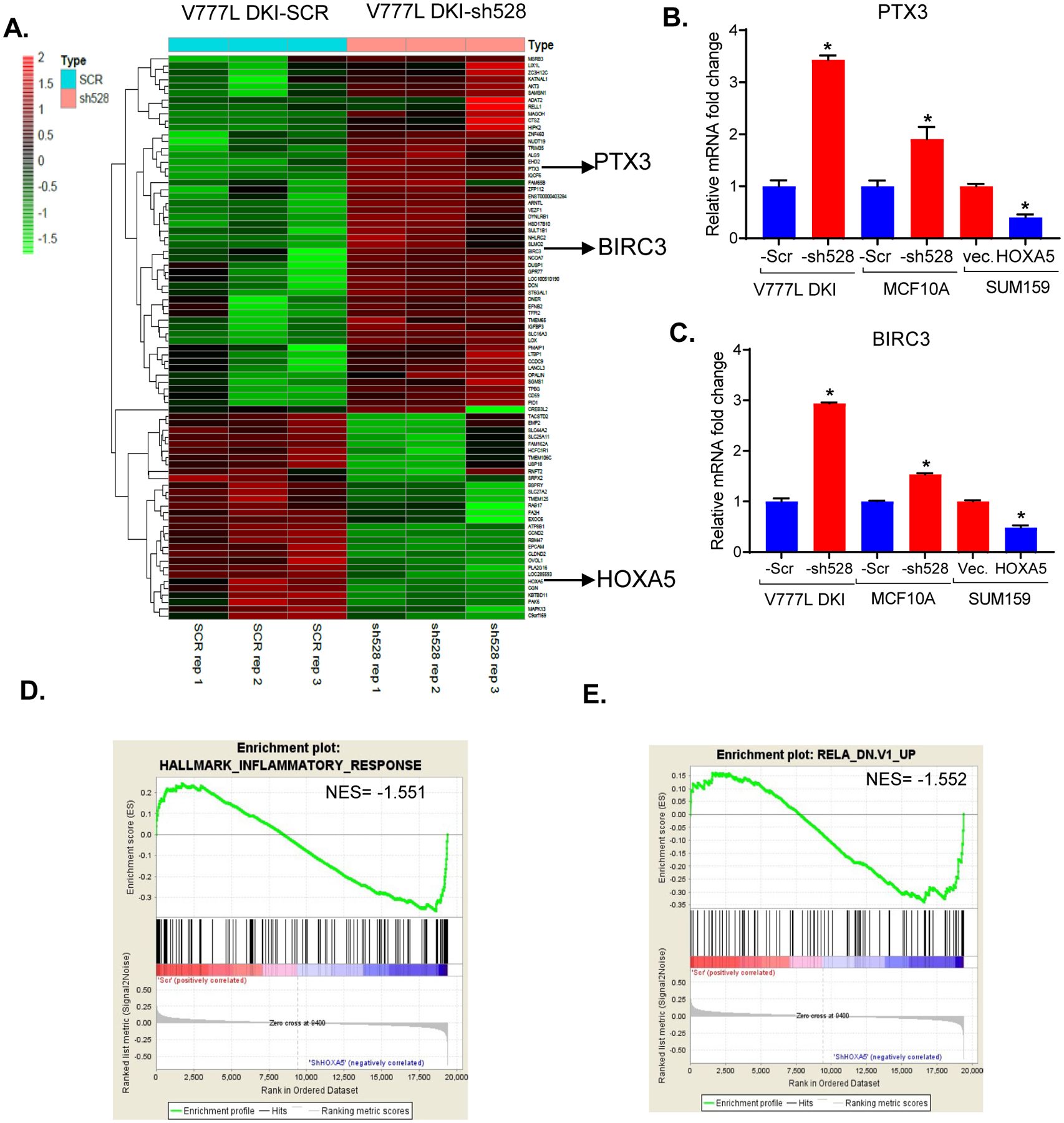
Agilent Human v2, 4x 44K two-color expression array was performed with V777L DKI-Scr and -sh528 cells (n=3 biological replicates). (A) Semi-supervised clustering heat map shows top significantly altered transcripts (p value < 0.05, fold change > 1.5) in V777L DKI- Scr and -sh528 (n=3 biological replicates). PTX3 and BIRC3, two genes, are highlighted. (B) qPCR validation of PTX3 and (C) BIRC3, NF-κB target transcripts identified as being significantly altered in cell lines with HOXA5 knock-down- V777L DKI, MCF10A and over-expression- SUM159, * p value <0.05. Gene set enrichment analysis (GSEA) for MC10A Scr and sh-528 shows that (D) inflammatory response genes and (E) RelA (p65) related genes are enriched in the -sh528 cohort.
Table 1.
