

Abstract
Traumatic brain injury (TBI) is a significant worldwide cause of morbidity and mortality. A chronic neurologic disease bearing the moniker of “the silent epidemic”, TBI currently has no targeted therapies to ameliorate cellular loss or enhance functional recovery. Compared to those of astrocytes, microglia, and peripheral immune cells, the functions and mechanisms of NG2-glia following TBI are far less understood, despite NG2-glia comprising the largest population of regenerative cells in the mature cortex. Here, we synthesize the results from multiple rodent models of TBI, with a focus on cortical NG2-glia proliferation and lineage potential, and propose future avenues for glia researchers to address this unique cell type in TBI. As the molecular mechanisms that regulate NG2-glia regenerative potential are uncovered, we posit that future therapeutic strategies may exploit cortical NG2-glia to augment local cellular recovery following TBI.
1. INTRODUCTION
Having earned the moniker of “the silent epidemic”, traumatic brain injury (TBI) afflicts an estimated 69 million individuals worldwide each year1. This has significant global impact. For instance, in the United States, 2.8 million people suffer TBI yearly, including 630,000 children2,3. It is the leading cause of death in 1-44 years olds2–4, and its economic cost totals $102bn yearly at current day value2. As TBI causes persistent physical, cognitive, and psychological disabilities in survivors, it is appropriately considered a chronic neurologic disease; in fact, TBI and Alzheimer’s Disease (AD) may be pathophysiologically linked5,6. However, no targeted therapies exist for TBI: all patients, young and old, can only be provided with supportive care and rehabilitation. Therefore, there is a critical need to uncover post-injury cellular mechanisms that may have therapeutic targets to aid in recovery from TBI7.
Compared to the post-TBI cellular response of resident glial cells (e.g. microglia, astrocytes) and members of the innate (e.g. neutrophils, monocytes) and adaptive immune systems (e.g. T-cells)8–10, the response of NG2-glia, also commonly referred to as oligodendrocyte precursor cells (OPCs), is far less researched. At 4-8% of the total number of cells, NG2-glia comprise the largest population of regenerative cells in the adult CNS11, accounting for 70-80% of active cellular proliferation in a naïve, uninjured brain12,13. NG2-glia also likely serve in cellular regeneration following TBI, demonstrating response kinetics for polarization, migration, and proliferation within 24 hours after an injury and persisting for at least one month12,14,15, therefore bridging the multiple phases following an injury: the “acute” or “early” phase (e.g. 0-2 days after injury), the “subacute” or “intermediate” phase (e.g. days to weeks after injury), and the “chronic”, “late”, or “delayed” phase (e.g. months to years after injury)16.
NG2-glia in the mature brain are perhaps most well known as a contributor to repair mechanisms in demyelinating diseases (e.g. multiple sclerosis17–19) and other white matter injuries (e.g. neonatal hypoxic ischemic encephalopathy20). This is justified by the proclivity of white matter NG2-glia to differentiate into oligodendrocytes, which contribute to remyelination21. Similarly, as TBI induces diffuse white matter injury with significant clinical implications22,23, there is interest in investigating the role of NG2-glia in post-TBI remyelination as well24,25. However, TBI also encompasses a wide range of focal cortical pathologies (e.g. skull fracture, parenchymal laceration, contusion, hemorrhage, focal edema)26 which have been associated with early disease progression and long-term neurologic outcomes27–30. Because NG2-glia demonstrate motility, proliferative capacity, and potential for multipotency soon after injury11, they present a unique lens by which to evaluate the cellular mechanisms contributing to the evolution of cortical trauma. Additionally, as conventional imaging techniques employed in the diagnosis of acute TBI (i.e. computed tomography) are more likely to identify focal cortical pathologies, especially if accompanied by hemorrhagic features, than other diffuse or white matter injuries, there is a seductive possibility that early exploitation of cortical NG2-glia endogenous regenerative could serve as a therapeutic strategy in the future. It is our goal here to review the relevant literature on the post-TBI responses of cortical NG2-glia, specifically, and the known molecular mechanisms that underly these cellular functions.
2. CORTICAL NG2-GLIA: A FUNCTIONALLY DISTINCT POPULATION
A relatively recent discovery, NG2-glia were initially identified as non-neuronal cells lacking the usual structural components of the three other major glial classes: astrocytes, oligodendrocytes (OLs), and microglia31. Today, this population is most often characterized by the co-expression of OL markers (e.g. Olig2, Sox10)13,32 in addition to two proteins not expressed by mature OLs: platelet derived growth factor receptor alpha (PDGFRA) and neuron-glial antigen 2 (NG2, also called chondroitin sulfate proteoglycan 4 [CSPG4])33. Expression of PDGFRA precedes that of NG2 in NG2-glia, while expression of both markers decreases as these cells differentiate into mature OLs33,34. Of note, NG2, an integral membrane chondroitin sulfate proteoglycan35,36, is expressed by CNS cell types, such as pericytes, microglia, and macrophages, especially in the setting of pathology37–40, while PDGFRA is also expressed by certain vascular and leptomeningeal cells41.
NG2-glia are found at seemingly comparable densities throughout the brain, including cortical gray matter, white matter, and deeper structures42. As they can self-renew and are multipotent, they also populate neurogenic areas such as the subventricular zone (SVZ) and dentate gyrus. Although NG2-glia are widespread, it has become increasingly apparent that they do not comprise a homogenous population, rather, demonstrate different properties and functions dependent on spatial location within the brain and the developmental stage of the organism11. For instance, the cell fates of NG2-glia isolated from white matter differ from those isolated from gray matter, regardless if they are heterotopically transplanted to gray matter and white matter, respectively43. While an initial study could not differentiate distinct subpopulations of NG2-glia between gray and white matter41, a more recent study has uncovered different proportions of NG2-glia subpopulations both at rest and following injuries, including the upregulation of specific genes in cortical NG2-glia versus their gray matter counterparts (Wnt signaling pathway: Fzd8, Axin2, AMPA receptor subunit: Gria2)44. Therefore, findings from injury models involving white matter NG2-glia15,17,19,45–47 may not be generalizable to cortical NG2-glia and vice versa. Similarly, progenitors from the SVZ are likely another unique population of cells that can migrate towards an injury site and contribute to the perilesional cellular response. However, the kinetics of their arrival to the injury site are thought to be slower than the immediate responses of perilesional cortical NG2-glia and are complicated by whether the SVZ and rostral migratory streams are also affected by the injury, themselves48–50. For the purpose of this review, we will focus primarily on the role of cortical NG2-glia in the response to TBI.
There are a number of functional characteristics that make NG2-glia a unique population of cells in the mature cortex. The first is that they are capable of ongoing proliferation and differentiation into other cell types. However, unlike their white matter counterparts, of which 80-90% will eventually differentiate into mature OLs, cortical NG2-glia most often continue to generate more NG2-glia, both in naïve and injured brains51,52. They also do not readily differentiate into astrocytes as seen during development53–55. In the mature, naïve animal, the majority of cortical NG2-glia (>96%) reside in a prolonged G1 phase, with only a miniscule percentage demonstrating proliferation at a rate that closely balances the rate of NG2-glia differentiation (e.g. into mature oligodendrocytes) and apoptosis, permitting a constant NG2-glia cell density over time14,52. Conversely, in the setting of injury or other environmental stimuli (e.g. exercise), cortical NG2-glia can respond quickly, either by re-entering the cell cycle or exiting the cell cycle and differentiating, respectively14,52.
A second noteworthy feature of cortical NG2-glia is that these cells are structurally dynamic. At a baseline state, they have a stellate configuration, with processes that extend/retract over the course of minutes14. They are also motile and migrate approximately 2 mm per day14. This motility is essential for maintaining an even density throughout the naïve cortex, as NG2-glia appear to divide and migrate towards areas in which other NG2-glia may have undergone apoptosis or differentiation14. Furthermore, upon acute application of a destructive stimulus (e.g. laser ablation, stab wound), NG2-glia demonstrate acute hypertrophy and polarization with process extension and migration towards the injury on a time course of several hours14,56. The NG2-glia responses following injury is perhaps most similar to the those of microglia, which activate, hyperpolarize, and migrate towards the injury on the order of minutes, and proliferate within 24 hours10,57. Conversely, astrocytes, which demonstrate significant astrogliosis and have functions in regulating blood-brain-barrier integrity and immune signaling following brain injury, demonstrate minimal gross migration following injury10,58.
