

Abstract
Adenine phosphoribosyltransferase (APRT) deficiency is a rare autosomal recessive disorder that leads to the accumulation of poorly soluble 2,8-dihydroxyadenine (DHA) in the kidneys, resulting in a variety of renal presentations including nephrolithiasis, acute kidney injury, and chronic kidney disease (CKD) caused by crystal nephropathy. Here, we report a case of a 43-year-old man with 2,8-DHA crystalline nephropathy caused by APRT deficiency strongly suspected by renal biopsy results and definitively diagnosed by a urine gas chromatography–mass spectrometry (GC/MS)-based plasma metabolomic assessment. This case represents the importance of awareness and recognition of the signs and symptoms of this rare condition and its progression to CKD, which can be prevented by the early administration of xanthine oxidoreductase inhibitors.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13730-022-00768-1.
Keywords: Adenine phosphoribosyltransferase deficiency; 2,8-Dihydroxyadenine crystal nephropathy; Xanthine oxidoreductase
Introduction
Adenine phosphoribosyltransferase (APRT) deficiency is a rare autosomal recessive disorder of adenine metabolism with more than 40 known mutations and an estimated prevalence of 1 in 50,000–100,000 persons [1–6]. Adenine accumulates in patients with APRT deficiency and is converted to large amounts of dihydroxyadenine (DHA) by xanthine oxidoreductase (XOR; xanthine dehydrogenase/oxidase) [1–6].
As a result, APRT deficiency may show various presentations, most commonly, kidney stones comprising 2,8-DHA crystals in the urine observed by microscopy, acute kidney injury (AKI), and progressive chronic kidney disease (CKD) caused by DHA crystal nephropathy. When 2,8-DHA excretion in urine continues and crystal deposition occurs in the tubules and interstitium, this can lead to tubulointerstitial inflammation and fibrosis, resulting in progression of kidney dysfunction [1–7]. Many individuals remain asymptomatic for years, and at least 15–20% of reported adult patients have already developed end-stage renal disease at the time of diagnosis [1, 5, 8–10]. Recently, XOR inhibitors have been considered effective against 2,8-DHA crystalline nephropathy progression [1, 4, 10]; therefore, early diagnosis and treatment intervention with XOR inhibitors should be performed. However, because of the variability in the clinical characteristics of APRT deficiency, the optimal diagnostic strategy for APRT deficiency remains unknown.
We report a case of 2,8-DHA crystalline nephropathy caused by APRT deficiency strongly suspected by renal biopsy results and definitively diagnosed by urine gas chromatography–mass spectrometry (GC/MS)-based plasma metabolomic assessment results, regardless of the absence of urine 2,8-DHA crystals. This case indicates the importance of early diagnosis of APRT deficiency in optimizing the management and prevention of the progression of kidney dysfunction.
Case report
The patient was a 43-year-old man. Four days before admission, he started experiencing frequent vomiting, diarrhea, and general fatigue; he presented with these symptoms to the local physician. He was prescribed probiotics, and his gastrointestinal symptoms improved; however, his severe general fatigue continued. Therefore, he visited our hospital. He was diagnosed with severe anemia (hemoglobin, 6.9 g/dL) and severe kidney dysfunction (serum creatinine, 9.35 mg/dL) and was admitted. Upper endoscopy revealed no gastrointestinal bleeding.
Although he had not received regular health examinations and no previous laboratory data had been obtained, he had a history of urinary tract stones during childhood and in his 30 s. The cause of the stones was not, however, further evaluated. Of his four brothers, one also had one episode of urinary tract stones during childhood. His grandfather had received hemodialysis for end-stage kidney disease caused by diabetic kidney disease.
