

Abstract
Pharmacogenomics (PGx) investigates the genetic influence on drug response and is an integral part of precision medicine. While PGx testing is becoming more common in clinical practice and may be reimbursed by Medicare/Medicaid and commercial insurance, interpreting PGx testing results for clinical decision support is still a challenge. The Pharmacogenomics Clinical Annotation Tool (PharmCAT) has been designed to tackle the need for transparent, automatic interpretations of patient genetic data. PharmCAT incorporates a patient’s genotypes, annotates PGx information (allele, genotype, and phenotype), and generates a report with PGx guideline recommendations from the Clinical Pharmacogenetics Implementation Consortium (CPIC) and/or the Dutch Pharmacogenetics Working Group (DPWG). PharmCAT has introduced new features in the last two years, including a VCF preprocessor, the inclusion of DPWG guidelines, and functionalities for PGx research. For example, researchers can use the VCF preprocessor to prepare biobank-scale data for PharmCAT. In addition, PharmCAT enables the assessment of novel partial and combination alleles that are composed of known PGx variants and can call CYP2D6 genotypes based on SNPs and INDELs in the input VCF file. This tutorial provides materials and detailed step-by-step instructions for how to use PharmCAT in a versatile way that can be tailored to users’ individual needs.
Keywords: Pharmacogenomics, pharmacogenetics, bioinformatics, genomics, precision medicine, translational medicine, utilization
INTRODUCTION
Pharmacogenomics (PGx) investigates the genetic influence on drug response and is an integral part of precision medicine. PGx improves health care outcomes by providing preemptive medication recommendations on an individual basis and preventing unnecessary health and economic burdens.1,2 The development of the Pharmacogenomics Clinical Annotation Tool (PharmCAT, https://pharmcat.org) is dedicated to facilitating the implementation of PGx in routine clinical practice.3,4
The clinical implementation of PGx is enabled by increasing guidance in expert-reviewed drug prescribing recommendation guidelines and agency-approved drug labels. For more than two decades, the Pharmacogenomics Knowledgebase (PharmGKB) has been the leading public source of expert-curated PGx knowledge.5,6 PharmGKB provides curated PGx content from published literature, international clinical guidelines, and drug labels containing PGx information. Guideline annotations from the Clinical Pharmacogenetics Implementation Consortium (CPIC)7,8 and the Dutch Pharmacogenetics Working Group (DPWG)9,10 include the functionality to retrieve genotype-level guidance. These drug prescribing recommendation guidelines need an annotation tool that links the clinical PGx testing results (genetic variations) to gene-drug recommendations to reduce barriers to implementation in clinical care and allow clinicians to easily interpret them.
In this tutorial, we present PharmCAT, a transparent, open-source, and customizable PGx annotation tool, which helps overcome the PGx implementation barrier of translating genetic data to gene-drug recommendations.
General overview of the PharmCAT modules
PharmCAT (https://pharmcat.org) has been developed to extract PGx-relevant variants from a VCF file derived from sequencing or genotyping technologies, determine genotype/diplotypes and infer corresponding phenotypes, and connect those with clinical or prescribing recommendations.3,4
PharmCAT packages pharmacogene allele definitions, function assignments, phenotype mappings, and prescribing recommendations provided by CPIC which are pulled directly from the CPIC database and DPWG which are pulled from the PharmGKB database.5–10 Information about the CPIC guideline development process11 and PharmGKB curation process for PGx guidelines6, as well as the details of the allele definitions, function assignments, phenotype mapping, and prescribing recommendations PharmCAT uses, are available online on the CPIC and PharmGKB websites. In some cases, PharmCAT slightly modifies the allele definitions or mappings provided by CPIC/PharmGKB as described in the online documentation (https://pharmcat.org/methods/Gene-Definition-Exceptions/). In the context of PGx and this tutorial, a PGx allele (sometimes referred to as a haplotype) indicates an alternative form of a gene that is made up of a specific combination of genetic variations, while the term variant refers to a single nucleotide polymorphism (SNP), small indel or repeat at a given position. PharmCAT has a modular design illustrated in Figure 1 consisting of the VCF preprocessor, the Named Allele Matcher, the Phenotyper, and the Reporter. Depending on the needs of users, PharmCAT can be run as a single application that takes in a VCF file and optional outside PGx genotype or phenotype data and generates a detailed report connecting the determined subject-specific genotypes to PGx prescribing recommendation; or each module can be run independently, as each module can produce intermediate output files.
Figure 1.
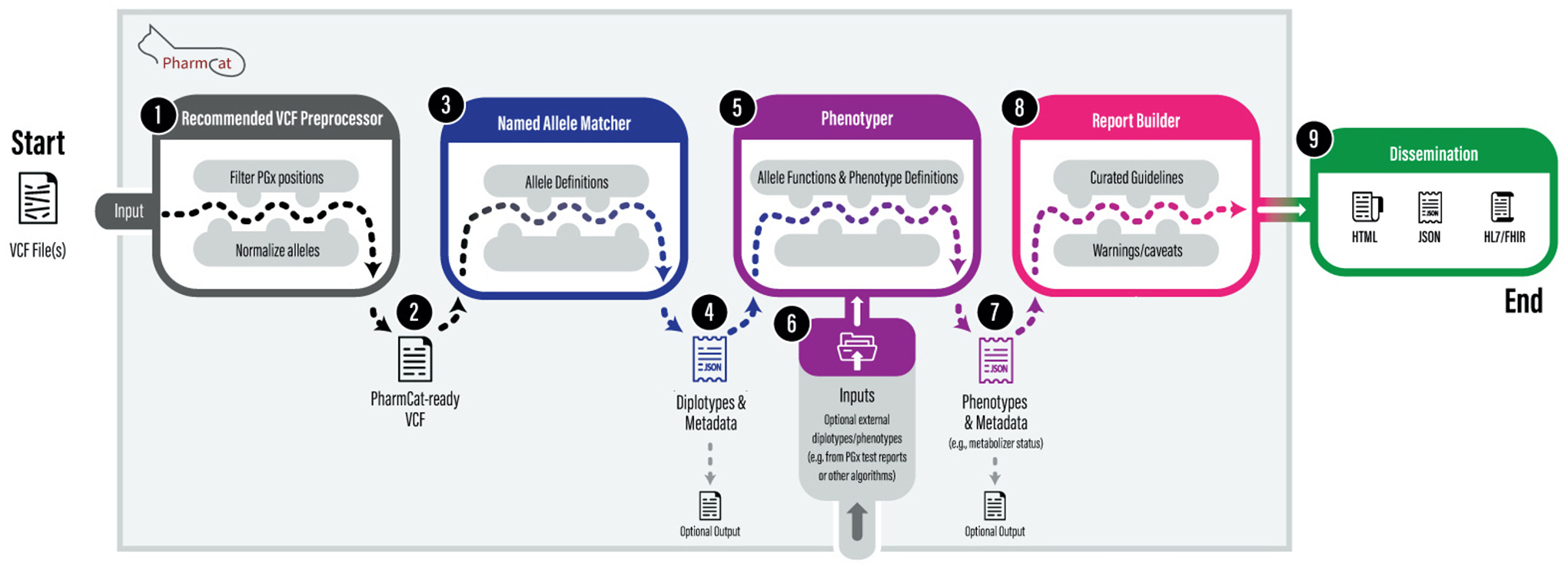
PharmCAT workflow. PharmCAT has a modular design: it can be run as a single application tool or individual modules. PharmCAT expects a VCF input from users and we highly recommend using the PharmCAT VCF preprocessor for standardizing and normalizing the input VCF file before PharmCAT. PharmCAT takes outside PGx genotype or phenotype data, which need to be formatted as a tab-delimited value (TSV) file. Allele definitions and recommendations (extracted from PGx guidelines) are combined with additional curated information such as notes, caveats, and warnings. HL7/FHIR is currently under development.