A third unique NG2-glia characteristic, and one that is perhaps the least explored, is that these cells appear to play a role in cell-to-cell signaling, as they express a variety of voltage-dependent and ligand-dependent ion channels and receive excitatory and inhibitory inputs from neurons59–61. These cortical neuron-NG2-glial synapses are critical in regulating NG2-glia functions and differentiation62–65, however their role in the adult cortex and in response to TBI is unexplored.
3. ANIMAL MODELS OF TBI
TBI is not a single disease entity, rather, an umbrella diagnosis that encompasses multiple types of brain pathology following head trauma. Within TBI are focal (e.g. contusions, intraparenchymal hemorrhage) and diffuse (e.g. traumatic/diffuse axonal injury, hypoxic ischemic injury, microvascular injury) pathologies26,66,67, which can present singly or in combination. These pathologies are most dramatic and often found together in severe TBI26, but even mild TBI is characterized by focal, injury-adjacent68,69 and deep white matter70,71 pathologies. Multiple animal models have been developed to study TBI, each recapitulating a unique combination of the injury characteristics seen in human neurotrauma72–75, including stab wound injury, open head injury, closed head injury, and rotational injury models (Figure 1). However, as cortical NG2-glia have been studied in various rodent TBI models with different pathological features (Supplemental Table 1), findings from one experimental setup may neither be applicable to other models nor generalizable to human TBI.
Figure 1. Models of Rodent TBI.
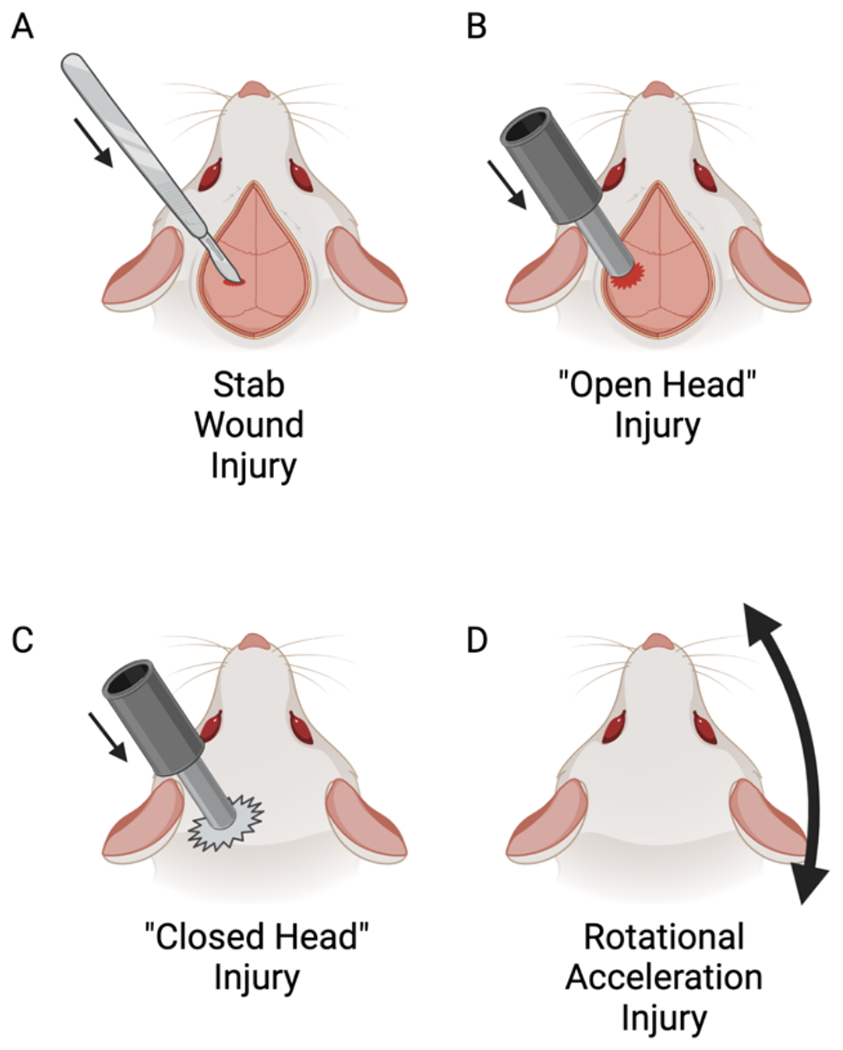
(A) In the stab wound injury (SWI) model, a sharp blade or needle is precisely inserted through a surgically created craniotomy into cortex (or cerebellum) and withdrawn. While the injury is dependent on the size and depth of the blade, this is most often a focal, mild TBI.
(B) In “open head” injury models (e.g. controlled cortical impact, weight drop, fluid percussion injury), a mechanical force is directly applied to the brain parenchyma through a surgically created craniotomy.
(C) In “closed head” injury models, the mechanical force is applied to the skull, either with or without a scalp incision. While more closely resembling clinical TBI, this model more often provides more heterogenous injuries than the open model (B).
(D) Rotational acceleration injury models do not direct force towards the brain/skull, rather, rotate the brain relative to the skull around a fixed axis. Some models require a mount affixed directly to the skull, while others are capable of rotation of a freely mobile head. The result is tissue shear and axonal injury, with minimal direct contusional pathology.
Created with BioRender.com
In the cortical stab wound model of TBI, the cranial vault is surgically breached (e.g. burr hole, craniectomy), and a sharp blade or needle is precisely inserted into cortex (or cerebellum) and withdrawn. This produces neuronal and vascular injuries that most closely resemble the direct tissue injury of a neurosurgical procedure; this is in contrast to the higher energy direct, indirect, and rotational forces of other penetrating, closed head, or rotational TBI models. This model results in a highly reproducible focal cortical injury that is most similar to “mild” injury induced by controlled cortical impact models discussed below76.
“Open head” TBI describes animal models in which a high energy force directly injures the brain through a surgical craniotomy, with or without replacement of the skull flap. Prime among these is the controlled cortical impact (CCI) model, which utilizes a mechanical piston to provide a direct parenchymal injury with precise control of impact diameter, velocity, depth, and dwell time 77,78. This produces intracranial hemorrhage, cortical contusion and tissue loss, and blood-brain barrier dysfunction, which are seen in human TBI72. Other commonly employed open head injuries include fluid percussion injury (FPI), in which energy is transmitted to the brain via a column of fluid79, and falling weight TBI, in which an object of a specified mass is dropped from a specified height to produce injury80. These models have been used to study both gray and white matter pathologies following TBI, although the pathological focus is often cortex with some consideration for subcortical hippocampus76.
In contrast to open head TBI models, “closed head” models involve the application of force directly to an intact skull, with indirect transmission of energy to the underlying brain parenchyma. Controlled cranial impact via a piston and weight drop have both been adopted for closed TBI models73,81,82. The benefits of closed TBI models includes less anesthesia exposure due to forgoing a surgical craniotomy, as well as closer approximation to human TBI. A drawback, however, is that closed models have more heterogeneity of brain injury, also mimicking the human clinical experience. By varying the energy of the impact, this model has been used for both moderate/severe TBI, with gray and white matter injuries, and mild TBI, producing predominantly white matter injury15,47,83.