Analysis of publicly available gene expression datasets shows negative correlation of HOXA5 expression with the NF-kB pathway
| Dataset* | Size** | Genes correlating# | Genes total## | KEGG NF-kB pathway representation† | P value†† |
|---|---|---|---|---|---|
| Yu | 683 | 50 | 88 | Under | 0.03 |
| Sotiriou | 198 | 15 | 78 | Under | 7.3e-03 |
| Miller | 251 | 29 | 77 | Under | 0.03 |
| Black | 1097 | 22 | 86 | Under | 0.05 |
Four publicly available datasets used to determine correlation of HOXA5 transcript with the NF-ΚB pathway (the NF-KB pathway as defined by the KEGG Pathway database https://www.genome.jp/kegg/pathway.html). Genome-wide expression profiling datasets of breast cancer patients available in the public domain were analyzed using the R2 platform (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi).
the number of tissue samples in the dataset
the number of genes correlating with HOXA5
the total number of genes in the KEGG NF-KB pathway expressed in the dataset
shows whether the correlation demonstrates an under- or over-representation of the NF-KB pathway genes in the samples expressing HOXA5 transcript. Under-representation is indicated by a negative correlation of the NF-KB pathway with HOXA5 mRNA in this cohort of samples.
p-value represents the significance of the under-representation of NF-KB genes in relation to HOXA5 mRNA in the dataset (significance cut-off p< 0.05).
HOXA5 loss increases expression of NF-kB inflammatory target genes.
Since our expression array results suggested an interaction between HOXA5 and NF-kB signaling, we analyzed mRNA expression of NF-kB target genes by RT-qPCR. There was a significant increase in expression of IL8, IL6 and COX-2 in MCF10A and V777L DKI upon HOXA5 KD (Fig. 4A–C) and in the breast cancer cells, MCF10A Kras, SUM159 and LM2, a lung metastasis derivative of MDA-MB-231 (Fig. S4A). For further validation, we used doxycycline (dox) inducible MCF10A-tet-sh528 cells (Fig. 4D) (9). A cytokine array comprising cytokines and chemokines regulated by NF-kB was performed with cell supernatant derived from MCF10A (control) and MCF10A-tet-sh528 cells treated with doxycycline (100 nM) for 48 hours (Fig. 4E). A detailed map of cytokines in the array and quantification of the pixel densities for altered cytokines is shown in Fig. S4B, C. Dox-treated MCF10A-tet-sh528 showed increased levels of TGF β1, TNF β, CCL23, CXCL13, CXCL6, CCL13, GDNF, IGFBP-1, CCL20 and PARC (Fig. 4E). Increased expression of three of the cytokines, LIF, CXCL6 and CXCL13, in HOXA5 KD V777L-DKI cells was validated by RT-qPCR (Fig. 4F).
Figure 4: HOXA5 perturbs NF-κB target genes by suppressing canonical NF-κB signaling.
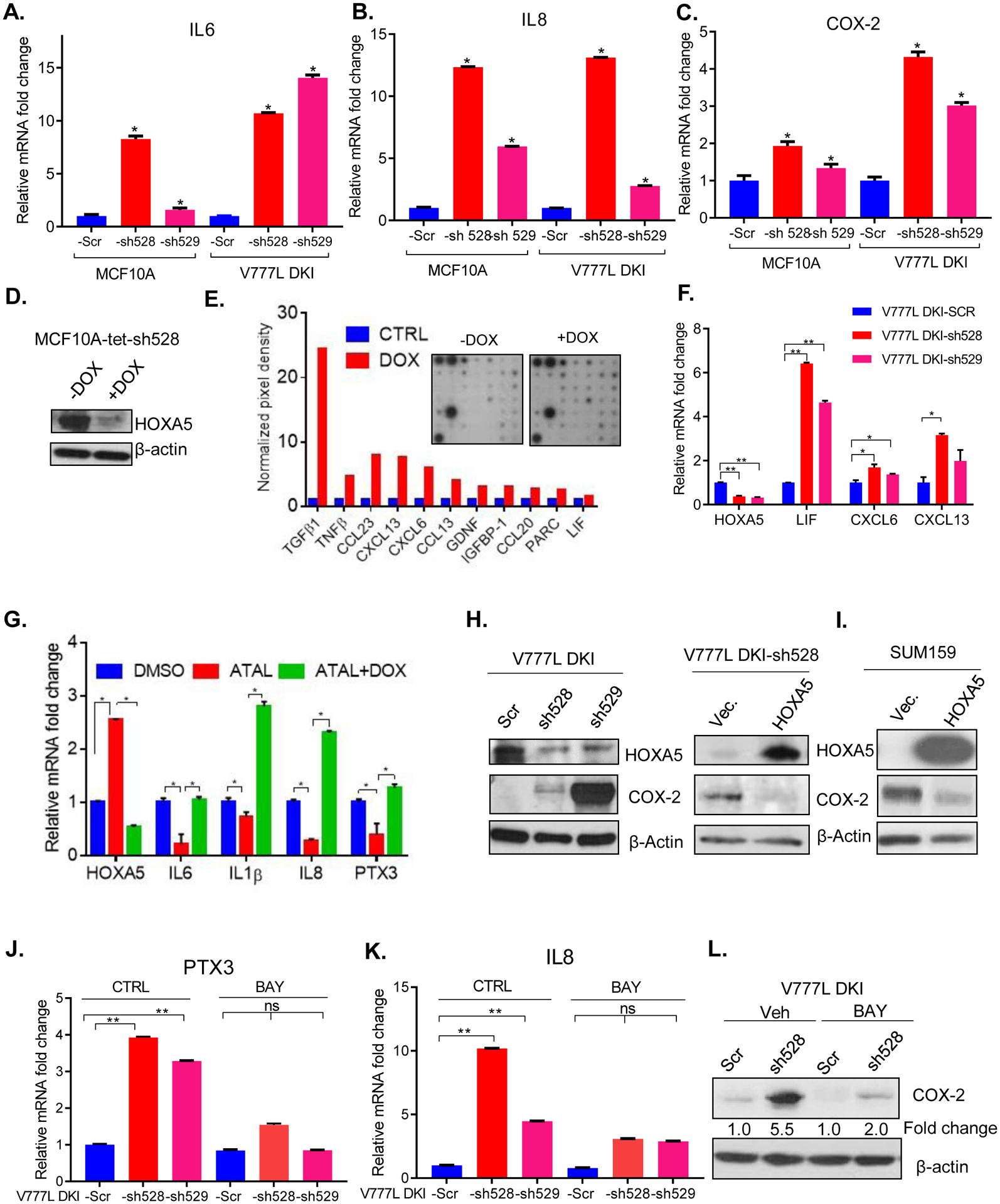
(A-C) qPCR shows that NF-κB target genes IL6, IL8 and COX-2 levels are increased upon ablation of HOXA5 in V777L DKI, MCF10A. (D) Western blotting confirms knock-down of HOXA5 in MCF10A-tet-sh528 cells (expressing a Dox-inducible sh528 hairpin) upon treatment with doxycycline (100 nM) for 48 hours. (E) Cytokine array of MCF10A-tet-sh528 cells (inset) showed that several cytokines were up-regulated. Quantification of dot intensity performed using ImageJ is shown. (F) qPCR validation in V777L DKI-Scr, -sh528 and -sh529 cells of 3 cytokines up-regulated in