Genotypes or phenotypes that are derived from other PGx tools or provided by laboratory genetic test reports are referred to as “outside” data in this manuscript as they are not generated by PharmCAT. User-provided outside data is incorporated into the PharmCAT report when such content is provided by the user.
The VCF preprocessor assists in formatting a user’s VCF file to comply with PharmCAT input requirements by normalizing and standardizing the variant representation. The Named Allele Matcher relies on a specific variant representation using GRCh38 that is documented in pharmcat_positions.vcf, which is included in each release. This file summarizes all genetic positions used by PharmCAT together with expected reference and alternative allele representation, which is important for insertions and deletions (INDELs). Any genetic positions not included in the input VCF file are assumed to be missing positions and will not be interpreted as reference alleles. The VCF preprocessor produces PharmCAT-ready VCF files and a report of missing PGx positions in the input VCF file. Since PharmCAT requires single-sample VCF files, the preprocessor will also separate multi-sample VCF files into multiple single-sample VCF files.
The Named Allele Matcher module predicts haplotypes and genotypes based on the provided allele definitions. Further information about the variant matching algorithm is available in the PharmCAT documentation (https://pharmcat.org/methods/) and the prior PharmCAT publication.4 PharmCAT supports unphased and phased data.
The Phenotyper module maps the alleles of the genotype to function terms and subsequently to the corresponding phenotypes. The underlying data for that process are CPIC allele function and phenotype files and PharmGKB-annotated DPWG gene information.5–10
The Reporter connects phenotypes to prescribing recommendations from CPIC and the PharmGKB-annotated DPWG guidelines and combines them with additional gene information and disclaimers/warnings. The reporter has two output file formats, JSON or HTML.
Structure of report
The PharmCAT report consists of four parts: a Genotype Summary table, Prescribing Recommendations, Allele Matching Details, and a Disclaimers section (Figure 2). The header of the report captures the date and version information of the PharmCAT software and data used to generate the report. The Genotype Summary table includes genes for which the input VCF file has at least one variant or genes that have outside PGx genotype or phenotype data provided. A count of the total number of genes with information vs the total number of genes with recommendations in PharmCAT is available at top of the table (Section 1 in Figure 2). Currently, the genes are listed in alphabetical order together with the determined genotype, the resulting allele functionality, and phenotype, in addition to the drugs for which gene-specific recommendations might be available. The right-most column of the table shows a “yes” if the genotype is determined based on missing information for one or more of the PharmCAT-interrogated genetic positions.
Figure 2.

A snapshot of PharmCAT report sections for the GeT-RM sample NA18526. The PharmCAT report consists of four parts: (1) Genotype Summary table, (2) Prescribing Recommendations section sorted by the drug in alphabetical order, (3) Allele Matching Details about the interrogated variants for each gene, and (4) Disclaimers.
The Prescribing Recommendations section provides genotype-specific CPIC and PharmGKB-annotated DPWG guideline recommendations, links to the guidelines, and if applicable specific warnings (Section 2 in Figure 2).5–10 The Allele Matching Details section lists all variant positions per gene that are part of the allele definitions used to match the input VCF file (Section 3 in Figure 2). Missing genetic positions or variants not considered due to input errors are highlighted.
The following sections provide examples of how to run PharmCAT in different ways, complete with specific command lines and tutorial materials.
Details on how to run PharmCAT
To run PharmCAT, make sure you have downloaded the Quick download list below. Follow the Download and set up and read the PharmCAT VCF requirements.
Users can jump-start with Running PharmCAT. However, we highly recommend running the PharmCAT VCF preprocessor (see PharmCAT VCF preprocessor).
For advanced uses of PharmCAT, please proceed to the later sections for explanations and examples of how to run individual PharmCAT modules, incorporate outside PGx genotype or phenotype data, perform multi-sample analysis, and use auxiliary research options.
Quick download list
Java 17 or newer, e.g., OpenJDK by Adoptium (https://adoptium.net)
Pre-compiled PharmCAT Jar file from either the PharmCAT website (https://pharmcat.org/) or the PharmCAT GitHub repository releases page
- Compressed tar file for the PharmCAT VCF preprocessor from the same PharmCAT GitHub repository releases page
- Python 3.9 or higher and necessary python packages
- bcftools and bgzip 1.16 or higher
Tutorial materials based on the GeT-RM samples from the PharmCAT Tutorial GitHub (https://github.com/PharmGKB/PharmCAT-tutorial)
Steps 1–3 can be replaced by using the docker image for PharmCAT (https://pharmcat.org/using/PharmCAT-in-Docker/), which saves one’s effort in downloading the dependencies and setting up the computing environment for PharmCAT. A docker image and a step-by-step tutorial are available on the PharmCAT tutorial GitHub.
Download and set up
The PharmCAT homepage (https://pharmcat.org/) has a “Download” button which directs users straight to the latest release of the PharmCAT at GitHub.com (https://github.com/PharmGKB/PharmCAT/releases/latest). This tutorial is based on PharmCAT v2.0 and above.
Users can download the latest release of PharmCAT as a pre-compiled Jar file (pharmcat-<latest-release>-all.jar), which requires Java 17 or newer. Users will need to install Java 17.
PharmCAT does not require an internet connection. All data needed by PharmCAT is either supplied by users on the command line or self-contained.
The PharmCAT VCF preprocessor is also available on the PharmCAT releases page and is written in python 3. The zip file contains scripts and accessory files that users will need for running the PharmCAT VCF preprocessor and preparing the input VCF file(s) for PharmCAT. Also included in the zip file are the VCF files needed for the PharmCAT VCF Preprocessor. The same VCFs are also listed on the releases page.