Rotational injury models provide a distinct type of TBI that differs from the stab wound and percussive injuries. Also a closed head injury model, rotational acceleration replicates the destruction that can be seen in road accidents and sports injuries where the brain and skull move at different velocities, rupturing vasculature and causing tissue shear between brain areas of different densities84. Prominent features of this injury model include diffuse axonal injury and sometimes subdural bleeding rather than parenchymal contusions84. While older models require skull fixation in order to deliver a reproducible rotational force, newer methods permit free movement of the head and reproduce significant white matter injuries without gray matter involvement85,86. As this injury favors white matter injury, cortical NG2-glia have not been the focus of much research87.
4. THE CORTICAL NG2-GLIAL RESPONSE TO TBI
4.1. Stab Wound Injury
The largest body of work examining the cortical NG2-glia response to TBI has utilized the stab wound injury (SWI) model. While cellular proliferation after a stab wound was first described in 197088, the proliferative response of NG2-glia, specifically, was detailed in the 1990’s12: NG2 gene and protein expression as well as NG2+ cell density increased in areas adjacent to the injury tract, reaching a peak density at 7 days post-injury and regressing12,89. Nevertheless, even at 30 days after injury, a small population of cells with increased NG2 decoration remained around the remnant injury site12. Follow-up studies utilizing pulse-chase methods of following mitotic activity, found that relative to the total rate of cellular proliferation following SWI (i.e. 12- to 22-fold increase12,13), NG2-glia comprised the largest percentage of the acute response (i.e. 1-2 days), regardless of experimental method (i.e. ~25-50% of BrdU+ cells13,90–92, 82% of 3H-thymidine containing cells12, 51% of retroviral tracer expressing cells73). Following the early peak of NG2+ cell proliferation, other cell types (e.g. microglia, monocytes, astrocytes) predominate such that at 1-2 weeks after injury, only 20-50% of actively proliferating cells express NG212,13,90,91. Of note, it is currently unclear whether all cortical NG2-glia are capable of reacting to TBI equally, or if injury response is yet another property subject to NG2-glia heterogeneity. For instance, expression of G-protein coupled receptor 17 (GPR17)93, which was initially thought to indicate maturation of NG2-glia from a precursor to a pre-myelinating phenotype94, may denote a unique pool of NG2-glia with limited capability to further differentiate in the naïve animal, but maintain the capacity to expand and differentiate following a large ischemic insult95.
On a very basic level, the proliferation of cortical NG2-glia following injury appears to play a significant role in wound healing. For instance, genetically impairing proliferation of NG2-glia leads to a larger lesion size following identical stab wounds as early as 4 days post-injury, suggesting that the response of NG2-glia is critical for acute wound closure56. The precise mechanism by which NG2-glia affects wound healing, however, is unknown. One possibility is that production of the NG2 proteoglycan, itself, plays a significant role. The size and density of the NG2-immunoreactive “plaque” surrounding SWI appears to peak at around 7-10 days after injury, following the peak of NG2+ cell counts12 and consistent with ongoing NG2 deposition at the site of the injury96. However, several studies using various models of surgical CNS injury (most often in spinal cord) have produced disparate conclusions regarding the regenerative or inhibitory role of NG2 on the regrowth of axons97–100, and their generalizability to cortex is not guaranteed. While no genetic NG2 knockout (KO) studies have been performed in SWI as in other TBI models (below), at the very least NG2, itself, appears to play a significant role in the polarization and migration of cortical NG2-glia towards the injury site101.
One potential contribution of NG2-glia to TBI recovery is to replace lost cells. The cellular fates of NG2-glia following TBI has been the subject of intense study, as few NG2-glia undergo apoptosis or necrosis following TBI51,91,92. It is generally accepted that following TBI/SWI, NG2-glia predominantly proliferate and give rise to additional NG2-glia51,52, with only a minority of NG2-glia expressing mature OL markers (e.g. GalC, O4), and not until weeks after injury91,92. This is not unique to TBI, as a similar lineage restriction is seen in cortical NG2-glia after ischemic stroke as well102. While most agree that NG2-glia do not differentiate into microglia following TBI39, interpretation of data supporting the potential for NG2-glia to give rise to astrocytes following injury has evolved over the years. Of note, NG2-glia-derived astrocytes are commonly referred to as Type 2 astrocytes, which differentiates them from Type 1 astrocytes derived from glial progenitors of the ventricular zone103. The post-injury appearance of cells co-expressing NG2 and GFAP90,92,104 or markers of immature astrocytes (e.g. nestin, vimentin)90 initially supported an astrocytic fate for cortical NG2-glia, as can be induced in vitro91. In hindsight, these early experiments attempting to trace proliferating cells were likely complicated by not being able to account for proliferative cells lacking obvious lineage markers (e.g. an astrocyte precursor) that could contribute to the astrocyte population. Subsequent lineage mapping studies using transgenic approaches (e.g. Ng2-Cre, Olig2-Cre) were less supportive of a significant contribution of NG2-glia to post-TBI astrogliosis. Ng2-dependent lineage tracing found that the vast majority (~85%) of Ng2-derived cells remained NG2+ following SWI, a small population expressed mature OL markers (<4%), and a minority transiently expressed GFAP (~6%), peaking around 10 days after injury. Of note, these Ng2-derived GFAP+ cells comprised a small percentage of total peri-lesional reactive astrocytes (<10%), and some continued to express NG2 and were more morphologically similar to NG2-glia than astrocytes105. A complementary Olig2-dependent lineage tracing strategy produced similar results, as 75-80% of cortical Olig2-derived progeny post-injury were NG2+, with under 5% transiently co-expressing astrocyte markers at 3 days post-injury, and the remainder expressing a mature OL marker51. When these studies were placed in context of additional fate-mapping efforts that characterized astrocyte and astrocyte precursor proliferation following injury106,107, likely accompanied by transient expression of oligodendrocyte lineage marker Olig2108, it is very unlikely that NG2-glia are a significant source of new astrocytes following SWI. Single-cell sorting has permitted a more sophisticated manner by which to characterize NG2-glia (i.e. by gene expression), including NG2-glia populations that more closely resemble oligodendrocytes and astrocytes. However, SWI does not appear to induce as strong expression of astrocyte-related markers, unlike focal ischemic injury44. It is possible that a TBI model with a more severe injury could feature a component of focal ischemia that could induce the appearance of this population of cells.
While the lineage potential of cortical NG2-glia appears to be restricted in the normal response to SWI, several efforts have been made to uncover key molecular checkpoints that could serve as therapeutic targets to permit NG2-glia to repopulate multiple cell types lost in TBI. Olig2, a critical OL lineage transcription factor, was initially implicated as a key molecule in determining NG2-glia fate, with nuclear retention of OLIG2 in NG2-glia being associated with OL fate choice, and cytoplasmic translocation portending progression to GFAP+ astrocytes91,92. However, given the astrocyte lineage mapping study above105, the lack of additional studies appreciating cytoplasmic localization of OLIG2 in NG2-glia105,108, the identification of an OLIG2-expressing population of astrocytes109, and a failure to effect astrocyte proliferation following NG2-glia-specific Olig2 KO105, it is less likely that NG2-glial expression of Olig2, itself, plays a determining role in shifting NG2-glia lineage potential towards astrocytes.
The bone morphogenetic protein (BMP) pathway, however, has been more consistently implicated in determining NG2-glia astrocytic lineage potential. In vitro, BMP signaling induces progression of NG2-glia to astrocytes, and BMP inhibition restricts lineage potential towards OLs104,110. In vivo experiments may also support this hypothesis. While not a strict lineage-tracing experiment, BMP pathway dis-inhibition via anti-noggin antibodies in the setting of SWI appears to produce a more vigorous NG2-glia response and the appearance of NG2-expressing, Type 2 astrocytes104. Similarly, surgical implantation of a parenchymal infusion cannula (i.e. a trauma most similar to a chronic cortical stab wound) with chronic administration of exogenous fibrinogen, thought to activate BMP signaling pathways in NG2-glia, induced cannula-adjacent NG2-derived cells to express GFAP, consistent with a shift in lineage towards astrocytes111. This latter finding is particularly interesting as hemorrhage and extravascular deposition of fibrin are features of SWI and TBI in general. The effects of cleavage or blockade112,113 of endogenous extravasated fibrin on in vivo NG2-glia lineage potential following TBI has not yet been explored, although at least one study supports such a link114.
