

Abstract
Sepsis remains a worldwide public health problem. This study aims to explore the role and mechanism of transcriptional factors (TFs) in sepsis‐induced myocardial injury. Firstly, TF KLF13 was selected to explore its role in sepsis‐induced myocardial injury. The caecal ligation and puncture (CLP) ‐induced sepsis mouse model was established and the septic mice were examined using standard histopathological methods. KLF13 expression was detected in the septic mouse heart and was also seen in a lipoploysaccharide (LPS) ‐induced cellular inflammation model. To explore this further both pro‐apoptotic cleaved‐caspase3/caspase3 and Bax levels and anti‐apoptotic Bcl2 levels were examined, also in both models, In addition inflammatory cytokine (IL‐1β, TNF‐α, IL‐8 and MCP‐1) production and IκB‐α protein level and p65 phosphorylation were examined in both septic mice and LPS‐induced cells. Thus three parameters ‐ cardiomyocyte apoptosis, inflammatory response and NF‐κB pathway activation were evaluated under similar conditions. The septic mice showed significant oedema, disordered myofilament arrangement and degradation and necrosis to varying degrees in the myocardial cells. KLF13 was downregulated in both the septic mouse heart and the LPS‐induced cellular inflammation model. Furthermore, both models showed abnormally increased cardiomyocyte apoptosis (increased cleaved‐caspase3/caspase and Bax protein levels and decreased Bcl2 level), elevated inflammation (increased production of inflammatory cytokines) and the activated NF‐κB pathway (increased p65 phosphorylation and decreased IκB‐α protein level). KLF13 overexpression notably ameliorated sepsis‐induced myocardial injury in vivo and in vitro. KLF13 overexpression protected against sepsis‐induced myocardial injury and LPS‐induced cellular inflammation and apoptosis via inhibiting the inflammatory pathways (especially NF‐κB signalling) and cardiomyocyte apoptosis.
Keywords: cardiomyocyte apoptosis, inflammation, KLF13, myocardial injury, sepsis, transcriptional factors
1. INTRODUCTION
Sepsis, validated as a worldwide public health care problem, is frequently caused by infection, with a mortality rate of more than 25%. 1 , 2 Sepsis is characterized by the maladjusted response of the host to infection and a sustained systemic inflammatory state, causing severe dysfunction of multiple organs and even death. 3 , 4 Early identification of sepsis is still the main goal of sepsis treatment. Nevertheless, conventional sepsis diagnostic tools (such as cultures) take a long time and are often accompanied by false‐negative results. 5 Therefore, an effective system is urgently needed for sepsis therapy. It has been pointed out that cardiac involvement frequently occurs during sepsis, and a set of changes are induced, leading to the malfunction of the myocardium. 6 Myocardial injury, validated as a serious complication of sepsis, makes substantial contributions to the death of sepsis patients. 7 However, despite multiple studies on sepsis, there is still a lack of appropriate markers for assessing sepsis severity. 8
An increasing number of studies have proposed that multiple mechanisms, such as inflammatory responses and cardiomyocyte apoptosis, are tightly implicated in sepsis‐induced myocardial dysfunction. 9 , 10 , 11 Moreover, as has been documented, NF‐κB plays a critical role in diverse sepsis‐related organ failure and systemic inflammation. 12 Hence, it would be of great value to investigate and analyse underlying molecular mechanisms of sepsis‐induced cardiac dysfunction involving inflammatory responses and cardiomyocyte apoptosis.
It is well‐established that transcriptional factors (TFs) are critical regulators of gene expression and biological processes, which can activate or inhibit the transcription process via binding to specific sequences in DNA. 13 A previous study has shown that multiple TFs, such as NF‐κB, play crucial roles in the pathophysiology of sepsis. 14 TFs also show close involvement in complex morphogenesis events for heart development. 15 It has been proposed that TF activation is tightly associated with early cardiomyocyte apoptosis, exhibiting a mechanistic role in septic myocardial dysfunction. 16 Aberrant TFs have been evidenced as key drivers of inflammatory conditions and various diseases. 17 However, little is known about the role of TFs in sepsis and sepsis‐induced myocardial injury involving inflammatory response and cardiomyocyte apoptosis.
Therefore, it was reasonable to hypothesize that deregulated TFs in sepsis and sepsis‐induced myocardial injury may exert an effect on inflammatory pathways (especially the NF‐κB signalling) and cardiomyocytes, thereby affecting disease progression. Consequently, we performed a series of histological and molecular experiments to identify the regulatory mechanism of TFs in sepsis‐induced myocardial injury, with the purpose of providing novel preventive and therapeutic targets for the treatment of sepsis‐induced myocardial injury.
2. MATERIALS AND METHODS
2.1. Animals
All the animal experimental procedures were performed with the approval of the Ethics Committee of the Second Xiangya Hospital and followed the NIH guidelines (Guide for the Care and Use of Laboratory Animals). Twenty‐four male C57BL/6 mice were purchased at ages 6–7 weeks from Hunan SJA Laboratory Animal Co., Ltd. All animals were housed at a 12:12 h light–dark cycle in a pathogen‐free environment set with the following conditions: temperature at 23 ± 3°C, relative humidity at 55 ± 10%.
