


Keywords: liver regeneration, mitochondria, mitochondrial bioenergetics, mitochondrial therapy, partial hepatectomy
Abstract
Treatment of advanced liver disease using surgical modalities is possible due to the liver’s innate ability to regenerate following resection. Several key cellular events in the regenerative process converge at the mitochondria, implicating their crucial roles in liver regeneration. Mitochondria enable the regenerating liver to meet massive metabolic demands by coordinating energy production to drive cellular proliferative processes and vital homeostatic functions. Mitochondria are also involved in terminating the regenerative process by mediating apoptosis. Studies have shown that attenuation of mitochondrial activity results in delayed liver regeneration, and liver failure following resection is associated with mitochondrial dysfunction. Emerging mitochondria therapy (i.e., mitotherapy) strategies involve isolating healthy donor mitochondria for transplantation into diseased organs to promote regeneration. This review highlights mitochondria’s inherent role in liver regeneration.
INTRODUCTION
Liver cancer causes more than 800,000 deaths per year, while an estimated 25% of the world’s population carries risk factors for liver disease, which includes viral infection, alcohol abuse, and nonalcoholic steatohepatitis (1). Fortunately, those with advanced segmental liver disease or hepatic malignancies can be treated using targeted surgical interventions such as partial hepatectomy (PH) and living donor liver transplantation (LDLT) with favorable clinical outcomes due to the liver’s intrinsic ability to regenerate following injury and cell loss. However, the partial liver must sustain its immense metabolic demands following PH. Not only does the remnant liver require energy for synthesizing the cellular components needed for regeneration, but it must also continue to support the entire body during the regenerative process by maintaining its intracellular energy homeostasis since the liver serves as the main metabolic organ (2). At the level of hepatocyte, mitochondria fulfill the energetic and biosynthetic demands of the cell by orchestrating and integrating all the major biochemical, hormonal, and inflammatory signaling pathways. However, mitochondrial activity can be attenuated during liver regeneration due to acute physiological strains associated with surgery. PH causes an immediate overflow of the portal venous system, and consequently, the hepatic arterial buffer response counteracts the portal overflow by inducing spontaneous arterial vasoconstriction. This induces a relatively hypoxic environment following PH, damaging liver mitochondria during a time when metabolic demands are particularly high (3). The critical roles of functional mitochondria during regeneration are demonstrated in a recent study that reported an association between mitochondrial dysfunction and an increased mortality rate secondary to postoperative liver failure in animals or patients following PH (4). Therefore, mitochondria play key roles during liver regeneration, and this review article highlights previously published reports and observations demonstrating how this organelle orchestrates the crucial regenerative processes in diseased livers.
CELL CYCLE AND LIVER REGENERATION
The liver is composed of parenchymal and nonparenchymal tissue consisting of several different cell types, such as hepatocytes, cholangiocytes, hepatic stellate cells, endothelial cells, and macrophages. Hepatocytes within the parenchyma are the major hepatic cell type, constitute 70% of the liver mass, and serve as the main drivers of liver regeneration. In a healthy liver, these cells remain mostly dormant in the G0 phase without replication. After injury or resection, remnants of hepatocytes enter the cell cycle, and the process of compensatory hyperplasia, or regeneration, proceeds until the original mass of the degenerated liver is restored. Previous reports have concluded that liver regeneration can be classified into three critical phases: priming, proliferation, and termination (5). The priming phase begins immediately after insult to the liver and is mediated by proinflammatory cytokines TNF-α and IL-6, which induce hepatocytes for transition from G0 phase to the G1 phase. The contribution of these proinflammatory cytokines has been established by studies using knockout mouse models of IL-6 and the receptor for TNF-α, which experienced high mortality rates and significant delay in regeneration following PH. However, those abnormalities are reversed by exogenous IL-6 administration (6). Interestingly, it has been shown that exogenous IL-6 also significantly increases ATP synthase activity within the mitochondria (7).
The proliferation phase encompasses progression of the cell cycle from G1 phase to its completion at M phase (i.e., mitosis). This phase is mediated by mitogens like hepatocyte growth factor (HGF), epidermal growth factor (EGF), and transforming growth factor (TGF-α), which are released by hepatocytes and resident macrophages within nonparenchymal tissue such as Kupffer cells. Cell cycle phase-specific cyclins associated with cyclin-dependent kinases (CDKs) are also required for proper progression through the cell cycle. Cyclin D1 interacts with CDK4 and promotes progression through the G1 phase, while binding of Cyclin E1 to CDK2 is essential to advance through the G1/S phase check point (8). Cyclin A2 is then synthesized during S phase and interacts with CDK2 to initiate DNA replication, and the typical onset of S phase is ∼36 h after PH in mice (9). After DNA replication, the cell enters the G2 phase to prepare for mitosis. The dissolution of the nuclear envelope initiates mitosis due to the phosphorylative effects of Cyclin B1/CDK1 (10). As discussed later, mitochondria adopt cell cycle phase-specific morphologies, which closely correlate with the function of the various cyclins (Fig. 1).
Figure 1.
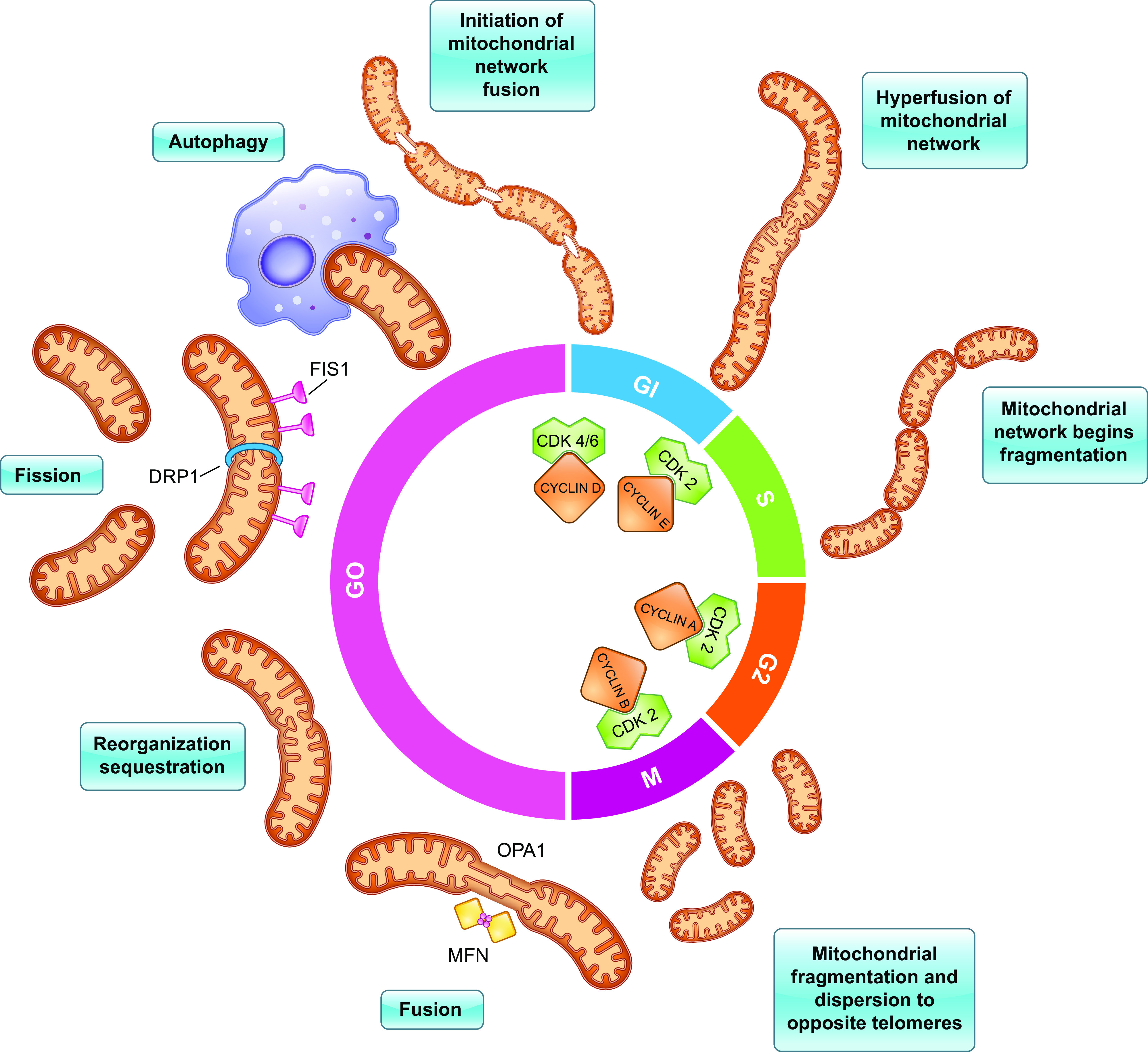
Mitochondria present two predominating morphologies throughout the cell cycle: a hyperfused reticulum and multiple fragmented mitochondria. This occurs in coordination with activation of phase-specific CDKs that are required for advancement through the cell cycle. Cyclin D1 first associates with CDK4 and promotes progression through G1 phase. During late G1 phase, fusion of the mitochondrial network is initiated by Mfn 1, Mfn 2, and Opa1. The reticulum is associated with enhanced oxidative capacity which induces accumulation of Cyclin E. Association of Cyclin E1 with CDK2 is essential to advance through the G1/S-phase check point. The reticulum begins to fragment via fission at the onset of S phase, which is mediated by Drp1, Fis1, Mff, MiD51, and MiD49. Cyclin A2 is also synthesized during S phase, which interacts with CDK2 to initiate DNA replication. After the S phase, the cell enters G2 phase to prepare for mitosis (i.e., M phase). Fission continues through M phase to promote equal distribution of mitochondria between the resulting daughter cells. The phosphorylative effects of Cyclin B1/CDK1 result in the dissolution of the nuclear envelope initiating mitosis. CDK, cyclin-dependent kinase; Fis1, fission 1; Mff, mitochondrial fission factor; Mfn1, mitofusin 1; Mfn2, mitofusin 2; Opa1, optic atrophy 1.
