

Summary
Background
Olutasidenib (FT-2102) is a potent, selective, oral, small-molecule inhibitor of mutant isocitrate dehydrogenase 1 (IDH1). The aims for phase 1 of this phase 1/2 study were to assess the safety, pharmacokinetics, pharmacodynamics, and clinical activity of olutasidenib, as monotherapy or in combination with azacitidine, in patients with acute myeloid leukaemia or myelodysplastic syndrome, harbouring mutant IDH1.
Methods
In this phase 1/2, multicentre, open-label clinical trial, we enrolled patients aged 18 years or older with acute myeloid leukaemia or intermediate, high, or very high risk myelodysplastic syndrome harbouring mutant IDH1 at 18 study sites in the USA, Australia, France, and Spain. Other key eligibility criteria included Eastern Cooperative Oncology Group performance status 0–2 with adequate liver and renal function. The primary outcomes were dose-limiting toxicities and the maximum tolerated dose, maximum evaluated dose, and the recommended phase 2 dose of olutasidenib. Olutasidenib was administered orally in doses of 150 mg once daily, 150 mg twice per day, and 300 mg once daily. Azacitidine (75 mg/m2) was administered subcutaneously or intravenously daily for 7 days on, 21 days off. The study was ongoing at the data cutoff (Oct 2, 2019) and is registered with ClinicalTrials.gov, NCT02719574.
Findings
Patients were enrolled between Aug 8, 2016, and Nov 14, 2018. 78 patients received olutasidenib as monotherapy (n=32) or in combination with azacitidine (n=46). The median follow-up was 8·3 months (IQR 3·1–13·3) for monotherapy and 10·1 months (4·2–15·3) for combination therapy. 16 (50%) of 32 patients in the monotherapy group and 24 (52%) of 46 patients in the combination therapy group were women. Most patients were White (26 [81%] for monotherapy and 31 [67%] for combination therapy). No dose-limiting toxicities were reported in the dose-escalation cohorts and 150 mg twice per day was declared the recommended phase 2 dose on the basis of safety, pharmacokinetics and pharmacodynamics, and clinical activity. The most common (≥20%) grade 3–4 treatment-emergent adverse events with monotherapy were thrombocytopenia (nine [28%] of 32 patients), febrile neutropenia (seven [22%] of 32), and anaemia (seven [22%] of 32); and with combination therapy were thrombocytopenia (19 [41%] of 46), febrile neutropenia (13 [28%] of 46), neutropenia (13 [28%] of 46), and anaemia (nine [20%] of 46). 11 (34%) of 32 patients in the monotherapy group and nine (20%) of 46 patients in the combination therapy group died (most commonly from disease progression [three (9%) of 32 and four (9%) of 46]). No deaths were considered study-drug related. For patients with relapsed or refractory acute myeloid leukaemia, 41% (95% CI 21–64; nine of 22) receiving monotherapy and 46% (27–67; 12 of 26) receiving combination therapy had an overall response. For treatment-naive patients with acute myeloid leukaemia, 25% (1–81; one of four) receiving monotherapy and 77% (46–95; ten of 13) receiving combination therapy had an overall response.
Interpretation
Olutasidenib, with or without azacitidine, was well tolerated and showed meaningful clinical activity in patients with IDH1-mutated acute myeloid leukaemia. The results of this phase 1 study provide rationale for the continued evaluation of olutasidenib in multiple patient populations with myeloid malignancies.
Funding
Forma Therapeutics.
Introduction
Mutation of the isocitrate dehydrogenase 1 gene (IDH1) occurs in 7–14% of patients with acute myeloid leukaemia and 3–4% of patients with myelodysplastic syndrome.1-3 IDH1 catalyses oxidative decarboxylation of isocitrate to α-ketoglutarate. IDH1 mutations confer neomorphic enzymatic activity, promoting conversion of α-ketoglutarate to the oncometabolite 2-hydroxyglutarate. Aberrant accumulation of 2-hydroxyglutarate leads to competitive inhibition of α-ketoglutarate dependent dioxygenases crucial to DNA and histone demethylation, and hence might block normal differentiation of stem and progenitor cells, thereby promoting neoplastic transformation.4-6 Inhibition of mutant IDH1 in tumour cells and the subsequent reduction in 2-hydroxyglutarate production can restore normal cellular differentiation and provide therapeutic benefit in IDH1-mutated cancers. Inhibitors of mutant IDH1 have shown efficacy in acute myeloid leukaemia and are currently being studied in patients with myelodysplastic syndrome, cholangiocarcinoma, and glioma, both as single-agent treatments, and in combination with other targeted drugs that might act synergistically.7-12
Olutasidenib (FT-2102) is a potent, selective, oral small-molecule inhibitor of mutant IDH1 with the therapeutic potential to restore normal cellular differentiation.12 Olutasidenib is a quinolinone-based, allosteric, noncompetitive inhibitor of mutant IDH1 that binds in a hydrophobic pocket situated near the IDH1 homodimer interface.13,14 This first-in-human study of olutasidenib assessed its safety, pharmacokinetic and pharmacodynamic profile, and clinical activity, both as monotherapy and in combination with the hypomethylating agent azacitidine, in patients with treatment-naive or relapsed or refractory acute myeloid leukaemia or myelodysplastic syndrome harbouring mutant IDH1.
Methods
Study design and participants
This study is a phase 1/2, open-label clinical trial, undertaken at 18 study sites in the USA, Australia, France, and Spain. The trial protocol is included in the appendix (pp 28-137). This Article reports on the phase 1 portion of the study (additional analyses are underway for the phase 2 portion of the study and will be reported separately). Patients aged 18 years or older with pathologically proven acute myeloid leukaemia or intermediate, high, or very high risk myelodysplastic syndrome (as defined by the Revised International Prognostic Scoring System) harbouring an IDH1 mutation were eligible for inclusion. There were no predefined dates for patient enrolment. Patients with acute myeloid leukaemia or myelodysplastic syndrome that had relapsed or was refractory to standard therapy, or for whom standard therapy was considered inappropriate, were eligible for olutasidenib monotherapy. Patients who met the monotherapy criteria and were candidates for azacitidine treatment were eligible for combination therapy (appendix p 19). Other key eligibility criteria included Eastern Cooperative Oncology Group performance status 0–2 with adequate liver and renal function, baseline corrected QT interval by Fridericia of 450 ms or less, negative serum pregnancy test if a woman of childbearing potential, and agreement to use highly effective contraception for the duration of study participation and 90 days after the last dose of study drug. Patients were excluded if they had a history of previous malignancy (unless disease-free for ≥12 months or considered surgically cured), symptomatic central nervous system metastases or other tumour location, previous allogeneic haemopoietic stem-cell transplantation (HSCT) within 100 days of screening, active acute or chronic graft versus host disease (GVHD) or were receiving immunosuppressive therapy as treatment or prophylaxis against GVHD, recent treatment with radiation therapy, chemotherapy, small molecule anticancer therapeutic, anticancer therapeutic antibody, or other experimental therapies, congestive heart failure (New York Heart Association Class III or IV) or unstable angina pectoris, previous history of myocardial infarction within 1 year before study entry, uncontrolled hypertension or uncontrolled arrhythmias, family history of QT prolongation, recent initiation of treatment with concomitant medications known to cause Torsades de Pointes, concurrent treatment with chronic corticosteroids (except if chronic treatment with <20 mg of methylprednisolone daily or equivalent), known HIV infection or active, uncontrolled bacterial, viral, or fungal infections requiring systemic therapy (prophylactic systemic antimicrobials was permitted), uncontrolled disease-related metabolic disorder or serious non-malignant disease that could compromise protocol objectives, any condition that could compromise the patient’s ability to understand the patient information, give informed consent, comply with the study protocol, or complete the study, or a history of severe allergic reaction to azacitidine (for patients enrolling into a combination cohort). Excess blasts were not required. Previous use of ivosidenib was allowed. To avoid a reduction in systemic exposure to olutasidenib, its coadministration with strong CYP3A4 inducers was not recommended; accordingly, investigators considered alternative concomitant medications.
The study was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. The protocol was approved by the institutional review board or independent ethics committee at each participating site. No deviations from the protocol affected the conduct of the study and all protocol amendments were approved by the institutional review board or independent ethics committee at each clinical site. All patients provided written informed consent before enrolment.
Procedures
Olutasidenib was administered orally during 28-day cycles as monotherapy or in combination with azacitidine. The first part of the study evaluated escalating doses of olutasidenib with or without azacitidine using a traditional 3 + 3 design, starting with olutasidenib 150 mg once daily monotherapy. Evaluation of olutasidenib plus azacitidine combination therapy was initiated after 150 mg once daily monotherapy was deemed safe. During dose escalation, sequential monotherapy cohorts received olutasidenib doses of 300 mg once daily or 150 mg twice per day. Combination patients received olutasidenib 150 mg once daily or twice per day plus azacitidine (75 mg/m2 administered subcutaneously or intravenously daily for 7 days on, 21 days off). Patients were subsequently enrolled into dose-expansion cohorts to evaluate monotherapy or combination therapy at doses of olutasidenib up to the maximum tolerated dose, or maximum evaluated dose. The recommended phase 2 dose for each regimen was determined by the safety and pharmacokinetic profile, the normalisation of plasma 2-hydroxyglutarate concentrations, and signs of clinical activity.
