

Abstract
Objective
To understand the phenotypic and genotypic spectrum of genetic forms of rickets in 10 families.
Methods
Detailed clinical, radiographic, and biochemical evaluation of 10 families with phenotypes suggestive of a genetic cause of rickets was performed. Molecular testing using exome sequencing aided in the diagnosis of six different forms of known genetic causes.
Results
Eleven disease-causing variants including five previously reported variants (CYP27B1:c.1319_1325dup, p.(Phe443Profs*24), VDR:c.1171C>T, p.(Arg391Cys), PHEX: c.1586_1586+1del, PHEX: c.1482+5G>C, PHEX: c.58C>T, p.(Arg20*)) and six novel variants (CYP27B1:c.974C>T, p.(Thr325Met), CYP27B1: c.1376G>A, p.(Arg459His), CYP2R1: c.595C>T, p.(Arg199*), CYP2R1:c.1330G>C, p.(Gly444Arg),SLC34A3:c.1336-11_1336-1del, SLC2A2: c.589G>C, p.(Val197Leu)) in the genes known to cause monogenic rickets were identified.
Conclusion
The authors hereby report a case series of individuals from India with a molecular diagnosis of rickets and provide the literature review which would help in enhancing the clinical and molecular profile for rapid and differential diagnosis of rickets.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12098-022-04393-9.
Keywords: Rickets, Vitamin-D-dependent rickets, Hypophosphatemic rickets, Exome sequencing
Introduction
Rickets is a disorder of growing bone caused by a deficiency of calcium/phosphorous/vitamin D or defects in their metabolism. Defective mineralization and widening of the cartilaginous growth plates are the characteristic features of rickets [1]. Since the first description of the term “rickets” in the medical literature [2], nutritional deficiency has been the most prevalent cause of rickets worldwide. Among nutritional deficiencies, vitamin D deficiency is reported as a common etiology [3]. In India, the prevalence rate ranges from 22%–92% in infants and 14%–24% in older children [4]. However, there are other non-nutritional causes of rickets including genetic causes, rickets due to drug ingestion, secondary rickets due to liver and kidney diseases, and malabsorption syndromes [5].
Hereditary forms of rickets are very rare and account for only about 13% [6]. Based on the underlying pathophysiology, they have been broadly classified into two categories, vitamin-D-dependent rickets (VDDR) and hypophosphatemic rickets (HR). VDDR is further subclassified into four different forms including VDDR1A, VDDR1B, VDDR2A, and VDDR2B, whereas hypophosphatemic rickets consists of 15 distinct disorders which are grouped into FGF-23-dependent and FGF-23-independent hypophosphatemic rickets [7]. More recently VDDR type 3 (VDDR3) caused by disease-causing variants in CYP3A4 has been reported [8]. Hitherto, 19 genes are known to cause monogenic forms of rickets [7, 9]. The overlap of the clinical spectrum of hereditary rickets with nutritional deficiency poses a challenge to diagnosis [9]. Therefore, molecular diagnosis is crucial for rickets persisting after adequate treatment of underlying nutritional deficiency and thereby initiating appropriate treatment. In this study, the authors detail the clinical and mutational spectrum of VDDR and hypophosphatemic rickets in 10 Indian families along with their laboratory findings. This condition is not well studied in this population. Understanding the landscape of phenotype and genetic etiology would help in differential diagnosis at an early stage.
Materials and Methods
Ten unrelated individuals from different families with suspected hereditary rickets were recruited for the study. A detailed history of illness, clinical photographs, and radiographs were obtained after receiving written informed consent from the families. An appropriate biochemical investigation was performed on all the subjects and values along with reference ranges were recorded. The Institutional Ethics Committee (IEC) of Kasturba Medical College and Hospital, Manipal granted ethical clearance (IEC:921/2018) for this study.
Exome sequencing (ES) was carried out in probands of all the families (Supplementary Fig. S1) using multiple sequencing platforms and capture kits as previously described [10]. Variant analysis was performed using a well-defined in-house strategy and pathogenicity assessment was based on parameters including type of variant, genomic location, effect on protein, patterns of inheritance, clinical correlation, allele frequency in population databases (gnomAD, and ExAC), in-house database (2155 exomes), and prediction scores of multiple in silico tools (including MutationTaster, REVEL, M-CAP, SIFT, Splice AI). Guidelines and criteria issued by the American College of Medical Genetics and Genomics and the Association of Molecular Pathologists (ACMG-AMP) were followed for classifying the disease-causing variants [11].