Finally, the termination phase begins once the regenerating liver achieves its original mass, which is mediated by antiproliferative and proapoptotic factors, such as TGF-β and other TGF-β-related proteins (5). The termination phase is essential for controlling the rate of regeneration, preventing the liver from undergoing tumorigenesis and proliferating excessively beyond its optimal mass, and ensuring that the liver proliferates in the appropriate direction and size. Moreover, many of these mechanisms that contribute to the termination phase converge at the mitochondria, highlighting the key roles of mitochondria during the entire regenerative process.
MITOCHONDRIA-ASSOCIATED METABOLIC CHANGES DURING LIVER REGENERATION
Cell Cycle Phase-Specific Changes to Mitochondrial Morphology Enhance Oxidative Capacity
Mitochondria exhibit two predominating morphologies throughout the cell cycle: a reticulum (i.e., a hyperfused state into a single tubular element) and multiple fragmented mitochondria of various sizes ranging from 0.5 to 5.0 μm in length (11, 12) (Fig. 1). During the late G1 phase, the cell’s mitochondrial network transiently adopts this hyperfused morphology, which begins to fragment during early S phase. Apart from the context of cellular proliferation, a fragmented mitochondrial network is typically associated with a nutrient-rich environment, while starving the cell of nutrients signals mitochondria to elongate into the reticular form (13). As hyperfused morphology is rarely observed in cells under physiological conditions, its appearance during this narrow window of G1-S transition suggests that mitochondrial hyperfusion plays a critical role in the cell cycle and energy metabolism, as ATP demand is significantly higher during G1-S compared with the other cell cycle phases (11). Furthermore, increased production and secretion of ATP help promote immune response by activating Kupffer cells (14). In an in vitro study, researchers treated various macrophage cell lines with extracellular ATP and noted activation of hepatic Kupffer cells, which demonstrated an upregulation of EGF (14).
Fusion of the outer mitochondrial membrane (OMM) is mediated by the GTPases, Mitofusin 1 (Mfn1), and Mitofusin 2 (Mfn2), whereas fusion of the inner mitochondrial membrane (IMM) involves optic atrophy 1 (Opa1) (15). Fusion of the OMM and IMM gives rise to a matrix continuum that allows various cellular components such as small proteins, lipids, and mitochondrial DNA (MtDNA) to diffuse freely between disparate regions of the mitochondrial network (11). Diffusion of these intramitochondrial components within the reticulum allows for repair of defective mitochondria by optimal use of available substrates and population of damaged molecules, which protects mitochondria from engulfment by autophagosomes during conditions of nutrient depletion (15). Fusion also yields an electrically continuous IMM, which promotes rapid energy transmission through propagation of the IMM potential (ΔΨm). ΔΨm is generated by both the proton gradient across the membrane and the electric potential created by charge separation that serves as an intermediate form of energy storage used by ATP synthase to generate ATP. Enhanced ΔΨm is observed following PH and is closely associated with increased phosphorylative activity and ATP production, which can be explained by the formation of the reticulum (16). As a result, the elongated reticulum would trigger one part of the cell to be dedicated to generating energy. In contrast, the other part could be used for other vital homeostatic functions such as reparatory or biosynthetic processes. Thus, the reticulum predominates in G1-S phase because mitochondrial metabolism centers on energy production and protein and lipid synthesis to prepare the cell for the following mitotic division (11, 15). Furthermore, the increased ATP production allows the cell to overcome AMPK-mediated cell cycle arrest at the G1-S ccheckpoint which is activated during settings of energy shortage.
At the onset of S phase, mitochondria undergo the fission process (i.e., become fragmented) and begin to establish a perinuclear localization (12). Fission of the mitochondrial tubular network is initiated by mobilization of cytosolic GTPase, dynamin 1-like protein (Drp1), to the OMM, which then promotes recruitment of other factors involved in the fission process such as fission 1 (Fis1), mitochondrial fission factor (Mff), MiD51, and MiD49 (15). Fission allows the cell to sort dysfunctional mitochondria to be eliminated via autophagy, thereby preventing generation of excessive reactive oxygen species (ROS) or inefficient ATP production. However, ROS can also activate the immune system by recruiting B cells, T cells, NK cells, and macrophages; for T cells and the NK cells activation, Drp1 is necessary for the mitochondria to translocate to the immune synapse (17). Interestingly, ROS increases the activation of Drp1, leading to fragmentation of mitochondria and phagocytosis (18). Fragmentation of mitochondria during the S, G2, and M phases also promotes equal distribution of mitochondria between the resulting daughter cells (15). This is of particular significance as MtDNA in humans encodes 13 polypeptides that are components of the OXPHOS system, and unequal distribution of MtDNA can result in various mitochondrial disorders (12)
Studies have shown that formation of a hyperfused mitochondrial network at G1-S is necessary for progression to S phase, not only for allowing the cell to meet the high metabolic demands of cellular proliferation but also due to its involvement with Cyclin E1 regulation (11). Mfn1 and Mfn2 knockouts demonstrate the same phenotypic effects seen in Cyclin E1 knockouts, including cell cycle arrest at the G1-S checkpoint and downregulation of cellular machinery required for initiating DNA replication in S-phase (11, 19). Interestingly, overexpression of Cyclin E1 has been shown to overcome the arrest at G1-S resulting from functional mitochondrial deficiency, implicating that the reticulum stimulates accumulation of Cyclin E1 in late G1 (11).
Taken together, mitochondria comprise repeating units that sustain the metabolic and biosynthetic functions of the organelle, and these units fuse into a reticulum during late G1 phase to maximize cellular bioenergetics that permits progression into the S phase. The enhanced oxidative capacity and ATP production inhibit the AMPK-mediated cell cycle arrest at the G1/S-phase checkpoint and promote accumulation of Cyclin E1 (11, 12). The repeating units can also readily come apart via fission to optimize inheritance of the MtDNA in a cell cycle-dependent fashion and prevent excessive ROS generation (15).
Increased Fatty Acid Oxidation Occurs during the Early Phases of Liver Regeneration
Transient microsteatosis is widely observed in the remnant liver shortly after PH. This has been described as a physiological response to PH and an essential process for optimal liver regeneration. Immediately after PH, the body enters an acute hypoglycemic state, triggering lipolysis in peripheral adipose tissue. The released lipids accumulate in the remnant liver, which can be used as an energy source via mitochondrial fatty acid oxidation (FAOX) (20). Energy demands are increased as the liver must maintain its regenerative capacity and homeostatic functions. Therefore, development of microsteatosis is postulated to be a physiological adaptation to facilitate robust hepatocyte proliferation with minimal impairment of the liver’s innate metabolic function. This event also aligns with the formation of the mitochondrial hyperfused reticulum detailed earlier, as the reticulum can use the lipids as substrates for generating energy.
However, lipids not only represent a potentially significant energy source but also serve as essential components for the formation of cellular membranes and are involved in the regulation of cell proliferation as secondary messengers. In a previous study, the importance of lipids was demonstrated by impaired regeneration resulting from the administration of leptin, which delays accumulation of fat in the liver, following PH (6). The mevalonate pathway, which regulates cholesterol levels in the liver, also yields prenylated intermediates that provide scaffolding for important signaling proteins involved in cellular proliferation. It has been shown that during liver regeneration, the activity of the rate-limiting enzyme in the mevalonate pathway, HMG-CoA reductase, accelerates while the production of cholesterol is diminished, implicating that the mevalonate pathway shifts away from cholesterol biosynthesis and toward the production of prenylated intermediates (6).
Peroxisome proliferator-activated receptor-α (PPAR-α) plays an integral role in enhancing cellular bioenergetics via interactions with mitochondria. PPAR-α is a transcription factor normally activated under conditions of energy deprivation and regulates lipid metabolism by promoting uptake, utilization, and catabolism of fatty acids within the liver by controlling constitutive expression of genes involved in mitochondrial FAOX. These target genes include carnitine palmitoyl transferase-1 (CPT-1) and carnitine palmitoyl transferase-2 (CPT-2), which mediate the transport of long-chain fatty acids through the OMM and IMM, respectively. PPAR-α also upregulates expression of acyl-CoA dehydrogenases (ACADs) that perform the first of four repeated catabolic steps during FAOX. The mitochondrial trifunctional protein (MTP), a large protein complex anchored to the IMM that possesses enoyl-CoA hydratase, l-3-hydroxyacyl-CoA dehydrogenase, and 3-ketoacyl-CoA thiolase catalytic functions, performs the remaining three steps. The four sequential steps are repeated to yield one acetyl-CoA molecule with each cycle, and expression of the MTP subunits is also controlled by PPAR-α (21, 22). The generated acetyl-CoA molecules are then used as substrates for the TCA cycle, which yields the reducing equivalents, NADH and FADH2, that are used in the mitochondrial electron transport chain (ETC) to produce ATP.
To maintain energy expenditure via FAOX, PPAR-α also inhibits the lipogenic pathway by inducing degradation of malonyl-CoA that inhibits CPT-1 and serves as a precursor for fatty acid synthesis (22). Enhanced FAOX also allows PPAR-α to shunt lipid metabolism away from the lipid peroxidation pathway, indirectly modulating the cell’s response to oxidative stress and reducing ROS burden (23). Thus, PPAR-α activation not only promotes a net bioenergetic gain within hepatocytes but also elicits anti-inflammatory effects. PPAR-α has been shown to be a vital component for the regenerative process as Ppar-α null mice demonstrate impaired liver regeneration following PH (24, 25). The inability of Ppar-α null mice to regenerate hepatic mass following PH suggests that PPAR-α’s role as a critical modulator of energy influx is crucial for optimizing the repair of liver damage and the regenerative process (21). Ppar-α null mice also display altered expression of genes involved in cell cycle control and cytokine signaling, with administration of PPAR-α agonists upregulating Cyclin D1, c-Myc, and receptors for IL-6 (21).