The PharmCAT tutorial GitHub repository provides materials for running PharmCAT on different types of input VCF files that users may have. It offers three types of input VCF files under the data directory, including a single-sample VCF file for each of the three tutorial GeT-RM samples (e.g., PharmCAT_tutorial_get-rm_wgs_30x_grch38.NA18526.vcf.bgz), multi-sample VCF file for all three samples (PharmCAT_tutorial_get-rm_wgs_30x_grch38.vcf.bgz), and VCF files divided by chromosome (e.g., PharmCAT_tutorial_get-rm_wgs_30x_grch38_chr1.vcf.bgz).12,13 These samples were part of the 30x whole-genome sequencing data from the 1000 Genomes Project sample collection.14
Users can use the following commands to clone the PharmCAT tutorial GitHub repository to their local computing environment and download other necessary tools:
# go to the desired location to clone/download the GitHub repository # “/Users/xyz/” as a hypothetical directory path cd /Users/xyz/ git clone git@github.com:PharmGKB/PharmCAT-tutorial.git # The GitHub repository should be available at /Users/xyz/PharmCAT-tutorial/ cd /Users/xyz/PharmCAT-tutorial/ # Please replace “<latest-release>“ with the latest release number # Download the latest PharmCAT release wget https://github.com/PharmGKB/PharmCAT/releases/download/v<latest-release>/pharmcat-<latest-release>-all.jar # Download the VCF preprocessor wget https://github.com/PharmGKB/PharmCAT/releases/download/v<latest-release>/pharmcat-preprocessor-<latest-release>.tar.gz # Unzip tar -xvf pharmcat-preprocessor-<latest-release>.tar.gz
The rest of this tutorial assumes this layout. All commands, if not otherwise specified, should be executed in the PharmCAT-tutorial/ directory in a command line terminal.
PharmCAT VCF requirements
PharmCAT has listed its requirements for the input VCF file in a detailed manner, which can be also found on the PharmCAT website (https://pharmcat.org/), to prevent ambiguity in the results and free users from avoidable trial-and-error test runs:
First and foremost, as an annotation tool, PharmCAT expects users to perform quality control on the input VCF file on their own. PharmCAT uses all PGx allele-defining genetic positions and does not consider the information in the QUAL and FILTER columns in the input VCF file. It is the responsibility of the users to remove data not meeting quality criteria before passing it to PharmCAT.
The input VCF should include all available PGx allele-defining genetic positions even for the genetic positions that are homozygous reference across all samples and harbor no alternative alleles. Any genetic positions not included in the input VCF file are assumed to be missing positions and will not be interpreted as reference alleles.
The input VCF file should not be compressed.
The input VCF file should only be of a single sample. If multiple samples are present, only the first sample in the input VCF file will be used by the PharmCAT. Use the PharmCAT VCF preprocessor to help convert a multi-sample VCF file to multiple single-sample VCF files. A tutorial on how to use PharmCAT to annotate for more than one sample can be found in a later section of this paper.
PharmCAT requires the input VCF file to follow the VCF specifications (>=4.1) widely used and commonly accepted by many bioinformatics tools (https://github.com/samtools/hts-specs).
The input VCF file can be either phased or unphased. PharmCAT can recognize both and handle them appropriately.
Chromosome positions of genetic variants must be aligned to GRCh38/hg38 as PharmCAT currently does not support other builds. For users with a VCF file on any genome build other than GRCh38/hg38, we provide an example of lift-over on one of the UK Biobank genetic datasets using the GATK LiftoverVcf15 on the PharmCAT website. Please note that this example is not meant to be a comprehensive documentation of solutions for all lift-over issues. Lift-over may require additional data cleaning or preparation steps that are specific to the user’s genomic data.
PharmCAT adopts the parsimonious, left-aligned variant representation format as discussed in Unified Representation of Genetic Variants by Tan, Abecasis, and Kang16 to avoid ambiguity in the representation of genetic variants for bioinformatics analysis.
Related to point #8, insertions, deletions, or INDELs must be represented with an anchoring base pair.
The CHROM field must be in the format of chr##, e.g., chr1, chr22, chrX, and chrM.
PharmCAT provides a VCF preprocessor to help users prepare VCF files to meet all the above VCF requirements.
PharmCAT VCF preprocessor
The PharmCAT VCF preprocessor, written in python 3, makes sure the input VCF file(s) meet PharmCAT’s VCF requirements. The PharmCAT VCF preprocessor has a few prerequisites. Users are expected to use python 3 (>= 3.9), have bcftools (>= 1.16) and htslib (>1.16), and install necessary python libraries. The complete list of required python libraries can be found in the PharmCAT VCF Preprocessor zip file. For convenience, one can install all the necessary libraries at one time using the following command in a Linux environment or a terminal or import the requirements to a conda environment.
# Install the required python libraries pip3 install -r preprocessor/requirements.txt
The only mandatory input for the PharmCAT VCF preprocessor is a VCF file, so the minimal command to run the PharmCAT VCF preprocessor is as follows:
python3 pharmcat_vcf_preprocessor.py -vcf <path_to_vcf(.gz)>
The input to “-vcf” can be a single-sample VCF file, a multi-sample VCF file, or a list file of VCF file paths. VCF files can be either bgzip-compressed or uncompressed.
In the example of the minimal command, the PharmCAT VCF preprocessor produces two outputs. The first output is one or more single-sample PharmCAT-ready VCF files by default named “<sample_id_1>.preprocessed.vcf”, “<sample_id_2>.preprocessed.vcf”, and the rest in like manner if applicable. The second output is a report of missing PGx positions in the input VCF file for the user’s reference, a bgzip-compressed VCF file named “<input_file_basename>.missing_pgx_var.vcf.bgz”.
All outputs are defaulted to the directory where the input VCF file is located.
List of optional arguments and flags:
-refFna, --reference-genome <path_to_file> = path to indexed human reference genome sequence on GRCh38/hg38. The reference genome sequence file can be either compressed or non-compressed, but it must be indexed.
-refVcf, --reference-pgx-vcf <path_to_file> = by default, the pharmcat_positions.vcf.bgz under the current working directory and used as the list of reference PGx positions to be extracted from the input.
-S, --sample-file <path_to_file> = path to a file of sample names which allows users to retain and preprocess a selective subset of samples.
-bcftools, --path_to_bcftools <path_to_ program> = the path to the executable bcftools which grants users the ability to designate a specific version of bcftools for analysis.
-bgzip, --path_to_bgzip <path_to_ program> = similar as the “-bcftools”; the path to the executable bgzip.