The possibility that NG2-glia have the capacity to differentiate into neurons, both in physiologic conditions and after injury, is controversial and well-reviewed11,115. As noted in the above lineage tracing studies, no neurons have been found as progeny of NG2-glia following in vivo SWI51,105, casting significant doubt on the capacity for neuronal repopulation by NG2-glia in physiologic conditions. However, manipulation of certain key molecules have been reported to elicit a neuronal linage potential. Specifically, viral vector-mediated suppression of Olig213 or expression of transcription factors NeuroD1116 or Sox2117 in NG2-glia were capable of inducing neuronal fates in vivo. Nevertheless, corroborative reports using complementary approaches to validate these findings in cortex are still required. As mentioned above, NG2-glia-specific Olig2 knockout failed to produce neuronal cell fates105, once again introducing uncertainty in its role in altering cell fate determination. Meanwhile, targeted Sox2 overexpression did show promise in a spinal cord injury model by inducing NG2-derived neurogenesis118. While there is doubt that NG2-glia can repopulate lost neurons following TBI absent a molecular intervention, the potential for such a strategy should continue to motivate NG2-glia researchers to continue to identify differentiation checkpoints and further probe whether NG2-glia-derived neurons are incorporated into functional circuits following brain injury.
4.2. Open Head Injury
The second largest body of literature investigating NG2-glia following TBI is in the open head controlled cortical impact model of TBI. The fluid percussion model has also been used, but with a focus on NG2-glia in white matter injuries119. Similar to that seen in SWI, NG2-glia comprise the largest population of proliferative cells immediately following open TBI, with a peak at 1-7 days following injury, and a more severe injury causing a later peak120–122. Interestingly, control mice undergoing a “sham” injury (i.e. a surgical craniotomy without impact) also demonstrate significant NG2-glia response with a proliferative cellular density similar to those having undergone impact120, suggesting that breach of the cranial vault, despite leaving the dura intact, is traumatic enough to produce a localized proliferative response. This bears significant implications for future NG2-glia research in that study design will have to be deliberate in the selection of an appropriate “control” condition depending on the research question of interest and sensitivity of techniques to detect the cellular/molecular consequences of the “sham” injury.
As in SWI, open TBI induces significant NG2 deposition at the site of injury that persisted for up to 28 days40. Genetic Cspg4 (i.e. Ng2) KO in open TBI models exacerbates lesion severity and functional recovery, further lending a role for NG2-glia beyond cell replacement123. Of note, the Ng2 KO condition also induced a more robust astrogliosis and innate immune response, suggesting that NG2-glia, via NG2 deposition, may serve to direct the actions of other cell types neighboring the injury123. Interestingly, the authors did not observe a difference in the density of PDGFRA-immunoreactive cells following injury in the Ng2 KO condition, although the presence of disparate injury severities between the two groups complicates interpretation of these data123.
Open TBI models have been used to probe the engagement of specific molecular pathways by NG2-glia following TBI. One group correlated post-TBI NG2-glia proliferation with the activation of β-catenin signaling, a downstream effector of the Wnt pathway that was rarely active in the naïve state (~2% of NG2-glia)124. Furthermore, the increased β-catenin activity may remain elevated for several days in peri-injury NG2-glia following proliferation, suggestive of prolonged activation of downstream targets124. However, the necessity and sufficiency of β-catenin activity for precise post-injury functions, such as proliferation and/or differentiation, is unknown. Interestingly, upregulation β-catenin activity using the same reporter mice was not seen in a spinal cord injury model, suggesting a differential response between NG2-glia in cortex and versus the cord124,125.
Another group discovered that the majority (~85%) of peri-injury NG2-glia demonstrate upregulation of nuclear factor erythroid 2-related factor (Nrf2) in a time course that mirrors the peak in NG2-glia proliferation (i.e. peak at 3 days post-injury)122. As Nrf2 is thought to activated by oxidative stress to initiate protective mechanisms126, it is also elevated in several cell types following TBI, including neurons, astrocytes, and microglia122. While the authors briefly mentioned the existence of data in which TBI in a Nrf2 global knockout condition increased NG2-glia proliferation, it is unclear whether the result was due to a role for Nrf2 in NG2-glia, specifically.
4.3. Closed Head Injury
While closed head injury models can induce cortical NG2-glia proliferation similar to that seen in open head models, they are more commonly used to evaluate white matter NG2-glial processes post-TBI15,45–47,119. Using a closed head TBI model127, our laboratory investigated the role of the circadian clock in governing NG2-glia proliferation, both at rest and following neurotrauma128. Interestingly, NG2-glia expressed the circadian clock gene Bmal1 both, during the time of day of peak basal proliferation as well as acutely following TBI. Additionally, utilizing a NG2-glia-specific Bmal1 conditional KO strategy, we found that Bmal1 was necessary for cortical NG2-glia proliferation, both at rest and following TBI. These data suggest that the BMAL1 transcription factor may serve as part of a common pathway by which to engage cellular proliferation, either at rest or following injury. However, while the role of Bmal1 in the mammalian circadian clock is well characterized129, the upstream regulators and downstream effectors of Bmal1 beyond molecular clock components in NG2-glia have not yet been identified. Thus, a defined molecular pathway connecting Bmal1 to proliferation, and its interaction with the circadian clock and TBI is unknown.
5. DISCUSSION AND FUTURE DIRECTIONS
There have been many descriptions of NG2-glia proliferation and differentiation following TBI, however, researchers have only begun to identify the molecular mechanisms regulating these processes, with most focusing on individual molecular candidates (Figure 2). Given the acceleration in cell-type-specific next generation molecular profiling, we anticipate that future studies will be better suited to elucidate entire molecular pathways responsible for the NG2-glial post-TBI response, which could provide potential targets for therapeutic manipulations. We also expect that these studies will divine which pathways bear some specificity for NG2-glia, as several of the molecular candidates discussed above are fairly ubiquitous (Wnt/ β-catenin, Nrf2, Bmal1). Likewise, as we continue to take advantage of studies with single-cell resolution, we expect to completely redefine the NG2-glia population beyond the use of markers such as NG2 and PDGFRA, much like has been recently done for microglia130, as there is increasing evidence that even NG2-glia within a particular neuroanatomic location may be functionally distinct44,95.
Figure 2. Molecular pathways controlling cortical NG2-glia proliferation and differentiation.