2.2. Cecal ligation and puncture (CLP)‐induced sepsis model in mice
The CLP‐induced sepsis model was established following the methods described before. 18 Briefly, animals were anaesthetised using isoflurane inhalation and a 1‐cm midline abdominal incision was made. The cecum was then exposed and ligated. Next, the cecum was punctured once with a 23‐Gauge needle to induce CLP. Simple running sutures and metallic clips were used to close the abdominal musculature and skin respectively. Sham‐operated mice underwent the same surgical procedure, without CLP. Forty‐eight hours later, the cardiac tissues were collected.
2.3. Lentivirus transduction
In vivo injection of Lv‐KLF13 (OBIO Biotech) by tail vein at a dose of 1 × 107 TU in 100 μl volumes. The injection was performed 5 days before CLP.
2.4. Histopathological analyses and immunohistochemistry (IHC) staining
The mice were euthanized under anaesthesia and cardiac tissues were collected. Tissue samples were incubated overnight in 10% buffered formaldehyde, embedded in paraffin and cut into slices of around 4 μm in thickness. Following deparaffinization, the tissue slides were stained using haematoxylin and eosin (H&E) staining by standard methods.
IHC examination was carried out as previously mentioned. 19 , 20 Once the tissue sections had been rehydrated, 3% hydrogen peroxide was used to quench the endogenous peroxidase activity. Next, sections were treated overnight at 4°C with primary antibodies against KLF13 (18352‐1‐AP, Proteintech). Sections were rinsed in PBS and treated with horseradish peroxidase (HRP)‐conjugated goat anti‐rabbit antibody (Beyotime) for 30 min at room temperature. Thereafter, sections were stained using a DAB kit (Sigma) and examined under a microscope (Olympus).
2.5. Immunoblotting
Protein samples were obtained from target tissue or cell samples and the concentration was evaluated by a BCA Protein Assay Kit (Nacalai Tesque). After electrophoresis with 10%–14% SDS‐PAGE, the separated proteins were transferred onto the PVDF membrane. The membrane was blocked using Odyssey blocking buffer (LI‐COR Bioscience) for 1 h at room temperature and incubated in primary antibody solution overnight at 4°C. The primary antibodies were against IκB‐α, p‐p65, p65, Bax and Bcl2. Primary antibody detection was performed with a horseradish peroxidase‐linked goat anti‐rabbit immunoglobulin G (IgG) (SC 1004, Santa Cruz Biotechnology) or goat anti‐mouse or rabbit IgG (SA00001‐1, SA00001‐2, Proteintech), visualized using the Enhanced Chemiluminescence (ECL) detection system (Promega).
2.6. qRT‐PCR
An RNeasy Mini Kit (Qiagen) and TRIzol (Invitrogen Life Technologies) were employed to isolate total RNA from target tissue or cell samples, and qRT‐PCR was carried out to examine the expression levels of target factors.
2.7. Enzyme‐linked immunosorbent assay (ELISA)
The cell culture supernatant and cardiac tissues were collected for measuring the secretion of cytokines. ELISA kits (Abcam) were employed to detect the levels of tumour necrosis factor‐alpha (TNF‐α), interleukin 1‐beta (IL‐1β), interleukin‐8 (IL‐8) and monocyte chemoattractant protein‐1 (MCP‐1) according to the protocol. A microplate reader was used to measure the absorbance at 450 nm (OD450).
2.8. Cell lineage, cell transfection and treatment
Mouse cardiomyocyte cell line HL‐1 was procured from Sigma‐Aldrich and grown in Claycomb Medium (Sigma‐Aldrich) containing 10% FBS (Invitrogen) in a 95% air 5% CO2 environment at 37°C. Using Lipofectamine 3000 Reagent (Thermo Fisher Scientific), the KLF13‐overexpressing vector (KLF13) was transfected to achieve KLF13 overexpression in HL‐1 cells. After 24 h, the transfected cells were further exposed to LPS (10 μg/ml, Sigma) for 48 h.
2.9. CCK‐8 assay detecting cell viability
Cell Counting Kit‐8 (CCK‐8, MedChemExpress, Monmouth Junction, NJ, USA) was used to detect cell viability. Cells were seeded into a 96‐well plate and then exposed to LPS for 48 h. Next, 10 μl CCK‐8 solution was added to each well, followed by incubation for 3 hours at 37°C. A microplate reader (Bio‐Rad, Hercules, USA) was used to detect the absorbance at 450 nm (OD450).