Study drug was taken orally and daily (either once daily or twice per day) in continuous 28-day cycles from day 1 until treatment discontinuation criteria were met. Patients were followed for safety for 28 days after treatment discontinuation, or until resolution or stabilisation of adverse events, except for patients who withdrew consent. Patients who discontinued for reasons other than disease progression, disease relapse, or withdrawal of consent were followed for response until progression or relapse occurred. Patients could be discontinued from the study at any time if considered medically necessary by the investigator or if it was the wish of the patient.
Adverse events and laboratory assessments were monitored throughout the study. Dose-limiting toxicities were defined as any event occurring during cycle one that fulfilled at least one dose-limiting toxicities criterion (appendix p 1). Adverse events and dose-limiting toxicities were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03. Relatedness of adverse events, including deaths, to the study treatment was assessed by the site investigator, informed by their clinical knowledge of the specifics of the patient’s case.
Olutasidenib dose reductions and interruptions for toxicity were permitted by the study protocol. For cases of differentiation syndrome, olutasidenib treatment was to be resumed as soon as differentiation syndrome symptoms and the patient’s clinical condition improved, and after a minimum of 3 days. In case of reappearance of differentiation syndrome symptoms, olutasidenib dosing was to again be held. If resumption was indicated, then olutasidenib was to be reduced one dose level for a minimum of the first 7 days after the disappearance of the differentiation syndrome. Thereafter, in absence of worsening of the previous toxicity, olutasidenib could be resumed at full dosage (see also appendix p 2).
Response, transfusion independence, and pharmacokinetics were assessed routinely over the course of the study as described in the study protocol (appendix pp 28-137). Survival follow-up was performed for up to 12 months from the first dose of study drug or for up to 28 days after the last dose (whichever was longer).
Outcomes
The primary outcomes were dose-limiting toxicities and the maximum tolerated dose, maximum evaluated dose, and the recommended phase 2 dose of olutasidenib, both as monotherapy and in combination with azacitidine. Primary safety analyses included the incidence and severity of adverse events, clinical laboratory abnormalities, and changes in electrocardiogram (ECG) parameters. Secondary outcomes were to determine the pharmacokinetic profile of olutasidenib as monotherapy and in combination with azacitidine, and to observe patients for any evidence of antileukaemic or antimyelodysplastic activity, including clinical response and transfusion independence. Determination of clinical response was based on the investigator’s assessment using disease-specific criteria derived from the International Working Group (IWG) and modified IWG criteria for acute myeloid leukaemia (2003; appendix p 4), and myelodysplastic syndrome (2006; appendix p 5).15-17 For patients with acute myeloid leukaemia, rates of overall response (defined as complete response, complete response with partial haematological recovery, complete response with incomplete blood count recovery, morphological leukaemia-free state, or partial response), complete response or complete response with partial haematological recovery, and complete response were assessed. For patients with myelodysplastic syndrome, overall response was defined as complete response plus marrow complete response plus partial response. Transfusion independence was also assessed. Patients were classified as dependent or independent at baseline within each category of platelets, packed red blood cells, and both platelets and packed red blood cells, based on whether they received a transfusion within 8 weeks before the first dose of olutasidenib. Transfusion independence was defined as the proportion of previously transfusion-dependent patients without a transfusion for at least a 56-day period on treatment. Time to response, duration of response, and overall survival were also used to evaluate clinical activity. Exploratory outcomes were to assess the on-target activity of olutasidenib (as determined by changes in plasma 2-hydroxyglutarate), to determine the frequency of cancer-associated mutations and genetic alterations in responding and non-responding patients, and to evaluate the pharmacokinetic, pharmacodynamic, and clinical response relationships (see appendix p 2 for additional supplemental methods).
Statistical analyses
The 3 + 3 design is not formally powered and is designed to select the maximum tolerated dose, defined as a dose at which up to 33% of patients had dose-limiting toxicities.18 Dose expansion group size was empirically driven to confirm the safety and tolerability of the maximum tolerated dose before initiation of phase 2. Safety analyses were conducted on the safety population, which included all patients who received at least one dose of olutasidenib. Activity analyses were conducted on the activity population, which included patients who had at least one post-baseline assessment, were on the study for three or more cycles, or discontinued before an assessment due to progression, death, or adverse events. Duration of olutasidenib exposure was calculated as the date of first dose to the date of last dose (data cutoff date) using Kaplan-Meier analysis. Time-to-event endpoints were estimated using Kaplan-Meier analysis. Descriptive statistics were used for all other clinical, laboratory, and pharmacokinetic and pharmacodynamic variables. All analyses were performed with SAS (Version 9.4). This trial is registered with ClinicalTrials.gov, NCT02719574.
Role of the funding source
The study sponsor collaborated with the clinical investigators to design and conduct the study design and collect, analyse, and interpret the data.
Results
78 patients were enrolled between Aug 8, 2016 and Nov 14, 2018 and received study treatment (figure 1). No patients were excluded from the analysis populations. The median age was 72 years (IQR 66–77) for the 32 patients who received monotherapy and 67 (58–72) years for the 46 patients who received combination therapy (table 1). Most patients had a diagnosis of acute myeloid leukaemia (monotherapy, 26 [81%] of 32; combination, 39 [85%] of 46) classified as primary (monotherapy, 21 [81%] of 26; combination therapy, 23 [59%] of 39), and relapsed or refractory (monotherapy, 22 [85%] of 26; combination therapy, 26 [67%] of 39). Three (8%) of 39 patients with acute myeloid leukaemia allocated to combination therapy had received previous ivosidenib.
Figure 1: Study profile.
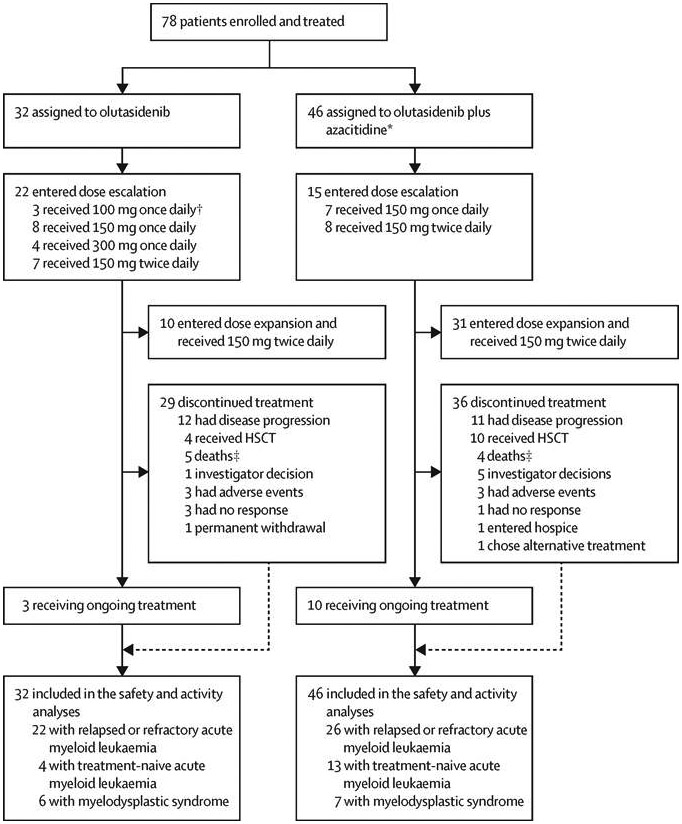
HSCT=haematopoietic stem-cell transplantation. *Standard dose of azacitidine (75 mg/m2). †Olutasidenib 100 mg once daily was received by a subgroup of patients (n=3) in a fed state. ‡For the nine patients with a primary reason of discontinuation of death, three patients had reached a complete response with incomplete hematologic recovery (two olutasidenib, one olutasidenib plus azacitidine) and two patients (both olutasidenib) had lost the response by the time of death but were still receiving study drug due to investigators’ assessment of ongoing clinical benefit, two patients had stable disease (both olutasidenib), and one patient had progressive disease (olutasidenib plus azacitidine); three patients died before day 28 (and before response assessment) of subdural haematoma (one olutasidenib, one olutasidenib plus azacitidine) and sepsis (one olutasidenib).