Results
Ten unrelated Indian individuals (P1 to P10), their parents, and unaffected siblings from 10 families were recruited for the study. Consanguinity was observed in 7 families. Their ages at clinical diagnosis ranged from 2–23 y. Detailed clinical and molecular information of affected individuals and families along with their laboratory investigation results are provided in Supplementary Material S1, Supplementary Table S1 and S2, and Table 1. All the variants identified in this study were submitted to ClinVar database (accession numbers: SUB10956971,SUB10957212, SUB10957214, SUB10957222, SUB10957254, SUB10957263,SUB10957267, SCV002053826.1, SCV002054009, SCV002053849.1). The summary of clinical, biochemical, and molecular findings of all the individuals based on the final diagnosis are enumerated below:
Table 1.
Summary of affected individuals with molecular diagnosis of rickets
| Family ID | Family 1 | Family 2 | Family 3 | Family 4 | Family 5 | Family 6 | Family 7 | Family 8 | Family 9 | Family 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Subject ID | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 |
| Subject characteristics | ||||||||||
| Age | 4 y | 4 y | 2 y | 3 y 6 mo | 4 y | 22 y | 10 y | 23 y | 13 y | 8 y |
| Gender | Male | Female | Female | Female | Male | Female | Female | Female | Male | Female |
| Consanguinity | + | + | − | + | + | + | − | − | + | + |
| Ethnicity | Asian Indian | Asian Indian | Asian Indian | Asian Indian | Asian Indian | Asian Indian | Asian Indian | Asian Indian | Asian Indian | Asian Indian |
| Height in cm (SDS) | 71 (-7 SDS) | 87 (-2.5) | 68 (−3) | NA | 76 (−6) | 145.7 (−2.6) | 124.5 (−2.45) | 127 (−5.2) | 128 (−3.5) | 80 (−9.6) |
| Weight in kg (SDS) | 8.5 (-7 SDS) | 11 (-2.2) | 6.38 (−3) | NA | − | 44 (−2.14) | 24 (−2) | 39 (−2.9) | 30 (−4) | 11.3 (−3.675) |
| Occipitofrontal circumference in cm (SDS) | 47 (-3 SDS) | 48.5 (-1.1) | 44 (−2.5) | NA | 47 (−2.4) | 53.5 (−1) | 52 (−1.33) | 52.5 (−1.6) | 50.5 (−3) | 45.5 (−3) |
| Clinical features | ||||||||||
| Wrist widening | + | + | + | + | + | − | + | + | + | + |
| Pectus carinatum | - | + | + | − | − | − | − | − | − | − |
| Pigeon chest | - | - | + | − | + | − | − | − | − | − |
| Harisson sulcus | - | - | + | − | + | − | − | − | − | − |
| Rachitic rosary | - | - | + | − | + | − | − | − | − | − |
| Pot belly | + | + | + | − | + | − | − | − | − | − |
| Double malleoli | + | - | + | − | + | − | − | − | − | − |
| Genu varum | + | - | + | + | + | + | + | + | − | − |
| Genu valgum | - | + | + | − | − | − | − | − | + | + |
| Pes planus | + | + | − | + | NA | + | + | + | + | NA |
| Radiological features | ||||||||||
| Osteopenia | - | + | + | + | + | + | + | + | + | + |
| Delayed carpal bone ossification | + | - | + | − | + | − | − | − | − | − |
| Small epiphyses | + | + | + | + | + | − | − | − | − | − |
| Frayed and/or irregular metaphyses | - | + | + | + | + | + | + | + | + | + |
| Bowing of lower limbs | + | + | − | + | + | + | + | + | + | − |
| Bowing of upper limbs | - | - | − | NA | + | − | − | − | − | + |
| Additional features | Square shape face, broad nasal bridge, maxillary prominence, coarse facies, hypertelorism and oligodontia | Dolichocephaly | Bronchopneumonia, bilateral lower limb hypotonia | None | Carious teeth, sparse eyebrows, joint laxity, and lumbar kyphosis | Telecanthus, bushy eyebrows, synorphis, low set ears, esotropia, central corneal opacity with iridotomy in eyes, unilateral vision loss, bilateral hearing loss, and everted left foot | None | None | None | Mild blue sclearae, sparse eyebrows, multiple sites of fracture |
| Molecular testing details | ||||||||||
| Gene(s) | CYP27B1 | CYP27B1 | CYP27B1 | CYP2R1 | VDR | PHEX | PHEX | PHEX | SLC34A3 | SLC2A2 |
| Transcript ID | NM_000785.4 | NM_000785.4 | NM_000785.4 | NM_024514.4 | NM_000376.3 | NM_000444.6 | NM_000444.6 | NM_000444.6 | NC_000009.12 | NM_000340.2 |