Though microsteatosis is essential for liver regeneration, a chronic and pathological accumulation of lipids within the liver, such as those seen in patients with nonalcoholic fatty liver disease (NAFLD) and chronic alcoholism, is paradoxically associated with blunted liver regeneration and propensity for more intra- and postoperative complications (26). Studies have shown that patients with fatty livers have higher rates of intraoperative blood loss and postoperative liver injury and insufficiency, as characterized by elevated AST, ALT, and bilirubin levels and impaired prothrombin time. Major complications such as bilioma, empyema, small bowel obstruction, and renal failure are also more frequent in patients who underwent PH with a fatty liver (27). This is particularly important for improving hepatic surgical outcomes as NAFLD is a common risk factor for obesity, and two-thirds of the American population is classified as obese. As an adaptive response for hepatocytes to survive chronic exposure to oxidative stress, NAFLD results in altered metabolic signaling, such as profound inhibition of the nuclear factor-kappa B (NF-κB) pathway (26). NF-κB is a transcription factor necessary for liver cell proliferation and survival as it upregulates expression of Cyclin D1. Fatty livers are associated with increased levels of inhibitor of NF-κB α (IκBα), perhaps due to impaired mitochondrial function in fatty livers. Dysfunctional mitochondria translate to reduced energy stores that precipitate failure in degrading IκBα (28). Furthermore, a deficiency in ATP and Cyclin D1 arrests the steatotic hepatocyte in the G1 phase (28). Therefore, deterioration in mitochondrial functions is critical in the pathological processes underlying intra- and postoperative complications in fatty livers that underwent PH.
Upstream Coordination by mTOR is Involved in the Regenerative Process
The mechanistic target of rapamycin (mTOR) is the master regulator of mRNA translation and global protein synthesis to promote cellular proliferation and has been shown to coordinate mitochondrial energy production during this process. In response to mitogens, mTOR complex 1 (mTORC1) is activated via the PI3K/AKT pathway and stimulates the expression and translation of various nuclear-encoded mitochondrial genes such as mitochondrial transcription factor A (TFAM), mitochondrial ribosomal proteins, and components of complexes I and V of the ETC (29). mTOR also inhibits autophagy, which can contribute to eliminating mitochondria (29).
It has been reported that inhibition of mTOR with rapamycin suppresses liver regeneration (30). However, rapamycin used to be an immunosuppressant of choice to prevent graft rejection after organ transplantation. As mTOR plays a crucial role in regulating cellular proliferation (31, 32), its inhibition raises concerns for possible adverse posttransplant outcomes such as small for size syndrome (SFSS), a condition in which impaired regeneration results in the grafted liver being too small to achieve adequate function and can occur in either the donor or recipient. Therefore, the use of rapamycin has become a highly deliberative issue, while the precise role of mTOR signaling following LDLT has yet to be more comprehensively elucidated.
EARLY MITOCHONDRIAL ANTIOXIDATION MAINTAINS OXIDATIVE CAPACITY AT THE ONSET OF LIVER REGENERATION
Studies have shown that hepatic stores of glutathione (GSH) play a critical role in preserving mitochondrial function during liver regeneration. Within mitochondria, GSH serves as the primary defense against ROS generated by cellular respiration or exogenous toxins. This is crucial for the functioning liver as it is continuously exposed to xenobiotics and, therefore, requires robust detoxification (33). In rats with PH, a decrease in the rate of ATP synthesis was observed 24 h following PH, which was found to be associated with a corresponding decrease in the β-F1 subunit of the ATP synthase complex. Similarly, mitochondrial GSH concentration also decreased, reaching a nadir at 24 h following PH. Based on these findings, it has been suggested that ROS generated by stress due to liver resection results in overconsumption of GSH, allowing excess ROS to inactivate the ATP synthase complex and compromise energy production (34).
The significance of GSH was further affirmed when the previous study was replicated using GSH-deficient rats, which presented with delayed liver regeneration and attenuated mitochondrial function following PH. The same study also demonstrated that hypothyroidism is associated with impaired liver regeneration, and administration of exogenous GSH in hypothyroid rats mitigated the regenerative response, which was associated with restoration of mitochondrial function (35). Hence, changes to the mitochondrial concentration of GSH have profound effects on the liver’s response to injury as it is closely linked to maintaining mitochondrial oxidative capacity by serving as its predominant antioxidant.
MODULATION OF MITOCHONDRIAL DNA DURING LIVER REGENERATION
In the early 1990s, mtDNA was implicated to play an essential role in liver regeneration. Using nonradioactive probes, it was revealed that mtDNA content iterates in an age-dependent manner (36). In rats, the total mtDNA gradually decreased upon aging to ∼15% or less compared with young ones. However, they also reported an increase in mtDNA content following PH, even in rats of advanced age. Studies investigating mitochondrial gene expression during rat liver regeneration noted an increase in mitochondrial DNA content 24 h following PH (37). Their observations suggested that the energy demand for liver regeneration is achieved by enhancing mitochondrial DNA replication and transcription, in which the mitochondrial DNA-binding proteins play critical regulatory roles. PH has also been shown to induce upregulation in mtDNA-binding proteins and mitochondrial mRNA levels (38). mtDNA replication and transcription occurred in the initial phase of liver regeneration, resulting in an increase in mitochondrial respiratory function within the remnant livers in rats following PH (39). Transcription of mtDNA also revealed a vital role in regeneration of the liver in the setting of chronic liver disease (40).
Adequate mtDNA copy number is vital to maintaining mitochondrial function and ATP production during cell proliferation (41). Mitochondrial division is linked to active mitochondrial synthesis, and mtDNA replication in mammals involves proteins distinct from those used for nuclear DNA replication.
Topoisomerases are essential for mtDNA replication and transcription. These are ubiquitous enzymes that relieve the DNA topological stress generated during replication and transcription by rapidly and reversibly breaking and rejoining the phosphodiester DNA backbone (42). Top1mt is a mitochondria-specific topoisomerase enzyme that regulates mtDNA replication (43), transcription (44), and mtDNA integrity (45). Top1mt-deficient mice have reduced liver regeneration, decreased replication of mtDNA, and defective mitochondrial functions that debilitate their energy-producing capacity (46).
Liver regeneration following PH or CCl4-induced liver injury in rats is stimulated by a hepatic mitogen called augmenter of liver regeneration (ALR), and inhibition of ALR expression attenuates regeneration due to weakened mtDNA synthesis and biogenesis (47).
Although the role of mtDNA in liver regeneration is well established, the detailed mechanism still remains elusive. The molecular mechanisms and the genes associated with mtDNA synthesis are yet to be explored and elucidated, which will help us gain deeper insights toward strategies for liver regeneration.
INTRAMITOCHONDRIAL SIGNALS FOR APOPTOSIS IN TERMINATING LIVER REGENERATION
The induction of apoptosis by various antiproliferation factors, most notably TGF-β and its related proteins, is essential for controlling the rate of regeneration, terminating regeneration once the original liver mass is achieved, and ensuring that regeneration proceeds in the appropriate direction. Furthermore, a small surge of apoptosis is observed at the end of DNA synthesis in regenerating livers, suggesting that the mechanism remained active past the regenerative process (48, 49). Moreover, studies have revealed that apoptosis promotes liver regeneration by releasing growth signals that stimulate proliferation, with impaired regeneration seen in mice lacking caspase 3 or caspase 7, two key executioners of apoptosis (50).
TGF-β, specifically TGF-β1, is expressed by hepatocytes and functions in a paracrine manner, resulting in its upregulation during the G1-phase and induction of cellular apoptosis via activation of various signaling pathways that converge at the mitochondria (5). Besides hepatocytes, TGF-β1 is also synthesized in extrahepatic tissues, such as platelets and the spleen. Spleen secretes TGF-β1 to promote termination of liver regeneration as splenectomy results in a significantly increased number of proliferating cells 48 h following PH. The spleen has also been shown to downregulate HGF and its receptor c-Met to inhibit cellular proliferation, indicating that the spleen can remotely influence and regulate the regenerative process in injured liver (5).
In the canonical pathway, binding of TGF-β1 to its cell surface receptor results in activation of suppressor of mothers against decapentaplegic protein 2/3 (SMAD2/3), allowing it to form a protein complex with SMAD4. The SMAD2/3-SMAD4 complex then localizes to the cell nucleus to upregulate proapoptotic genes and downregulate antiapoptotic genes. One particular protein upregulated in the canonical pathway is Bcl-2-interacting mediator of cell death (Bim), confirmed by studies demonstrating that accumulation of Bim within the mitochondria occurs downstream of TGF-β1 (51). Bim then sequesters the antiapoptotic Bcl-XL, allowing proapoptotic factors Bax and Bak to undergo a conformational change that forms proteolipid pores in the OMM. The increased permeability of the OMM permits the release of cytochrome c from the mitochondria. Cytochrome c associates with procaspase-9 in the cytosol to form the apoptosome, thereby initiating the caspase cascade. Formation of the apoptosome facilitates autoactivation of procaspase-9 into caspase-9. Caspase-9 then activates the executioner caspases-3, -6, and -7, resulting in cleavage of cellular proteins and culminating in cell death (50, 51). However, in vitro studies have revealed an alternative transcription-independent pathway that diverges from the canonical pathway.