Table 1:
Baseline characteristics
| Olutasidenib group (n=32) |
Olutasidenib plus azacitidine group (n=46)* |
|
|---|---|---|
| Whole cohort | ||
| Age, years | 72 (66–77) | 67 (58–72) |
| <65 | 7 (22%) | 16 (35%) |
| 65–74 | 13 (41%) | 20 (43%) |
| ≥75 | 12 (38%) | 10 (22%) |
| Sex | ||
| Female | 16 (50%) | 24 (52%) |
| Male | 16 (50%) | 22 (48%) |
| Race | ||
| Asian | 1 (3%) | 2 (4%) |
| Black or African American | 4 (13%) | 3 (7%) |
| White | 26 (81%) | 31 (67%) |
| Other | 1 (3%) | 1 (2%) |
| Not reported | 0 | 9 (20%) |
| ECOG performance status | ||
| 0 | 9 (28%) | 13 (28%) |
| 1 | 16 (50%) | 26 (57%) |
| 2 | 7 (22%) | 7 (15%) |
| Acute myeloid leukaemia | ||
| n (%) | 26 (81%) | 39 (85%) |
| Primary (de novo) | 21/26 (81%) | 23/39 (59%) |
| Secondary | 5/26 (19%) | 16/39 (41%) |
| Disease state | ||
| Treatment-naive | 4/26 (15%) | 13/39 (33%) |
| Relapsed | 14/26 (54%) | 11/39 (28%) |
| Refractory | 8/26 (31%) | 15/39 (39%) |
| Cytogenetic risk | ||
| Intermediate | 9/26 (35%) | 22/39 (56%) |
| Poor | 7/26 (27%) | 8/39 (21%) |
| Unknown | 10/26 (38%) | 9/39 (23%) |
| Previous therapies, mean (SD) | 2·0 (1·9) | 2·0 (1·6) |
| Hypomethylating agent—azacitidine or decitabine | 12/26 (46%) | 10/39 (26%) |
| Mutated IDH1 inhibitor | 0/26 | 3/39 (8%) |
| Allogeneic haematopoietic stem-cell transplantation | 2/26 (8%) | 3/39 (8%) |
| Intensive chemotherapy | 17/26 (65%) | 23/39 (59%) |
| Myelodysplastic syndrome | ||
| n (%) | 6 (19%) | 7 (15%) |
| Disease state | ||
| Treatment-naive | 2/6 (33%) | 5/7 (71%) |
| Relapsed or refractory | 4/6 (67%) | 2/7 (29%) |
| Previous therapies, mean (SD) | 1·0 (1·5) | 1·0 (1·5) |
| Hypomethylating agent—azacitidine or decitabine | 4/6 (67%) | 2/7 (29%) |
| WHO classification | ||
| Refractory anaemia with ring sideroblasts | 1/6 (17%) | 0/7 |
| Refractory cytopenia with multilineage dysplasia | 1/6 (17%) | 2/7 (29%) |
| Refractory anaemia with excess blasts—1 | 1/6 (17%) | 1/7 (14%) |
| Refractory anaemia with excess blasts—2 | 1/6 (17%) | 4/7 (57%) |
| Myelodysplastic syndrome—unclassified | 2/6 (33%) | 0/7 |
| Revised International Prognostic Scoring System risk category | ||
| Intermediate | 0/6 | 1/7 (14%) |
| High | 5/6 (83%) | 5/7 (71%) |
| Very high | 1/6 (17%) | 1/7 (14%) |
Data are n (%), n/N (%), or median (IQR), unless otherwise specified. HMA=hypomethylating agent. HSCT=haemopoietic stem cell transplantation. IPSS-R=Revised International Prognostic Scoring System. *Standard dose of azacitidine (75 mg/m2).
At the data cutoff (Oct 2, 2019), study treatment was ongoing for three (9%) of 32 patients with monotherapy and ten (22%) of 46 with combination therapy (figure 1). Median time on treatment was 4·2 months (IQR 1·0–6·7) for monotherapy and 4·7 months (2·1–15·0) for combination therapy. The most common reason for treatment discontinuation was disease progression (monotherapy, 12 [38%] of 32; combination therapy, 11 [24%] of 46). Four (13%) of 32 monotherapy patients and ten (22%) of 46 combination patients discontinued to undergo haematopoietic stem-cell transplantation (HSCT; figure 1).
No dose-limiting toxicities were reported in dose-escalation cohorts. Olutasidenib 150 mg twice per day was selected as the recommended phase 2 dose for monotherapy and combination dose expansion cohorts on the basis of acceptable safety, preliminary evidence of clinical activity, pharmacodynamic data showing activity at all evaluated doses, and pharmacokinetic data showing that twice per day dosing provided more consistent exposure.
All 78 patients reported at least one treatment-emergent adverse event during the study. Grade 1–2 non-haematological events occurring in at least 25% of the 32 patients who received monotherapy were nausea (15 [47%]), fatigue (13 [41%]), pyrexia (ten [31%]), and vomiting (eight [25%]; table 2). For combination therapy, grade 1–2 non-haematological events occurring in at least 25% of the 46 patients were nausea (30 [65%]), constipation (27 [59%]), diarrhoea (20 [43%]), coughing (17 [37%]), vomiting (17 [37%]), fatigue (14 [30%]), headache (14 [30%]), hypokalaemia (13 [28%]), and dyspnoea (13 [28%]). Grade 3–4 haematological events occurring in at least 20% of patients were thrombocytopenia (nine [28%] of 32 on monotherapy and 19 [41%] of 46 on combination therapy), febrile neutropenia (seven [22%] and 13 [28%]), anaemia (seven [22%] and nine [20%]), and neutropenia (two [6%] and 13 [28%]; table 2).
Table 2:
Summary of treatment-emergent adverse events (regardless of causality)
| Olutasidenib group (n=32) | Olutasidenib plus azacitidine group* (n=46) | |||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Grade 1–2 | Grade 3 | Grade 4 | Grade 5 | Grade 1–2 | Grade 3 | Grade 4 | Grade 5 | |
| Blood and lymphatic system disorders | 1 (3%) | 7 (22%) | 0 | 0 | 7 (15%) | 13 (28%) | 0 | 0 |
| Febrile neutropenia | 0 | 7 (22%) | 0 | 0 | 2 (4%) | 13 (28%) | 0 | 0 |
| Cardiac disorders | 10 (31%) | 4 (13%) | 0 | 1 (3%) | 13 (28%) | 3 (7%) | 1 (2%) | 0 |
| Cardiac arrest | 0 | 0 | 0 | 1 (3%)† | 0 | 0 | 1 (2%) | 0 |
| Gastrointestinal disorders | 26 (81%) | 4 (13%) | 0 | 0 | 44 (96%) | 8 (17%) | 0 | 2 (4%) |
| Abdominal pain | 1 (3%) | 0 | 0 | 0 | 9 (20%) | 1 (2%) | 0 | 0 |
| Constipation | 6 (19%) | 1 (3%) | 0 | 0 | 27 (59%) | 1 (2%) | 0 | 0 |
| Diarrhoea | 7 (22%) | 1 (3%) | 0 | 0 | 20 (43%) | 2 (4%) | 0 | 0 |
| Gastrointestinal fistula | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2%) |
| Large intestinal obstruction | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2%) |
| Nausea | 15 (47%) | 0 | 0 | 0 | 30 (65%) | 4 (9%) | 0 | 0 |
| Vomiting | 8 (25%) | 0 | 0 | 0 | 17 (37%) | 1 (2%) | 0 | 0 |
| General disorders and administration site conditions | 25 (78%) | 2 (6%) | 0 | 5 (16%) | 36 (78%) | 10 (22%) | 0 | 4 (9%) |
| Asthenia | 1 (3%) | 0 | 0 | 0 | 10 (22%) | 2 (4%) | 0 | 0 |
| Death | 0 | 0 | 0 | 1 (3%)† | 0 | 0 | 0 | 0 |
| Disease progression | 0 | 0 | 0 | 3 (9%) | 0 | 0 | 0 | 4 (9%) |
| Fatigue | 13 (41%) | 2 (6%) | 0 | 0 | 14 (30%) | 8 (17%) | 0 | 0 |
| Multiple organ dysfunction syndrome | 0 | 0 | 0 | 1 (3%)† | 0 | 0 | 0 | 0 |
| Oedema peripheral | 2 (6%) | 0 | 0 | 0 | 10 (22%) | 0 | 0 | 0 |
| Pyrexia | 10 (31%) | 2 (6%) | 0 | 0 | 11 (24%) | 0 | 0 | 0 |
| Infections and infestations | 6 (19%) | 9 (28%) | 0 | 3 (9%) | 27 (59%) | 11 (24%) | 3 (7%) | 1 (2%) |
| Mucormycosis | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2%) | 0 |
| Pneumonia | 2 (6%) | 5 (16%) | 0 | 1 (3%) | 4 (9%) | 4 (9%) | 0 | 1 (2%) |
| Sepsis | 0 | 0 | 0 | 0 | 0 | 0 | 2 (4%) | 0 |
| Septic shock | 0 | 0 | 0 | 2 (6%) | 0 | 0 | 0 | 0 |
| Injury, poisoning, and procedural complications | 11 (34%) | 0 | 0 | 1 (3%) | 12 (26%) | 1 (2%) | 0 | 1 (2%) |
| Subdural haematoma | 0 | 0 | 0 | 1 (3%) | 0 | 0 | 0 | 1 (2%) |
| Investigations | 18 (56%) | 8 (25%) | 11 (34%) | 0 | 29 (63%) | 14 (30%) | 22 (48%) | 0 |
| Anaemia | 3 (9%) | 7 (22%) | 0 | 0 | 5 (11%) | 9 (20%) | 0 | 0 |
| Leukocytosis | 4 (13%) | 3 (9%) | 1 (3%) | 0 | 7 (15%) | 6 (13%) | 1 (2%) | 0 |
| Leukopenia | 0 | 0 | 2 (6%) | 0 | 4 (9%) | 4 (9%) | 3 (7%) | 0 |
| Lymphocyte count decreased | 0 | 1 (3%) | 2 (6%) | 0 | 3 (7%) | 1 (2%) | 1 (2%) | 0 |
| Neutropenia | 1 (3%) | 0 | 2 (6%) | 0 | 3 (7%) | 3 (7%) | 10 (22%) | 0 |
| Thrombocytopenia | 3 (9%) | 3 (9%) | 6 (19%) | 0 | 8 (17%) | 5 (11%) | 14 (30%) | 0 |
| Metabolism and nutrition disorders | 22 (69%) | 8 (25%) | 0 | 0 | 31 (67%) | 13 (28%) | 0 | 0 |
| Decreased appetite | 7 (22%) | 0 | 0 | 0 | 11 (24%) | 1 (2%) | 0 | 0 |
| Hypokalaemia | 5 (16%) | 2 (6%) | 0 | 0 | 13 (28%) | 3 (7%) | 0 | 0 |
| Musculoskeletal and connective tissue disorders | 16 (50%) | 4 (13%) | 0 | 0 | 22 (48%) | 4 (9%) | 0 | 0 |
| Arthralgia | 3 (9%) | 2 (6%) | 0 | 0 | 9 (20%) | 0 | 0 | 0 |
| Nervous system disorders | 13 (41%) | 3 (9%) | 0 | 1 (3%) | 29 (63%) | 2 (4%) | 1 (2%) | 1 (2%) |
| Cerebral haemorrhage | 0 | 0 | 0 | 1 (3%) | 0 | 0 | 0 | 0 |
| Dizziness | 7 (22%) | 1 (3%) | 0 | 0 | 11 (24%) | 1 (2%) | 0 | 0 |
| Haemorrhage intracranial | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2%) | 0 |