-o, --output-dir <path_to_dir> = directory path to write the result files to. By default, outputs will be saved to the same directory as the “-vcf” input file.
-bf, --base-filename <output_name> = the prefix for output files, e.g., “<base_filename>.<sample_id>.preprocessed.vcf”.
-k, --keep_intermediate_files = an option to keep intermediate files. This can be useful when users want to keep intermediate files for further analyses.
-0, --missing_to_ref = an option [-number zero] to add missing PGx positions as reference to the output. The missing genetic positions are defined as those whose genotypes are missing “./.” in every single sample or not present in the input VCF files. This option will not convert “./.” to “0/0” if any other sample has a non-missing genotype at this genetic position. This should only be used if the user is sure the genotype is the reference at the missing genetic positions instead of unreadable/uncallable. Running PharmCAT with genetic positions as missing vs reference can lead to different results.
PharmCAT’s VCF preprocessor requires a copy of the GRCh38/hg38 reference human genome sequence, which can be supplied by “-refFna”. If “-refFna” is not provided by users, the PharmCAT VCF preprocessor will automatically download the necessary human reference genome assembly and its index. In this case, an internet connection is required.
PharmCAT’s VCF preprocessor can handle large-scale genetic datasets. The “-vcf” argument takes either a single VCF file or a sorted list of chromosome VCF files which are commonly seen for large genetic study cohorts. The list of chromosome VCF files needs to be supplied as a file with one VCF file per line sorted according to chromosomes or genetic positions; all chromosome VCF files should have the same set of samples. This functionality is designed to accommodate the analytic needs of large-scale genetic cohorts that commonly split the genetic data by chromosome or more granular consecutive genomic regions into multiple VCF files with the same set of samples, such as the UK Biobank.
Case 1. Single-sample VCF file
We highly recommend that users run the PharmCAT VCF file preprocessor even for their single-sample VCF file to ensure that the genetic variation in the file is presented in the way that PharmCAT expects. The following command runs the single-sample VCF file from a GeT-RM sample NA18526.
python3 preprocessor/pharmcat_vcf_preprocessor.py -vcf data/PharmCAT_tutorial_get-rm_wgs_30x_grch38.NA18526.vcf.bgz -refVcf preprocessor/pharmcat_positions.vcf.bgz -o results/pharmcat_ready/
Case 2. Multi-sample VCF file
Population- or biobank-scale VCF files most likely come in multi-sample format. The PharmCAT VCF preprocessor is designed to help users with this case and produce multiple single-sample VCF files that PharmCAT requires.
python3 preprocessor/pharmcat_vcf_preprocessor.py -vcf data/PharmCAT_tutorial_get-rm_wgs_30x_grch38.vcf.bgz -refVcf preprocessor/pharmcat_positions.vcf.bgz -o results/pharmcat_ready/
Case 3. Multiple VCFs divided into non-overlapping genetic blocks
Large-scale genetic data sometimes is divided into multiple by-chromosome VCF files or VCF files with consecutive genetic blocks. The PharmCAT VCF preprocessor can manage this type of genetic data sets by taking a list of VCF files as the input. The input of the VCF file list must be sorted based on chromosome position.
python3 preprocessor/pharmcat_vcf_preprocessor.py -vcf data/input_vcf_list.txt -refVcf preprocessor/pharmcat_positions.vcf.bgz -o results/pharmcat_ready/
Running PharmCAT
We strongly recommend users run the PharmCAT VCF preprocessor before running PharmCAT to avoid ambiguity of variant representation formats commonly seen in VCF files generated from different data sources. Users should have PharmCAT-ready single-sample VCF file(s) after running the PharmCAT VCF preprocessor following the previous sections, e.g., NA18526.preprocessed.vcf.
The minimal command to run PharmCAT from the command line is as follows:
java -jar <path_to_the_latest_pharmcat_jar> -vcf <sample_file>
Running PharmCAT means that the user sequentially runs all the individual PharmCAT modules: the Named Allele Matcher, Phenotyper, and Reporter. By default, PharmCAT saves all outputs to the directory where the input VCF file is located. The default outputs are JSON files from the Named Allele Matcher and the Phenotyper, and a PGx report in HTML format which contains sections of a Genotype Summary table, Prescribing Recommendations, Allele Matching Details, and Disclaimers (Figure 2).
The full command-line syntax of PharmCAT is as follows:
java -jar <path_to_the_latest_pharmcat_jar> -vcf <sample_file> [(optional) -o <output_dir> -bf <base_output_file_name> -del]
List of required arguments:
-jar <path_to_jar_file> = the compiled PharmCAT Jar file.
-vcf <sample_file> = a non-compressed, single-sample VCF file, which must comply with PharmCAT’s VCF requirements. If multiple samples are present, only the first one will be analyzed.
List of optional arguments and flags:
-o, --output-dir <output_dir> = directory path to write the result files to. By default, outputs will be saved to the same directory as the input VCF file.
-bf, --base-filename <output_name> = the prefix for output files (e.g., <output_name>.html).
-del, --delete-intermediary-files = a flag to remove the intermediate outputs and save only the HTML report.
It is noteworthy that optional arguments and flags introduced here are also effective in individual PharmCAT modules. For example, “-bf” can be used to change the base output file name (without file extension) for the Named Allele Matcher outputs.
To provide a concrete example, the following command runs PharmCAT on one of the GeT-RM samples:
java -jar pharmcat-<latest_release>-all.jar -vcf results/pharmcat_ready/NA18526.preprocessed.vcf -o results/pharmcat_all/
These commands generate an intermediate Named Allele Matcher JSON file (NA18526.preprocessed.match.json), an intermediate Phenotyper JSON file (NA18526.preprocessed.phenotype.json), and a PharmCAT PGx report (NA18526.preprocessed.report.html).
Full details can be found on the PharmCAT website (https://pharmcat.org/).
Running individual PharmCAT modules
Users can run individual modules of PharmCAT, which is useful for obtaining specific PGx annotations (alleles, haplotypes, genotypes/diplotypes, allele functions, phenotypes, etc.).
Named Allele Matcher
The PharmCAT Named Allele Matcher takes the input VCF file, matches the genetic data to the PGx allele definitions in PharmCAT, and infers the sample’s genotypes/diplotypes if available.
The command syntax of the PharmCAT Named Allele Matcher is as follows:
java -jar <path_to_the_latest_pharmcat_jar> -matcher -vcf <vcf_file> [(optional) -md <json_definitions_dir> -ma -matcherHtml]
List of required argument or flag:
-matcher = run the PharmCAT Named Allele Matcher.
-vcf <vcf_file> = the same input VCF file that users will use for running the PharmCAT.