MCF10A-tet-sh528, per the cytokine array- LIF, CXCL6, CXCL13. (G) RNA from flow-sorted CD24−, MCF10A-tet-sh528 cells treated with DMSO, 1 μM retinal (ATAL) or ATAL+ doxycycline (DOX), cultured for 1 week was used for RT-qPCR analysis of IL6, IL1β, IL8, PTX3. (H) Western blot analysis of a canonical NF-ΚB target gene, COX-2, upon knock-down (V777L DKI-sh528, -sh529, left panel) and re-expression of HOXA5 (right panel). (I) COX 2 expression in SUM159 overexpressing HOXA5 cells. RT-qPCR of mRNA, from V777L DKI Scr and -sh528, -sh529 treated with BAY 11–7085 (20 μM, 17 h), for levels of NF-κB target genes (J) PTX-3, and (K) IL8. (L) Western blot of the V777L DKI-sh528 cells shows reduced induction of COX2 from 5.5-fold to 2-fold upon treatment with BAY 11–7085 (20 μM, 17 h). * p value <0.05, ** p value <0.01, three replicates per experiment.
Retinoids are known transcriptional up-regulators of HOXA5 expression (13). ATAL treatment of MCF10A-tet-sh528 cells for a week increased HOXA5 expression; Dox treatment of these cells activated the tet-shRNA and suppressed HOXA5 levels (9). The transcript levels of NF-kB target genes, IL6, IL8, IL1B and PTX-3 were reduced in flow-sorted CD24-negative, HOXA5-KD cells upon ATAL treatment, and increased upon tet-induction of the shRNA (Fig. 4G). In addition, the expression of COX-2 protein, a prototypical NF-κB target gene, was increased in V777L DKI-sh528 and -sh529 (Fig. 4H). Conversely, restoration of HOXA5 with an expression vector that lacks susceptibility to -sh528 caused significantly decreased COX-2 protein (Fig. 4H) and COX-2, IL6 and IL1B mRNA (Fig. S4D). HOXA5-overexpressing SUM159 cells also expressed lower COX-2 compared to vector controls (Fig. 4I). Similar results were obtained with MCF10A-KRAS cells, where HOXA5 upregulation by ATRA caused IL6 and PTX3 mRNA repression, which was reversed with HOXA5 KD (Fig. S4E). Thus, both by pharmacological upregulation using retinoids, and by forced expression, HOXA5 suppressed NF-κB targets.
To determine if HOXA5 affects upstream molecules in the NF-κB pathway, we treated cells with TNF-α. We observed that increases in IL8, IL1B and PTX-3 expression were bolstered 2- to 4-fold in HOXA5 KD cells in the presence of TNF-α (Fig. S4F, G). Further, pharmacological inhibition of IκB degradation by BAY 11-7085 in the V777L DKI cell line reduced the effect of HOXA5 KD on PTX-3 and IL8 by more than 2-fold (Fig. 4 J, K). BAY treatment also led to the blunting of the increase of COX-2 upon HOXA5 KD from 5.5-fold to 2-fold (Fig. 4L). Based upon this data, we concluded that HOXA5 likely mediates some of its tumor suppressor properties via the canonical NF-κB pathway.
Mechanism of HOXA5 suppression of NF-ΚB transcriptional activity
To investigate whether NF-κB pathway activity is modulated by HOXA5, we performed a NF-κB reporter-luciferase assay using Dox-inducible MCF10A-tet-sh528 cells transfected with an NF-κB consensus binding sequence-driven luciferase plasmid. There was a significant increase in NF-κB reporter activity 48 and 72 hours following -sh528 induction (Fig. 5A), which was enhanced further by TNFα (Fig. S5A). These results led us to conclude that HOXA5 suppressed NF-ΚB transcriptional activity.
Figure 5: HOXA5 suppresses canonical NF-κB signaling.