| Zygosity | Homozygous | Homozygous | Compound heterozygous | Compound heterozygous | Homozygous | Heterozygous | Heterozygous | Heterozygous | Homozygous | Homozygous |
| Disease-causing variants | c.974C>T | c.1319_1325dup | c.1376G>A; c.1319_1325dup | c.595C> T;c.1330G>C | c.1171C>T | c.1586_1586+1del | c.1482+5G > C | c.58C>T | c.1336-11_1336-1del | c.589G>C |
| Protein change | p.(Thr325Met) | p.(Phe443Profs*24) | p.(Arg459His); p.(Phe443Profs*24) | p.(Arg199*);p.(Gly444Arg) | p.(Arg391Cys) | − | − | p.(Arg20*) | − | p.(Val197Leu) |
| Location | Exon 6 | Exon 8 | Exon 8 | Exon 3; Exon 4 | Exon 11 | Intron 14 | Intron 13 | Exon 1 | Intron 12 | Exon 5 |
| Variant status | Novel | Known | 1 novel variant | 2 novel variants | Known | Known | Known | Known | Novel | Novel |
| OMIM disease | Vitamin-D dependent rickets type 1A | Vitamin-D dependent rickets type 1A | Vitamin-D dependent rickets type 1A | Vitamin-D dependent rickets type 1B | Vitamin-D dependent rickets type 2A | X-linked dominant hypophosphatemic rickets | X-linked dominant hypophosphatemic rickets | X-linked dominant hypophosphatemic rickets | Hypophosphatemic rickets with hypercalciuria | Fanconi–Bickel syndrome |
| ACMG classification (assertation criteria) | Variant of uncertain significance (PM2, PP3, PP4) | Pathogenic (PVS1, PM2, PP3, PP4, PP5) | Likely pathogenic (PM2, PM5, PP3, PP5) Pathogenic (PVS1, PM2, PP3, PP4, PP5) | Variant of uncertain significance (PM2, PP3, PP4) | Pathogenic (PS3, PM1, PM5, PM2, PP3, PP4) | Pathogenic (PVS1, PM2, PP5) | Likely pathogenic (PM2, PM4, PP4, PP5) | Pathogenic (PVS1, PM2, PP5) | Pathogenic (PVS1, PM2, PP4) | Likely pathogenic (PM2, PM5, PP3, PP4) |
(+): Present, (−): Absent
NA Not available, SDS Standard deviation score
Vitamin-D-dependent rickets type 1A (VDDR1A): Three affected individuals (P1, P2, and P3) from unrelated families had clinical and radiological features suggestive of VDDR1A. P1 had a severe spectrum of disease (Fig. 1) with early age of onset as compared to P2 and P3. The complete biochemical investigations were available for P1 and P3, which were consistent with the findings of VDDR1A. P3 had normal levels of vitamin D and calcium owing to the treatment with vitamin-D supplements. ES helped in the identification of variants in the homozygous state, c.974C>T in CYP27B1 in P1, c.1319_1325dup in CYP27B1 in P2, and variants in the compound heterozygous state, c.1376G>A, and c.1319_1325dup in CYP27B1 in P3. Thus, a molecular diagnosis of VDDR1A was ascertained.
Fig. 1.

Radiographic profile of an individual with vitamin-D-dependent rickets type I. P1 (age: 4 y) shows delayed tooth eruption at 8 y 9 mo (a), delayed carpal ossification (b), metaphyseal dysplasia at the ends of long bones (c–e): small epiphyses at the knee (c), bending of long bones (c, d), and small capital femoral epiphysis (e). The radiographic appearance was affected by his treatment
Vitamin-D-dependent rickets type 1B (VDDR1B): One affected individual (P4) was presented with bowing of legs and had radiological features suggestive of rickets. Her biochemical investigation revealed low serum calcium and phosphate levels with increased ALP values. Molecular analysis aided in the identification of two novel variants in the compound heterozygous state c.595C>T and c.1330G>C in CYP2R1, thus, a diagnosis of VDDR1B was ascertained.
Vitamin-D-dependent rickets type 2A (VDDR2A): An affected individual (P5) was presented with a leg deformity and wrist widening (Fig. 2). He had a severe phenotype of rickets along with alopecia which was noticed since birth. His biochemical profile showed hypocalcemia, hypophosphatemia, and elevated ALP levels. Analysis of exome sequencing data led to the identification of a known missense variant, c.1171C>T in the homozygous state in VDR thereby asserting the diagnosis of VDDR2A.
Fig. 2.