List of optional arguments and flags:
-ma, --matcher-all-results = provide to request the PharmCAT Named Allele Matcher to return all possible PGx calls, not just the most possible call(s).
-matcherHtml, --matcher-save-html = provide to also save the results in HTML format.
-md, --matcher-definitions-dir <json_definitions_dir> = a directory containing the allele definitions in JSON format for PharmCAT to use instead of the default packaged allele definitions.
By default, the PharmCAT Named Allele Matcher saves the output in a JSON file.
The command to run the PharmCAT Named Allele Matcher on one of the GeT-RM samples is as follows:
java -jar pharmcat-<latest_release>-all.jar -matcher -vcf results/pharmcat_ready/NA18526.preprocessed.vcf -matcherHtml -o results/pharmcat_matcher/
Phenotyper
The PharmCAT Phenotyper provides allele function and predicted phenotype information of a sample by ingesting the genotypes/diplotypes identified by the PharmCAT Name Allele Matcher.
The command-line syntax for running the PharmCAT Phenotyper is as follows:
java -jar <path_to_the_latest_pharmcat_jar> -phenotyper -pi <matcher_json_file> [(optional) -po <path_to_an_outside_call_file.tsv>]
List of required arguments:
-phenotyper = run the PharmCAT Phenotyper.
-pi <json_file> = the JSON file generated by the Named Allele Matcher; cannot be used with -vcf.
List of optional arguments:
-po, --phenotyper-outside-call-file <path_to_outside_call> = a tab-delimited file of outside PGx genotype or phenotype data for the sample (see Incorporating outside PGx calls).
The PharmCAT Phenotyper expects only the Named Allele Matcher JSON file as the input. To obtain the Phenotyper output using a VCF file, users can run a combination of both the Named Allele Matcher and the Phenotyper (see the example below).
The command to run the PharmCAT Phenotyper on one of the GeT-RM samples is as follows:
# Use the Named Allele Matcher JSON java -jar pharmcat-<latest_release>-all.jar -phenotyper -pi results/pharmcat_matcher/NA18526.preprocessed.match.json -o results/pharmcat_phenotyper/ # Use a VCF file and run both the Named Allele Matcher and the Phenotyper java -jar pharmcat-<latest_release>-all.jar -matcher -vcf results/pharmcat_ready/NA18526.preprocessed.vcf -phenotyper -o results/pharmcat_phenotyper/
Reporter
The PharmCAT Reporter takes the genotype/diplotype and phenotype data from the Phenotyper, interprets the data of relevant drug annotation, includes appropriate warnings and caveats, and generates a comprehensive, reader-friendly report.
The command-line syntax for running the PharmCAT Reporter is as follows:
java -jar <path_to_the_latest_pharmcat_jar> -reporter -ri <phenotyper_json_file> [(optional) -rt “Report title goes here” -reporterJson]
List of required arguments:
-ri <phenotyper_json_file> = the JSON file generated by the Phenotyper.
List of optional arguments:
-rt “Report title goes here” = the PharmCAT report title, by default no title line
-reporterJson = provide to also save the results in JSON format.
The HTML output can be easily rendered into a PDF by many other available tools, including web browsers.
The command to run the PharmCAT Reporter on one of the GeT-RM samples is as follows:
java -jar pharmcat-<latest_release>-all.jar -reporter -ri results/pharmcat_phenotyper/NA18526.preprocessed.phenotype.json -o results/pharmcat_reporter/ -rt ‘Report for NA18526’
Incorporating outside PGx calls
PharmCAT allows users to supply genotypes or phenotypes that are derived from other means, referred to as “outside” data as they are not generated by PharmCAT. For example, outside genotypes may be called by other PGx tools17–21, or outside genotypes or phenotypes may be provided from a laboratory genetic test report. The outside genotype and phenotype data will take precedence over the input VCF file when both offer information for a certain pharmacogene. In such cases, PharmCAT will use the outside genotype or phenotype calls in the final report but issue a warning to users in the Allele Matching Details section. Currently, users only need to provide outside genotype or phenotype data for CYP2D6, MT-RNR1, and F5 for which the PharmCAT currently does not call PGx alleles. PharmCAT does not support PGx annotations at this time for these genes due to various reasons including, for example, the difficulty of the VCF file format to handle structural variation (SV) and copy number variation (CNV) for calling CYP2D6 alleles. (To call CYP2D6 alleles using only SNPs and INDELs, see Inferring CYP2D6 based on SNPs and INDELs.) PharmCAT will, however, include these outside data in the Phenotyper and Reporter and thus they will be included in the PharmCAT reports.
Outside genotype or phenotype data need to be provided to PharmCAT using a tab-delimited plain text file. An outside genotype or phenotype data file can only include content for one individual. Each line of the file is a gene-genotype pair with up to four fields:
a gene denoted by its HUGO Gene Nomenclature Committee (HGNC) gene symbol,
diplotype (e.g., CYP2D6*1/*3) or a single-allele call (e.g., 1555A>G for MT-RNR1), or
a phenotype or other gene result (e.g., *57:01 positive for HLA-B), or
activity score, such as for CYP2D6.
Each field must be separated by a tab. An example of the content in an outside genotype or phenotype data file can be found in Table S1. Or see the relevant outside call format page on the PharmCAT website.
Users can provide the outside PGx genotype or phenotype data file to PharmCAT by using the argument “-po <tsv_file>“.
The command-line syntax for running PharmCAT as a single application with an outside call file is as follows:
java -jar <path_to_the_latest_pharmcat_jar> -vcf <sample_file> -o <output_dir> -po <path_to_an_outside_call_file.tsv>
Take NA18526 for example:
java -jar pharmcat-<latest_release>-all.jar -vcf results/pharmcat_ready/NA18526.preprocessed.vcf -po data/outside_calls_from_get-rm.NA18526.txt -o results/pharmcat_all/
The command-line syntax for running the PharmCAT Phenotyper with an outside call file is as follows:
java -jar <path_to_the_latest_pharmcat_jar> -phenotyper -pi <matcher_json_file> -po <path_to_outside_call>
Take NA18526 for example:
java -jar pharmcat-<latest_release>-all.jar -phenotyper -pi results/pharmcat_matcher/NA18526.preprocessed.match.json -po data/outside_calls_from_get-rm.NA18526.txt -o results/pharmcat_phenotyper
Batch-annotation on multiple samples
PharmCAT handles each single input VCF file and generates a PGx report in seconds, which enables PharmCAT to process a data set of biobank scale in an efficient manner, with help from as few as one simple additional script (https://pharmcat.org/using/Multi-Sample-Analysis/) (Table 1).
Table 1.