| Haemorrhagic stroke | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2%) |
| Headache | 6 (19%) | 0 | 0 | 0 | 14 (30%) | 1 (2%) | 0 | 0 |
| Respiratory, thoracic, and mediastinal disorders | 15 (47%) | 5 (16%) | 2 (6%) | 0 | 36 (78%) | 8 (17%) | 2 (4%) | 0 |
| Acute respiratory distress syndrome | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2%) | 0 |
| Acute respiratory failure | 0 | 0 | 1 (3%) | 0 | 0 | 0 | 0 | 0 |
| Cough | 5 (16%) | 0 | 0 | 0 | 17 (37%) | 1 (2%) | 0 | 0 |
| Dyspnoea | 7 (22%) | 0 | 0 | 0 | 13 (28%) | 1 (2%) | 0 | 0 |
| Hypoxia | 0 | 1 (3%) | 1 (3%) | 0 | 2 (4%) | 1 (2%) | 0 | 0 |
| Pharyngeal haemorrhage | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2%) | 0 |
| Skin and subcutaneous tissue disorders | 13 (41%) | 1 (3%) | 0 | 0 | 23 (50%) | 3 (7%) | 0 | 0 |
| Pruritus | 4 (13%) | 0 | 0 | 0 | 9 (20%) | 0 | 0 | 0 |
| Vascular disorders | 10 (31%) | 7 (22%) | 0 | 0 | 7 (15%) | 11 (24%) | 0 | 0 |
| Hypertension | 1 (3%) | 3 (9%) | 0 | 0 | 2 (4%) | 8 (17%) | 0 | 0 |
| Patients with any treatment-emergent adverse event | 31 (97%) | 13 (41%) | 8 (25%) | 11 (34%) | 46 (100%) | 10 (22%) | 24 (52%) | 9 (20%) |
Data are n (%). Grade 1–2 events occurring in 20% or more patients in either group, grade 3 events in 10% or more patients in either group, and all grade 4–5 events are reported. Grade 1–2 events include all patients with any grade 1–2 event, whereas grade 3–5 events reflect the maximum grade reported per patient (ie, a patient was counted once at the maximum severity for each system organ class and preferred term). No deaths were considered to be related to the study drug. A summary of grade 1–2 events occurring in 10% or more patients in either group and all reported grade 3–5 events is reported in the appendix (pp 8-11). Treatment-emergent adverse events included any events that emerged during treatment, that were absent pretreatment, or worsened relative to the pretreated state. Adverse events were coded using the Medical Dictionary for Drug Regulatory Affairs, version 19.1. * Standard dose of azacitidine (75 mg/m2). † Event occurred in the context of pneumonia (suspected or confirmed) and in the context of relapse or progressive disease with no evidence of isocitrate dehydrogenase differentiation syndrome or QT prolongation.
Serious adverse events were reported for 24 (75%) of 32 patients in the monotherapy group and 35 (76%) of 46 patients in the combination therapy group, including febrile neutropenia, pneumonia, and leukocytosis (appendix p 12).
Adverse events of special interest included differentiation syndrome, liver function test abnormalities, and QT interval prolongation on ECG. Differentiation syndrome was reported in four (13%) of 32 patients in the monotherapy group and six (13%) of 46 patients in the combination therapy group, with a maximum severity of grade 3 in six patients (three [9%] of 32 on monotherapy; three [7%] of 46 on combination therapy), grade 2 in two patients (one [3%]; one [2%]), and grade 1 in two (4%) of 46 patients on combination therapy. Differentiation syndrome events were mostly observed during cycle 1 (seven events: one grade 1; two grade 2; four grade 3), with two events in cycle 2 (one grade 1; one grade 3) and one event in cycle 5 (grade 3). All cases resolved with treatment interruption or dose reduction, dexamethasone, or supportive treatment, without recurrences. Two (50%) of four patients on monotherapy and five (83%) of six patients on combination therapy who developed differentiation syndrome had a response. Grade 3 or higher liver function test abnormalities of alanine aminotransferase, aspartate aminotransferase, or total bilirubin were reported in five (16%) of 32 patients on monotherapy and five (11%) of 46 patients on combination therapy. Two patients discontinued olutasidenib due to grade 3 liver function events, which subsequently resolved; one was receiving monotherapy and one combination therapy. In all other patients, treatment continued after temporary interruptions or dose adjustments.
Adverse events of QT prolongation were reported only in patients who received combination therapy (three [7%] of 46, grades 2–3) and were taking concomitant medications associated with QT interval prolongation; no events were considered serious, and all events were transient. Patients resumed treatment after interruption for 1 day (n=1), or continued treatment after no action (n=2), without recurrence.
Adverse events leading to treatment discontinuation occurred in nine (28%) of 32 patients in the monotherapy group, including two due to pneumonia (one grade 3 and one death) and two due to fatigue (one grade 1, one grade 2), and in eight (17%) of 46 patients in the combination therapy group, including two due to leukocytosis (one grade 2, one grade 3). Olutasidenib dose reductions occurred in five (16%) of 32 patients in the monotherapy group and eight (17%) of 46 patients in the combination therapy group. Fatal adverse events were reported for 11 (34%) of 32 patients in the monotherapy group and nine (20%) of 46 patients in the combination therapy group; the most common fatal event was disease progression in three (9%) of 32 patients in the monotherapy group and four (9%) of 46 patients in the combination therapy group (table 2). No deaths were considered related to the study drug.
Plasma olutasidenib concentrations increased until pharmokinetic steady-state was reached, after which consistent exposure over time (based on trough concentrations on day 1 of subsequent cycles) was observed for olutasidenib 150 mg twice per day, with plasma concentrations more than those predicted by in-vivo models13 to result in more than 90% reduction of 2-hydroxyglutarate, and less than those predicted to pose a corrected QT interval prolongation risk in monkeys (appendix p 20). Addition of azacitidine did not alter the pharmacokinetic profile (appendix p 13). The degree of interpatient pharmacokinetic variability observed following single and multiple doses of olutasidenib was moderate to high, with coefficient of variation percentages ranging from 26% to 79% for the maximum plasma concentration and 34% to 52% for area under the plasma concentration curve (AUC) from time zero to 24 h (appendix p 13).
After a single oral dose of olutasidenib under modified-fasting conditions (≥2 h postprandially or ≥30 mins before the next meal), time to maximum plasma concentration ranged from 2 h to 24 h (appendix p 13). The maximum plasma concentration of olutasidenib and AUC increased in a less-than-dose-proportional manner between the 150 mg and 300 mg once-daily doses. Mean olutasidenib elimination half-life estimates ranged from 52 h to 72 h and steady state was reached on day 15 of cycle 1 (appendix p 14).
The pharmacokinetic and pharmacodynamic relationship between individual patient plasma olutasidenib levels and 2-hydroxyglutarate concentration across treatment cohorts was evaluated irrespective of time on treatment (appendix p 21). Maximal reduction in 2-hydroxyglutarate concentrations occurred at the median steady-state concentration of olutasidenib 150 mg twice per day. Sustained reductions in plasma 2-hydroxyglutarate concentrations were observed across all dose cohorts for patients who received monotherapy or combination therapy (appendix p 22).
Among patients with relapsed or refractory acute myeloid leukaemia, nine (41% [95% CI 21–64]) of 22 patients receiving olutasidenib monotherapy and 12 (46% [27–67]) of 26 patients receiving combination therapy had an overall response (figure 2, appendix p 15). Among patients with treatment-naive acute myeloid leukaemia, one (25%) of four receiving monotherapy and ten (77%) of 13 receiving combination therapy had an overall response. For patients with relapsed or refractory acute myeloid leukaemia previously treated with a hypomethylating agent, four (33%) of 12 receiving monotherapy and one (10%) of ten receiving combination therapy had an overall response.