Upon activation of the TGF-β1 receptor, a portion of the SMAD2/3-SMAD4 complex translocates to the mitochondria and associates with the mtDNA-encoded cytochrome c oxidase subunit 2 (COXII), a highly conserved subunit of the cytochrome c oxidase enzyme that catalyzes the reduction of water to oxygen within the mitochondrial respiratory chain, to induce apoptosis. Although the exact mechanism remains unclear, the results suggest that this transcription-independent event potentiates apoptosis rather than precedes it (52).
Another component of TGF-β1-mediated cell death involves the generation of ROS within mitochondria, followed by the release of cytochrome c and caspase activation. TGF-β1 is also shown to negatively regulate antioxidant genes and increase the expression of NADPH oxidase 4 (NOX4), which is localized to the IMM. NOX4 is required for the proapoptotic activity of TGF-β1 in hepatocytes, and dysfunction of this enzyme can confer resistance to apoptosis in hepatocellular carcinoma (53). Excessive ROS yields many effects, all of which facilitate initiation of apoptosis, such as direct stimulation of tumor suppressor p53, release of cytochrome c from the IMM via oxidation of mitochondrial cardiolipin, and direct disruption of the IMM and OMM to accelerate the apoptotic effect of TGF-β1 (54) Furthermore, mitochondrial morphology remains pertinent in the propagation of mitochondria-mediated apoptosis as fragmented morphology correlates with the release of cytochrome c, while the hyperfused reticulum is protective against apoptosis (11).
In conjunction with TGF-β1, studies have shown that mitochondrial Ca2+ is also implicated in the regenerative process through its role in regulating apoptosis (54, 55). Conversely, members of the Bcl-2 family, the key regulators of apoptosis, directly influence excessive intramitochondrial Ca2+ accumulation via their various interregulatory processes that contribute to apoptosis. Proapoptotic members of the Bcl-2 family enhance the release of Ca2+ from the endoplasmic reticulum and facilitate entry of Ca2+ into the mitochondria, whereas the antiapoptotic members reduce the entry of Ca2+ into the mitochondria (55). Overloading intramitochondrial Ca2+ concentration opens the mitochondrial permeability transition pore (MPTP) complex in the IMM. In addition, elevated intramitochondrial Ca2+ concentration results in osmotic swelling, which further permeabilizes the mitochondrial membrane and allows the escape of Cytochrome c into the cytosol (54). The significance of mitochondrial Ca2+ in the context of liver regeneration is highlighted by a study that investigated the effects of administering parvalbumin, a Ca2+ buffer. The study shows that treatment in rats with parvalbumin following PH produced decreased activity of caspases 3 and 9, decreased expression of Bax, increased expression of Bcl-XL, and enhanced hepatocyte proliferation via increased proliferating cell nuclear antigen (PCNA) labeling, indicating that attenuation of mitochondrial Ca2+ levels accelerate the rate of liver regeneration via inhibition of apoptosis (55).
MITOCHONDRIA-CENTRIC STRATEGIES FOR ACCELERATING LIVER REGENERATION
As presented, mitochondria play a vital role in liver regeneration and preservation of its functional and structural integrity is critical for optimal regeneration of the liver, which ensures favorable outcomes after liver transplants. The role of mitochondria and its relevance in transplantation has been explored in other organ transplants as well, such as lung (56), heart (57, 58), and bone marrow-derived mesenchymal stem cells (MSCs) (59). For solid organ transplants, there is often an unavoidable long interval between organ procurement and transplantation of the donor organ into the recipient. During this critical time of ex vivo preservation, the harvested organs are kept on ice and subjected to cold ischemia, which predisposes the ischemic organ to undergo metabolic changes while adapting to these nonphysiological conditions. After transplantation, the organ is reperfused with oxygenated blood, which paradoxically damages the organ via ischemia-reperfusion injury (IRI). IRI is caused by the release of ROS from the mitochondrial respiratory chain, initiating a cascade of tissue damage (60, 61). These produce significant oxidative damage to mitochondria, resulting in the opening of the MPTP and impending cell death (62, 63). Cell death following the opening of the MPTP leads to remodeling and scarring of tissue via fibrosis, i.e., cirrhosis in transplanted liver (60, 64), which inevitably contributes to graft dysfunction and rejection of transplanted organs. Moreover, cell death is associated with the release of mtDNA and metabolites, such as succinate, into the circulation, which activates inflammatory immune responses (65, 66). Thus, regulating mitochondrial metabolic homeostasis during transplantation is critical for ensuring optimal postoperative graft function and surgical outcomes. There are nonpharmacological strategies that optimize tactical expediency to reduce ischemia time. These measures do not directly target mitochondria but shortening the intervening period of ischemia and optimizing the cold storage media composition collectively attenuate organ damage and underlying mitochondrial dysfunctions. However, numerous therapeutic interventions and strategies that target mitochondria can be favorably exploited to improve posttransplant outcomes (Table 1).
Table 1.
Agents and their effect on mitochondria that contribute to regulation of liver regeneration in experimental models
| Mitochondria-Centered Strategies for Accelerating Liver Regeneration | ||
|---|---|---|
| Strategies | Agents | References |
| Antioxidation |
|
(35) |
|
(67) | |
|
(68) | |
|
(69) | |
|
(70) | |
|
(71) | |
|
(72) | |
|
(72) | |
|
(73) | |
| Calcium Dyshomeostasis |
|
(55) |
|
(74) | |
|
(74) | |
|
(75) | |
| Metabolic Conditioning |
|
(76) |
|
(77) | |
|
(78) | |
|
(14) | |
| Cellular |
|
(79, 80) |
Antioxidants
Certain pharmacological agents selectively aim to prevent the metabolic changes associated with ischemia (74). This review has already highlighted the use of exogenous GSH to restore liver regeneration in hypothyroid and GSH-deficient rats, as antioxidation with GSH during the early stages of the regenerative process is essential for maintaining mitochondrial oxidative capacity (35). N-acetylcysteine (NAC) is a potent GSH precursor and is typically used to treat early acetaminophen (APAP) toxicity by increasing GSH levels in hepatocytes (81). It has also been shown that preoperative administration of NAC can accelerate liver regeneration in rats with NAFLD following PH. In addition, the rats with NAFLD that were treated with NAC had higher levels of GSH compared with their saline-treated counterparts (67). Other antioxidants have also been studied that function similarly to GSH by ameliorating the oxidative damage caused by IRI. Administration of allopurinol, or hypoxanthine, to the grafted liver inhibits xanthine oxidoreductase and succinate dehydrogenase (82), which increases the NAD+ pool via NAD+ precursors such as nicotinamide ribosides (68), thereby decreasing succinate accumulation during ischemia, respectively. These mechanisms collectively lower mitochondrial ROS production following reperfusion (66, 83). Mangafodipir trisodium is a contrast agent used for imaging of the liver, and its administration in the donor liver before LDLT prevents mitochondrial injury, oxidative stress, and hepatic apoptosis in an isolated perfused rat liver model (69, 74).
Resveratrol, a naturally occurring phenolic compound, improves liver regeneration and restores the quantity and quality of mitochondria in mice with APAP-induced liver damage (70, 74). Other herbal antioxidants, such as green tea catechins, also revealed similar salutary effects, owing to their anti-inflammatory properties, when administered preoperatively in rats (71, 74). Likewise, other naturally occurring antioxidants such as Silybum marinaum (i.e., Milk thistle) and vitamin E confer favorable results when administered intraperitoneally following PH (72, 74). Melatonin is also known to have potent antioxidative properties and can modulate the degree of oxidative stress that hepatic mitochondria experience following PH, in addition to promoting mitochondrial proliferation. This is further supported by the studies that demonstrate that removal of the pineal gland, the body’s source of melatonin, resulted in delayed liver regeneration in rats (73, 74).
Calcium Dyshomeostasis
As previously discussed, excessive intramitochondrial Ca2+ accumulation plays a key role in mediating apoptosis by triggering the opening of the MPTP, which releases cytochrome c, and preventing early cell death by decreasing Ca2+ levels using parvalbumin accelerates liver regeneration (55). Calcium channel blockers such as amlodipine and 2-aminoethoxydiphenyl borate function similarly by attenuating the dysregulated rise in intramitochondrial Ca2+ concentration that occurs as a consequence of IRI (74). Cyclosporine A inhibits the opening of the MPTP to prevent cell death and reduces the generation of excessive ROS (75).
Metabolic Conditioning
In general, PPARs are transcription factors involved in cellular differentiation, development, and metabolism, and the role of PPAR-α in promoting mitochondrial FAOX during the regenerative process has been discussed (21). Use of Wy-14,643, a PPAR-α agonist, exerts hepatoprotective effects by reducing hepatic injury in mice with NAFLD that are subjected to IRI and also promoted accelerated progression through the G1/S-phase checkpoint by upregulating Cyclin D1 (76). PPAR-γ is an isoform of PPAR-α with similar anti-inflammatory and antiobesity properties but not as widely distributed in the liver as PPAR-α (21). However, it may be involved in the liver’s response to injury, since preoperative intragastric injections of Pioglitazone, a PPAR-γ agonist, improved liver regeneration in mice with NAFLD (74, 77). Polyethylene glycol, a nontoxic polymer used as an additive in organ preservation solutions before transplantation, is shown to reduce damage caused by IRI by protecting the structural integrity of the mitochondria and upregulating the prosurvival AKT pathway (78). Methylation-controlled J protein (MCJ) is a negative regulator of mitochondrial respiration, which results in decreased ATP production, and inhibition of MCJ with small interfering RNA molecules restores liver regeneration in mice by enhancing mitochondrial activity (14). Furthermore, the increased ATP production due to MCJ inhibition causes early activation of Kupffer cells, which release TNF-α, IL-6, and EGF that promote an earlier onset of the priming phase and entry into the cell cycle (14).