PharmCAT runtimes for reference. The PharmCAT runtime and computing speed estimates are variables depending on the input sample size and variant size as well as the computing environment. VCF input to the PharmCAT has gone through the VCF preprocessor.
| Test sample Size | Computing environment | Speed (sec/sample) | |
|---|---|---|---|
| VCF Preprocessor | 50K | High-performance computing cluster* | 1.2 |
| PharmCAT | 50K | High-performance computing cluster* | 3.6 |
| PharmCAT | 50K | MacOS (2.9 GHz Intel Core i7) | 2.1 |
For more information about the Stanford Sherlock high-performance computing cluster, please go to https://www.sherlock.stanford.edu/.
This tutorial section uses the VCF files of three GeT-RM samples (NA18526, NA18565, and NA18861)14; and is based on Stanford University’s Sherlock, a High-Performance Computing (HPC) cluster. The scripts to run the PharmCAT on the Stanford Sherlock are written in shell language, which can be easily adapted to another HPC environment supported by a different job scheduler.
Assuming users have run the PharmCAT VCF preprocessor to generate multiple single-sample VCF files, such as NA18526.preprocessed.vcf.
# obtain the sample list bcftools query -l data/PharmCAT_tutorial_get-rm_wgs_30x_grch38.vcf.bgz > data/test_get-rm_samples.txt # run PharmCAT for SINGLE_SAMPLE in $(cat data/test_get-rm_samples.txt) do java -jar pharmcat-<latest_release>-all.jar -vcf results/pharmcat_ready/”$SINGLE_SAMPLE”.preprocessed.vcf -o results/pharmcat_all/ done
The output is the PGx reports in HTML format for each of the three GeT-RM samples, NA18526.preprocessed.report.html, NA18565.preprocessed.report.html, and NA18861.preprocessed.report.html.
Batch outside calls
To incorporate outside genotype or phenotype calls with PharmCAT for multiple samples, users must prepare the outside genotype or phenotype data of each sample in separate files and supply the individual outside data file to PharmCAT along with the sample’s VCF file.
# data for NA18526: data/outside_calls_from_get-rm.NA18526.txt # data for NA18565: data/outside_calls_from_get-rm.NA18565.txt # data for NA18861: data/outside_calls_from_get-rm.NA18861.txt # run PharmCAT for SINGLE_SAMPLE in $(cat data/test_get-rm_samples.txt) do java -jar pharmcat-<latest_release>-all.jar -vcf results/pharmcat_ready/”$SINGLE_SAMPLE”.preprocessed.vcf -po data/outside_calls_from_get-rm.”$SINGLE_SAMPLE”.txt -o results/pharmcat_all/ done
Running individual modules
Similarly, users can run the modules of the PharmCAT on multiple samples in a similar manner to how one runs the whole PharmCAT. This behavior is desirable by researchers who are interested in understanding population PGx and specifically obtaining PGx frequencies (alleles, diplotypes, or phenotypes) for their population cohort. To obtain such statistics, one can run the following commands substituted with specific content:
# PharmCAT - Named Allele Matcher for SINGLE_SAMPLE in $(cat data/test_get-rm_samples.txt) do java -jar pharmcat-<latest_release>-all.jar -matcher -vcf “$SINGLE_SAMPLE”.preprocessed.vcf -o results/pharmcat_matcher/ done # PharmCAT - Phenotyper for SINGLE_SAMPLE in $(cat data/test_get-rm_samples.txt) do java -jar pharmcat-<latest_release>-all.jar -phenotyper -pi “$SINGLE_SAMPLE”.preprocessed.match.json -o results/pharmcat_phenotyper/ done # PharmCAT - Reporter for SINGLE_SAMPLE in $(cat data/test_get-rm_samples.txt) do java -jar pharmcat-<latest_release>-all.jar -reporter -ri “$SINGLE_SAMPLE”.preprocessed.phenotype.json -o results/pharmcat_reporter/ done
These commands yield sample-specific results with a Named Allele Matcher JSON file, a Phenotyper JSON file, and a Reporter HTML. We encourage users to explore and perform data analysis using the rich content in these JSON files, which can be easily achieved using auxiliary JSON libraries and data analysis packages in R or python.
Extracting the PharmCAT JSON data into TSV
We also provide accessory R scripts that organize and extract content from the Named Allele Matcher or Reporter JSON outputs into tab-separated values (TSV) files, which is a format commonly used in data analysis. In the TSV output, each row is a sample-gene pair with sample ID, gene name, PGx annotations, and missing allele-defining positions if applicable. The commands are as follows:
# Extract the allele matching details from the PharmCAT Named Allele Matcher JSON files Rscript src/json2tsv_pharmcat_named_allele_matcher.R --input-dir results/pharmcat_matcher/ --input-file-pattern *match.json --output-dir ./ # Extract the phenotype details that are used for drug prescribing recommendations from the PharmCAT Reporter JSON files Rscript src/json2tsv_pharmcat_report.R --input-dir results/pharmcat_all/ --input-file-pattern *report.json --output-dir ./
You can find these utility R scripts and also another example, which is applied to the Penn Medicine BioBank data22 and isabout how to convert JSON data into a CSV file using python, on the PharmCAT website (https://pharmcat.org/using/Multi-Sample-Analysis/).
PharmCAT for research
Starting with v.2.0.0, PharmCAT offers two new options for research. One is to call novel PGx genotypes that are combinations of known PGx variants or alleles, and the other is to call CYP2D6 alleles based on SNPs and INDELs available in the input VCF file.
Calling combination or partial alleles
PharmCAT will try to determine the combination and partial alleles if an exact match to any single defined allele cannot be found. Without the research flag, these samples will have a ‘not called’ output from the Named Allele Matcher.
This option addresses variant combinations not cataloged by PharmVar (pharmvar.org) or other nomenclature sites. It does not consider novel variants; it only considers variants included in existing allele definitions found in novel combinations. Providing these calls can highlight alleles of functional consequence. For example, an individual carries both the CYP2C19*2 no function allele and *7 no function allele on one haplotype. The default PharmCAT behavior, without the option of calling combinations, will result in a “not called” output for this individual as this variant combination is not defined as a CYP2C19 allele in PharmVar; on the other hand, with the option of calling combinations, this individual will be identified as CYP2C19[*2 + *7]. It can aid in further expanding the knowledge of the nomenclature repositories to improve the translation of genetic information into standardized phenotype terms that can be connected to clinical guidance.
Partial alleles are one or more genetic variants that make up part of a PGx allele, e.g., CYP2C19*2/[*17+chr10.g.94781859G>A]. In this case, this specific sample carries a CYP2C19*2, a no function allele on one haplotype. And the genetic variants on the other haplotype make up CYP2C19*17, an increased function allele, plus chr10.g.94781859G>A, a splicing defect variation that is part of the *2 allele definition.