2.10. Flow cytometry detecting cell apoptosis
Following transfection and/or treatment, HL‐1 cells were digested and collected using trypsin (trypsin) without ethylenediaminetetraacetic acid at room temperature for 1 min. The digestion was stopped by the addition of DMEM (Corning) added with 10% FBS. Cells were harvested by centrifugation at 1000 × g for 3 min at room temperature which was followed by removing of the supernatant. Cells were rinsed twice with pre‐cooled PBS, and 1 × binding buffer was added to resuspended cells. Next, 5 μl Annexin V‐FITC and 5 μl propidium iodide (PI) were added to the buffer and incubated for 10 min at dark. Following the protocols of the Annexin V‐FITC cell apoptosis detection kit (cat. no. K201‐100, BioVision), flow cytometry (Novocyte, Agilent) was used to measure the cell apoptosis rate.
2.11. Statistical analysis
Data are expressed as mean ± standard deviations (SD). To deal with data that were not normally distributed, an ANOVA with a Tukey's post‐test or a Kruskal–Wallis test was used to analyse data among more than two groups. Data between the two groups were analysed using a Student's t test. All statistical analyses were carried out using GraphPad Prism 7 software. If p value was less than .05, the null hypothesis was rejected.
3. RESULTS
3.1. Selecting TF correlated to sepsis‐induced myocardial injury
Given the key role of TFs in gene regulation 21 , 22 and sepsis aetiology, 23 , 24 the transcription factor list (GO:0003700, transcription factor activity) was firstly extracted from Ensembl (http://asia.ensembl.org/), and 1351 TF genes were obtained. The expression of these TFs was analysed in normal and septic hearts based on GSE171546. Among these TFs, 19 were downregulated and 24 were upregulated (thresholds: logFC >0.56 or <−0.56, adjusted p value <0.05). The top 10 most significantly differentially expressed TFs were shown in the volcano plots and hierarchical clustering (Figure 1A,B). Among the top 10 deregulated TFs, KLF13 has been found to play an important role in cardiac development 25 and is necessary for cardiomyocyte proliferation. 26 Downregulation of KLF13 in cardiomyocytes inhibits cell activity and accelerates apoptosis. 26 Moreover, based on online data sets GSE171546 and GSE79962, KLF13 expression is significantly downregulated in septic hearts compared with that in normal hearts (Figure 1C), suggesting that KLF13 downregulation in septic hearts might affect cardiomyocytes proliferation and apoptosis. For confirming the involvement of KLF13 in sepsis‐induced myocardial injury, genes correlated with KLF13 were analysed based on GSE171546 using Pearson correlation coefficient analysis; a total of 419 genes were found to correlate to KLF13 and further Pathway & Process Enrichment (Min Enrichment =1.5, p value cut‐off: 0.01, Min Overlap: 3) in metascape (https://metascape.org/) indicated that these genes were significantly enriched in signalling in interleukins, positive regulation of cytokine production and regulation of hemopoiesis (Figure 1D), which reflects the pathological state of cardiac tissue after sepsis. Thus, KLF13 was selected for further experiments.
FIGURE 1.
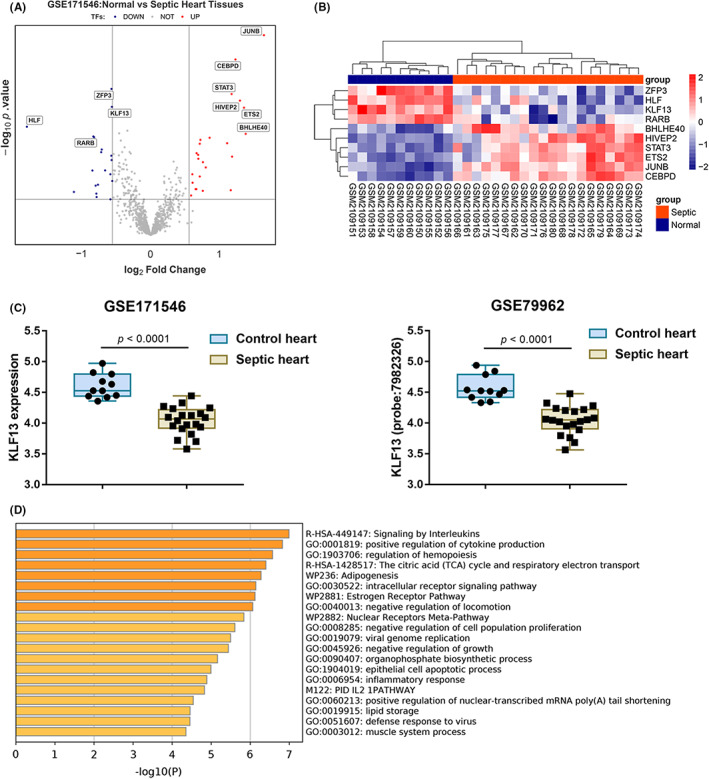
Selecting transcriptional factor (TF) correlated to sepsis‐induced myocardial injury. (A‐B) The TF list (GO:0003700, TF activity) was extracted from Ensembl (http://asia.ensembl.org/), and 1351 TF genes were obtained. Expression of these TFs was analysed in normal and septic hearts based on GSE171546. The top 10 most significantly differentially expressed TFs were shown in the volcano plots and hierarchical clustering. (C) The expression of KLF13 in normal and septic hearts is based on data from GSE171546 and GSE79962. (D) A total of 419 genes correlated to KLF13 were applied for Gene Ontology (GO) functional enrichment annotation in metascape (https://metascape.org/)
3.2. KLF13 is downregulated in septic mouse heart
For the investigation of KLF13 function on sepsis‐induced myocardial injury, the CLP‐induced sepsis model was established in mice and confirmed for histopathological changes by H&E staining; in the sepsis group, myocardial cells showed significant oedema, disordered myofilament arrangement and degradation and necrosis to varying degrees (Figure 2A). In septic hearts, the levels of KLF13 were decreased compared with those in normal hearts, as revealed by IHC staining (Figure 2B). Consistently, KLF13 mRNA was downregulated in septic heart tissues compared with that in normal heart tissues (Figure 2C). Moreover, the ratio of pro‐apoptotic cleaved‐caspase3/caspase3 and Bax protein levels were significantly increased in septic hearts and anti‐apoptotic Bcl2 was decreased (Figure 2D), indicating an increase in apoptotic cardiomyocytes.