A physical examination was performed at admission and indicated the following: height, 179.8 cm; weight, 63.2 kg; blood pressure, 109/68 mmHg; pulse, 89 beats/minute; and body temperature, 36.3 °C. Pallor was observed in the conjunctiva of the eyelid, but there were no other abnormalities. Upon admission, laboratory tests showed a serum creatinine level of 9.35 mg/dL, uric acid level of 9.6 mg/dL, the urinary protein level of 0.41 g/day (without albuminemia but with a high degree of β2 microglobulin), white blood cell count of 10 to 19/high power field, and no hematuria (Table 1). Fractional excretion of urea nitrogen was 60.5%, and size of inferior vena cava was 20 mm without respiratory collapse, suggesting that there were no obvious evidence of volume depletion. The hemoglobin level was 6.9 g/dL without hemolysis or iron deficiency. The erythropoietin level was low. The results of the stool blood test were negative, suggesting that his anemia was caused by renal dysfunction. The immunological examination showed no abnormal findings other than slightly elevated IgG4 levels. Computed tomography (CT) showed a single small stone in the right kidney (Fig. 1); however, no hydronephrosis or mild cortical atrophy was observed (the sizes of the kidney were 85 mm × 37 mm in the right, and 90 × 46 mm in left kidney). No abnormal findings were observed on ultrasound cardiography. Aggressive rehydration was performed in addition to administering erythropoiesis-stimulating agents (darbepoetin alfa 30 U/month) and two units of red blood cells were transfused at a time; however, severe renal dysfunction persisted. Therefore, an echo-guided renal biopsy was performed on day 9.
Table 1.
Main laboratory data on admission
| Variables | Normal values | Data |
|---|---|---|
| Hematological parameters | ||
| White blood cells (/μL) | 3300–8600 | 4070 |
| Hemoglobin (g/dL) | 11.6–14.8 | 6.9 |
| Schistocytes | Negative | Negative |
| Haptoglobin (mg/dL) | 15–116 | 87 |
| Platelets (/uL) | 158,000–348,000 | 218,000 |
| Biochemical parameters | ||
| TP (g/dL) | 6.6–8.1 | 7.2 |
| Alb (g/dL) | 4.1–5.1 | 4.0 |
| Na (mEq/L) | 135–140 | 137 |
| K (mEq/L) | 3.5–4.5 | 4.4 |
| Cl (mEq/L) | 101–108 | 105 |
| BUN (mg/dL) | 8.0–20 | 84.3 |
| Cre (mg/dL) | 0.46–0.79 | 9.35 |
| eGFR (mL/min/1.73 m2) | 5.8 | |
| Ca (mg/dL) | 8.5–10.0 | 9.8 |
| P (mg/dL) | 2.5–4.5 | 5.9 |
| UA (mg/dL) | 3.5–7.2 | 9.6 |
| LDH (U/L) | 124–222 | 166 |
| CRP (mg/dL) | 0.0–0.14 | 3.44 |
| Erythropoietin (mIU/mL) | 2.6–18.5 | 4.0 |
| Fe (µg/dL) | 70–160 | 121 |
| Ferritin (ng/mL) | 20–250 | 381 |
| Intact-PTH (pg/mL) | 10–65 | 146 |
| Hemoglobin A1c (%) | < 6.5 | 5.7 |
| Immunologic parameters | ||
| IgG (mg/dL) | 861–1747 | 1376 |
| IgA (mg/dL) | 93–393 | 299 |
| IgM (mg/dL) | 50–269 | 106 |
| IgG4 (mg/dL) | < 135 | 158 |
| C3 (mg/dL) | 86–160 | 132 |
| C4 (mg/dL) | 17–45 | 49 |
| CH50 (U/mL) | 30–40 | 49.4 |
| MPO-ANCA (U/mL) | < 3.5 | < 1.0 |
| PR3-ANCA (U/mL) | < 3.5 | < 1.0 |
| ANA | < 40 | < 40 |
| RF (U/mL) | < 15 | < 4.0 |
| Anti SS-A antibody (U/mL) | < 10 | < 1.0 |
| Venous blood gas analysis | ||
| pH | 7.35–7.45 | 7.270 |
| HCO3− (mmol/L) | 23–28 | 15.9 |
| Urinalysis test | ||
| Urine red blood cell (/HPF) | 1–4 | |
| Urine white blood cell (/HPF) | 10–19 | |
| Urinary protein (g/day) | < 0.15 | 0.41 |
| Urinary β2-MG (µg/L) | < 289 | 34,800 |
| Fecal occult blood test | Negative | Negative |
TP total protein, Alb albumin, BUN blood urea nitrogen, Cre creatine, eGFR estimated glomerular filtration rate, Ca calcium, P phosphate, UA uric acid, LDH lactate dehydrogenase, CRP C-reactive protein, PTH parathyroid hormone, MPO myeloperoxidase, PR3 proteinase-3, ANCA anti-neutrophil cytoplasmic antibody, ANA antinuclear antibodies, RF rheumatoid factor, β2-MG β-2 microglobulin
Fig. 1.