(A) MCF10A tet-sh528 was stably transfected with an NF-κB reporter luciferase plasmid pHAGE NF-κB-TA-LUC-UBC-GFP-W and was treated with doxycycline (100 nM) for the indicated durations, followed by luciferase analysis. Experiments were performed in triplicate, repeated thrice. * p value <0.05, ** p value <0.01 (B) Cytoplasmic and nuclear fractionation of SUM159-vector and -HOXA5 cells was performed, followed by western blotting for the indicated proteins. (C) A significant increase in nuclear IKB-α was observed upon HOXA5 expression. * p value <0.05, ** p value <0.01, three replicates per experiment. Western blotting showed reduced IKB-α levels upon HOXA5 knockout (D) V777L DKI-Sham and -A5 KO, or knockdown (E) MCF10A Scr, -sh528, -sh529 in whole cell extracts, (F) in flow-sorted CD24− population of MCF10A-tet-sh528 treated with DMSO, retinal (ATAL) or (G) ATAL+ doxycycline (DOX), cultured for 1 week and in (H) SUM159 vec. and SUM159 cells overexpressing HOXA5. (I) RT-qPCR of HOXA5 -KO, -KD, and overexpression shows lack of correlation between HOXA5 and NFΚBIA (IKB-α) mRNA.
To determine the mechanism of suppression of the NF-κB pathway by HOXA5, we tested cytoplasmic and nuclear extracts of SUM159-vector and SUM159-HOXA5 cells. Protein levels of NF-κB pathway components, p65, IKK-α, and pIKK-α (S176/180) were not significantly altered upon HOXA5 over-expression. However, IκB-α levels increased in the presence of HOXA5; a more pronounced increase was observed in the nuclear extract (Fig. 5B, C, Fig. S5B). This positive correlation between HOXA5 and IκB-α protein was observed in multiple cell lines: V777L DKI, MCF10A (Fig. 5D, E), and flow-sorted MCF10A-tet-sh528, CD24-negative cells, expressing high levels of endogenous HOXA5 by exposure to ATAL, or with Dox-induced HOXA5 KD (Fig. 5F, G). Conversely, in SUM159, HOXA5 over-expression resulted in increased levels of IκB-α (Fig. 5H). Thus, HOXA5-depleted cells had reduced IκB-α protein, while HOXA5 overexpression increased IκB-α. Interestingly, in these cells mRNA levels IKB-α (NFKBIA) did not correlate with the observed changes in protein levels (Fig. 5I). Similarly, no direct correlation between HOXA5 and NFKBIA was seen in an mRNA expression array of 50 breast cell lines (44) (Fig. S5C). These findings demonstrate that, while ruling out transcriptional control of NFKBIA by HOXA5, HOXA5 likely suppressed the NF-κB pathway through an interaction with IκB-α, thus suggesting non-transcriptional mechanisms of regulation.
HOXA5 stabilizes and increases nuclear IKB-α protein through direct protein-protein interaction
The half-life of IκB-α has been reported to be approximately 2 hours (45). To test IκB-α stability in breast cancer cells, we transiently transfected SUM159 cells with full length HOXA5. Protein stability was tested by treating cells with 10 μM cycloheximide for 0–4 hours. Following treatment, IκB-α protein was stable for up to 2 h in vector-control cells, compared to 4 h in the HOXA5-expressing SUM159 cells (Fig. 6A, Fig. S6A). These results were also confirmed in the MCF10A-tet-sh528 cells. Here, cycloheximide treated HOXA5 KD cells showed less stable IκB-α (Fig. 6B, Fig. S6B). Treatment of the SUM159 cells with MG132 (10 uM), an inhibitor of proteasomal degradation (Fig. 6C), showed that at 0 hours of MG132 treatment, SUM159-HOXA5 expressed higher levels of IκB-α than SUM159-vector cells. However, at the 4-hour timepoint, it appears that there was no significant decrease relative to the 0-hour time point in either cell line.
Figure 6: HOXA5 stabilizes IKB-α protein in the nucleus via a protein-protein interaction.