Figure 2: Duration of treatment and best overall response in patients with acute myeloid leukaemia.
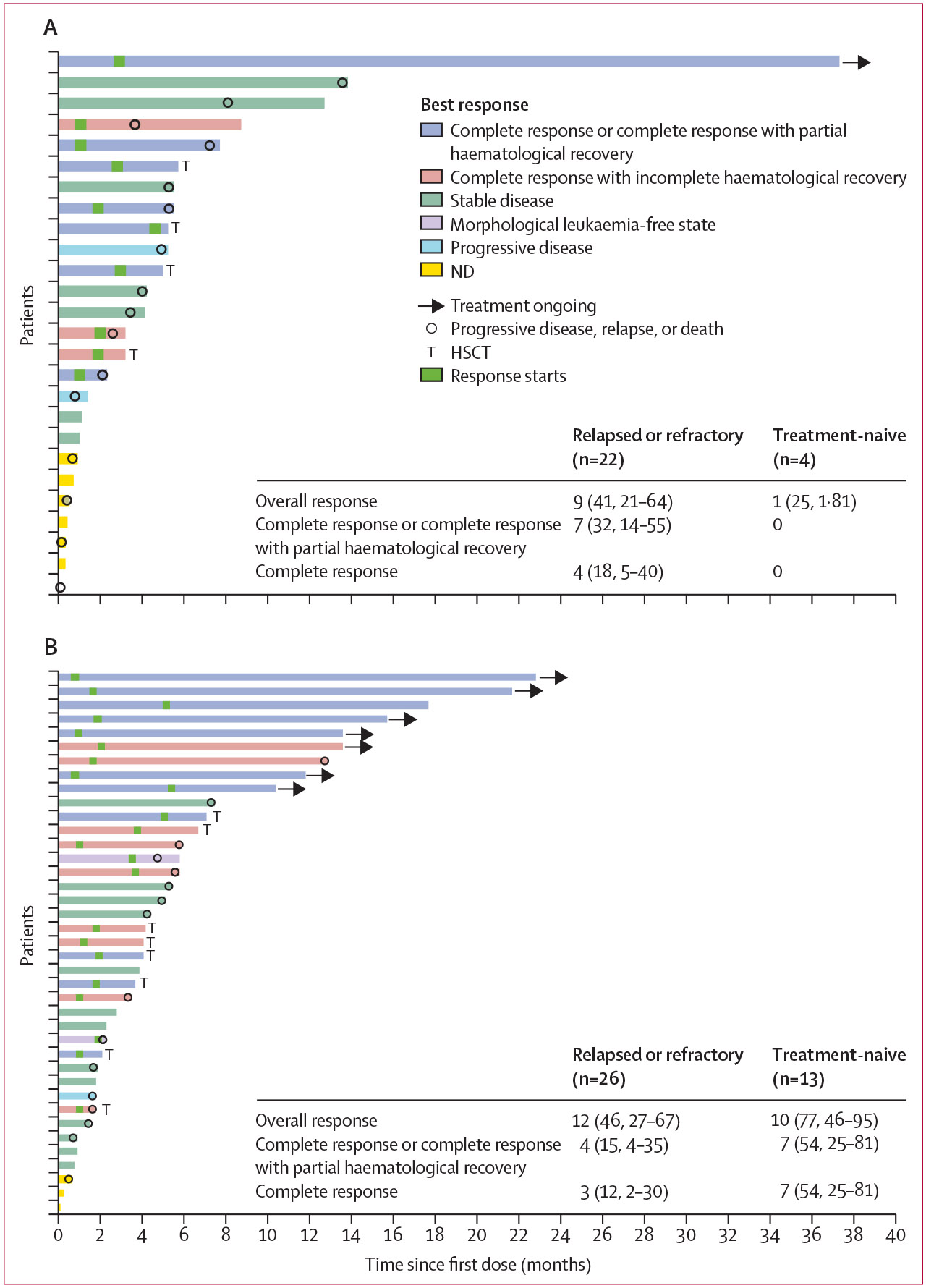
(A) Duration of treatment and best overall response in patients in patients receiving olutasidenib. (B) Duration of treatment and best overall response in patients receiving olutasidenib plus azacitidine. For patients with a best overall response of ND who did not have progressive disease, relapse, or death, the reasons for the ND assessment included study discontinuation due to adverse event (two monotherapy, two combination therapy) and investigator decision (one monotherapy). HSCT=haemopoietic stem cell transplantation. ND=not done.
Among patients with acute myeloid leukaemia, median time to first overall response was similar for those receiving either therapy regimen (1·9 months [IQR 1·0–2·9] vs 1·9 months [IQR 1·0–3·7]; appendix p 15). For patients with relapsed or refractory acute myeloid leukaemia, median durations of all response categories were similar for monotherapy and combination therapy, although the maximum durations were generally longer for monotherapy, including patients with complete response (possibly reflecting more patients proceeding to transplant in the combination group; appendix p 15). In patients with treatment-naive acute myeloid leukaemia who received combination therapy, median duration of response was not reached, and the maximum durations were long, including 20·3 months for overall response and 19·3 months both for complete response or complete response with partial haematological recovery and for complete response.
For patients with acute myeloid leukaemia who were transfusion-dependent at baseline, 21 (36%) of 59 had 56-day transfusion independence (appendix p 23). In patients with an abnormal absolute neutrophil count at baseline, most had improvements on treatment (15 [65%] of 23 on monotherapy and 26 [70%] of 37 on combination therapy), with a median recovery time of 44 days (95% CI 15–148) on monotherapy and 29 (15–85) days on combination therapy (appendix p 16).
Median follow-up was 6·7 months (IQR 2·5–12·4) for monotherapy and 9·3 months (3·0–14·2) for combination, with one (4%) of 25 patients on monotherapy and seven (18%) of 39 patients on combination therapy ongoing at data cutoff. The Kaplan-Meier estimate of median overall survival for patients with relapsed or refractory acute myeloid leukaemia was 8·7 months (95% CI 2·5–not estimable) for monotherapy (13 [59%] of 22 patients had an event) and 12·1 (4·2–not estimable) months for combination therapy (12 [46%] of 26 patients had an event; figure 3). In treatment-naive acute myeloid leukaemia, median overall survival was 8·8 months (0·6–14·5) for monotherapy (three [75%] of four patients had an event) and not reached (9·6 months–not estimable) for combination therapy (four [31%] of 13 patients had an event). The estimated 12-month overall survival was lower for monotherapy in acute myeloid leukaemia (relapsed or refractory, 44% [95% CI 23–64]; treatment-naive, 50% [6–85]) than for combination therapy (relapsed or refractory, 54% [32–71]; treatment-naive, 75% [41–91]). The Kaplan-Meier estimate of median progression-free survival for patients with relapsed or refractory acute myeloid leukaemia was 16·3 weeks (95% CI 5·1–24·0) for monotherapy, with 18 (82%) of 22 patients having an event. Median progression-free survival for combination therapy was 19·6 weeks (11·1–23·7) with 19 (73%) of 26 patients having an event (appendix p 24).
Figure 3: Overall survival in patients with acute myeloid leukaemia.