FIGURE 2.
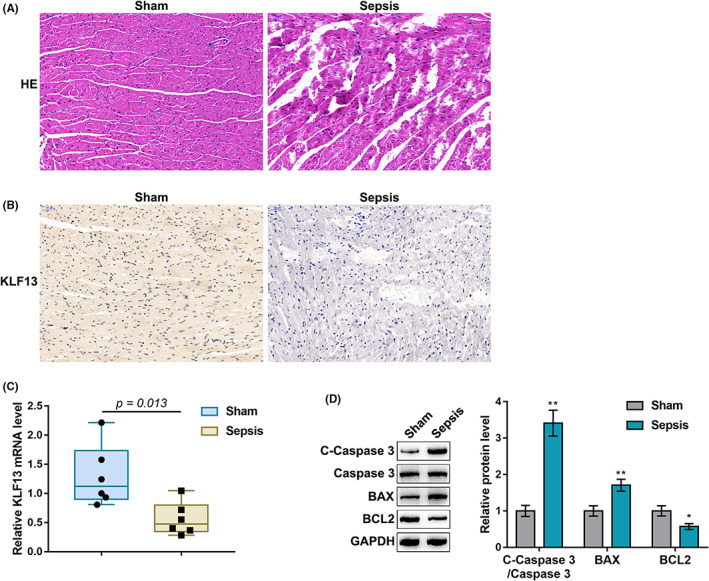
KLF13 is downregulated in septic mouse hearts. Cecal ligation and puncture (CLP)‐induced sepsis model was established in mice and examined for histopathological changes of normal and septic mouse hearts by H&E staining (A); the levels and distribution of KLF13 in normal and septic mouse hearts by immunohistochemical (IHC) staining (B); (C); mRNA expression levels of KLF13 in normal and septic mouse heart by qRT‐PCR (D); the protein levels of cleaved‐caspase3, caspase3, Bax and Bcl2 in normal and septic mouse heart by Immunoblotting
3.3. In vivo effects of KLF13 on septic mouse heart
Since KLF13 is downregulated in septic heart tissues, mice were injected with lentivirus overexpressing KLF13 (Lv‐KLF13) and then received CLP 5 days later. The overexpression of KLF13 in heart tissues was confirmed by qRT‐PCR and Immunoblotting (Figure 3A,B). The specific effects of KLF13 overexpression on sepsis‐induced myocardial injury were subsequently investigated. Mice were assigned to four groups: sham, sepsis model, sham injected with Lv‐KLF13 and sepsis model injected with Lv‐KLF13. In the sepsis group, cleaved‐caspase3/caspase3 and Bax levels were increased and Bcl2 was decreased compared with those in the sham group, whereas KLF13 overexpression exerted opposite effects on these factors. In septic mice injected with Lv‐KLF13, sepsis‐induced changes in these factors were partially eliminated (Figure 3C). Sepsis increased, whereas KLF13 overexpression decreased the production of cytokines including IL‐1β, TNF‐α, IL‐8 and MCP‐1; in septic mice injected with Lv‐KLF13, sepsis‐induced production of IL‐1β, TNF‐α, IL‐8 and MCP‐1 was partially attenuated by KLF13 overexpression (Figure 3D). Lastly, sepsis decreased the protein levels of IκB‐α but increased p65 phosphorylation, whereas KLF13 overexpression exerted opposite effects on them; in septic mice injected with Lv‐KLF13, KLF13 overexpression partially reversed sepsis effects on IκB‐α level and p65 phosphorylation (Figure 3 E). These data indicate that KLF13 overexpression ameliorates sepsis‐induced myocardial injury.
FIGURE 3.