(A) SUM159 cells were transiently transfected with HOXA5 for 48 hours and subsequently treated with Cycloheximide (2 μg/μl) or DMSO for 0, 1, 2 or 4 hours and lysed. Western blotting was used to examine the stability of IKB- α. (B) Western blotting was performed to examine the stability of IκB- α in MCF10A-Scr and -sh528 cells treated with cycloheximide (1 μg/μl) or DMSO for 0, 1, 2, 3, 4, or 5 hours and lysed. (C) SUM159 cells were transiently transfected with HOXA5 for 48 hours and subsequently treated with MG132 (10 μM) or DMSO for 0, 1, 2 or 4 hours and lysed. Western blotting was used to examine the stability of IκB- α. (D) Western blot of the nuclear extracts of SUM159 cells transiently transfected with 0.5, 1 or 2 μg HOXA5 plasmid, showed greater enhancement in nuclear IKB-α levels. (E) Nuclear extract of HEK293T cells transfected with 2 μg HOXA5 plasmid showed similar results. (F) Immunofluorescence for HOXA5 (green) and IκB-α (red) was performed in SUM159 stably transfected with empty vector and HOXA5. Co-localization was observed in the nucleus. (G) Publicly available datasets for ChIP-seq: HOXA5 (GEO accession no. GSM1239461) and IκB-α (GEO accession no. GSM744581) showed peak overlap for 528 putative targets of combined HOXA5 and IκB- α action. (H). IP using a pull down with (a) IκB-a antibody or (b) a HOXA5-directed Myc-tag antibody in whole cell lysates of HEK293T-HOXA5 showed presence of both HOXA5 in the IκB-a PD, and IκB-α protein the HOXA5/myc PD. (I) A pictorial representation of the findings of this study
Next, we investigated whether HOXA5 stabilizes IκB-α through a physical interaction. In SUM159 and HEK293T cell lines, nuclear IκB-α protein levels increased upon HOXA5 expression (Fig. 6 D, E). We transfected SUM159 with 0, 0.5, 1 or 2 μg HOXA5. There was a marked increase of nuclear IκB-α following HOXA5 expression, suggesting that even a relatively small increase in HOXA5 was sufficient for its effect on IκB-α. In both cell lines, there was no change in cytosolic IκB-α levels (Fig. S6 C, D). Nuclear colocalization of HOXA5 and IκB-α was confirmed by immunofluorescence in SUM159 and HEK293T cells transfected with HOXA5 (Fig. 6 F). Based upon this observation, publicly available datasets of ChIP-seq for HOXA5 (GEO accession no. GSM1239461) and ChIP-seq for IΚB-α (GEO accession no. GSM744581) were compared for peak overlaps. A significant overlap of binding sites was found in 528 genes suggesting that HOXA5 and IκB-α enrichment could potentially occur at these genomic sites (Fig. 6 G). To test if there was protein/protein interaction between HOXA5 and IκB-α, we performed co-immunoprecipitation of Myc-tagged-HOXA5 and IκB-α in HEK293T. Interaction was observed between IκB-α and HOXA5 (Fig. 6 H).
Through a series of experiments both in cell culture and in vivo, we have provided evidence that HOXA5 stabilizes nuclear IκB-α. This might result in fine tuning the suppressive function of IκB-α on the transcription of oncogenic NF-κB target genes. Our findings are summarized pictorially in Fig. 6 I.
Given the tumor suppressive function of both HOXA5 and IKB-α, mRNA expression of both genes could be predictive of a higher probability of recurrence-free-survival (RFS) and overall survival (OS) in breast cancer. We analyzed gene expression data downloaded from publicly available GEO, EGA and TCGA databases. Using the K-M plotter program (https://kmplot.com/analysis/), we observed that the combination of HOXA5 and NFKBIA was predictive of significantly improved RFS and OS in breast cancer (Fig. S7A, B), and better RFS in the basal subtype of breast cancer (Fig. S7C).
DISCUSSION
The goal of this study was to understand the significance of HOXA5 loss in the context of other commonly known activating and suppressive mutations that occur in breast cancer. Here, we have studied HOXA5 loss in a diploid non- tumorigenic mammary epithelial cell line, MCF10A, in the context of the presence of a common PI3K mutation in breast cancer, and a less well studied HER2 mutation. We report that HOXA5 loss potentiated the tumorigenic properties of MCF10A cells harboring mutations in both HER2 and PIK3CA. The tumor inhibitory activity of HOXA5 is mediated, in this system, through its ability to stabilize IKB-α in the nucleus, thus prolonging its ability to suppress expression of the inflammatory pro-tumorigenic NFκB-target genes. In addition, an intriguing interaction possibly occurring through direct transcriptional regulation of this key oncogenic target of NF-kB by HOXA5.
Our group has previously found that HOXA5 thwarts breast tumor development and promotes differentiation via numerous mechanisms, such as transcriptional up-regulation of p53 (14), the progesterone receptor (15), and E-cadherin and CD24 (9), as well as through apoptosis by p53-dependent and p53-independent mechanisms (16). HOXA5 is upregulated by retinoic acid contributing to its pro-apoptotic effect (13). In this study, we corroborated our previous findings (9) that HOXA5 loss contributes to EMT, and conversely, when over-expressed, it impedes mesenchymal properties of cells. Additionally, we have shown the loss of HOXA5 in the V777L DKI cells accelerates aggressive tumor formation in mice. Our expression array analysis of V777L DKI-HOXA5 KD showed that the loss of HOXA5 enhanced the expression of inflammatory transcripts which are targets of the NF-κB pathway. The interaction between HOXA5 and the NF-κB pathway has been hinted at in prior studies. A study by Lee et al reported that HOXA5 caused p65 sequestration in the nucleolus without affecting p65 transcriptional function (28). In this study, we did not find any increase of p65 nuclear localization upon HOXA5 loss/gain. In yet another study, accumulation of methylglyoxal (a byproduct of glycolysis in diabetes mellitus) in mouse microvascular coronary endothelial cells resulted in higher levels of HOXA5 expression through direct transcriptional upregulation of HOXA5 by NF-ĸB-p65. The authors concluded that upregulation of HOXA5, which acts as an antiangiogenic factor, might be responsible for vascular impairment observed in diabetes mellitus (46). Our work does not support a similar function for NF-ĸB-p65 in HOXA5 regulation in breast cancer cells.
We observed a direct interaction between HOXA5 and IKB-α, suggesting a stabilizing function for HOXA5 that increases the half-life of IκB-α. Interestingly, we found that HOXA5 did not alter the RNA levels of IκB-α (NFKBIA), suggesting that HOXA5 does not act as a transcription factor in this context. Such an interaction has been reported with MEOX2, a homeodomain protein that interacts with IκB-β and p65 (47).