Computed tomography image. The kidney stone is indicated by the arrow
Renal pathology findings are shown in Figs. 2, 3 and 4. Four of 34 glomeruli were sclerotic, and no significant pathological alterations were observed on light microscopy. Although the tubulointerstitium was largely injured with tubular atrophy (Fig. 2), no storiform fibrosis or plasma cell infiltration suggestive of IgG4-related kidney disease was observed [11]. There was a green/brown crystalline material within the tubular lumina, and epithelial cells eliciting a multinucleate giant cell reaction were often observed (Fig. 3). Polarized light microscopy revealed that the crystals were refractile (Fig. 4). All immunofluorescence results were negative (Supplementary Fig. 1), and no obvious abnormal findings were observed by electron microscopy. Based on the characteristic findings of the crystals, tubulointerstitial nephropathy caused by 2,8-DHA crystals was suspected. Before obtaining the renal biopsy results, tubulointerstitial nephritis was suspected and steroid treatment was started from day 11. However, because 2,8-DHA crystals were suspected based on the renal pathology findings, steroids were tapered quickly, and febuxostat (10 mg/day) was started. Subsequently, the febuxostat dosage was increased to 30 mg/day on day 28, and the serum creatinine level improved to 4 mg/dL at 2 months. Further examination was performed by analyzing the urine metabolome using GC/MS at the Japan Clinical Metabolomics Institute (http://www.jc-metabolomics.com/index_e.html) on day 44 of admission (Fig. 5). The results showed a marked increase in urinary 2,8-DHA (9.1SD), 8-hydroxyadenine (7.7 SD), and adenine (8.4 SD); therefore, a chemical diagnosis of APRT deficiency-induced 2,8-DHA crystalline nephropathy was confirmed. Subsequently, the patient’s kidney function continued to be stable, with a creatinine level of 4.0 mg/dL and a uric acid level of 6.2 mg/dL, at 1 year after renal biopsy (Fig. 6).
Fig. 2.
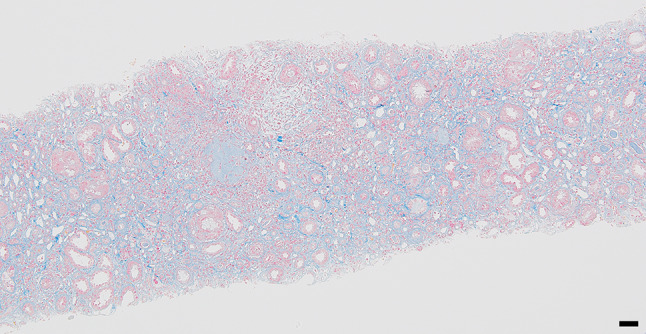
Masson-trichrome staining showing a high degree of interstitial fibrosis within the renal capsule (scale bar, 50 μm)
Fig. 3.
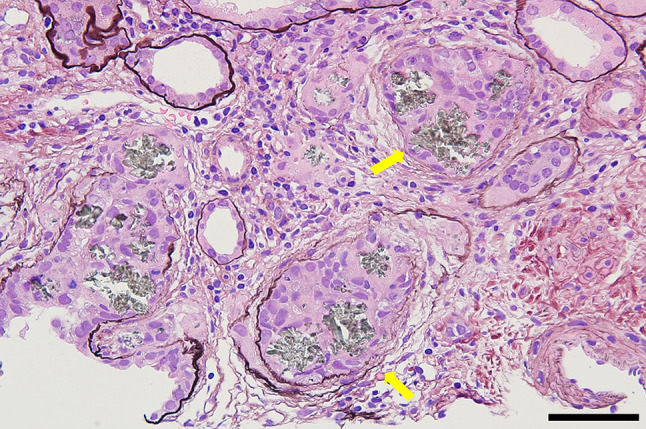
Periodic acid-methenamine-silver staining showing green/brown crystalline material within the tubular lumina and epithelial cells, eliciting a multinucleate giant cell reaction (arrows) (scale bar, 50 μm)
Fig. 4.