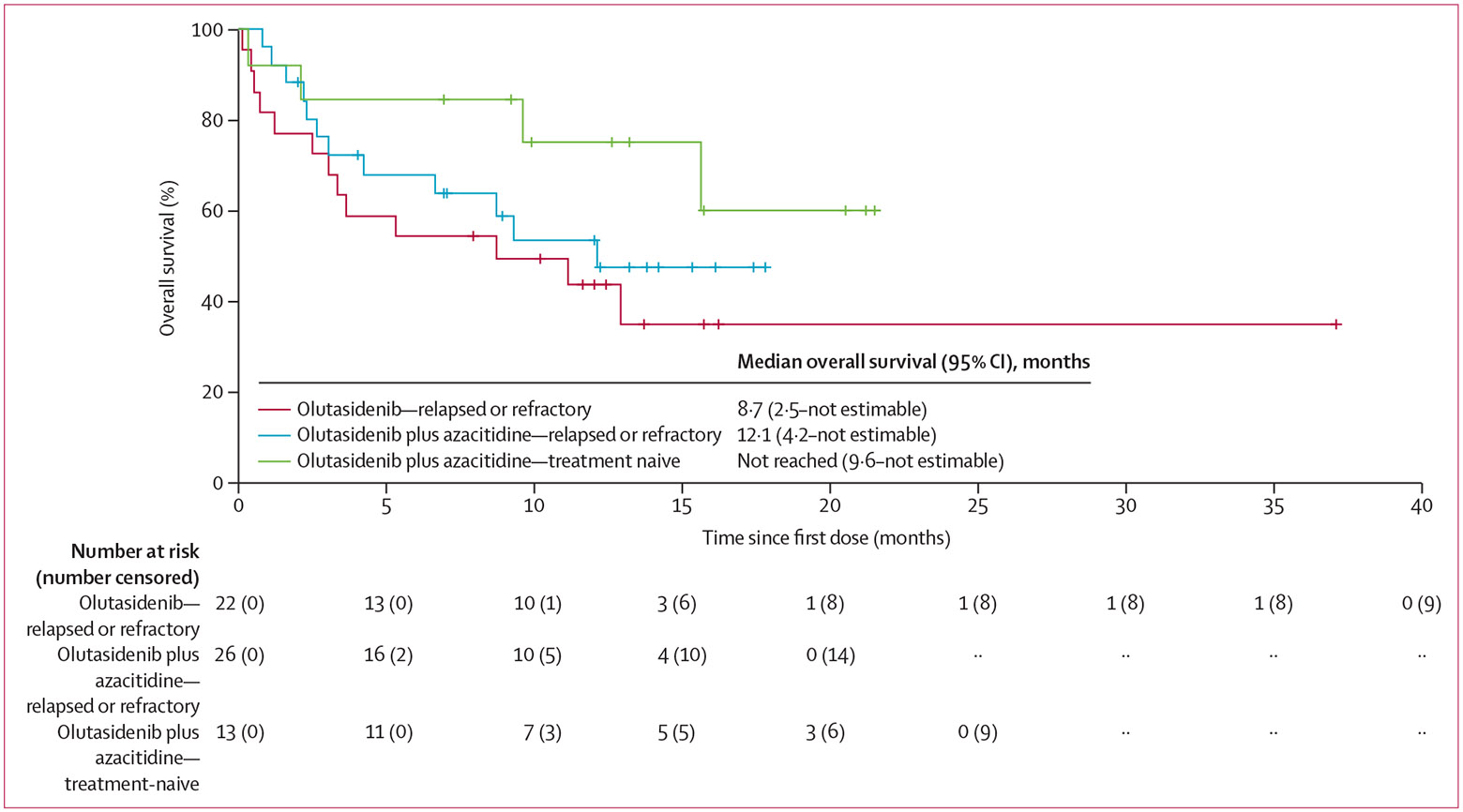
The Kaplan-Meier curve shows the survival probability of patients with relapsed or refractory acute myeloid leukaemia who received olutasidenib or olutasidenib plus azacitidine and patients with treatment-naive acute myeloid leukaemia who received olutasidenib plus azacitidine. There were too few patients at risk with treatment-naive acute myeloid leukaemia who received olutasidenib (n=4) for the results to be reliably meaningful, so this cohort was not included.
For patients with myelodysplastic syndrome, median follow-up was 12·5 months (IQR 6·9–17·8) for monotherapy, with two (33%) of six patients ongoing at data cutoff, and 15·2 months (10·2–17·5) for combination therapy, with three (43%) of seven patients ongoing. For patients in the monotherapy group (two treatment-naive; four relapsed or refractory [all four relapsed or refractory previously treated with a hypomethylating agent]), two (33%) of six patients had an overall response, with one (17%) reaching complete response (appendix p 17). For patients receiving combination therapy (five treatment-naive; two relapsed or refractory [both relapsed or refractory previously treated with a hypomethylating agent]), six (86%) of seven had an overall response, with four (57%) reaching complete response. For patients with relapsed or refractory myelodysplastic syndrome previously treated with a hypomethylating agent, one (25%) of four patients receiving monotherapy and one (50%) of two patients receiving combination therapy had an overall response. Median duration of response, in months, was not reached for monotherapy (95% CI 6·7–not reached) or combination therapy (12·8–not reached; appendix p 17). All responders with myelodysplastic syndrome (one [50%] of two for monotherapy and four [67%] of six for combination therapy) had a complete response, except one patient in the monotherapy group and two patients in the combination therapy group who had a best response of marrow complete response.
Central testing confirmed the presence of mutant IDH1 at study entry in 56 (95%) of 59 patients with both local and central results available (appendix p 25). There was strong correlation between blood and bone marrow aspirate droplet digital PCR (ddPCR) results (r2=0·76, n=16), as well as ddPCR and next generation sequencing results (r2=0·93, n=216 for all timepoints collected for all patients; appendix p 26). Co-mutation analysis using next-generation sequencing was performed in 58 patients. Genes mutated in more than 10% of patients with acute myeloid leukaemia included DNMT3A, NPM1, SRSF2, NRAS, RUNX1, ASXL1, FLT3, BCOR, PTPN11, and TET2 (appendix p 18). For patients with myelodysplastic syndrome, the reported co-mutations were similar to those observed in acute myeloid leukaemia, although with different frequencies (appendix p 27).
Analysis of variant allele frequency (VAF) across treatment cycles in 31 patients with acute myeloid leukaemia showed a transient increase in IDH1 VAF after initiation of treatment, with a subsequent decrease. Ten (40%) of 25 patients with acute myeloid leukaemia with an overall response, and three (50%) of six patients with stable disease had a clinically significant reduction in VAF (reduction to <1%; appendix p 25). Four (44%) of nine patients with myelodysplastic syndrome and an overall response had VAF reduction to less than 1%.
Discussion
In this first-in-human study, olutasidenib was well tolerated in patients with acute myeloid leukaemia and myelodysplastic syndrome, either as monotherapy or in combination with azacitidine. No dose-limiting toxicities occurred in the dose escalation cohorts; hence olutasidenib 150 mg twice per day is the recommended phase 2 dose based on an acceptable safety profile, optimal pharmacokinetic exposure, a robust 2-hydroxyglutarate response, and early signs of clinical activity.
Overall, adverse events were similar for olutasidenib monotherapy and combination therapy, apart from more frequent thrombocytopenia and neutropenia with the combination. This difference might result from the increased risk of haematological toxicities associated with hypomethylating agents such as azacitidine.19 In our study, the incidence of differentiation syndrome was 13% both for olutasidenib monotherapy and combination therapy, which is similar to commercially available IDH inhibitors.7,20-22 Azacitidine did not appear to mitigate or worsen the frequency or severity of differentiation syndrome. Transient liver toxicity was observed in 10 (13%) of 78 patients, with most patients able to continue therapy following dose interruption or modification. Transient QT prolongation was observed in only three (4%) of 78 patients, all of whom were taking concomitant medications associated with QT prolongation and were able to continue on study.
Olutasidenib showed meaningful clinical activity in patients with IDH1-mutated acute myeloid leukaemia. Although combination of IDH1 inhibitor therapy with azacitidine has been previously reported in treatment-naive patients with acute myeloid leukaemia,23,24 to our knowledge, our study provides the first assessment of this combination in patients with relapsed or refractory acute myeloid leukaemia. Among patients receiving combination therapy, response rates were higher in treatment-naive acute myeloid leukaemia (77% had an overall response, 54% had a complete response or complete response with partial haematological recovery) than in relapsed or refractory acute myeloid leukaemia (46% had an overall response, 15% a complete response or complete response with partial haematological recovery). These results, when taken together with the established benefit of the venetoclax–azacitidine combination in treatment-naive acute myeloid leukaemia,25 provide a strong rationale for future evaluation of either sequential or triplet therapy in this setting. Olutasidenib, both alone and in combination with azacitidine, also produced durable responses in patients with relapsed or refractory acute myeloid leukaemia, thereby showing the therapeutic potential of IDH1 inhibition in this poor-prognosis population, as previously reported for ivosidenib monotherapy.7 Although response rates were similar for olutasidenib monotherapy and for the combination in relapsed or refractory acute myeloid leukaemia, it is noteworthy that the number of patients bridged to transplant, and the median overall survival were higher with the combination.
Although median duration of response was similar for monotherapy and the combination, maximum durations were generally longer with monotherapy. However, duration of response assessments ended comparatively early in patients undergoing allogeneic transplantation, and more so for patients in the combination group (four monotherapy; eight combination therapy). Resuming olutasidenib therapy post-transplantation to mitigate the risk of relapse could also be a viable option for patients undergoing HSCT after reaching a response, particularly for those with relapsed or refractory disease. This option is being prospectively investigated.
For patients with relapsed or refractory acute myeloid leukaemia who received olutasidenib monotherapy, 32% had a complete response or complete response with partial haematological recovery and median overall survival was 8·7 months (95% CI 2·5–not estimable). These results are comparable with outcomes reported for the IDH1 inhibitor ivosidenib in patients with relapsed or refractory acute myeloid leukaemia—30% had a complete response or complete response with partial haematological recovery and the median overall survival was 8·8 months (95% CI 6·7–10·2).7 Similarly, activity results for the olutasidenib and azacitidine combination (77% reaching overall response; 54% reaching complete response or complete response with partial haematological recovery) are comparable with those reported for ivosidenib and azacitidine in patients with treatment-naive acute myeloid leukaemia (78% reaching overall response; 61% reaching complete response).23 Olutasidenib appeared to be minimally myelosuppressive, and comparable numbers of patients reached transfusion independence in monotherapy and combination groups. Moreover, in relapsed or refractory acute myeloid leukaemia, the median overall survival of 12·1 months (4·2–not estimable) for patients who received combination therapy compares favourably with historical controls and warrants further exploration.
Further evidence of olutasidenib activity is provided by a clinically significant VAF reduction (<1%) in 40% of responders with acute myeloid leukaemia, and in 50% of those with stable disease. Overall, patient numbers were insufficient to conclude whether specific co-mutations, VAF, or mutation burden correlated with response.