IκB-α appears to function as tumor suppressor protein in cancer. Nfκbia knock-out mice show enhanced neonatal mortality and constitutive NF-κB pathway activation (48). While the chief mechanism of NFΚBIA tumor suppressive action is through its inhibitory action on the NF-κB pathway, its interaction with other proteins and signaling pathways also appears to play a role in cancer. The product of NFΚBIA, IκB-α, and p53 have been reported to stabilize each other in the cytosol via formation of a ternary complex with BCR-ABL in leukemia (49). This leads to nuclear exclusion and inactivation of p53, lending credence to the notion that IκB-α can act as both a tumor suppressor and oncogene (50). However, we observed a marked increase in IκB-α in nuclear extracts, suggesting that IκB-α is not excluded from the nucleus. This suggests that IκB-α acts as a tumor suppressor protein in breast cancer cells. The presence of high molecular weight phosphorylated and sumoylated IκB-α in the nucleus has been reported in squamous cell carcinoma (51).
Considering the tumor suppressive functions of both HOXA5 and IκB-α, it is unsurprising that expression of both genes together is predictive of overall- and regression-free survival in breast cancer. The NF-κB pathway is more active in the basal and triple negative subtypes (52), where it has been shown to confer a worse prognosis (53) and is responsible for drug resistance (54) HOXA5 and NFΚBIA genes were predictive of a better prognosis in this subtype.
Our findings tie in with HOXA5’s role as a tumor suppressor in breast cancer. Interestingly, IκB-α has also been shown to regulate genes associated with differentiation, including HOX genes (51) independent of its role as an NF-κB suppressor. Whether IκB-α regulates the expression of HOXA5 in breast cancer is unknown. HOXA5 loss and NF-κB pathway activation likely work in concert to increase the expression of EMT-related genes in breast cancer, since the NF-κB pathway has been shown to up-regulate EMT genes such as Slug and Snail (55). We found changes in mRNA levels of many NF-κB target genes. Future work will reveal if some of the key NF-κB targets, such as IL6, IL8 and COX2 are also direct transcriptional targets of HOXA5. Thus, in addition to mediating its action through protein- protein interaction, HOXA5 may exert its prototypical function as a transcriptional regulator of gene expression.
In conclusion, we report for the first time that HOXA5 can suppress the oncogenic NF-κB pathway by stabilizing IκB-α, an important inhibitor of the pathway. We also show that HOXA5 and IκB-α colocalize and interact directly in the nucleus.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Loss of HOXA5 reduces IκB-α stability and increases NF-κB signaling to exacerbate breast cancer aggressiveness, providing new insights into the tumor suppressor functions of HOXA5.
ACKNOWLEDGEMENTS
We would like to thank Dr. Ben Ho Park for generously gifting us the various MCF10A cell line derivatives.
This work was supported by an Avon Foundation for Women, Grant # 02-2016-013 to SS
Abbreviations:
- BC
breast cancer
- EMT
epithelial to mesenchymal transition
- TNF
tumor necrosis factor
- DKI
double knock-in
- KD
knock-down
- ChIP
chromatin immunoprecipitation
Footnotes
Conflict of Interest: None
REFERENCES
- 1.Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer 2010;10:361–71 [DOI] [PubMed] [Google Scholar]
- 2.Bhatlekar S, Fields JZ, Boman BM. HOX genes and their role in the development of human cancers. J Mol Med (Berl) 2014;92:811–23 [DOI] [PubMed] [Google Scholar]
- 3.de Bessa Garcia SA, Araujo M, Pereira T, Mouta J, Freitas R. HOX genes function in Breast Cancer development. Biochim Biophys Acta Rev Cancer 2020;1873:188358. [DOI] [PubMed] [Google Scholar]
- 4.Lewis MT. Homeobox genes in mammary gland development and neoplasia. Breast Cancer Res 2000;2:158–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeannotte L, Gotti F, Landry-Truchon K. Hoxa5: A Key Player in Development and Disease. J Dev Biol 2016;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landry-Truchon K, Houde N, Boucherat O, Joncas FH, Dasen JS, Philippidou P, et al. HOXA5 plays tissue-specific roles in the developing respiratory system. Development 2017;144:3547–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pineault KM, Wellik DM. Hox genes and limb musculoskeletal development. Curr Osteoporos Rep 2014;12:420–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henderson GS, van Diest PJ, Burger H, Russo J, Raman V. Expression pattern of a homeotic gene, HOXA5, in normal breast and in breast tumors. Cell Oncol 2006;28:305–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teo WW, Merino VF, Cho S, Korangath P, Liang X, Wu RC, et al. HOXA5 determines cell fate transition and impedes tumor initiation and progression in breast cancer through regulation of E-cadherin and CD24. Oncogene 2016;35:5539–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bagadi SA, Prasad CP, Kaur J, Srivastava A, Prashad R, Gupta SD, et al. Clinical significance of promoter hypermethylation of RASSF1A, RARbeta2, BRCA1 and HOXA5 in breast cancers of Indian patients. Life Sci 2008;82:1288–92 [DOI] [PubMed] [Google Scholar]