Combination alleles are more than one PGx allele on a haplotype, e.g., CYP2C9*1/[*2+*33], which is a *1 on one haplotype and [*2+*33] on the other haplotype.
PharmCAT’s syntax for combination calls uses square brackets to reflect that it is a variation on one gene copy and to distinguish it from gene duplications, e.g., tandem arrangements in CYP2D6*36+*10.
Users need to supply the “-research combinations” flag to turn on the function. The command-line syntax for the function is as follows:
java -jar <path_to_the_latest_pharmcat_jar> -vcf <sample_file> -o <output_dir> -research combinations
The command on one of the GeT-RM samples is as follows:
java -jar pharmcat-<latest_release>-all.jar -vcf results/pharmcat_ready/NA18861.preprocessed.vcf -o results/pharmcat_for_research/ -bf pharmcat_tutorial.NA18861.combinations -research combinations
Inferring CYP2D6 based on SNPs and INDELs
The other new PharmCAT option allows users to call CYP2D6 alleles using only SNPs and INDELs present in an input VCF file.
This option is discouraged if users have access to whole genome sequencing (WGS) CRAM/BAM files. Please refer to our documentation about CYP2D6 calling (https://pharmcat.org/using/Calling-CYP2D6/).
CYP2D6 contains SV and CNV which have a significant influence on inferring CYP2D6 phenotypes. These classes of genetic variation are beyond the scope of what can be called given SNPs or INDELs in a VCF file. For example, CYP2D6 ultrarapid metabolizer prediction entirely depends on the ability to detect CNVs. Utilizing just VCF file input has the potential to misrepresent a sample’s genotype due to missing input.
In the case where a sample has the whole gene deletion (*5) on one CYP2D6 allele and variants on the other CYP2D6 allele, these hemizygous variants will be falsely presented as homozygous in the VCF file, e.g., a CYP2D6*5/*29 will be detected and misrepresented as *29/*29 in a VCF file. This is another reminder that calling CYP2D6 from VCF is provided only under research mode and should be used at the user’s own risk. Calling CYP2D6 from VCF should not be used for clinical purposes.
The command-line syntax to call CYP2D6 based on SNPs and INDELs in a VCF file is as follows:
java -jar <path_to_the_latest_pharmcat_jar> -vcf <sample_file> -o <output_dir> -research cyp2d6
Take NA18861 for example, the command to call CYP2D6 is as follows:
java -jar pharmcat-<latest_release>-all.jar -vcf results/pharmcat_ready/NA18861.preprocessed.vcf -o results/pharmcat_for_research/ -bf NA18861.cyp2d6 -research cyp2d6
Discussion
In this tutorial, we present detailed guides and instructions for PharmCAT, an open-source, customizable PGx annotation tool.3,4 PharmCAT incorporates a patient’s genotypes, annotates PGx information, and generates a report with gene-drug recommendations from expert-reviewed guidelines.
PharmCAT is under active development and PharmCAT versions are released when new content or development updates are available. PharmCAT uses git as its version control system (managed through GitHub) which includes code branches. Releases of PharmCAT are based on the main branch of the PharmCAT codebase. Active updates that have not been included in the releases can be found under the development branch. Users have the option to build a fresh copy of the Jar file for the PharmCAT, which this tutorial will not cover but the detailed explanation can be found at the PharmCAT GitHub wiki. More features are being developed, such as multiprocessing support.
PGx annotations in this tutorial may be different from the GeT-RM reports due to various reasons. This tutorial calls NA18861 with SLCO1B1*1/*43, rather than GeT-RM’s SLCO1B1*1/*14 because the *43 allele was added to PharmVar’s SLCO1B1 nomenclature after GeT-RM tested the sample.
PharmCAT has several decision-making steps before it returns the matched PGx alleles. For example,
(1) Allele-defining genetic positions that are absent from the input VCF file are assumed missing and excluded from the allele-calling process. If the missing genetic position(s) affect an entire allele definition, the star allele is excluded from the matching process. If an allele is defined by additional genetic positions for which information is provided in the VCF file, the allele is considered for the allele-calling process based on the remaining genetic positions.
(2) Information in the QUAL and FILTER columns in an input VCF file is not considered in PharmCAT.
(3) For unphased data, PharmCAT will generate all possible combinations of genotypes, representing potential diplotypes, given the provided PGx positions of interest. A diplotype is considered valid if both haplotypes match PGx allele definitions (default mode). The haplotype variation(s) is assumed on different gene copies (trans), e.g., CYP2C19*2/*17 but with CYP2C19*1/[*2+*17] not being an optional result for unphased data in the default mode. The diplotype with the longest valid haplotype match is returned by the Named Allele Matcher (ties are also returned); this is PharmCAT’s inferred diplotype based on unphased data, e.g., a sample heterozygous for TPMT rs1142345 T>C and rs1800460 C>T is reported as TPMT*1/*3A with *3A being the longer haplotype match compared to *3B/*3C. With unphased data, the exact diplotype cannot be verified. The diplotype with the longest haplotype match may or may not be the same as the diplotype from phased data. Different diplotypes can result in different phenotypes and ultimately different prescribing recommendations. Users have the option to include all potential diplotypes, regardless of the length of a match, in the PharmCAT output.
(4) For phased data, PharmCAT will compare the reported genetic positions with the PGx allele definitions and if there is a match, PharmCAT reports the diplotype. Otherwise, PharmCAT returns a “not called” output.
(5) If provided, outside genotypes or phenotypes will take precedence over what is presented in the input VCF file.
(6) In some cases, PharmCAT follows the corresponding drug prescribing recommendation guidelines and modifies how diplotypes are used for phenotype assignment and prescribing recommendations, as described in the online documentation (https://pharmcat.org/methods/Gene-Definition-Exceptions/). For example, if no PGx diplotype can be determined for SLCO1B1 by the Named Allele Matcher but the rs4149056 genotypes are available, PharmCAT will follow the CPIC statin guideline to provide prescribing recommendations based on the rs4149056 genotype alone.23
PharmCAT has several limitations. It cannot account for genotype errors introduced by sequencing or genotyping technologies, phasing, or imputation. Genotype error by sequencing or genotyping technologies can be due to various reasons, such as sample preparation, polymerase errors, difficulty with complex genetic regions, etc.24,25 PharmCAT cannot account for missing or low-quality data and is not designed to perform quality control of the VCF file. The responsibility of quality control is the responsibility of the user before an input VCF file is fed into PharmCAT. Complex SV is not represented in VCF file formats, so PharmCAT cannot account for this type of variation. If users have access to next-generation sequencing (NGS) CRAM/BAM files for genes with complex SV, such as CYP2D6, they can use other bioinformatics tools that leverage this data to call diplotypes in PGx genes (e.g., Stargazer17, StellarPGx18, Aldy19, Cyrius20, or Astrolabe21). While these tools can handle complex SV from NGS, they cannot be used with genotyping results as PharmCAT can. None of these tools map to prescribing recommendations. Diplotype outputs from these tools can be incorporated into PharmCAT reports with guideline-based drug prescribing recommendations using the outside calls option. PharmCAT will return “not called” if the input genotypes do not match the PGx allele definitions in default mode. This includes cases where no PGx positions from the allele definitions are present in the input VCF for a PGx gene. It also includes cases where novel combinations of known PGx positions are present in the input VCF file, though these can be identified by using the research option. PharmCAT cannot detect novel variations in genetic positions that are not present in the PGx allele definitions; it only considers genetic positions in the allele definition files. However, no prescribing recommendations from CPIC or DPWG guidelines exist for novel combinations of known PGx variations.