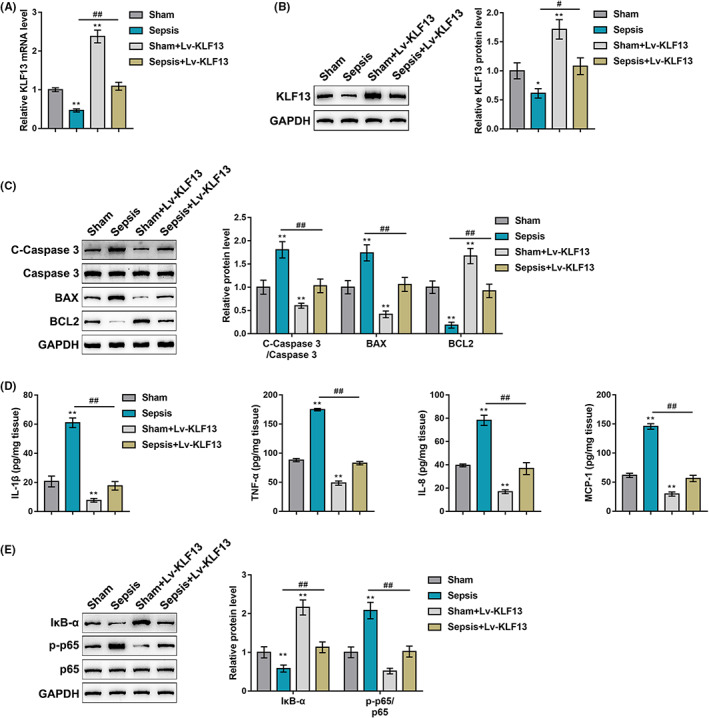
In vivo effects of KLF13 on septic mouse heart. (A‐B) Mice were injected with lentivirus overexpressing KLF13 (Lv‐KLF13) and then received CLP 5 days later. KLF13 overexpression in cardiac tissues was confirmed by qRT‐PCR and immunoblotting. Mice were assigned to four groups: sham, sepsis model, sham injected with Lv‐KLF13 and sepsis model injected with Lv‐KLF13. Mice in each group received the injection, underwent surgery accordingly and examined for the protein levels of cleaved‐caspase3, caspase3, Bax and Bcl2 in normal and septic mouse heart by immunoblotting (C); levels of cytokines including IL‐1β, TNF‐α, IL‐8 and MCP‐1 in normal and septic mouse heart by ELISA (D); protein levels of IκB‐α, p‐p65 and p65 by Immunoblotting (E)
3.4. In vitro effects of KLF13 on the LPS‐induced cellular inflammation model
Considering the key role of KLF13 in cardiomyocyte proliferation, 26 the effects of KLF13 overexpression on the LPS‐induced cellular inflammation model were investigated. HL‐1 mouse cardiomyocytes were transfected or non‐transfected with KLF13‐overexpressing vector, treated or non‐treated with 10 μg/mL LPS for 48 h and examined for KLF13 mRNA expression and protein levels. Figure 4A,B showed that LPS stimulation significantly downregulated KLF13 expression and decreased KLF13 protein levels, whereas KLF13 transfection partially rescued KLF13 mRNA expression and protein levels. Regarding cellular functions, LPS stimulation inhibited cell viability and promoted cell apoptosis, whereas KLF13 transfection exerted opposite effects (Figure 4C,D); LPS‐induced alterations in cell phenotypes were partially reversed by KLF13 overexpression (Figure 4C,D). Consistent with in vivo results, LPS stimulation significantly increased, whereas KLF13 overexpression decreased the levels of cytokines including IL‐1β, TNF‐α, IL‐8 and MCP‐1; LPS‐induced increases in these cytokines were partially attenuated by KLF13 overexpression (Figure 4 E). Regarding the inflammatory and apoptotic pathways, LPS stimulation increased p65 phosphorylation and Bax protein level but decreased IκB‐α and Bcl2 protein levels, whereas KLF13 overexpression exerted opposite effects on these factors; similarly, LPS‐induced alteration in these factors was partially abolished by KLF13 overexpression (Figure 4F). Thus, KLF13 overexpression could protect LPS‐induced apoptosis and inflammation in cardiomyocytes.
FIGURE 4.