- 11.Conway K, Edmiston SN, May R, Kuan PF, Chu H, Bryant C, et al. DNA methylation profiling in the Carolina Breast Cancer Study defines cancer subclasses differing in clinicopathologic characteristics and survival. Breast Cancer Res 2014;16:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abba MC, Gong T, Lu Y, Lee J, Zhong Y, Lacunza E, et al. A Molecular Portrait of High-Grade Ductal Carcinoma In Situ. Cancer Res 2015;75:3980–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H, Zhang H, Lee J, Liang X, Wu X, Zhu T, et al. HOXA5 acts directly downstream of retinoic acid receptor beta and contributes to retinoic acid-induced apoptosis and growth inhibition. Cancer Res 2007;67:8007–13 [DOI] [PubMed] [Google Scholar]
- 14.Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, et al. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature 2000;405:974–8 [DOI] [PubMed] [Google Scholar]
- 15.Raman V, Tamori A, Vali M, Zeller K, Korz D, Sukumar S. HOXA5 regulates expression of the progesterone receptor. J Biol Chem 2000;275:26551–5 [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Chung S, Sukumar S. HOXA5-induced apoptosis in breast cancer cells is mediated by caspases 2 and 8. Mol Cell Biol 2004;24:924–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aubin J, Lemieux M, Tremblay M, Behringer RR, Jeannotte L. Transcriptional interferences at the Hoxa4/Hoxa5 locus: importance of correct Hoxa5 expression for the proper specification of the axial skeleton. Dev Dyn 1998;212:141–56 [DOI] [PubMed] [Google Scholar]
- 18.Gendronneau G, Lemieux M, Morneau M, Paradis J, Tetu B, Frenette N, et al. Influence of Hoxa5 on p53 tumorigenic outcome in mice. Am J Pathol 2010;176:995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zabransky DJ, Yankaskas CL, Cochran RL, Wong HY, Croessmann S, Chu D, et al. HER2 missense mutations have distinct effects on oncogenic signaling and migration. Proc Natl Acad Sci U S A 2015;112:E6205–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia Y, Shen S, Verma IM. NF-kappaB, an active player in human cancers. Cancer Immunol Res 2014;2:823–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell 2008;132:344–62 [DOI] [PubMed] [Google Scholar]
- 22.Brantley DM, Chen CL, Muraoka RS, Bushdid PB, Bradberry JL, Kittrell F, et al. Nuclear factor-kappaB (NF-kappaB) regulates proliferation and branching in mouse mammary epithelium. Mol Biol Cell 2001;12:1445–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shostak K, Chariot A. NF-kappaB, stem cells and breast cancer: the links get stronger. Breast Cancer Res 2011;13:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Connelly L, Barham W, Onishko HM, Sherrill T, Chodosh LA, Blackwell TS, et al. Inhibition of NF-kappa B activity in mammary epithelium increases tumor latency and decreases tumor burden. Oncogene 2011;30:1402–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gordon AH, O’Keefe RJ, Schwarz EM, Rosier RN, Puzas JE. Nuclear factor-kappaB-dependent mechanisms in breast cancer cells regulate tumor burden and osteolysis in bone. Cancer Res 2005;65:3209–17 [DOI] [PubMed] [Google Scholar]
- 26.Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, et al. Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest 1997;100:2952–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pai P, Sukumar S. HOX genes and the NF-kappaB pathway: A convergence of developmental biology, inflammation and cancer biology. Biochim Biophys Acta Rev Cancer 2020;1874:188450. [DOI] [PubMed] [Google Scholar]
- 28.Lee DH, Forscher C, Di Vizio D, Koeffler HP. Induction of p53-independent apoptosis by ectopic expression of HOXA5 in human liposarcomas. Sci Rep 2015;5:12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taminiau A, Draime A, Tys J, Lambert B, Vandeputte J, Nguyen N, et al. HOXA1 binds RBCK1/HOIL-1 and TRAF2 and modulates the TNF/NF-kappaB pathway in a transcription-independent manner. Nucleic Acids Res 2016;44:7331–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim YR, Kim IJ, Kang TW, Choi C, Kim KK, Kim MS, et al. HOXB13 downregulates intracellular zinc and increases NF-kappaB signaling to promote prostate cancer metastasis. Oncogene 2014;33:4558–67 [DOI] [PubMed] [Google Scholar]
- 31.Murata S, Kominsky SL, Vali M, Zhang Z, Garrett-Mayer E, Korz D, et al. Ductal access for prevention and therapy of mammary tumors. Cancer Res 2006;66:638–45 [DOI] [PubMed] [Google Scholar]
- 32.Wang G, Kumar A, Ding W, Korangath P, Bera T, Wei J, et al. Intraductal administration of transferrin receptor-targeted immunotoxin clears ductal carcinoma in situ in mouse models of breast cancer-a preclinical study. Proc Natl Acad Sci U S A 2022;119:e2200200119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stasinopoulos IA, Mironchik Y, Raman A, Wildes F, Winnard P Jr., Raman V. HOXA5-twist interaction alters p53 homeostasis in breast cancer cells. J Biol Chem 2005;280:2294–9 [DOI] [PubMed] [Google Scholar]