Polarized light microscopy showing the clearly refractile crystals (scale bar, 50 μm)
Fig. 5.

Results of the urine metabolome by GC/MS. The retention time of 8-hydroxyadenine, 2, 8-DHA, and adenine are indicated by an arrow
Fig. 6.

Clinical course. Cre creatinine, UA uric acid
Discussion
Our patient had APRT deficiency caused by 2,8-DHA crystalline nephropathy that was diagnosed by renal biopsy and urine metabolomic assessment results. The administration of XOR inhibitors could slow the progression of CKD to a stage that requires renal replacement therapy.
The first step in diagnosing APRT deficiency includes observing kidney stones and DHA crystals in the urine, especially during childhood [4, 5, 12, 13], although the sensitivity for urinary 2,8-DHA crystal detection was 64% [1]. Even though the nature of the stones was not further evaluated and urine 2,8-DHA crystals by microscopy were also not observed, in our case, the history of urinary stones during childhood and kidney stones observed on CT might have been caused by 2,8-DHA crystals.
In general, renal biopsy is not required for diagnosis if 2,8-DHA crystals are identified in the urine. However, renal biopsy (as well as polarizing microscopy) frequently leads to the diagnosis of DHA nephropathy in cases with unexplained renal failure, such as in our case. This is true even when urine 2,8-DHA crystals are not visible by microscopy because DHA nephropathy typically manifests as severe tubular injury and precipitation of crystals within the tubular lumen and in the renal interstitium [14].
Deficient erythrocyte APRT enzyme activity and mutations known to abolish APRT enzyme activity by genetic testing have been considered useful for confirming the diagnosis [8, 15]. However, since the patient refused to undergo genetic analyses, we could not obtain the results of the same.
The utility of GC/MS-based metabolomic assessment for diagnosing APRT deficiency has also been previously reported [16, 17]. The metabolomic assessment is an omics analysis method that comprehensively analyzes more than several hundred metabolites. The advantage of the metabolomic assessment is that it can quickly and easily provide a chemical diagnosis of even rare diseases, and can be used to obtain highly accurate biological information noninvasively using a small specimen.
In the present case, the metabolomic assessment of urine samples revealed significantly high levels of 2,8-DHA, 8-hydroxyadenine and adenine, which was consistent with previous studies [16, 17], leading to the firm conclusion that the patient had 2,8-DHA crystalline disease caused by APRT deficiency, even though the patient refused to undergo further examinations, and gene analyses were not performed.
Regarding the treatment of APRT deficiency, XOR inhibitors (allopurinol, febuxostat, or topiroxostat) effectively reduce urinary DHA excretion, leading to the suppression of 2,8-DHA crystal-induced kidney dysfunction [1, 4, 10, 18]. In our case, after the administration of febuxostat, kidney function gradually recovered. Although we could not definitely conclude that the administration of only this drug could improve kidney function, febuxostat might help reduce 2,8-DHA crystal formation, resulting in the clearance of tubular crystal obstruction. and leading to renal recovery and the avoidance of dialysis. If the patient had been diagnosed with kidney stones during childhood and started XOR inhibitors, the renal prognosis of may improve.
In our case, 30 mg/day of febuxostat was used, following which the uric acid levels decreased to the normal range. However, no optimal dose of XOR inhibitors has been identified, or therapeutic milestone achieved, because the appropriate monitoring pharmacotherapy, which is necessary to ensure adequate dosing and adherence, has not been identified. Recently, it was observed that urinary DHA assays using ultra high-performance liquid chromatography–tandem mass spectrometry can facilitate both the clinical diagnosis and therapeutic monitoring of pharmacotherapy for patients with APRT deficiency [19]. Furthermore, one study [10] revealed that patients with APRT deficiency showed a remarkable reduction in amount of urine DHA excretion after administration of allopurinol or febuxostat, which were once validated and are promising and more accessible. The generalizability should be validated by additional studies because the previous studies relied on observational registry data.