In the myelodysplastic syndrome cohort, four of nine responding patients also had mutation clearance. Complete responses were observed with both monotherapy and combination therapy, with 86% of patients having an overall response and 57% a complete response in the combination group. To our knowledge, our dataset provides the first evaluation of an IDH1 inhibitor with azacitidine in patients with myelodysplastic syndrome, and the first evaluation showing clinically significant activity of IDH inhibitor monotherapy in myelodysplastic syndrome.26
One limitation of our study is the open-label design, whereby patients were not randomly assigned to treatment, thus the observed response rates could be affected by selection bias. More specifically, the activity of combination therapy might consequently appear to be more encouraging than that of monotherapy if healthier or younger patients were allocated to treatment with olutasidenib and azacitidine, instead of olutasidenib alone. A further limitation of the study is the small sample size, with relatively few patients with treatment-naive acute myeloid leukaemia or myelodysplastic syndrome enrolled. More specifically, with only four patients with treatment-naive acute myeloid leukaemia treated with olutasidenib monotherapy, it is difficult to assess the true response rate in this patient population and thus additional studies are required. Similarly, although our initial findings for patients with myelodysplastic syndrome are encouraging, additional studies in this population are warranted.
In summary, the results of this phase 1 study show that olutasidenib is a promising drug, both alone and in combination with azacitidine, for patients with IDH1-mutated acute myeloid leukaemia and myelodysplastic syndrome. The overall safety profile and preliminary evidence of clinical activity, including durable responses in all treatment cohorts and a clinically significant reduction in VAF, provide rationale for the continued evaluation of olutasidenib in multiple patient populations with myeloid malignancies.
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed with no date or language restrictions using the terms “ivosidenib”, “isocitrate dehydrogenase 1 (IDH1) inhibitor”, “clinical trial”, “acute myeloid leukaemia”, and “myelodysplastic syndrome”. There were no restrictions to the search and all studies were examined. Two key reports emerged showing that the mutant isocitrate dehydrogenase 1 (IDH1) inhibitor ivosidenib has shown meaningful clinical activity as a single agent in relapsed or refractory acute myeloid leukaemia, and as a monotherapy or in combination with azacitidine in treatment-naive patients with acute myeloid leukaemia.
Added value of this study
This study showed that olutasidenib has the potential to provide an additional treatment option for IDH1-mutated acute myeloid leukaemia that might have a different therapeutic and safety profile than currently available treatments. To our knowledge, our study is the first to test an IDH1 inhibitor (olutasidenib) in combination with azacitidine in relapsed or refractory acute myeloid leukaemia, showing high response rates and a median overall survival of 12·1 months. To our knowledge, we also report the first available data on patients with myelodysplastic syndrome treated with an IDH1 inhibitor plus azacitidine, where we observed a strong preliminary activity signal. We also show clinical activity in relapsed or refractory acute myeloid leukaemia with olutasidenib monotherapy, and in treatment-naive acute myeloid leukaemia with olutasidenib and azacitidine combination therapy.
Implications of all the available evidence
Mutant IDH1 inhibitors (ivosidenib and olutasidenib) are well tolerated and show meaningful clinical activity as monotherapy in treatment-naive or relapsed or refractory acute myeloid leukaemia and in combination with azacitidine in treatment-naive, and now relapsed or refractory acute myeloid leukaemia. Our Article expands the purview of IDH1 inhibitors, where we show clinical activity with combination therapy in relapsed or refractory acute myeloid leukaemia and with monotherapy and combination therapy in myelodysplastic syndrome, thus warranting further clinical investigation to understand the potential of olutasidenib as a future treatment option for multiple patient populations with myeloid malignancies. Different IDH1 inhibitors might have unique safety and pharmacokinetic profiles, which allow for patient-specific treatment options based on underlying comorbidities and concomitant medications. Doublet or triplet combination strategies with venetoclax, azacitidine, and an IDH1 inhibitor in acute myeloid leukaemia look to be particularly promising. More studies are needed in patients with myelodysplastic syndrome, for whom early results are highly encouraging with combination therapy.
Acknowledgments
This study was funded by Forma Therapeutics. Medical writing support was provided by Patrick Barry (Acumen Medical Communications) and was funded by Forma Therapeutics. Data analysis support was provided by Olga Polyanskaya.
Declaration of interests
JMW has received research funding from and was a board/advisory committee member for Takeda; has received research funding from Immune System Key Ltd and Takeda; and was a board/advisory committee member for Genentech, Rafael Pharma, Reven Pharma, and Celgene/Bristol-Myers Squib (BMS). MRB has received research funding for her institution from AbbVie, Forma Therapeutics, Kite, Kura, and Takeda. JY has received research funding from Seattle Genetics, Janssen, AROG, Loxo Oncology, and Agios. TP is a consultant to Curios and Daiichi; and has received research funding, is a consultant to, and has recently become an employee of BMS. SL is a consultant to AstraZeneca, BMS, Helsinn, Innate Pharma, and PIN Pharma, and has recently become an employee of Janssen Research and Development. GJS has received research funding, honoraria, is on the speakers’ bureau for, and is a board/advisory committee member for Agios, Gamida, Gilead, and Incyte; has received research funding, honoraria, is on the speakers’ bureau for, holds stock in, and is a board/advisory committee member for Amgen and BMS; has received research funding, honoraria, and is a board/advisory committee member for Novartis, Ono Pharma, and AVM Biotech; has received honoraria, is on the speakers’ bureau for, and is a board/advisory committee member for GlaxoSmithKline (GSK); has received research funding and holds stock in Janssen/Johnson & Johnson; has received research funding and is on the speakers’ bureau for AbbVie, Astellas, Celgene, Karyopharm, and Stemline; has received honoraria and is a board/advisory committee member for AstraZeneca; and has received research funding from Actinium, Actuate, Ambit, Cellectis, Cyclacel, Constellation, Daiichi-Sankyo, Deciphera, DeltaFly, Forma Therapeutics, FujiFilm, Genentech/Roche, Geron, Glycomimetics, Kura Oncology, Mateon, Medimmune, Millennium; Onconova, Pfizer, PrECOG, RegImmune, Sangamo, Samus, Sellas, Tolero, and Trovagene. SND has no conflicts to disclose. AP has received honoraria from and is a consultant to AbbVie; is a consultant to Gilead; and has received honoraria from Astellas, Agios, Pfizer, and Jazz Pharmaceuticals. PM has received research funding from, is a consultant to, and is on the speakers’ bureau for BMS; and is a consultant to Forma Therapeutics, Syndax, and Kura Oncology.
ESW is a consultant to and a board/advisory committee member for AbbVie and Gilead; is a consultant to, and is on the speakers’ bureau for Astellas, Pfizer, and Stemline; is a board/advisory committee member for Rafael Pharmaceuticals; is a consultant to Amgen, BMS, GSK, Janssen, Jazz Pharmaceuticals, Kite, Mana Therapeutics, Novartis, PharmaEssentia; and is on the speakers’ bureau for Kura Oncology and Dava Oncology. KPS has received research funding and honoraria from, is a consultant to, and is on the speakers’ bureau of Jazz Pharmaceuticals; has received honoraria from, is a consultant to, and is on the speakers’ bureau for Novartis; has received honoraria from and is on the speakers’ bureau for Incyte; and has received research funding from Rafael, Glycomimetics, and Celgene. AHW has received research funding, honoraria, is a consultant to, is on the speakers’ bureau for, and is a board/advisory committee member for Astellas; has received research funding, honoraria, is on the speakers’ bureau for, and is a board/advisory committee member for AbbVie/Genentech, Amgen, Celgene/BMS, and Novartis; has received research funding, honoraria, is a consultant to, is a board/advisory committee member for Servier and Syndax; has received honoraria, is a consultant to, and is a board/advisory committee member for Janssen and Gilead; has received honoraria, and is a board/advisory committee member for MacroGenetics and Pfizer; has received research funding and honoraria from, and is a board/advisory committee member for AstraZeneca; has received research funding from Astex; and is an employee of the Walter and Eliza Hall Institute and is eligible for a fraction of the royalty stream related to venetoclax. SDB has received honoraria and research funding from Forma Therapeutics, has received honoraria, research funding, and is a consultant to Agios; has received honoraria, is a consultant to, and is on the speakers’ bureau for Celgene; has received honoraria and is a consult to Astellas, Daiichi Sankyo, Syros, AbbVie, Bayer, and Janssen; has received honoraria from Seattle Genetics; and is a consultant to Pierre Fabre, Novartis, Pfizer, and Servier. MA is a consultant to and a board/advisory committee member for BMS-Celgene and Novartis; and is a consultant to Astellas, Jazz Pharmaceuticals, and Pfizer. WD is a consultant to Janssen, BMS, and BeiGene. APS has received honoraria from AbbVie, Novartis, and Amgen; and is a board/advisory committee member for Pfizer. CR has received research funding from MaaT Pharma; has received honoraria from Incyte and Janssen; has received honoraria and has membership on an entity’s board of directors or advisory committee for MacroGenics, Pfizer, and Takeda; has received honoraria, research funding, and has membership on an entity’s board of directors or advisory committee for Amgen, Astellas, BMS/Celgene and Roche; and has received honoraria, research funding, is a consultant to, and has membership on an entity’s board of directors or advisory committee for AbbVie, Daiichi Sankyo, Jazz Pharmaceuticals, and Novartis. BAJ has received research funding to his institution, is a consultant to, and is a board/advisory committee member for AbbVie, BMS, Genentech/Roche, Jazz Pharmaceuticals, Pfizer, and Treadwell; has received research funding to his institution, is a consultant to, and is a data monitoring committee member for Gilead; has received research funding to his institution, is a consultant to, and is a protocol steering committee member for GlycoMimetics; is a consultant to and a board/advisory committee member for Servier, Takeda, and Tolero; has received travel reimbursement from AbbVie; has received research funding to his institution from 47, Accelerated Medical Diagnostics, Amgen, AROG, Celgene, Daiichi Sankyo, F. Hoffman-La-Roche, Forma Therapeutics, Hanmi, Immune-Onc, Incyte, Loxo Oncology, LP Therapeutics, Pharmacyclics, and Sigma Tau. PBF has received research funding from Forma Therapeutics, Astex Pharmaceuticals, and Incyte. CM has received honoraria from Astellas, BMS, and Celgene. PK, JS, SF, SMG, and JB are employees of and hold stock in Forma Therapeutic. PH was an employee of and holds stock in Forma Therapeutics; and is an employee of and holds stock in Kymera Therapeutics. HM was an employee of and holds stock in Forma Therapeutics. JEC has received research funding for his institution from, and is a consultant to BMS, Novartis, Pfizer, Takeda, Daiichi, Jazz Pharmaceuticals, Merus, and Forma Therapeutics; has received research funding for his institution from Astellas and Amphivena; and is a consultant to BiolineRx and Bioptah. All authors had access to and had the opportunity to review the study data and are responsible for data analysis and interpretation. All authors participated in writing of the report (including original draft, review, and editing). All authors attest to study completeness, and accuracy of the data and data analysis. JMW and JEC participated in analysis of the data, and directly accessed and verified the underlying data reported in the manuscript. JMW had final responsibility for the decision to submit for publication.