- 34.Chang CJ, Chen YL, Hsieh CH, Liu YJ, Yu SL, Chen JJW, et al. HOXA5 and p53 cooperate to suppress lung cancer cell invasion and serve as good prognostic factors in non-small cell lung cancer. J Cancer 2017;8:1071–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konishi H, Karakas B, Abukhdeir AM, Lauring J, Gustin JP, Garay JP, et al. Knock-in of mutant K-ras in nontumorigenic human epithelial cells as a new model for studying K-ras mediated transformation. Cancer Res 2007;67:8460–7 [DOI] [PubMed] [Google Scholar]
- 36.Croessmann S, Wong HY, Zabransky DJ, Chu D, Rosen DM, Cidado J, et al. PIK3CA mutations and TP53 alterations cooperate to increase cancerous phenotypes and tumor heterogeneity. Breast Cancer Res Treat 2017;162:451–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cochran RL, Cidado J, Kim M, Zabransky DJ, Croessmann S, Chu D, et al. Functional isogenic modeling of BRCA1 alleles reveals distinct carrier phenotypes. Oncotarget 2015;6:25240–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Croessmann S, Wong HY, Zabransky DJ, Chu D, Mendonca J, Sharma A, et al. NDRG1 links p53 with proliferation-mediated centrosome homeostasis and genome stability. Proc Natl Acad Sci U S A 2015;112:11583–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiss MB, Vitolo MI, Mohseni M, Rosen DM, Denmeade SR, Park BH, et al. Deletion of p53 in human mammary epithelial cells causes chromosomal instability and altered therapeutic response. Oncogene 2010;29:4715–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Cello F, Flowers VL, Li H, Vecchio-Pagan B, Gordon B, Harbom K, et al. Cigarette smoke induces epithelial to mesenchymal transition and increases the metastatic ability of breast cancer cells. Mol Cancer 2013;12:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allard J, Li K, Lopez XM, Blanchard S, Barbot P, Rorive S, et al. Immunohistochemical toolkit for tracking and quantifying xenotransplanted human stem cells. Regen Med 2014;9:437–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Basile A, Sica A, d’Aniello E, Breviario F, Garrido G, Castellano M, et al. Characterization of the promoter for the human long pentraxin PTX3. Role of NF-kappaB in tumor necrosis factor-alpha and interleukin-1beta regulation. J Biol Chem 1997;272:8172–8 [DOI] [PubMed] [Google Scholar]
- 43.Zhao X, Laver T, Hong SW, Twitty GB Jr., Devos A, Devos M, et al. An NF-kappaB p65-cIAP2 link is necessary for mediating resistance to TNF-alpha induced cell death in gliomas. J Neurooncol 2011;102:367–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006;10:515–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA. Kinetic enhancement of NF-kappaBxDNA dissociation by IkappaBalpha. Proc Natl Acad Sci U S A 2009;106:19328–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nigro C, Leone A, Longo M, Prevenzano I, Fleming TH, Nicolo A, et al. Methylglyoxal accumulation de-regulates HoxA5 expression, thereby impairing angiogenesis in glyoxalase 1 knock-down mouse aortic endothelial cells. Biochim Biophys Acta Mol Basis Dis 2019;1865:73–85 [DOI] [PubMed] [Google Scholar]
- 47.Chen Y, Rabson AB, Gorski DH. MEOX2 regulates nuclear factor-kappaB activity in vascular endothelial cells through interactions with p65 and IkappaBbeta. Cardiovasc Res 2010;87:723–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev 1995;9:2736–46 [DOI] [PubMed] [Google Scholar]
- 49.Crivellaro S, Panuzzo C, Carra G, Volpengo A, Crasto F, Gottardi E, et al. Non genomic loss of function of tumor suppressors in CML: BCR-ABL promotes IkappaBalpha mediated p53 nuclear exclusion. Oncotarget 2015;6:25217–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morotti A, Crivellaro S, Panuzzo C, Carra G, Guerrasio A, Saglio G. IkappaB-alpha: At the crossroad between oncogenic and tumor-suppressive signals. Oncol Lett 2017;13:531–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mulero MC, Ferres-Marco D, Islam A, Margalef P, Pecoraro M, Toll A, et al. Chromatin-bound IkappaBalpha regulates a subset of polycomb target genes in differentiation and cancer. Cancer Cell 2013;24:151–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamamoto M, Taguchi Y, Ito-Kureha T, Semba K, Yamaguchi N, Inoue J. NF-kappaB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat Commun 2013;4:2299. [DOI] [PubMed] [Google Scholar]
- 53.Kim JY, Jung HH, Ahn S, Bae S, Lee SK, Kim SW, et al. The relationship between nuclear factor (NF)-kappaB family gene expression and prognosis in triple-negative breast cancer (TNBC) patients receiving adjuvant doxorubicin treatment. Sci Rep 2016;6:31804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Darvishi B, Farahmand L, Eslami SZ, Majidzadeh AK. NF-kappaB as the main node of resistance to receptor tyrosine kinase inhibitors in triple-negative breast cancer. Tumour Biol 2017;39:1010428317706919 [DOI] [PubMed] [Google Scholar]
- 55.Pires BR, Mencalha AL, Ferreira GM, de Souza WF, Morgado-Diaz JA, Maia AM, et al. NF-kappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS One 2017;12:e0169622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All pertinent data is presented the main paper and in the Supplementary Data files of this paper. Raw data for this study were generated at SKCCC Microarray Core. Derived data supporting the findings of this study are available from the corresponding author upon request.