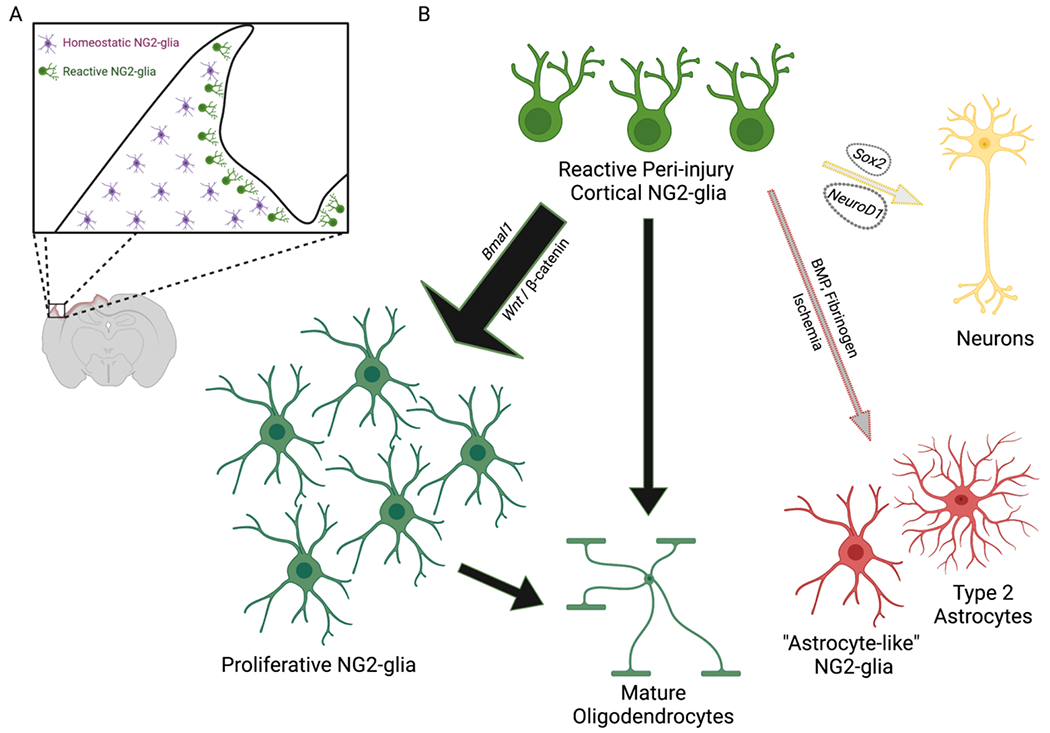
Cortical NG2-glia respond to neurotrauma with robust proliferation and differentiation. (A) Reactive NG2-glia (green) polarize and migrate towards the site of TBI, while NG2-glia remote from the injury (magenta) lack any apparent morphologic changes. (B) In the acute response to TBI, reactive cortical NG2-glia (green) most commonly proliferate, leading to additional NG2-glia (large black arrow). A smaller population of NG2-glia undergo differentiation to mature oligodendrocytes (small black arrow). While data from stab wound injury models suggest that only few NG2-glia may be able to differentiate into Type 2 astrocytes or “astrocyte-like NG2-glia” (gray arrow), it is possible that larger lesions with ischemia or more significant blood brain barrier compromise may promote an astrocytic lineage potential through activation of BMP signaling (e.g. fibrinogen). Although NG2-glia have not been observed to differentiate into neurons under physiologic conditions in vivo (yellow arrow), virus-mediated expression of specific transcription factors (e.g. Sox2, NeuroD1) may be able to induce such a fate following TBI.
Created with BioRender.com
Another knowledge gap to be addressed is the change in functions of NG2-glia over time following injury. While the majority of studies appear to be focused on the immediate response of NG2-glia within the first few weeks after injury (Supplemental Table 1), there is evidence of morphological changes approaching a month after neurotrauma12,14. Furthermore, literature from static white matter injury (e.g. HIE, TBI) suggest that there is a prolonged NG2-glial response in the chronic phase following a CNS injury24,131; the functions of cortical NG2-glia, and how they may contribute to brain healing, in the following months after TBI are entirely unknown.
Beyond the scientific advances that will improve resolution of NG2-glia molecular pathways, much attention moving forward should also be focused on the choice of TBI model. As shown here, investigations into the functions of NG2-glia following TBI are most complete in SWI models. This model, while offering the best experimental reproducibility, is limited in generalizability due to its inherent differences compared to the human condition and relatively mild degree of injury. Clinical TBI features cellular destruction, immune interaction, and blood vessel compromise, all of which contribute to focal ischemia, hemorrhage, and blood brain barrier dysfunction that may be more severe than in a focal stab wound. Even the use of a larger “stab” wound versus a smaller “punctate” wound appears to produce a more robust NG2-glial polarization and migration relative to the injury site56. Thus, there is increasing support for TBI researchers aiming to bridge the gap between bench and bedside to use multiple standardized experimental TBI models (including injuries in gyrencephalic species), with therapeutic “rescue” being defined as positive changes in several peripheral and central biomarkers and clinically relevant behavioral testing132,133. While the translation of molecular mechanisms to clinical therapies has been slow, if not impossible134, we are hopeful that NG2-glia present a unique cell type with the potential to enhance cellular recovery following TBI.
Supplementary Material
8. ACKNOWLEDGEMENTS:
This work was supported by the District of Columbia Intellectual and Developmental Disabilities Research Center award (DC-IDDRC U54HD090257 and P50HD105328) (V.G.) from the National Institute of Child Health and Human Development (NICHD). T.D. was supported by NICHD K12HD001399. This work was also supported by R37NS109478 (Javits Award to V.G.). The authors would like to thank members of T.D.’s K12 leadership committee (Dr. Robert Freishtat, Dr. Steven Teach) and scholarship oversight committee (Dr. Michael Shoykhet, Dr. Tarik Haydar, Dr. Michael Bell) for their guidance. Biorender.com was used for the composition of all associated figures.
7. FUNDING SOURCES:
TD: NICHD K12HD001399
VG: DC-IDDRC U54HD090257 and P50HD105328; R37NS109478
Footnotes
POTENTIAL CONFLICTS OF INTERESTS:
Authors report no conflict of interest.
Ethics Approval Statement: No new experiments or patient data are included in this review article.
Patient Consent Statement: No patient data are included in this review article
Permission to reproduce material: only figure produced in BioRender with appropriate subscription for journal publication.
9. Data Availability:
No new data are included in this review article.
REFERENCES
- 1.Dewan MC, Rattani A, Gupta S, et al. Estimating the global incidence of traumatic brain injury. J Neurosurg 2018: 1–18. [DOI] [PubMed] [Google Scholar]
- 2.CDC Grand Rouds: Reducing Severe Traumatic Brain Injury in the United States. MMWR 2013; 62(27): 549–52. [PMC free article] [PubMed] [Google Scholar]
- 3.Thurman DJ. The Epidemiology of Traumatic Brain Injury in Children and Youths: A Review of Research Since 1990. J Child Neurol 2016; 31(1): 20–7. [DOI] [PubMed] [Google Scholar]
- 4.Soreide K, Kruger AJ, Ellingsen CL, Tjosevik KE. Pediatric trauma deaths are predominated by severe head injuries during spring and summer. Scand J Trauma Resusc Emerg Med 2009; 17: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramos-Cejudo J, Wisniewski T, Marmar C, et al. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine 2018; 28: 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahoney SO, Chowdhury NF, Ngo V, Imms P, Irimia A. Mild Traumatic Brain Injury Results in Significant and Lasting Cortical Demyelination. Front Neurol 2022; 13: 854396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adams KL, Gallo V. The diversity and disparity of the glial scar. Nat Neurosci 2018; 21(1): 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alam A, Thelin EP, Tajsic T, et al. Cellular infiltration in traumatic brain injury. J Neuroinflammation 2020; 17(1): 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mira RG, Lira M, Cerpa W. Traumatic Brain Injury: Mechanisms of Glial Response. Front Physiol 2021; 12: 740939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017; 95(6): 1246–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dimou L, Gallo V. NG2-glia and their functions in the central nervous system. Glia 2015; 63(8): 1429–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levine JM. Increased expression of the NG2 chondroitin-sulfate proteoglycan after brain injury. J Neurosci 1994; 14(8): 4716–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buffo A, Vosko MR, Erturk D, et al. Expression pattern of the transcription factor Olig2 in response to brain injuries: implications for neuronal repair. Proc Natl Acad Sci U S A 2005; 102(50): 18183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes EG, Kang SH, Fukaya M, Bergles DE. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci 2013; 16(6): 668–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mierzwa AJ, Sullivan GM, Beer LA, Ahn S, Armstrong RC. Comparison of cortical and white matter traumatic brain injury models reveals differential effects in the subventricular zone and divergent Sonic hedgehog signaling pathways in neuroblasts and oligodendrocyte progenitors. ASN Neuro 2014; 6(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Algattas H, Huang JH. Traumatic Brain Injury pathophysiology and treatments: early, intermediate, and late phases post-injury. Int J Mol Sci 2013; 15(1): 309–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Psenicka MW, Smith BC, Tinkey RA, Williams JL. Connecting Neuroinflammation and Neurodegeneration in Multiple Sclerosis: Are Oligodendrocyte Precursor Cells a Nexus of Disease? Front Cell Neurosci 2021; 15: 654284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gruchot J, Weyers V, Gottle P, et al. The Molecular Basis for Remyelination Failure in Multiple Sclerosis. Cells 2019; 8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skaper SD. Oligodendrocyte precursor cells as a therapeutic target for demyelinating diseases. Prog Brain Res 2019; 245: 119–44. [DOI] [PubMed] [Google Scholar]