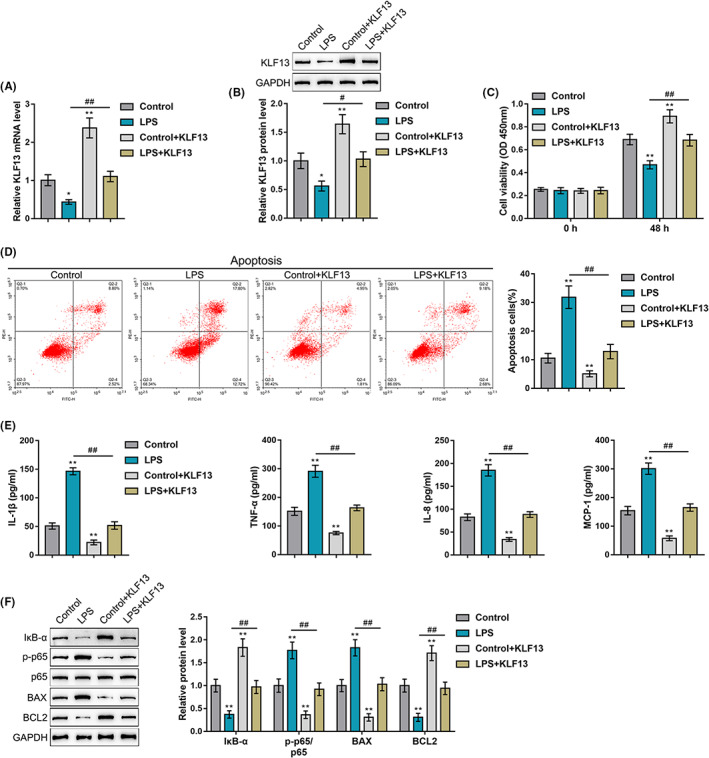
In vitro effects of KLF13 on LPS‐induced cellular inflammation model. HL‐1 mouse cardiomyocytes were transfected or non‐transfected with KLF13‐overexpressing vector, treated or non‐treated with 10 μg/mL LPS for 48 h and examined for KLF13 mRNA expression by qRT‐PCR and protein levels by Immunoblotting (A,B); cell viability by CCK‐8 assay (C); cell apoptosis by flow cytometry (D); levels of cytokines including IL‐1β, TNF‐α, IL‐8 and MCP‐1 in normal and septic mouse heart by ELISA (E); protein levels of IκB‐α, p‐p65, p65, Bax and Bcl2 by immunoblotting (F)
4. DISCUSSION
Sepsis is a highly fatal condition during which the heart is one of the most sensitive organs. 27 TFs have been shown to play crucial roles in governing cardiac development. 28 This study documented that TF KLF13 overexpression protected against sepsis‐induced myocardial injury by inhibiting the inflammatory pathways (especially the NF‐κB signalling) and cardiomyocyte apoptosis.
Previous work has indicated that about 50% of patients with severe sepsis suffer from cardiac dysfunction, and the recovery of cardiac function is considered to be crucial for the survival of sepsis patients. 29 To investigate this further in this study the first step was to establish the CLP‐induced sepsis mouse model. H&E staining revealed severe cardiac injury in the septic mice, as evidenced by significant oedema, disordered myofilaments, and degradation and necrosis in the cardiomyocytes. Similarly, as has been proposed previously, cardiac dysfunction was present in CLP‐induced sepsis. 30 Taken together, it was clear that sepsis contributed to the induction of cardiac injury.
Several current studies have provided evidence about the pathogenesis of sepsis‐induced multi‐faceted myocardial injuries. 31 , 32 , 33 Activation of inflammatory cells and inflammatory factors are critical events in sepsis‐induced myocardial injury. 34 , 35 , 36 It is well established that IL‐1β, IL‐8, IL‐10, TNF‐α and MCP‐1 are typical pro‐inflammatory cytokines. 28 In the present study, increased IL‐1β, TNF‐α, IL‐8 and MCP‐1 levels were observed in the septic mouse heart, suggesting activated inflammatory responses. Moreover, the NF‐κB pathway has been proposed to participate in modulating the production of pro‐inflammatory cytokines during sepsis. 37 In this study, septic mice exhibited decreased IκB‐α protein levels and increased p65 phosphorylation, suggesting NF‐κB signalling activation. In support of these results, a previous study has shown that NF‐κB, as a crucial mediator of inflammation, exerts an essential effect on the progression of sepsis. 37 Furthermore, in this study, increased cleaved‐caspase3/caspase3 and Bax levels and decreased Bcl2 levels were observed in septic mouse hearts, which indicated an increase in cardiomyocyte apoptosis. This is consistent with the previous findings that excessive cardiomyocyte apoptosis is closely associated with depressed cardiac function in the CLP‐induced sepsis model. 38 Thus three key components ‐ cytokines, the NF‐κB pathway, and cardiomyocyte apoptosis ‐ are ‐ implicated in the sepsis‐induced myocardial injury model.
Therefore in subsequent experiments we shifted to explore the underlying mechanism of sepsis‐induced myocardial injury. Previous work has pointed out that multiple TFs are implicated in the pathophysiology of sepsis. 14 KLF13, belonging to the KLF family, has been validated as a cardiac TF that can activate cardiac transcription via binding to regulatory elements on the cardiac promoter. 26 , 39 Also KLF13 has been recognized as a novel regulator of cardiac development, showing tight involvement in cardiomyocyte proliferation. 25 In the current study, KLF13 expression was identified as downregulated abnormally in septic mouse hearts. Next, KLF13 was overexpressed in septic mice to explore its potential in vivo effect on sepsis‐induced myocardial dysfunction. The results demonstrated that KLF13 overexpression protected against sepsis‐induced myocardial injury, as manifested by the reduced levels of inflammatory factors (IL‐1β, TNF‐α, IL‐8 and MCP‐1), the inhibited NF‐κB pathway (increased IκB‐α protein level and decreased p65 phosphorylation) and ameliorated apoptosis (decreased cleaved‐caspase3/caspase3 and Bax levels and increased Bcl2 level). Furthermore, the in vitro effect of KLF13 overexpression on an LPS‐induced cellular inflammation model of H9C2 rat cardiomyocytes was confirmed. The results showed that KLF13 overexpression protected against LPS‐induced cardiomyocyte injury, as shown by the enhanced cell viability and proliferation, inhibited cell apoptosis, decrease levels of pro‐inflammatory cytokines, and inhibited NF‐κB pathway. This is an original finding as there appear to have been no previous reports about an effect of KFL13 on sepsis‐induced myocardial injury.