This tutorial presents a detailed walkthrough of how to run PharmCAT for different purposes. The latest details and documents of PharmCAT are available on the website (https://pharmcat.org/). As PharmCAT bridges the technical gap between clinical genetic tests and drug prescribing recommendations, we aim to facilitate the implementation of PGx as part of precision medicine in standard clinical care.
Materials
This tutorial paper used the genetic dataset of the GeT-RM characterized Coriell samples, specifically, the high-coverage WGS performed by New York Genome Center (NYGC), funded by National Human Genome Research Institute.14 This collection of 30x WGS on GRCh38 has sequenced 3202 samples from the 1000 Genomes Project sample collection to 30x coverage. NYGC aligned the data to GRCh38 and those alignments are publicly available.
The collection of 30x WGS on GRCh38 is hosted on the International Genome Sample Resource (IGSR, https://academic.oup.com/nar/article/45/D1/D854/2770649). On May 4, 2022, we downloaded the genotype VCF files that contain SNV and INDELs (<50bp) were downloaded from the FTP site (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20201028_3202_raw_GT_with_annot/). The names of the VCF files and their index files followed the format 20201028_CCDG_14151_B01_GRM_WGS_2020-08-05_chr##.recalibrated_variants.vcf.gz(.tbi).
Only genetic variants in PGx regions that passed quality control were retained14. Genetic positions were retained only if they were within 1Mbp of the pharmacogenes included in PharmCAT and passed quality control (i.e., FILTER is PASS).
PharmCAT calls for the three GeT-RM samples used in this tutorial were compared to the consensus diplotype in a GeT-RM study.13
Supplementary Material
Funding:
PharmCAT is supported by the National Institutes of Health (NIH)/National Human Genome Research Institute (NHGRI: U24HG010862).
Footnotes
Conflicts of Interest:
The authors declared no competing interests in this work.
References
- 1.Elliott RA, Camacho E, Jankovic D, Sculpher MJ & Faria R Economic analysis of the prevalence and clinical and economic burden of medication error in England. BMJ Qual Saf 30, 96–105 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Sultana J, Cutroneo P & Trifirò G Clinical and economic burden of adverse drug reactions. J Pharmacol Pharmacother 4, S73–77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klein TE & Ritchie MD PharmCAT: A Pharmacogenomics Clinical Annotation Tool. Clin Pharmacol Ther 104, 19–22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sangkuhl K et al. Pharmacogenomics Clinical Annotation Tool (PharmCAT). Clin Pharmacol Ther 107, 203–210 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whirl-Carrillo M et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther 92, 414–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whirl-Carrillo M et al. An Evidence-Based Framework for Evaluating Pharmacogenomics Knowledge for Personalized Medicine. Clin Pharmacol Ther 110, 563–572 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Relling MV & Klein TE CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther 89, 464–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Relling MV et al. The Clinical Pharmacogenetics Implementation Consortium: 10 Years Later. Clin Pharmacol Ther 107, 171–175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swen JJ et al. Pharmacogenetics: from bench to byte. Clin Pharmacol Ther 83, 781–787 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Swen JJ et al. Pharmacogenetics: from bench to byte--an update of guidelines. Clin Pharmacol Ther 89, 662–673 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Caudle K et al. Incorporation of Pharmacogenomics into Routine Clinical Practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline Development Process. Current Drug Metabolism 15, 209–217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaedigk A et al. Characterization of Reference Materials for Genetic Testing of CYP2D6 Alleles: A GeT-RM Collaborative Project. J Mol Diagn 21, 1034–1052 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pratt VM et al. Characterization of 137 Genomic DNA Reference Materials for 28 Pharmacogenetic Genes: A GeT-RM Collaborative Project. J Mol Diagn 18, 109–123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrska-Bishop M et al. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. 2021.02.06.430068 Preprint at 10.1101/2021.02.06.430068 (2021). [DOI] [PMC free article] [PubMed]
- 15.Auwera G. A. V. de & O’Connor BD Genomics in the cloud: using Docker, GATK, and WDL in Terra. (O’Reilly, 2020).
- 16.Tan A, Abecasis GR & Kang HM Unified representation of genetic variants. Bioinformatics 31, 2202–2204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S-B, Wheeler MM, Thummel KE & Nickerson DA Calling Star Alleles With Stargazer in 28 Pharmacogenes With Whole Genome Sequences. Clin Pharmacol Ther 106, 1328–1337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Twesigomwe D et al. StellarPGx: A Nextflow Pipeline for Calling Star Alleles in Cytochrome P450 Genes. Clin Pharmacol Ther 110, 741–749 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Numanagić I et al. Allelic decomposition and exact genotyping of highly polymorphic and structurally variant genes. Nat Commun 9, 828 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X et al. Cyrius: accurate CYP2D6 genotyping using whole-genome sequencing data. Pharmacogenomics J 21, 251–261 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Twist GP et al. Constellation: a tool for rapid, automated phenotype assignment of a highly polymorphic pharmacogene, CYP2D6, from whole-genome sequences. NPJ Genom Med 1, 15007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verma SS et al. Evaluating the frequency and the impact of pharmacogenetic alleles in an ancestrally diverse Biobank population. 2022.08.26.22279261 Preprint at 10.1101/2022.08.26.22279261 (2022). [DOI] [PMC free article] [PubMed]
- 23.Cooper-DeHoff RM et al. The Clinical Pharmacogenetics Implementation Consortium Guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and Statin-Associated Musculoskeletal Symptoms. Clinical Pharmacology & Therapeutics 111, 1007–1021 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pompanon F, Bonin A, Bellemain E & Taberlet P Genotyping errors: causes, consequences and solutions. Nat Rev Genet 6, 847–859 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Salk JJ, Schmitt MW & Loeb LA Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet 19, 269–285 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.