- 20.van Tilborg E, de Theije CGM, van Hal M, et al. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 2018; 66(2): 221–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franklin RJM, Frisen J, Lyons DA. Revisiting remyelination: Towards a consensus on the regeneration of CNS myelin. Semin Cell Dev Biol 2021; 116: 3–9. [DOI] [PubMed] [Google Scholar]
- 22.Filley CM, Kelly JP. White Matter and Cognition in Traumatic Brain Injury. J Alzheimers Dis 2018; 65(2): 345–62. [DOI] [PubMed] [Google Scholar]
- 23.Spitz G, Alway Y, Gould KR, Ponsford JL. Disrupted White Matter Microstructure and Mood Disorders after Traumatic Brain Injury. J Neurotrauma 2017; 34(4): 807–15. [DOI] [PubMed] [Google Scholar]
- 24.Armstrong RC, Mierzwa AJ, Marion CM, Sullivan GM. White matter involvement after TBI: Clues to axon and myelin repair capacity. Exp Neurol 2016; 275 Pt 3: 328–33. [DOI] [PubMed] [Google Scholar]
- 25.Armstrong RC, Mierzwa AJ, Sullivan GM, Sanchez MA. Myelin and oligodendrocyte lineage cells in white matter pathology and plasticity after traumatic brain injury. Neuropharmacology 2016; 110(Pt B): 654–9. [DOI] [PubMed] [Google Scholar]
- 26.McKee AC, Daneshvar DH. The neuropathology of traumatic brain injury. Handb Clin Neurol 2015; 127: 45–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Juratli TA, Zang B, Litz RJ, et al. Early hemorrhagic progression of traumatic brain contusions: frequency, correlation with coagulation disorders, and patient outcome: a prospective study. J Neurotrauma 2014; 31(17): 1521–7. [DOI] [PubMed] [Google Scholar]
- 28.Cepeda S, Gomez PA, Castano-Leon AM, Munarriz PM, Paredes I, Lagares A. Contrecoup Traumatic Intracerebral Hemorrhage: A Geometric Study of the Impact Site and Association with Hemorrhagic Progression. J Neurotrauma 2016; 33(11): 1034–46. [DOI] [PubMed] [Google Scholar]
- 29.Qureshi AI, Malik AA, Adil MM, Defillo A, Sherr GT, Suri MF. Hematoma Enlargement Among Patients with Traumatic Brain Injury: Analysis of a Prospective Multicenter Clinical Trial. J Vasc Interv Neurol 2015; 8(3): 42–9. [PMC free article] [PubMed] [Google Scholar]
- 30.Adatia K, Newcombe VFJ, Menon DK. Contusion Progression Following Traumatic Brain Injury: A Review of Clinical and Radiological Predictors, and Influence on Outcome. Neurocrit Care 2021; 34(1): 312–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaughn JE, Peters A. A third neuroglial cell type. An electron microscopic study. J Comp Neurol 1968; 133(2): 269–88. [DOI] [PubMed] [Google Scholar]
- 32.Ligon KL, Kesari S, Kitada M, et al. Development of NG2 neural progenitor cells requires Olig gene function. Proc Natl Acad Sci U S A 2006; 103(20): 7853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB. Co-localization of NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells in the developing rat brain. J Neurosci Res 1996; 43(3): 299–314. [DOI] [PubMed] [Google Scholar]
- 34.Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB. Interaction between NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells is required for optimal response to PDGF. J Neurosci Res 1996; 43(3): 315–30. [DOI] [PubMed] [Google Scholar]
- 35.Levine JM, Card JP. Light and electron microscopic localization of a cell surface antigen (NG2) in the rat cerebellum: association with smooth protoplasmic astrocytes. J Neurosci 1987; 7(9): 2711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishiyama A, Dahlin KJ, Prince JT, Johnstone SR, Stallcup WB. The primary structure of NG2, a novel membrane-spanning proteoglycan. J Cell Biol 1991; 114(2): 359–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bu J, Akhtar N, Nishiyama A. Transient expression of the NG2 proteoglycan by a subpopulation of activated macrophages in an excitotoxic hippocampal lesion. Glia 2001; 34(4): 296–310. [DOI] [PubMed] [Google Scholar]
- 38.Ozerdem U, Grako KA, Dahlin-Huppe K, Monosov E, Stallcup WB. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn 2001; 222(2): 218–27. [DOI] [PubMed] [Google Scholar]
- 39.Huang W, Bai X, Meyer E, Scheller A. Acute brain injuries trigger microglia as an additional source of the proteoglycan NG2. Acta Neuropathol Commun 2020; 8(1): 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yi JH, Katagiri Y, Susarla B, Figge D, Symes AJ, Geller HM. Alterations in sulfated chondroitin glycosaminoglycans following controlled cortical impact injury in mice. J Comp Neurol 2012; 520(15): 3295–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marques S, Zeisel A, Codeluppi S, et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 2016; 352(6291): 1326–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishiyama A, Watanabe M, Yang Z, Bu J. Identity, distribution, and development of polydendrocytes: NG2-expressing glial cells. J Neurocytol 2002; 31(6-7): 437–55. [DOI] [PubMed] [Google Scholar]
- 43.Vigano F, Mobius W, Gotz M, Dimou L. Transplantation reveals regional differences in oligodendrocyte differentiation in the adult brain. Nat Neurosci 2013; 16(10): 1370–2. [DOI] [PubMed] [Google Scholar]
- 44.Kirdajova D, Valihrach L, Valny M, et al. Transient astrocyte-like NG2 glia subpopulation emerges solely following permanent brain ischemia. Glia 2021; 69(11): 2658–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sullivan GM, Mierzwa AJ, Kijpaisalratana N, et al. Oligodendrocyte lineage and subventricular zone response to traumatic axonal injury in the corpus callosum. J Neuropathol Exp Neurol 2013; 72(12): 1106–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marion CM, McDaniel DP, Armstrong RC. Sarm1 deletion reduces axon damage, demyelination, and white matter atrophy after experimental traumatic brain injury. Exp Neurol 2019; 321: 113040. [DOI] [PubMed] [Google Scholar]
- 47.Marion CM, Radomski KL, Cramer NP, Galdzicki Z, Armstrong RC. Experimental Traumatic Brain Injury Identifies Distinct Early and Late Phase Axonal Conduction Deficits of White Matter Pathophysiology, and Reveals Intervening Recovery. J Neurosci 2018; 38(41): 8723–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang EH, Adorjan I, Mundim MV, Sun B, Dizon ML, Szele FG. Traumatic Brain Injury Activation of the Adult Subventricular Zone Neurogenic Niche. Front Neurosci 2016; 10: 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.David-Bercholz J, Kuo CT, Deneen B. Astrocyte and Oligodendrocyte Responses From the Subventricular Zone After Injury. Front Cell Neurosci 2021; 15: 797553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pous L, Deshpande SS, Nath S, et al. Fibrinogen induces neural stem cell differentiation into astrocytes in the subventricular zone via BMP signaling. Nat Commun 2020; 11(1): 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dimou L, Simon C, Kirchhoff F, Takebayashi H, Gotz M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci 2008; 28(41): 10434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simon C, Gotz M, Dimou L. Progenitors in the adult cerebral cortex: cell cycle properties and regulation by physiological stimuli and injury. Glia 2011; 59(6): 869–81. [DOI] [PubMed] [Google Scholar]
- 53.Huang W, Zhao N, Bai X, et al. Novel NG2-CreERT2 knock-in mice demonstrate heterogeneous differentiation potential of NG2 glia during development. Glia 2014; 62(6): 896–913. [DOI] [PubMed] [Google Scholar]
- 54.Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development 2008; 135(1): 145–57. [DOI] [PubMed] [Google Scholar]
- 55.Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, Nishiyama A. Age-dependent fate and lineage restriction of single NG2 cells. Development 2011; 138(4): 745–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.von Streitberg A, Jakel S, Eugenin von Bernhardi J, et al. NG2-Glia Transiently Overcome Their Homeostatic Network and Contribute to Wound Closure After Brain Injury. Front Cell Dev Biol 2021; 9: 662056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Donat CK, Scott G, Gentleman SM, Sastre M. Microglial Activation in Traumatic Brain Injury. Front Aging Neurosci 2017; 9: 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bardehle S, Kruger M, Buggenthin F, et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci 2013; 16(5): 580–6. [DOI] [PubMed] [Google Scholar]