Thus in summary this study suggest that overexpression of KFL13 could protect from sepsis‐induced cardiac injury and LPS‐induced cellular inflammation and apoptosis. These findings imply that novel KLF13‐based therapy might be possible for septic patients with cardiac injury. Certainly further experiments are necessary before clinical applications, but taken in conjunction with previous observations they justify further serious consideration.
AUTHOR CONTRIBUTIONS
Ni Zeng designed the experiments and drafted the article. Feng Xiao revised the article critically for important intellectual content. Zaijin Jian and Wenxin Zhu contributed to cell and animal experiments. Junmei Xu and Yongmei Fan contributed to the analysis and manuscript preparation. All the authors read and approved the manuscript.
FUNDING INFORMATION
This study was supported by the National Natural Science Foundation of China (grant no. 81971819) and the Natural Science Foundation of Hunan Province (grant no. 2020JJ4777 and 2022JJ70132).
CONFLICT OF INTEREST
N/A
CONSENT FOR PUBLICATION
All the authors’ consent to the publication.
ACKNOWLEDGEMENTS
N/A
Zeng N, Jian Z, Zhu W, Xu J, Fan Y, Xiao F. KLF13 overexpression protects sepsis‐induced myocardial injury and LPS‐induced inflammation and apoptosis. Int J Exp Path. 2023;104:23‐32. doi: 10.1111/iep.12459
REFERENCES
- 1. Carey MG, Valcin EK, Lent D, White M. Nursing Care for the Initial Resuscitation of severe sepsis patients. Crit Care Nurs Clin North Am. 2021;33(3):263‐274. [DOI] [PubMed] [Google Scholar]
- 2. Niederman MS, Baron RM, Bouadma L, et al. Initial antimicrobial management of sepsis. Crit Care. 2021;25(1):307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moriyama K, Nishida O. Targeting cytokines, pathogen‐associated molecular patterns, and damage‐associated molecular patterns in sepsis via blood purification. Int J Mol Sci. 2021;22(16):8882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Appiah MG, Park EJ, Akama Y, et al. Cellular and Exosomal regulations of sepsis‐induced metabolic alterations. Int J Mol Sci. 2021;22(15):8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Piccioni A, Santoro MC, de Cunzo T, et al. Presepsin as early marker of sepsis in emergency department: A narrative review. Medicina (Kaunas). 2021;57(8):770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Poveda‐Jaramillo R. Heart dysfunction in sepsis. J Cardiothorac Vasc Anesth. 2021;35(1):298‐309. [DOI] [PubMed] [Google Scholar]
- 7. Wang X, Li XL, Qin LJ. The lncRNA XIST/miR‐150‐5p/c‐Fos axis regulates sepsis‐induced myocardial injury via TXNIP‐modulated pyroptosis. Lab Invest. 2021;101(9):1118‐1129. [DOI] [PubMed] [Google Scholar]
- 8. Qi Y, Chen X, Wu N, Ma C, Cui X, Liu Z. Identification of risk factors for sepsis‐associated mortality by gene expression profiling analysis. Mol Med Rep. 2018;17(4):5350‐5355. [DOI] [PubMed] [Google Scholar]
- 9. Liu YC, Yu MM, Shou ST, Chai YF. Sepsis‐induced cardiomyopathy: mechanisms and treatments. Front Immunol. 2017;8:1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Piper RD. Myocardial dysfunction in sepsis. Clin Exp Pharmacol Physiol. 1998;25(11):951‐954. [DOI] [PubMed] [Google Scholar]
- 11. Sharma AC. Sepsis‐induced myocardial dysfunction. Shock. 2007;28(3):265‐269. [DOI] [PubMed] [Google Scholar]
- 12. Matsuda N. Alert cell strategy in SIRS‐induced vasculitis: sepsis and endothelial cells. J Intensive Care. 2016;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Falak N, Imran QM, Hussain A, Yun BW. Transcription factors as the "blitzkrieg" of plant defense: a pragmatic view of nitric Oxide's role in gene regulation. Int J Mol Sci. 2021;22(2):522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hattori Y, Hattori K, Suzuki T, Palikhe S, Matsuda N. Nucleic‐acid based gene therapy approaches for sepsis. Eur J Pharmacol. 2018;833:403‐410. [DOI] [PubMed] [Google Scholar]
- 15. Kathiriya IS, Nora EP, Bruneau BG. Investigating the transcriptional control of cardiovascular development. Circ Res. 2015;116(4):700‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar A, Kumar A, Michael P, et al. Human serum from patients with septic shock activates transcription factors STAT1, IRF1, and NF‐kappaB and induces apoptosis in human cardiac myocytes. J Biol Chem. 2005;280(52):42619‐42626. [DOI] [PubMed] [Google Scholar]
- 17. Papavassiliou KA, Papavassiliou AG. Transcription factor drug targets. J Cell Biochem. 2016;117(12):2693‐2696. [DOI] [PubMed] [Google Scholar]