In conclusion, we encountered a case of 2,8-DHA crystalline nephropathy caused by APRT deficiency, as revealed by renal biopsy and urine metabolomic assessment results. Because of the variability in the clinical presentation of APRT deficiency, the diagnosis is complicated; however, XOR inhibitors may inhibit the progression of renal dysfunction. Therefore, it is necessary to consider APRT deficiency as one of the causes of urinary stones and progressive CKD; hence, early diagnosis and treatment can be achieved.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 Supplementary Fig. 1 Glomeruli under immunofluorescence microscopy. The glomeruli were stained for immunoglobulin IgG, IgA, IgM, C3, C4, C1q, κ, and λ, respectively. No positive staining was observed (TIF 17034 KB)
Acknowledgements
We thank all medical staff from our department. Furthermore, we thank the staff who gave us the metabolomic findings at the Japan Clinical Metabolomics Institute.
Data Availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Declarations
Conflict of interest
The authors have declared that no conflict of interest exists.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from the patient in this study.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Runolfsdottir HL, Palsson R, Agustsdottir IM, Indridason OS, Edvardsson VO. Kidney disease in adenine phosphoribosyltransferase deficiency. Am J Kidney Dis. 2016;67:431–438. doi: 10.1053/j.ajkd.2015.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edvardsson V, Palsson R, Olafsson I, Hjaltadottir G, Laxdal T. Clinical features and genotype of adenine phosphoribosyltransferase deficiency in Iceland. Am J Kidney Dis. 2001;38:473–480. doi: 10.1053/ajkd.2001.26826. [DOI] [PubMed] [Google Scholar]
- 3.Bollee G, Dollinger C, Boutaud L, Guillemot D, Bensman A, Harambat J, Deteix P, Daudon M, Knebelmann B. Ceballos-Picot I phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency. J Am Soc Nephrol. 2010;21:679–688. doi: 10.1681/ASN.2009080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Runolfsdottir HL, Palsson R, Agustsdottir IM, Indridason OS, Edvardsson VO. Long-term renal outcomes of APRT deficiency presenting in childhood. Pediatr Nephrol. 2019;34:435–442. doi: 10.1007/s00467-018-4109-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ceballos-Picot I, Saha A, Arora N, Kapoor K, Kaur M, Dhull RS, Goyal S. APRT deficiency due to novel mutation. Kidney Int Rep. 2019;4:624–628. doi: 10.1016/j.ekir.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huq A, Nand K, Juneja R, Winship I. APRT deficiency: the need for early diagnosis. BMJ Case Rep. 2018;2018:bcr2018225742. doi: 10.1136/bcr-2018-225742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizuno S. Urinary crystals with 2,8-dihydroxyadeninuria. Intern Med. 2021;60:963. doi: 10.2169/internalmedicine.5939-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaidan M, Palsson R, Meriequ E, Cornec-Le Gall E, Garstka A, Maggiore U, Deteix P, Battista M, Gagné ER, Ceballos-Picot I, Duong Van Huyen JP. Recurrent 2,8-dihydroxyadenine nephropathy: a rare but preventable cause of renal allograft failure. Am J Transplant. 2014;14:2623–2632. doi: 10.1111/ajt.12926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Shingde M, Nankivell BJ, Tchan MC, Bose B, Chapman JR, Kable K, Kim SK, Vucak-Dzumhur M, Wong G, Ranga GK. Adenine phosphoribosyltransferase deficiency: a potentially reversible cause of CKD. Kidney Int Rep. 2019;4:1161–1170. doi: 10.1016/j.ekir.2019.