Cellular Approaches
It has been reported that the use of MSCs increases survival, attenuates liver damage, prevents the mitochondrial cell death pathway, promotes antioxidation, and stimulates hepatocyte proliferation with APAP-induced liver damage (79, 80). Furthermore, transplanted stem cells were shown to remove dysfunctional mitochondria from damaged cells and replace them with their own healthy mitochondria to maintain the metabolic state of the cells (84). Though not directly related to liver regeneration, in vitro transfer of autologous functional mitochondria into recipient BMSCs that were transplanted into a cranial bone defect rat model demonstrated enhanced osteogenesis, ATP production, and bone healing (59). This raises the prospect for a potential strategy for optimizing stem cell therapy using mitochondria.
Mitochondrial Transfer
Another emerging approach, commonly known as “mitotherapy,” involves infusion of exogenous mitochondria into the transplanted organ. In mitotherapy, healthy mitochondria are isolated from cell lines or tissues, such as the liver, through several cycles of centrifugation and filtration; following isolation, the mitochondria are injected or infused into the recipient (85). If the mitochondria are labeled with a fluorescent protein, localization of the infused mitochondria can be visualized. There are several emerging theories that describe mitochondrial entry into the cells. Mitochondria have been implicated to move through the gap junctions between cells such as via donor cell exocytosis and recipient cell endocytosis (86). Mitochondria have also been suggested to travel along tunneling nanotubes between donor and recipient cells; tunneling nanotubes are cell membrane protuberances leading to direct interaction between cells (87).
Mitotherapy is reported to significantly improve lung function and aeromechanics during the reparative process attenuating lung tissue injury in an experimental murine model. Transplantation of mitochondria can protect lung tissue from IRI, which reduces morbidity and mortality following lung transplantation (56). Moreover, mitotherapy used in pretransplant BMSCs in vitro demonstrated improved bone defect repair in situ (59). Thus, the transfer of exogenous mitochondria to degenerating cells and solid organs via mitotherapy offers a promising therapeutic potential with significantly improved outcomes, which ought to be explored further for solid organ transplants such as liver and lung.
MECHANISMS IN MITOTHERAPY STRATEGIES IN VARIOUS LIVER DISEASES
Recent studies have focused on the mechanisms and salutary outcomes of mitochondrial transplantation in numerous liver pathologies. One study used mitotherapy to overcome the effect of CCl4 in a liver injury model in mice (88). It was shown that exogenously perfused mitochondria, isolated from healthy mice liver are rapidly internalized by damaged liver cells through the macropinocytosis pathway. This was demonstrated using amiloride, an inhibitor of macropinocytosis, which blocked mitochondrial internalization. The internalized mitochondria increased cell viability and ATP production during injury and reduced ROS generation, resulting in improved liver function and reduced liver fibrosis. They confirmed these findings using transcriptomics data, which showed an association with the unfolded protein response mitochondria-regulated pathways (88).
Massive resections that exceed the degree of surgical reduction of diseased liver in PH can be used for the treatment of extensive liver malignancies and certain LDLT. Unfortunately, this greatly increases the risk of postoperative liver failure such as SFSS. Massive liver resections are associated with major blood loss that exacerbates IRI, reduction of growth factors that attenuates regeneration, and induction of MPTP that promotes cell death, all of which collectively contribute to liver failure (89–91). However, the MPTP inhibitory effect of cyclosporin A and its nonimmunosuppressive derivative, NIM81, can be used in solution during cold ischemic preservation of liver grafts to prevent IRI, or for mitochondria during isolation and generation of cultured hepatocytes (92–95).
Furthermore, profound mitochondrial dysfunction is seen in livers along the NAFLD/nonalcoholic steatohepatitis (NASH) spectrum, which necessitates novel defatting strategies (96). Mitochondrial respiratory chain complexes in liver cells of patients with NASH are disrupted due to severe ROS production and endoplasmic reticulum stress (97). In addition, internalized excessive lipids by hepatocytes disrupt the dephosphorylation capacity of liver mitochondria by affecting the voltage-dependent anion channels. This results in permeabilization of the IMM, which leads to depolarization of the mitochondria, reduction in ATP synthesis, and limited antioxidative capacity (98–100). Mitotherapy can be used to restore cell viability and prevent disease progression in the liver (101). Infusion of isolated healthy mitochondria from donor hepatocytes can be internalized by the pathologic liver, which can mitigate lipid accumulation, facilitate cellular redox balance, and enhance energy production. These can contribute to the restoration of hepatocyte function in patients with metabolic diseases such as NASH, diabetes, or other liver injuries including IRI associated with surgery (102).
It is noteworthy that exogenous mitochondria were identified in different murine tissues including lungs, brain, muscle, and kidneys (103, 104). Strategies that target mitochondria in these tissues could prove beneficial in a damaged organ. In a recent study, coinjection of an endosomal release agent, AsOR-LLO, with exogenous mitochondria was demonstrated to be crucial for preventing the degradation of the transfused mitochondria after internalization in the hepatocytes of rats (105, 106).
Multiple manufacture kits are available for isolation of mitochondria, and it can be isolated from various tissues and cells; however primary sources are liver, muscle, and mesenchymal stem cells. Isolated mitochondria can be administered directly to tissue (i.e., heart) or systemically (intravenously or intraperitoneally). In addition, it can be coincubated with cells or chemicals before injection. Timing of administration can be important for effectiveness of isolated mitochondria. Generally, studies inject isolated mitochondria before or during the experiments as prophylactic and afterward as a treatment option depending on the experimental models. Along with timing, the doses of mitochondria administrated are also important for standardizing therapy. Isolated mitochondria are quantified before injection by either measuring protein concentrations or counting number of dye-labeled mitochondria using fluorescent microscope and hemacytometer or by using coulter particle counter (Beckman, multisizer4e) (107).
As metabolism differs among the various tissue types and proteomic capabilities can vary between nontumor and tumor-derived tissues, the sources, applications, conditions, dose, and timing of mitotherapy deserve further investigation.
CONCLUSIONS
A wide array of emerging literature convincingly demonstrates that mitochondria play key roles in liver regeneration. From reprogramming metabolic homeostasis to meet the energy demands for cellular proliferation to terminating liver regeneration via exquisite modulation of apoptosis, mitochondria are quintessential for sequential progression through the regenerative process and maintaining adequate liver function since mitochondrial damage or attenuation of mitochondrial functional capacity can result in deleterious consequences that negatively impact postoperative outcomes. Much work remains to be done in further elucidating the various cellular pathways that converge at the mitochondria and how those upstream modulators affect liver regeneration. Emerging mitotherapy appears to be a promising new horizon for clinicians to explore as novel strategies for improving surgical outcomes in LDLT and PH, whereas previous studies that demonstrated mitotherapy preserving other organs should serve as a guide for exploring mitotherapy as a potential intervention involving solid organ transplantation. Therefore, focusing on the role of mitochondria during liver regeneration may lead to an effective therapeutic strategy that will significantly advance the arts and sciences in solid organ transplantation and regenerative medicine.
GRANTS
Research reported in this work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (NIH) under award numbers DK117183 and DK132230 (to A.B.), institutional startup by the James D. Eason Transplant Institute (to A.B.), and UTHSC MSRF 2020 and Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship (to G.G.L.).
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
G.G.L., M.D., S.K.B., J.D.E., C.K., C.K., and A.B. conceived and designed research; G.G.L., M.D., and S.A. performed experiments; G.G.L., M.D., and A.B. analyzed data; G.G.L., M.D., P.S.P., S.A., S.K.B., and A.B. interpreted results of experiments; G.G.L., P.S.P., D.S.P., and A.B. prepared figures; G.G.L., M.D., D.S.P., and A.B. drafted manuscript; G.G.L., M.D., P.S.P., S.A., D.S.P., S.K.B., J.D.E., C.K., C.K., and A.B. edited and revised manuscript; G.G.L., M.D., P.S.P., S.K.B., J.D.E., C.K., C.K., and A.B. approved final version of manuscript.
ACKNOWLEDGMENTS
The graphical abstract figure was made in BioRender.