Contributor Information
Justin M Watts, University of Miami Sylvester Comprehensive Cancer Center, Miami, FL, USA.
Maria R Baer, University of Maryland Greenebaum Comprehensive Cancer Center, Baltimore, MD, USA.
Jay Yang, Karmanos Cancer Institute, Detroit, MI, USA.
Thomas Prebet, Department of Hematology, Yale University, New Haven, CT, USA.
Sangmin Lee, Department of Hematology and Oncology, Weill Cornell Medicine, New York, NY, USA.
Gary J Schiller, David Geffen School of Medicine at University of California, Los Angeles, CA, USA.
Shira N Dinner, Department of Hematology and Oncology, Northwestern University, Chicago, IL, USA.
Arnaud Pigneux, Centre Hospitalier Universitaire Bordeaux, Bordeaux, France.
Pau Montesinos, Hospital Universitari i Politècnic La Fe, Valencia, Spain.
Eunice S Wang, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA.
Karen P Seiter, New York Medical College, New York, NY, USA.
Andrew H Wei, The Alfred Hospital and Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia.
Stephane De Botton, Institut Gustave-Roussy, Villejuif, France.
Montserrat Arnan, Institut Català d’Oncologia-Hospital Duran i Reynals, IDIBELL, Hospitalet Llobregat, Barcelona, Spain.
Will Donnellan, Sarah Cannon Research Institute at Tennessee Oncology, Nashville, TN, USA.
Anthony P Schwarer, Eastern Health Monash University Clinical School and Austin Hospital, Melbourne, Australia.
Christian Récher, Centre Hospitalier Universitaire de Toulouse, Institut Universitaire du Cancer Toulouse—Oncopole, Toulouse, France.
Brian A Jonas, University of California, Davis Comprehensive Cancer Center, Sacramento, CA, USA.
P Brent Ferrell, Jr, Vanderbilt University, Nashville, TN, USA.
Christophe Marzac, Institut Gustave-Roussy, Villejuif, France.
Patrick Kelly, Forma Therapeutics, Watertown, MA, USA.
Jennifer Sweeney, Forma Therapeutics, Watertown, MA, USA.
Sanjeev Forsyth, Forma Therapeutics, Watertown, MA, USA.
Sylvie M Guichard, Forma Therapeutics, Watertown, MA, USA.
Julie Brevard, Forma Therapeutics, Watertown, MA, USA.
Patrick Henrick, Forma Therapeutics, Watertown, MA, USA.
Hesham Mohamed, Forma Therapeutics, Watertown, MA, USA.
Jorge E Cortes, Georgia Cancer Center, Augusta, GA, USA.
Data sharing
Data sharing requests will be considered on a case-by-case basis and may be sent to datasharing@rigel.com.
References
- 1.de Botton S, Watts J, Baer M, et al. PS1051 FT-2102, an IDH1m inhibitor, induces mutation clearance in patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) treated in phase 1 dose escalation and expansion study. HemaSphere 2019; 3: 475–76 (abstr). [Google Scholar]
- 2.DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol 2015; 90: 732–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 2010; 207: 339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov 2013; 3: 730–41. [DOI] [PubMed] [Google Scholar]
- 5.Losman JA, Looper RE, Koivunen P, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013; 339: 1621–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012; 483: 474–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med 2018; 378: 2386–98. [DOI] [PubMed] [Google Scholar]
- 8.Fan B, Le K, Manyak E, et al. Longitudinal pharmacokinetic/pharmacodynamic profile of AG-120, a potent inhibitor of the IDH1 mutant protein, in a phase 1 study of IDH1-mutant advanced hematologic malignancies. Blood 2015; 126: 1310. [Google Scholar]
- 9.de la Fuente MI, Colman H, Rosenthal M, et al. Olutasidenib (FT-2102) in patients with relapsed or refractory IDH1-mutant glioma: a multicenter, open-label, phase 1b/2 trial. Neuro-oncol 2022; noac139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abou-Alfa GK, Macarulla T, Javle MM, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2020; 21: 796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norsworthy KJ, Luo L, Hsu V, et al. FDA approval summary: ivosidenib for relapsed or refractory acute myeloid leukemia with an isocitrate dehydrogenase-1 mutation. Clin Cancer Res 2019; 25: 3205–09. [DOI] [PubMed] [Google Scholar]
- 12.Watts J, Baer MR, Yang J, et al. Phase 1 study of the IDH1m inhibitor FT-2102 as a single agent in patients with IDH1m acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS). Blood 2018; 132 (suppl 1): 1453 (abstr). [Google Scholar]
- 13.Caravella JA, Lin J, Diebold RB, et al. Structure-based design and identification of FT-2102 (olutasidenib), a potent mutant-selective IDH1 inhibitor. J Med Chem 2020; 63: 1612–23. [DOI] [PubMed] [Google Scholar]
- 14.Lin J, Lu W, Caravella JA, et al. Discovery and optimization of quinolinone derivatives as potent, selective, and orally bioavailable mutant isocitrate dehydrogenase 1 (mIDH1) inhibitors. J Med Chem 2019; 62: 6575–96. [DOI] [PubMed] [Google Scholar]
- 15.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol 2003; 21: 4642–49. [DOI] [PubMed] [Google Scholar]
- 16.Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006; 108: 419–25. [DOI] [PubMed] [Google Scholar]
- 17.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017; 130: 722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Storer BE. Design and analysis of phase I clinical trials. Biometrics 1989; 45: 925–37. [PubMed] [Google Scholar]
- 19.Gao C, Wang J, Li Y, et al. Incidence and risk of hematologic toxicities with hypomethylating agents in the treatment of myelodysplastic syndromes and acute myeloid leukopenia: a systematic review and meta-analysis. Medicine (Baltimore) 2018; 97: e11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol 2018; 4: 1106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Issa GC, DiNardo CD. Acute myeloid leukemia with IDH1 and IDH2 mutations: 2021 treatment algorithm. Blood Cancer J 2021; 11: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norsworthy KJ, Mulkey F, Scott EC, et al. Differentiation syndrome with ivosidenib and enasidenib treatment in patients with relapsed or refractory IDH-mutated AML: a U.S. Food and Drug Administration systematic analysis. Clin Cancer Res 2020; 26: 4280–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DiNardo CD, Stein AS, Stein EM, et al. Mutant isocitrate dehydrogenase 1 inhibitor ivosidenib in combination with azacitidine for newly diagnosed acute myeloid leukemia. J Clin Oncol 2021; 39: 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montesinos P, Recher C, Vives S, et al. Ivosidenib and azacitidine in IDH1-mutated acute myeloid leukemia. N Engl J Med 2022; 386: 1519–31. [DOI] [PubMed] [Google Scholar]
- 25.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med 2020; 383: 617–29. [DOI] [PubMed] [Google Scholar]
- 26.DiNardo CD, Hochhaus A, Frattini MG, et al. A phase 1 study of IDH305 in patients with IDH1R132-mutant acute myeloid leukemia or myelodysplastic syndrome. J Cancer Res Clin Oncol 2022; published online March 30. 10.1007/s00432-022-03983-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sharing requests will be considered on a case-by-case basis and may be sent to datasharing@rigel.com.