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edvardsson VO, Runolfsdottir HL, Thorsteinsdottir UA, Agustsdottir IMS, Oddsdottir GS, Eiriksson F, Goldfarb DS, Thorsteinsdottir M, Palsson R. Comparison of the effect of allopurinol and febuxostat on urinary 2,8-dihydroxyadenine excretion in patients with Adenine phosphoribosyltransferase deficiency (APRTd): a clinical trial. Eur J Intern Med. 2018;48:75–79. doi: 10.1016/j.ejim.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saeki T, Nishi S, Imai N, Ito T, Yamazaki H, Kawano M, Yamamoto M, Takahashi H, Matsui S, Nakada S, Origuchi T, Hirabayashi A, Homma N, Tsubata Y, Takata T, Wada Y, Saito A, Fukase S, Ishioka K, Miyazaki K, Masaki Y, Umehara H, Sugai S, Narita I. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010;78:1016–1023. doi: 10.1038/ki.2010.271. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Y, Guo L, Wang M, Chen J, Wang R. Recurrence of 2,8-dihydroxyadenine crystalline nephropathy in a kidney transplant recipient: a case report and literature review. Intern Med. 2021;60:2651–7. [DOI] [PMC free article] [PubMed]
- 13.Ikeda H, Watanabe T, Toyama D, Isoyama K. Use of LightCycler mutation analysis to detect type II adenine phosphoribosyltransferase deficiency in two patients with 2,8-dihydroxyadeninuria. CEN Case Rep. 2016;5:34–39. doi: 10.1007/s13730-015-0186-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Estepa-Maurice L, Hennequin C, Marfisi C, Bader C, Lacour B, Daudon M. Fourier transform infrared microscopy identification of crystal deposits in tissues: clinical importance in various pathologies. Am J Clin Pathol. 1996;105:576–582. doi: 10.1093/ajcp/105.5.576. [DOI] [PubMed] [Google Scholar]
- 15.Edvardsson VO, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani F, Milliner DS, Palsson R. Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol. 2013;28:1923–1942. doi: 10.1007/s00467-012-2329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki K, Kobayashi S, Kawamura K, Kuhara T, Tsugawa R. Family study of 2,8-dihydroxyadenine stone formation: report of two cases of a compound heterozygote for adenine phosphoribosyltransferase deficiency (APRT*J/APRT*Qo) Int J Urol. 1997;4:304–306. doi: 10.1111/j.1442-2042.1997.tb00195.x. [DOI] [PubMed] [Google Scholar]
- 17.Kimura T, Yasuda K, Obi Y, Kobayashi K, Kuhara T, Isaka Y, Imai E, Rakugi H, Hayashi T. Quiz page June 2009: worsening kidney function with a history of urolithiasis. Am J Kidney Dis. 2009;53:A37–A39. doi: 10.1053/j.ajkd.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 18.Kamijo-Ikemori A, Sugaya T, Hibi C, Nakamura T, Murase T, Oikawa T, Hoshino S, Hisamichi M, Hirata K, Kimura K, Shibagaki Y. Renoprotective effect of the xanthine oxidoreductase inhibitor topiroxostat on adenine-induced renal injury. Am J Physiol Renal Physiol. 2016;310:F1366–1376. doi: 10.1152/ajprenal.00517.2015. [DOI] [PubMed] [Google Scholar]
- 19.Thorsteinsdottir M, Thorsteinsdottir UA, Eiriksson FF, Runolfsdottir HL, Agustsdottir IM, Oddsdottir S, Sigurdsson BB, Hardarson HK, Kamble NR, Sigurdsson ST, Edvardsson VO, Palsson R. Quantitative UPLC-MS/MS assay of urinary 2,8-dihydroxyadenine for diagnosis and management of adenine phosphoribosyltransferase deficiency. J Chromatogr B Anal Technol Biomed Life Sci. 2016;1036–1037:170–177. doi: 10.1016/j.jchromb.2016.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file1 Supplementary Fig. 1 Glomeruli under immunofluorescence microscopy. The glomeruli were stained for immunoglobulin IgG, IgA, IgM, C3, C4, C1q, κ, and λ, respectively. No positive staining was observed (TIF 17034 KB)
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.