- 59.Kukley M, Nishiyama A, Dietrich D. The fate of synaptic input to NG2 glial cells: neurons specifically downregulate transmitter release onto differentiating oligodendroglial cells. J Neurosci 2010; 30(24): 8320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li R, Zhang P, Zhang M, Yao Z. The roles of neuron-NG2 glia synapses in promoting oligodendrocyte development and remyelination. Cell Tissue Res 2020; 381(1): 43–53. [DOI] [PubMed] [Google Scholar]
- 61.De Biase LM, Nishiyama A, Bergles DE. Excitability and synaptic communication within the oligodendrocyte lineage. J Neurosci 2010; 30(10): 3600–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mangin JM, Li P, Scafidi J, Gallo V. Experience-dependent regulation of NG2 progenitors in the developing barrel cortex. Nat Neurosci 2012; 15(9): 1192–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Orduz D, Maldonado PP, Balia M, et al. Interneurons and oligodendrocyte progenitors form a structured synaptic network in the developing neocortex. Elife 2015; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Velez-Fort M, Maldonado PP, Butt AM, Audinat E, Angulo MC. Postnatal switch from synaptic to extrasynaptic transmission between interneurons and NG2 cells. J Neurosci 2010; 30(20): 6921–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gibson EM, Purger D, Mount CW, et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 2014; 344(6183): 1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Lanerolle NC, Kim JH, Bandak FA. Neuropathology of traumatic brain injury: comparison of penetrating, nonpenetrating direct impact and explosive blast etiologies. Semin Neurol 2015; 35(1): 12–9. [DOI] [PubMed] [Google Scholar]
- 67.Andriessen TM, Jacobs B, Vos PE. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J Cell Mol Med 2010; 14(10): 2381–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mastorakos P, Mihelson N, Luby M, et al. Temporally distinct myeloid cell responses mediate damage and repair after cerebrovascular injury. Nat Neurosci 2021; 24(2): 245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Russo MV, Latour LL, McGavern DB. Distinct myeloid cell subsets promote meningeal remodeling and vascular repair after mild traumatic brain injury. Nat Immunol 2018; 19(5): 442–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abdullah AN, Ahmad AH, Zakaria R, et al. Disruption of white matter integrity and its relationship with cognitive function in non-severe traumatic brain injury. Front Neurol 2022; 13: 1011304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palacios EM, Yuh EL, Mac Donald CL, et al. Diffusion Tensor Imaging Reveals Elevated Diffusivity of White Matter Microstructure that Is Independently Associated with Long-Term Outcome after Mild Traumatic Brain Injury: A TRACK-TBI Study. J Neurotrauma 2022; 39(19–20): 1318–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci 2013; 14(2): 128–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bodnar CN, Roberts KN, Higgins EK, Bachstetter AD. A Systematic Review of Closed Head Injury Models of Mild Traumatic Brain Injury in Mice and Rats. J Neurotrauma 2019; 36(11): 1683–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma X, Aravind A, Pfister BJ, Chandra N, Haorah J. Animal Models of Traumatic Brain Injury and Assessment of Injury Severity. Mol Neurobiol 2019; 56(8): 5332–45. [DOI] [PubMed] [Google Scholar]
- 75.Cernak I. Animal models of head trauma. NeuroRx 2005; 2(3): 410–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Siebold L, Obenaus A, Goyal R. Criteria to define mild, moderate, and severe traumatic brain injury in the mouse controlled cortical impact model. Exp Neurol 2018; 310: 48–57. [DOI] [PubMed] [Google Scholar]
- 77.Lighthall JW. Controlled cortical impact: a new experimental brain injury model. J Neurotrauma 1988; 5(1): 1–15. [DOI] [PubMed] [Google Scholar]
- 78.Osier N, Dixon CE. Mini Review of Controlled Cortical Impact: A Well-Suited Device for Concussion Research. Brain Sci 2017; 7(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lyeth BG. Historical Review of the Fluid-Percussion TBI Model. Front Neurol 2016; 7: 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kalish BT, Whalen MJ. Weight Drop Models in Traumatic Brain Injury. Methods Mol Biol 2016; 1462: 193–209. [DOI] [PubMed] [Google Scholar]
- 81.Foda MA, Marmarou A. A new model of diffuse brain injury in rats. Part II: Morphological characterization. J Neurosurg 1994; 80(2): 301–13. [DOI] [PubMed] [Google Scholar]
- 82.Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J Neurosurg 1994; 80(2): 291–300. [DOI] [PubMed] [Google Scholar]
- 83.Yu F, Shukla DK, Armstrong RC, et al. Repetitive Model of Mild Traumatic Brain Injury Produces Cortical Abnormalities Detectable by Magnetic Resonance Diffusion Imaging, Histopathology, and Behavior. J Neurotrauma 2017; 34(7): 1364–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davidsson J, Risling M. A new model to produce sagittal plane rotational induced diffuse axonal injuries. Front Neurol 2011; 2: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Namjoshi DR, Cheng WH, McInnes KA, et al. Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model of traumatic brain injury. Mol Neurodegener 2014; 9: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McNamara EH, Grillakis AA, Tucker LB, McCabe JT. The closed-head impact model of engineered rotational acceleration (CHIMERA) as an application for traumatic brain injury pre-clinical research: A status report. Exp Neurol 2020; 333: 113409. [DOI] [PubMed] [Google Scholar]
- 87.Losurdo M, Davidsson J, Skold MK. Diffuse Axonal Injury in the Rat Brain: Axonal Injury and Oligodendrocyte Activity Following Rotational Injury. Brain Sci 2020; 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cavanagh JB. The proliferation of astrocytes around a needle wound in the rat brain. J Anat 1970; 106(Pt 3): 471–87. [PMC free article] [PubMed] [Google Scholar]
- 89.Rhodes KE, Moon LD, Fawcett JW. Inhibiting cell proliferation during formation of the glial scar: effects on axon regeneration in the CNS. Neuroscience 2003; 120(1): 41–56. [DOI] [PubMed] [Google Scholar]
- 90.Alonso G. NG2 proteoglycan-expressing cells of the adult rat brain: possible involvement in the formation of glial scar astrocytes following stab wound. Glia 2005; 49(3): 318–38. [DOI] [PubMed] [Google Scholar]
- 91.Magnus T, Coksaygan T, Korn T, et al. Evidence that nucleocytoplasmic Olig2 translocation mediates brain-injury-induced differentiation of glial precursors to astrocytes. J Neurosci Res 2007; 85(10): 2126–37. [DOI] [PubMed] [Google Scholar]
- 92.Zhao JW, Raha-Chowdhury R, Fawcett JW, Watts C. Astrocytes and oligodendrocytes can be generated from NG2+ progenitors after acute brain injury: intracellular localization of oligodendrocyte transcription factor 2 is associated with their fate choice. Eur J Neurosci 2009; 29(9): 1853–69. [DOI] [PubMed] [Google Scholar]
- 93.Franke H, Parravicini C, Lecca D, et al. Changes of the GPR17 receptor, a new target for neurorepair, in neurons and glial cells in patients with traumatic brain injury. Purinergic Signal 2013; 9(3): 451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Boda E, Vigano F, Rosa P, et al. The GPR17 receptor in NG2 expressing cells: focus on in vivo cell maturation and participation in acute trauma and chronic damage. Glia 2011; 59(12): 1958–73. [DOI] [PubMed] [Google Scholar]
- 95.Vigano F, Schneider S, Cimino M, et al. GPR17 expressing NG2-Glia: Oligodendrocyte progenitors serving as a reserve pool after injury. Glia 2016; 64(2): 287–99. [DOI] [PubMed] [Google Scholar]