REFERENCES
- 1. Dar WA, Sullivan E, Bynon JS, Eltzschig H, Ju C. Ischaemia reperfusion injury in liver transplantation: cellular and molecular mechanisms. Liver Int 39: 788–801, 2019. doi: 10.1111/liv.14091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mao SA, Glorioso JM, Nyberg SL. Liver regeneration. Transl Res 163: 352–362, 2014. doi: 10.1016/j.trsl.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexandrino H, Rolo A, Tralhão JG, Castro e Sousa F, Palmeira C. Mitochondria in liver regeneration: energy metabolism and posthepatectomy liver dysfunction. In: Mitochondrial Biology and Experimental Therapeutics. Cham, Switzerland: Springer, 2018. [Google Scholar]
- 4. Budai A, Horvath G, Tretter L, Radak Z, Koltai E, Bori Z, Torma F, Lukats A, Rohlich P, Szijarto A, Fulop A. Mitochondrial function after associating liver partition and portal vein ligation for staged hepatectomy in an experimental model. Br J Surg 106: 120–131, 2019. doi: 10.1002/bjs.10978. [DOI] [PubMed] [Google Scholar]
- 5. Tao Y, Wang M, Chen E, Tang H. Liver regeneration: analysis of the main relevant signaling molecules. Mediators Inflamm 2017: 4256352, 2017.doi: 10.1155/2017/4256352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Delgado-Coello B, Briones-Orta MA, Macías-Silva M, Mas-Oliva J. Cholesterol: recapitulation of its active role during liver regeneration. Liver Int 31: 1271–1284, 2011. doi: 10.1111/j.1478-3231.2011.02542.x. [DOI] [PubMed] [Google Scholar]
- 7. Berthiaume F, MacDonald AD, Kang YH, Yarmush ML. Control analysis of mitochondrial metabolism in intact hepatocytes: effect of interleukin-1β and interleukin-6. Metab Eng 5: 108–123, 2003. doi: 10.1016/s1096-7176(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 8. Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70: 993–1006, 1992. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- 9. Nevzorova YA, Tolba R, Trautwein C, Liedtke C. Partial hepatectomy in mice. Lab Anim 49: 81–88, 2015. doi: 10.1177/0023677215572000. [DOI] [PubMed] [Google Scholar]
- 10. Thyberg J, Moskalewski S. Role of microtubules in the organization of the Golgi complex. Exp Cell Res 246: 263–279, 1999. doi: 10.1006/excr.1998.4326. [DOI] [PubMed] [Google Scholar]
- 11. Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA 106: 11960–11965, 2009. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Margineantu DH, Gregory Cox W, Sundell L, Sherwood SW, Beechem JM, Capaldi RA. Cell cycle dependent morphology changes and associated mitochondrial DNA redistribution in mitochondria of human cell lines. Mitochondrion 1: 425–435, 2002. doi: 10.1016/s1567-7249(02)00006-5. [DOI] [PubMed] [Google Scholar]
- 13. Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol Cell 61: 683–694, 2016. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 14. Goikoetxea-Usandizaga N, Serrano-Macia M, Delgado TC, Simon J, Fernandez Ramos D, Barriales D, , et al. Mitochondrial bioenergetics boost macrophage activation, promoting liver regeneration in metabolically compromised animals. Hepatology 75: 550–566, 2022. [Erratum in Hepatology 76: 532, 2022]. doi: 10.1002/hep.32149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pendin D, Filadi R, Pizzo P. The concerted action of mitochondrial dynamics and positioning: new characters in cancer onset and progression. Front Oncol 7: 102, 2017. doi: 10.3389/fonc.2017.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nishihira T, Tanaka J, Nishikawa K, Jikko A, Taki Y, Morimoto T, Koizumi K, Kamiyama Y, Ozawa K, Tobe T. Biological significance of enhanced mitochondrial membrane potential in regenerating liver. Hepatology 6: 220–224, 1986. doi: 10.1002/hep.1840060211. [DOI] [PubMed] [Google Scholar]
- 17. Angajala A, Lim S, Phillips JB, Kim J-H, Yates C, You Z, Tan M. Diverse roles of mitochondria in immune responses: novel insights into immuno-metabolism. Front Immunol 9: 1605, 2018. doi: 10.3389/fimmu.2018.01605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Y, Subramanian M, Yurdagul A Jr, Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, Ryan TA, Nomura M, Maxfield FR, Tabas I. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell 171: 331–345.e22, 2017. doi: 10.1016/j.cell.2017.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, Sicinski P. Cyclin E ablation in the mouse. Cell 114: 431–443, 2003. doi: 10.1016/S0092-8674(03)00645-7. [DOI] [PubMed] [Google Scholar]
- 20. Abshagen K, Degenhardt B, Liebig M, Wendt A, Genz B, Schaeper U, Stumvoll M, Hofmann U, Frank M, Vollmar B, Klöting N. Liver-specific Repin1 deficiency impairs transient hepatic steatosis in liver regeneration. Sci Rep 8: 16858, 2018. doi: 10.1038/s41598-018-35325-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elias-Miró M, Jiménez-Castro MB, Mendes-Braz M, Casillas-Ramírez A, Peralta C. The current knowledge of the role of PPAR in hepatic ischemia-reperfusion injury. PPAR Res 2012: 802384, 2012. doi: 10.1155/2012/802384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mello T, Materozzi M, Galli A. PPARs and mitochondrial metabolism: from NAFLD to HCC. PPAR Res 2016: 7403230, 2016. doi: 10.1155/2016/7403230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Damrauer SM, Studer P, da Silva CG, Longo CR, Ramsey HE, Csizmadia E, Shrikhande GV, Scali ST, Libermann TA, Bhasin MK, Ferran C. A20 modulates lipid metabolism and energy production to promote liver regeneration. PLoS One 6: e17715, 2011. doi: 10.1371/journal.pone.0017715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie G, Yin S, Zhang Z, Qi D, Wang X, Kim D, Yagai T, Brocker CN, Wang Y, Gonzalez FJ, Wang H, Qu A. Hepatocyte peroxisome proliferator-activated receptor α enhances liver regeneration after partial hepatectomy in mice. Am J Pathol 189: 272–282, 2019. doi: 10.1016/j.ajpath.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kersten S. Integrated physiology and systems biology of PPARα. Mol Metab 3: 354–371, 2014. doi: 10.1016/j.molmet.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang SQ, Lin HZ, Mandal AK, Huang J, Diehl AM. Disrupted signaling and inhibited regeneration in obese mice with fatty livers: implications for nonalcoholic fatty liver disease pathophysiology. Hepatology 34: 694–706, 2001. doi: 10.1053/jhep.2001.28054. [DOI] [PubMed] [Google Scholar]
- 27. McCormack L, Petrowsky H, Jochum W, Furrer K, Clavien P-A. Hepatic steatosis is a risk factor for postoperative complications after major hepatectomy: a matched case-control study. Ann Surg 245: 923–930, 2007. doi: 10.1097/01.sla.0000251747.80025.b7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeAngelis RA, Markiewski MM, Taub R, Lambris JD. A high-fat diet impairs liver regeneration in C57BL/6 mice through overexpression of the NF-kappaB inhibitor, IkappaBalpha. Hepatology 42: 1148–1157, 2005. doi: 10.1002/hep.20879. [DOI] [PubMed] [Google Scholar]
- 29. Morita M, Gravel S-P, Hulea L, Larsson O, Pollak M, St-Pierre J, Topisirovic I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 14: 473–480, 2015. doi: 10.4161/15384101.2014.991572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fouraschen SM, de Ruiter PE, Kwekkeboom J, de Bruin RW, Kazemier G, Metselaar HJ, Tilanus HW, van der Laan LJ, de Jonge J. mTOR signaling in liver regeneration: rapamycin combined with growth factor treatment. World J Transplant 3: 36–47, 2013. doi: 10.5500/wjt.v3.i3.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haga S, Ozaki M, Inoue H, Okamoto Y, Ogawa W, Takeda K, Akira S, Todo S. The survival pathways phosphatidylinositol-3 kinase (PI3-K)/phosphoinositide-dependent protein kinase 1 (PDK1)/Akt modulate liver regeneration through hepatocyte size rather than proliferation. Hepatology 49: 204–214, 2009. doi: 10.1002/hep.22583. [DOI] [PubMed] [Google Scholar]
- 32. Chen P, Yan H, Chen Y, He Z. The variation of AkT/TSC1- TSC1/mTOR signal pathway in hepatocytes after partial hepatectomy in rats. Exp Mol Pathol 86: 101–107, 2009. doi: 10.1016/j.yexmp.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 33. Kretzschmar M. Regulation of hepatic glutathione metabolism and its role in hepatotoxicity. Exp Toxicol Pathol 48: 439–446, 1996. doi: 10.1016/S0940-2993(96)80054-6. [DOI] [PubMed] [Google Scholar]
- 34. Vendemiale G, Guerrieri F, Grattagliano I, Didonna D, Muolo L, Altomare E. Mitochondrial oxidative phosphorylation and intracellular glutathione compartmentation during rat liver regeneration. Hepatology 21: 1450–1454, 1995. [PubMed] [Google Scholar]
- 35. Grattagliano I, Lauterburg BH, Portincasa P, Caruso ML, Vendemiale G, Valentini AM, Palmieri VO, Palasciano G. Mitochondrial glutathione content determines the rate of liver regeneration after partial hepatectomy in eu- and hypothyroid rats. J Hepatol 39: 571–579, 2003. doi: 10.1016/s0168-8278(03)00317-9. [DOI] [PubMed] [Google Scholar]
- 36. Asano K, Nakamura M, Asano A, Sato T, Tauchi H. Quantitation of changes in mitochondrial DNA during aging and regeneration of rat liver using non-radioactive DNA probes. Mech Ageing Dev 64: 85–98, 1992. doi: 10.1016/0047-6374(92)90098-x. [DOI] [PubMed] [Google Scholar]
- 37. Shimizu Y, Suzuki H, Nimura Y, Onoue S, Nagino M, Tanaka M, Ozawa T. Elevated mitochondrial gene expression during rat liver regeneration after portal vein ligation. Hepatology 22: 1222–1229, 1995. doi: 10.1016/0270-9139(95)90632-0. [DOI] [PubMed] [Google Scholar]