- 18. Dejager L, Pinheiro I, Dejonckheere E, Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 2011;19(4):198‐208. [DOI] [PubMed] [Google Scholar]
- 19. Yamashita S, Suzuki T, Iguchi K, et al. Cardioprotective and functional effects of levosimendan and milrinone in mice with cecal ligation and puncture‐induced sepsis. Naunyn Schmiedebergs Arch Pharmacol. 2018;391(9):1021‐1032. [DOI] [PubMed] [Google Scholar]
- 20. Sakai M, Suzuki T, Tomita K, et al. Diminished responsiveness to dobutamine as an inotrope in mice with cecal ligation and puncture‐induced sepsis: attribution to phosphodiesterase 4 upregulation. Am J Physiol Heart Circ Physiol. 2017;312(6):H1224‐H1237. [DOI] [PubMed] [Google Scholar]
- 21. Arbibe L, Kim DW, Batsche E, et al. An injected bacterial effector targets chromatin access for transcription factor NF‐kappaB to alter transcription of host genes involved in immune responses. Nat Immunol. 2007;8(1):47‐56. [DOI] [PubMed] [Google Scholar]
- 22. Sullivan KE, Reddy ABM, Dietzmann K, et al. Epigenetic regulation of tumor necrosis factor alpha. Mol Cell Biol. 2007;27(14):5147‐5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dai XG, Li T, Huang WB, et al. Upregulation of mitochondrial transcription factor a promotes the repairment of renal tubular epithelial cells in sepsis by inhibiting reactive oxygen species‐mediated toll‐like receptor 4/p38MAPK signaling. Pathobiology. 2019;86(5–6):263‐273. [DOI] [PubMed] [Google Scholar]
- 24. El Gazzar M, Liu T, Yoza BK, CE MC. Dynamic and selective nucleosome repositioning during endotoxin tolerance. J Biol Chem. 2010;285(2):1259‐1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lavallee G, Andelfinger G, Nadeau M, et al. The Kruppel‐like transcription factor KLF13 is a novel regulator of heart development. EMBO J. 2006;25(21):5201‐5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nemer M, Horb ME. The KLF family of transcriptional regulators in cardiomyocyte proliferation and differentiation. Cell Cycle. 2007;6(2):117‐121. [DOI] [PubMed] [Google Scholar]
- 27. Potz BA, Sellke FW, Abid MR. Endothelial ROS and Impaired myocardial oxygen consumption in sepsis‐induced cardiac dysfunction. J Intensive Crit Care. 2016;2(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Whitcomb J, Gharibeh L, Nemer M. From embryogenesis to adulthood: critical role for GATA factors in heart development and function. IUBMB Life. 2020;72(1):53‐67. [DOI] [PubMed] [Google Scholar]
- 29. An R, Zhao L, Xi C, et al. Melatonin attenuates sepsis‐induced cardiac dysfunction via a PI3K/Akt‐dependent mechanism. Basic Res Cardiol. 2016;111(1):8. [DOI] [PubMed] [Google Scholar]
- 30. O'Riordan CE, Purvis GSD, Collotta D, et al. Bruton's tyrosine kinase inhibition attenuates the cardiac dysfunction caused by Cecal ligation and puncture in mice. Front Immunol. 2019;10:2129. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Kakihana Y, Ito T, Nakahara M, Yamaguchi K, Yasuda T. Sepsis‐induced myocardial dysfunction: pathophysiology and management. J Intensive Care. 2016;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Jet al. Clinical significance of plasma levels of brain natriuretic peptide and cardiac troponin T in patients with sepsis. Exp Ther Med. 2016;11(1):154‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Latini R, Caironi P, Masson S. Cardiac dysfunction and circulating cardiac markers during sepsis. Minerva Anestesiol. 2016;82(6):697‐710. [PubMed] [Google Scholar]
- 34. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71(4):549‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. 2016;118(6):1021‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225(3):631‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X, Zingarelli B, O′Connor M, et al. Overexpression of Hsp20 prevents endotoxin‐induced myocardial dysfunction and apoptosis via inhibition of NF‐kappaB activation. J Mol Cell Cardiol. 2009;47(3):382‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Han X, Chen D, Liufu N, et al. MG53 protects against sepsis‐induced myocardial dysfunction by upregulating peroxisome proliferator‐activated receptor‐alpha. Oxid Med Cell Longev. 2020;2020:7413693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li W, Li B, Li T, et al. Identification and analysis of KLF13 variants in patients with congenital heart disease. BMC Med Genet. 2020;21(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]