- 96.Dehn D, Burbach GJ, Schafer R, Deller T. NG2 upregulation in the denervated rat fascia dentata following unilateral entorhinal cortex lesion. Glia 2006; 53(5): 491–500. [DOI] [PubMed] [Google Scholar]
- 97.Hossain-Ibrahim MK, Rezajooi K, Stallcup WB, Lieberman AR, Anderson PN. Analysis of axonal regeneration in the central and peripheral nervous systems of the NG2-deficient mouse. BMC Neurosci 2007; 8: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Asher RA, Morgenstern DA, Properzi F, Nishiyama A, Levine JM, Fawcett JW. Two separate metalloproteinase activities are responsible for the shedding and processing of the NG2 proteoglycan in vitro. Mol Cell Neurosci 2005; 29(1): 82–96. [DOI] [PubMed] [Google Scholar]
- 99.de Castro R Jr., Tajrishi R, Claros J, Stallcup WB. Differential responses of spinal axons to transection: influence of the NG2 proteoglycan. Exp Neurol 2005; 192(2): 299–309. [DOI] [PubMed] [Google Scholar]
- 100.Dou CL, Levine JM. Inhibition of neurite growth by the NG2 chondroitin sulfate proteoglycan. J Neurosci 1994; 14(12): 7616–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Biname F, Sakry D, Dimou L, Jolivel V, Trotter J. NG2 regulates directional migration of oligodendrocyte precursor cells via Rho GTPases and polarity complex proteins. J Neurosci 2013; 33(26): 10858–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bonfanti E, Gelosa P, Fumagalli M, et al. The role of oligodendrocyte precursor cells expressing the GPR17 receptor in brain remodeling after stroke. Cell Death Dis 2017; 8(6): e2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tabata H. Diverse subtypes of astrocytes and their development during corticogenesis. Front Neurosci 2015; 9: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hampton DW, Asher RA, Kondo T, Steeves JD, Ramer MS, Fawcett JW. A potential role for bone morphogenetic protein signalling in glial cell fate determination following adult central nervous system injury in vivo. Eur J Neurosci 2007; 26(11): 3024–35. [DOI] [PubMed] [Google Scholar]
- 105.Komitova M, Serwanski DR, Lu QR, Nishiyama A. NG2 cells are not a major source of reactive astrocytes after neocortical stab wound injury. Glia 2011; 59(5): 800–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Buffo A, Rite I, Tripathi P, et al. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A 2008; 105(9): 3581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zawadzka M, Rivers LE, Fancy SP, et al. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 2010; 6(6): 578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen Y, Miles DK, Hoang T, et al. The basic helix-loop-helix transcription factor olig2 is critical for reactive astrocyte proliferation after cortical injury. J Neurosci 2008; 28(43): 10983–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang H, Xu L, Lai C, et al. Region-specific distribution of Olig2-expressing astrocytes in adult mouse brain and spinal cord. Mol Brain 2021; 14(1): 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mabie PC, Mehler MF, Marmur R, Papavasiliou A, Song Q, Kessler JA. Bone morphogenetic proteins induce astroglial differentiation of oligodendroglial-astroglial progenitor cells. J Neurosci 1997; 17(11): 4112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Petersen MA, Ryu JK, Chang KJ, et al. Fibrinogen Activates BMP Signaling in Oligodendrocyte Progenitor Cells and Inhibits Remyelination after Vascular Damage. Neuron 2017; 96(5): 1003–12 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ryu JK, Rafalski VA, Meyer-Franke A, et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat Immunol 2018; 19(11): 1212–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Adams RA, Bauer J, Flick MJ, et al. The fibrin-derived gamma377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med 2007; 204(3): 571–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xia Y, Pu H, Leak RK, et al. Tissue plasminogen activator promotes white matter integrity and functional recovery in a murine model of traumatic brain injury. Proc Natl Acad Sci U S A 2018; 115(39): E9230–E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rivers LE, Young KM, Rizzi M, et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci 2008; 11(12): 1392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Guo Z, Zhang L, Wu Z, Chen Y, Wang F, Chen G. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model. Cell Stem Cell 2014; 14(2): 188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Heinrich C, Bergami M, Gascon S, et al. Sox2-mediated conversion of NG2 glia into induced neurons in the injured adult cerebral cortex. Stem Cell Reports 2014; 3(6): 1000–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tai W, Wu W, Wang LL, et al. In vivo reprogramming of NG2 glia enables adult neurogenesis and functional recovery following spinal cord injury. Cell Stem Cell 2021; 28(5): 923–37 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Flygt J, Clausen F, Marklund N. Diffuse traumatic brain injury in the mouse induces a transient proliferation of oligodendrocyte progenitor cells in injured white matter tracts. Restor Neurol Neurosci 2017; 35(2): 251–63. [DOI] [PubMed] [Google Scholar]
- 120.Susarla BT, Villapol S, Yi JH, Geller HM, Symes AJ. Temporal patterns of cortical proliferation of glial cell populations after traumatic brain injury in mice. ASNNeuro 2014; 6(3): 159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen S, Pickard JD, Harris NG. Time course of cellular pathology after controlled cortical impact injury. Exp Neurol 2003; 182(1): 87–102. [DOI] [PubMed] [Google Scholar]
- 122.Dong W, Sun Y, Cheng H, et al. Dynamic cell type-specific expression of Nrf2 after traumatic brain injury in mice. Eur J Neurosci 2019; 50(2): 1981–93. [DOI] [PubMed] [Google Scholar]
- 123.Huang C, Sakry D, Menzel L, et al. Lack of NG2 exacerbates neurological outcome and modulates glial responses after traumatic brain injury. Glia 2016; 64(4): 507–23. [DOI] [PubMed] [Google Scholar]
- 124.White BD, Nathe RJ, Maris DO, et al. Beta-catenin signaling increases in proliferating NG2+ progenitors and astrocytes during post-traumatic gliogenesis in the adult brain. Stem Cells 2010; 28(2): 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hackett AR, Lee JK. Understanding the NG2 Glial Scar after Spinal Cord Injury. Front Neurol 2016; 7: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cuadrado A, Manda G, Hassan A, et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol Rev 2018; 70(2): 348–83. [DOI] [PubMed] [Google Scholar]
- 127.Ren Z, Iliff JJ, Yang L, et al. ‘Hit & Run’ model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J Cereb Blood Flow Metab 2013; 33(6): 834–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dean TKA, Goldstein E, Ghaemmaghami J, Gallo V. Endogenous circadian clock machinery regulates cortical NG2-glia proliferation. (submitted); 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet 2017; 18(3): 164–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022; 110(21): 3458–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Forbes TA, Goldstein EZ, Dupree JL, et al. Environmental enrichment ameliorates perinatal brain injury and promotes functional white matter recovery. Nat Commun 2020; 11(1): 964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.DeWitt DS, Hawkins BE, Dixon CE, et al. Pre-Clinical Testing of Therapies for Traumatic Brain Injury. J Neurotrauma 2018; 35(23): 2737–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kochanek PM, Dixon CE, Mondello S, et al. Multi-Center Pre-clinical Consortia to Enhance Translation of Therapies and Biomarkers for Traumatic Brain Injury: Operation Brain Trauma Therapy and Beyond. Front Neurol 2018; 9: 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kochanek PM, Jackson TC, Jha RM, et al. Paths to Successful Translation of New Therapies for Severe Traumatic Brain Injury in the Golden Age of Traumatic Brain Injury Research: A Pittsburgh Vision. J Neurotrauma 2020; 37(22): 2353–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new data are included in this review article.