- 38. Onoue S, Suzuki H, Nimura Y, Shimizu Y, Nagino M, Tanaka M, Ozawa T. Increase of mtDNA-binding proteins and mitochondrial mRNAs in regenerating liver. J Surg Res 62: 172–178, 1996. doi: 10.1006/jsre.1996.0191. [DOI] [PubMed] [Google Scholar]
- 39. Koyama H, Kurokawa T, Nonami T, Nakao A, Sugiyama S, Murakami T, Shimomura Y, Takagi H. Increases in the mitochondrial DNA replication and transcription in the remnant liver of rats. Biochem Biophys Res Commun 243: 858–861, 1998. doi: 10.1006/bbrc.1998.8174. [DOI] [PubMed] [Google Scholar]
- 40. Kotake K, Nonami T, Kurokawa T, Nakao A, Murakami T, Shimomura Y. Effects of chronic liver diseases on mitochondrial DNA transcription and replication in human liver. Life Sci 65: 557–563, 1999. doi: 10.1016/s0024-3205(99)00276-3. [DOI] [PubMed] [Google Scholar]
- 41. Lee S, Kim S, Sun X, Lee J-H, Cho H. Cell cycle-dependent mitochondrial biogenesis and dynamics in mammalian cells. Biochem Biophys Res Commun 357: 111–117, 2007. doi: 10.1016/j.bbrc.2007.03.091. [DOI] [PubMed] [Google Scholar]
- 42. Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 17: 421–433, 2010. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang H, Pommier Y. Mitochondrial topoisomerase I sites in the regulatory D-loop region of mitochondrial DNA. Biochemistry 47: 11196–11203, 2008. doi: 10.1021/bi800774b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sobek S, Dalla Rosa I, Pommier Y, Bornholz B, Kalfalah F, Zhang H, Wiesner RJ, von Kleist-Retzow J-C, Hillebrand F, Schaal H, Mielke C, Christensen MO, Boege F. Negative regulation of mitochondrial transcription by mitochondrial topoisomerase I. Nucleic Acids Res 41: 9848–9857, 2013. doi: 10.1093/nar/gkt768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Medikayala S, Piteo B, Zhao X, Edwards JG. Chronically elevated glucose compromises myocardial mitochondrial DNA integrity by alteration of mitochondrial topoisomerase function. Am J Physiol Cell Physiol 300: C338–C348, 2011. doi: 10.1152/ajpcell.00248.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khiati S, Baechler SA, Factor VM, Zhang H, Huang S-YN, Dalla Rosa I, Sourbier C, Neckers L, Thorgeirsson SS, Pommier Y. Lack of mitochondrial topoisomerase I (TOP1mt) impairs liver regeneration. Proc Natl Acad Sci USA 112: 11282–11287, 2015. doi: 10.1073/pnas.1511016112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Han L-H, Dong L-Y, Yu H, Sun G-Y, Wu Y, Gao J, Thasler W, An W. Deceleration of liver regeneration by knockdown of augmenter of liver regeneration gene is associated with impairment of mitochondrial DNA synthesis in mice. Am J Physiol Gastrointest Liver Physiol 309: G112–G122, 2015. doi: 10.1152/ajpgi.00435.2014. [DOI] [PubMed] [Google Scholar]
- 48. Michalopoulos GK. Liver regeneration. J Cell Physiol 213: 286–300, 2007. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu M, Chen P. Proliferation-inhibiting pathways in liver regeneration (Review). Mol Med Rep 16: 23–35, 2017. doi: 10.3892/mmr.2017.6613. [DOI] [PubMed] [Google Scholar]
- 50. Guicciardi ME, Malhi H, Mott JL, Gores GJ. Apoptosis and necrosis in the liver. Compr Physiol 3: 977–1010, 2013. doi: 10.1002/cphy.c120020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yu J, Zhang L, Chen A, Xiang G, Wang Y, Wu J, Mitchelson K, Cheng J, Zhou Y. Identification of the gene transcription and apoptosis mediated by TGF-beta-Smad2/3-Smad4 signaling. J Cell Physiol 215: 422–433, 2008. doi: 10.1002/jcp.21325. [DOI] [PubMed] [Google Scholar]
- 52. Pang L, Qiu T, Cao X, Wan M. Apoptotic role of TGF-β mediated by Smad4 mitochondria translocation and cytochrome c oxidase subunit II interaction. Exp Cell Res 317: 1608–1620, 2011. doi: 10.1016/j.yexcr.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 53. Carmona-Cuenca I, Roncero C, Sancho P, Caja L, Fausto N, Fernández M, Fabregat I. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J Hepatol 49: 965–976, 2008. doi: 10.1016/j.jhep.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 54. Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 1863: 2977–2992, 2016. doi: 10.1016/j.bbamcr.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 55. Guerra MT, Fonseca EA, Melo FM, Andrade VA, Aguiar CJ, Andrade LM, Pinheiro ACN, Casteluber MCF, Resende RR, Pinto MCX, Fernandes SOA, Cardoso VN, Souza-Fagundes EM, Menezes GB, de Paula AM, Nathanson MH, Leite MDF. Mitochondrial calcium regulates rat liver regeneration through the modulation of apoptosis. Hepatology 54: 296–306, 2011. doi: 10.1002/hep.24367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Moskowitzova K, Orfany A, Liu K, Ramirez-Barbieri G, Thedsanamoorthy JK, Yao R, Guariento A, Doulamis IP, Blitzer D, Shin B, Snay ER, Inkster JAH, Iken K, Packard AB, Cowan DB, Visner GA, Del Nido PJ, McCully JD. Mitochondrial transplantation enhances murine lung viability and recovery after ischemia-reperfusion injury. Am J Physiol Lung Cell Mol Physiol 318: L78–L88, 2020. doi: 10.1152/ajplung.00221.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dare AJ, Logan A, Prime TA, Rogatti S, Goddard M, Bolton EM, Bradley JA, Pettigrew GJ, Murphy MP, Saeb-Parsy K. The mitochondria-targeted anti-oxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. J Heart Lung Transplant 34: 1471–1480, 2015. doi: 10.1016/j.healun.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Saeb-Parsy K, Martin JL, Summers DM, Watson CJE, Krieg T, Murphy MP. Mitochondria as therapeutic targets in transplantation. Trends Mol Med 27: 185–198, 2021. [Erratum in Trends Mol Med 27: 512, 2021]. doi: 10.1016/j.molmed.2020.08.001. [DOI] [PubMed] [Google Scholar]
- 59. Guo Y, Chi X, Wang Y, Heng BC, Wei Y, Zhang X, Zhao H, Yin Y, Deng X. Mitochondria transfer enhances proliferation, migration, and osteogenic differentiation of bone marrow mesenchymal stem cell and promotes bone defect healing. Stem Cell Res Ther 11: 245, 2020. doi: 10.1186/s13287-020-01704-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab 23: 254–263, 2016. doi: 10.1016/j.cmet.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 61. Pell VR, Chouchani ET, Murphy MP, Brookes PS, Krieg T. Moving forwards by blocking back-flow: the yin and yang of MI therapy. Circ Res 118: 898–906, 2016. doi: 10.1161/CIRCRESAHA.115.306569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434: 652–658, 2005. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 63. Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA 102: 12005–12010, 2005. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lutz J, Thürmel K, Heemann U. Anti-inflammatory treatment strategies for ischemia/reperfusion injury in transplantation. J Inflamm (Lond) 7: 27, 2010. doi: 10.1186/1476-9255-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lin L, Xu H, Bishawi M, Feng F, Samy K, Truskey G, Barbas AS, Kirk AD, Brennan TV. Circulating mitochondria in organ donors promote allograft rejection. Am J Transplant 19: 1917–1929, 2019. doi: 10.1111/ajt.15309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Martin JL, Costa ASH, Gruszczyk AV, Beach TE, Allen FM, Prag HA, Hinchy EC, Mahbubani K, Hamed M, Tronci L, Nikitopoulou E, James AM, Krieg T, Robinson AJ, Huang MM, Caldwell ST, Logan A, Pala L, Hartley RC, Frezza C, Saeb-Parsy K, Murphy MP. Succinate accumulation drives ischaemia-reperfusion injury during organ transplantation. Nat Metab 1: 966–974, 2019. doi: 10.1038/s42255-019-0115-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Uzun MA, Koksal N, Kadioglu H, Gunerhan Y, Aktas S, Dursun N, Sehirli AO. Effects of N-acetylcysteine on regeneration following partial hepatectomy in rats with nonalcoholic fatty liver disease. Surg Today 39: 592–597, 2009. doi: 10.1007/s00595-008-3930-4. [DOI] [PubMed] [Google Scholar]
- 68. Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov 12: 465–483, 2013. doi: 10.1038/nrd4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ben Mosbah I, Mouchel Y, Pajaud J, Ribault C, Lucas C, Laurent A, Boudjema K, Morel F, Corlu A, Compagnon P. Pretreatment with mangafodipir improves liver graft tolerance to ischemia/reperfusion injury in rat. PLoS One 7: e50235, 2012. doi: 10.1371/journal.pone.0050235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. de Moraes ACN, de Andrade CBV, Ramos IPR, Dias ML, Batista CMP, Pimentel CF, de Carvalho JJ, Goldenberg RCDS. Resveratrol promotes liver regeneration in drug-induced liver disease in mice. Food Res Int 142: 110185, 2021. doi: 10.1016/j.foodres.2021.110185. [DOI] [PubMed] [Google Scholar]
- 71. Saito Y, Mori H, Takasu C, Komatsu M, Hanaoka J, Yamada S, Asanoma M, Ikemoto T, Imura S, Morine Y, Utsunomiya T, Shimada M. Beneficial effects of green tea catechin on massive hepatectomy model in rats. J Gastroenterol 49: 692–701, 2014. doi: 10.1007/s00535-013-0799-9. [DOI] [PubMed] [Google Scholar]
- 72. Yormaz S, Bulbuloglu E, Kurutas EB, Ciralik H, Yuzbasioglu MF, Yildiz H, Coskuner I, Silay E, Kantarceken B, Goksu M, Senoglu N, Kale IT. The comparison of the effects of hepatic regeneration after partial hepatectomy, silybum marinaum, propofol, N-acetylcysteine and vitamin E on liver. Bratisl Lek Listy 113: 145–151, 2012. doi: 10.4149/bll_2012_035. [DOI] [PubMed] [Google Scholar]
- 73. Majidinia M, Reiter RJ, Shakouri SK, Mohebbi I, Rastegar M, Kaviani M, Darband SG, Jahanban-Esfahlan R, Nabavi SM, Yousefi B. The multiple functions of melatonin in regenerative medicine. Ageing Res Rev 45: 33–52, 2018. doi: 10.1016/j.arr.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 74. Tara A, Dominic JL, Patel JN, Garg I, Yeon J, Memon MS, Gergal Gopalkrishna Rao SR, Bugazia S, Dhandapani TPM, Kannan A, Kantamaneni K, Win M, Went TR, Yanamala VL, Mostafa JA. Mitochondrial targeting therapy role in liver transplant preservation lines: mechanism and therapeutic strategies. Cureus 13: e16599, 2021. doi: 10.7759/cureus.16599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Martins RM, Teodoro JS, Furtado E, Rolo AP, Palmeira CM, Tralhão JG. Recent insights into mitochondrial targeting strategies in liver transplantation. Int J Med Sci 15: 248–256, 2018. doi: 10.7150/ijms.22891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Teoh NC, Williams J, Hartley J, Yu J, McCuskey RS, Farrell GC. Short-term therapy with peroxisome proliferation-activator receptor-α agonist Wy-14,643 protects murine fatty liver against ischemia–reperfusion injury. Hepatology 51: 996–1006, 2010. doi: 10.1002/hep.23420. [DOI] [PubMed] [Google Scholar]
- 77. Aoyama T, Ikejima K, Kon K, Okumura K, Arai K, Watanabe S. Pioglitazone promotes survival and prevents hepatic regeneration failure after partial hepatectomy in obese and diabetic KK-Ay mice. Hepatology 49: 1636–1644, 2009. doi: 10.1002/hep.22828. [DOI] [PubMed] [Google Scholar]
- 78. Pasut G, Panisello A, Folch-Puy E, Lopez A, Castro-Benítez C, Calvo M, Carbonell T, García-Gil A, Adam R, Roselló-Catafau J. Polyethylene glycols: an effective strategy for limiting liver ischemia reperfusion injury. World J Gastroenterol 22: 6501–6508, 2016. doi: 10.3748/wjg.v22.i28.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hu C, Zhao L, Wu Z, Li L. Transplantation of mesenchymal stem cells and their derivatives effectively promotes liver regeneration to attenuate acetaminophen-induced liver injury. Stem Cell Res Ther 11: 88, 2020. doi: 10.1186/s13287-020-01596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu Z, Meng F, Li C, Zhou X, Zeng X, He Y, Mrsny RJ, Liu M, Hu X, Hu J-F, Li T. Human umbilical cord mesenchymal stromal cells rescue mice from acetaminophen-induced acute liver failure. Cytotherapy 16: 1207–1219, 2014. doi: 10.1016/j.jcyt.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 81. Jones AL. Mechanism of action and value of N-acetylcysteine in the treatment of early and late acetaminophen poisoning: a critical review. J Toxicol Clin Toxicol 36: 277–285, 1998. doi: 10.3109/15563659809028022. [DOI] [PubMed] [Google Scholar]
- 82. Johnson TA, Jinnah HA, Kamatani N. Shortage of cellular ATP as a cause of diseases and strategies to enhance ATP. Front Pharmacol 10: 98, 2019. doi: 10.3389/fphar.2019.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu C-H, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515: 431–435, 2014. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu Z, Sun Y, Qi Z, Cao L, Ding S. Mitochondrial transfer/transplantation: an emerging therapeutic approach for multiple diseases. Cell Biosci 12: 66, 2022. doi: 10.1186/s13578-022-00805-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rousselle TV, Kuscu C, Kuscu C, Schlegel K, Huang L, Namwanje M, Eason JD, Makowski L, Maluf D, Mas V, Bajwa A. FTY720 regulates mitochondria biogenesis in dendritic cells to prevent kidney ischemic reperfusion injury. Front Immunol 11: 1278, 2020. doi: 10.3389/fimmu.2020.01278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zampieri LX, Silva-Almeida C, Rondeau JD, Sonveaux P. Mitochondrial transfer in cancer: a comprehensive review. Int J Mol Sci 22: 3245, 2021. doi: 10.3390/ijms22063245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jackson MV, Morrison TJ, Doherty DF, McAuley DF, Matthay MA, Kissenpfennig A, O'Kane CM, Krasnodembskaya AD. Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the in vitro and in vivo models of ARDS. Stem Cells 34: 2210–2223, 2016. doi: 10.1002/stem.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhao Z, Hou Y, Zhou W, Keerthiga R, Fu A. Mitochondrial transplantation therapy inhibit carbon tetrachloride-induced liver injury through scavenging free radicals and protecting hepatocytes. Bioeng Transl Med 6: e10209, 2021. doi: 10.1002/btm2.10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Helling TS. Liver failure following partial hepatectomy. HPB (Oxford) 8: 165–174, 2006. doi: 10.1080/13651820510035712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Takeda K, Togo S, Kunihiro O, Fujii Y, Kurosawa H, Tanaka K, Endo I, Takimoto A, Sekido H, Hara M, Shimada H. Clinicohistological features of liver failure after excessive hepatectomy. Hepatogastroenterology 49: 354–358, 2002. [PubMed] [Google Scholar]
- 91. Eguchi S, Kamlot A, Ljubimova J, Hewitt WR, Lebow LT, Demetriou AA, Rozga J. Fulminant hepatic failure in rats: survival and effect on blood chemistry and liver regeneration. Hepatology 24: 1452–1459, 1996. doi: 10.1002/hep.510240626. [DOI] [PubMed] [Google Scholar]
- 92. Waldmeier PC, Feldtrauer J-J, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol 62: 22–29, 2002. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- 93. Zhong Z, Ramshesh VK, Rehman H, Currin RT, Sridharan V, Theruvath TP, Kim I, Wright GL, Lemasters JJ. Activation of the oxygen-sensing signal cascade prevents mitochondrial injury after mouse liver ischemia-reperfusion. Am J Physiol Gastrointest Liver Physiol 295: G823–G832, 2008. doi: 10.1152/ajpgi.90287.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Theruvath TP, Zhong Z, Pediaditakis P, Ramshesh VK, Currin RT, Tikunov A, Holmuhamedov E, Lemasters JJ. Minocycline and N-methyl-4-isoleucine cyclosporin (NIM811) mitigate storage/reperfusion injury after rat liver transplantation through suppression of the mitochondrial permeability transition. Hepatology 47: 236–246, 2008. doi: 10.1002/hep.21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rehman H, Sun J, Shi Y, Ramshesh VK, Liu Q, Currin RT, Lemasters JJ, Zhong Z. NIM811 prevents mitochondrial dysfunction, attenuates liver injury, and stimulates liver regeneration after massive hepatectomy. Transplantation 91: 406–412, 2011. doi: 10.1097/TP.0b013e318204bdb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Young EN, Dogan M, Watkins C, Bajwa A, Eason JD, Kuscu C, Kuscu C. A review of defatting strategies for non-alcoholic fatty liver disease. Int J Mol Sci 23: 11805, 2022. doi: 10.3390/ijms231911805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pérez-Carreras M, Del Hoyo P, Martín MA, Rubio JC, Martín A, Castellano G, Colina F, Arenas J, Solis-Herruzo JA. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 38: 999–1007, 2003. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 98. Yin X, Zheng F, Pan Q, Zhang S, Yu D, Xu Z, Li H. Glucose fluctuation increased hepatocyte apoptosis under lipotoxicity and the involvement of mitochondrial permeability transition opening. J Mol Endocrinol 55: 169–181, 2015. doi: 10.1530/JME-15-0101. [DOI] [PubMed] [Google Scholar]
- 99. Navarro CDC, Figueira TR, Francisco A, Dal'Bó GA, Ronchi JA, Rovani JC, Escanhoela CAF, Oliveira HCF, Castilho RF, Vercesi AE. Redox imbalance due to the loss of mitochondrial NAD(P)-transhydrogenase markedly aggravates high fat diet-induced fatty liver disease in mice. Free Radic Biol Med 113: 190–202, 2017. doi: 10.1016/j.freeradbiomed.2017.09.026. [DOI] [PubMed] [Google Scholar]
- 100. King AL, Swain TM, Mao Z, Udoh US, Oliva CR, Betancourt AM, Griguer CE, Crowe DR, Lesort M, Bailey SM. Involvement of the mitochondrial permeability transition pore in chronic ethanol-mediated liver injury in mice. Am J Physiol Gastrointest Liver Physiol 306: G265–G277, 2014. doi: 10.1152/ajpgi.00278.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Fu A. Mitotherapy as a novel therapeutic strategy for mitochondrial diseases. Curr Mol Pharmacol 13: 41–49, 2020. doi: 10.2174/1874467212666190920144115. [DOI] [PubMed] [Google Scholar]
- 102. Pessayre D, Fromenty B. NASH: a mitochondrial disease. J Hepatol 42: 928–940, 2005. doi: 10.1016/j.jhep.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 103. Fu A, Shi X, Zhang H, Fu B. Mitotherapy for fatty liver by intravenous administration of exogenous mitochondria in male mice. Front Pharmacol 8: 241, 2017. doi: 10.3389/fphar.2017.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ajith TA. Role of mitochondria and mitochondria-targeted agents in non-alcoholic fatty liver disease. Clin Exp Pharmacol Physiol 45: 413–421, 2018. doi: 10.1111/1440-1681.12886. [DOI] [PubMed] [Google Scholar]
- 105. Gupta N, Wu CH, Wu GY. Targeted transplantation of mitochondria to hepatocytes. Hepat Med 8: 115–134, 2016. doi: 10.2147/HMER.S116852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Liu X, Khouri-Farah N, Wu CH, Wu GY. Targeted delivery of mitochondria to the liver in rats. J Gastroenterol Hepatol 35: 2241–2247, 2020. doi: 10.1111/jgh.15091. [DOI] [PubMed] [Google Scholar]
- 107. Kubat GB, Ulger O, Akin S. Requirements for successful mitochondrial transplantation. J Biochem Mol Toxicol 35: e22898, 2021. doi: 10.1002/jbt.22898. [DOI] [PubMed] [Google Scholar]