Radiographs of an individual with vitamin-D-dependent rickets type IIA. P5 (age 4 y) shows severe osteopenia, small epiphyses at the knee, irregular metaphyses, bending of the fibula (a), dorsal–lumbar kyphosis (b), and cupping of radius and ulna (c and d)
X-linked dominant hypophosphatemic rickets (XLDHR): Three individuals (P6, P7, and P8) with clinical features suggestive of hypophosphatemic rickets were presented with genu varum and abnormal gait. One of the individuals (P6) had a severe phenotype (Supplementary Material S1 and Fig. 3) with early onset of disease. The biochemical findings were available for P6; however, low levels of serum phosphate and increased values of ALP were observed in P7 and P8. On molecular analysis, one splice-site variant, c.1586_1586+1del in PHEX was observed in the heterozygous state in P6. The c.1482+5G>C in PHEX was observed in P7, whereas c.58C>T was observed in heterozygous in PHEX in P8. Thus, a diagnosis of X-linked dominant hypophosphatemic rickets in all three individuals was made.
Fig. 3.

Lower limb radiograph of P6 with X-linked hypophosphatemic rickets (age 22 y). Bowing of long bones, and osteopenia with frayed metaphyses are seen (a)
Hypophosphatemic rickets with hypercalciuria (HHRH): One individual (P9) was presented with short stature and difficulty in walking. His radiographs and biochemical findings (low phosphate levels with elevated ALP) were suggestive of a monogenic form of hypophosphatemic rickets. Analysis of ES data revealed a novel variant c.1336-11_1336-1del in the homozygous state in SLC34A3, thereby the molecular diagnosis was identified.
Fanconi–Bickel syndrome (FBS): One affected girl (P10) of 8 y of age was referred with complaints of multiple fractures. Generalized osteopenia and bowing of upper limbs were observed in her radiographs (Fig. 4). Low levels of glucose, phosphate, and elevated serum ALP were observed in her laboratory profile. A provisional diagnosis of osteogenesis imperfecta was made. However, exome sequencing helped in the identification of a novel missense variant, c.589G>C in SLC2A2, which led to the diagnosis of FBS.
Fig. 4.
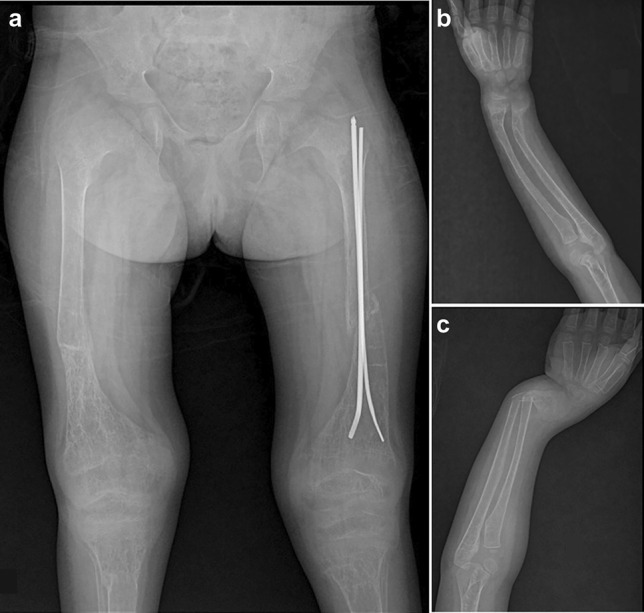
Radiographic profile of P10 (age 8 y) with Fanconi–Bickel syndrome. Multiple sites of fracture and osteopenia are seen in her upper and lower limbs (a–c)
Discussion
The interplay of multiple genetic factors maintains the metabolic homeostasis of vitamin D, calcium, and phosphate in the body, and the perturbation of this essential balance leads to different genetic forms of rickets. Disease-causing variants in CYP27B1 cause an autosomal recessive disease, vitamin-D-dependent rickets type 1A, VDDR1A (MIM # 264700) [12]. The three most reported variants in CYP27B1 are c.1319_1325dup, c.262delG, and c.195+2T>G [13]. The most common variant, c.1319_1325dup is also found in two individuals in the present cohort (P2 and P3). Genotype–phenotype correlation for VDDR1A is not well established. However, by statistical analysis of reported disease-causing variants and clinical features, a recent study has highlighted a few findings [13]. According to this study, c.195+2T>G leads to the most severe clinical presentation whereas in contrast patients with c.262delG variant present with the least severe phenotypic manifestation owing to age and height at evaluation. However, affected individuals with variant, c.1319_1325dup (as found in P2 and P3) had different phenotypic manifestations according to a study. One of the individuals had seizures along with the clinical presentation and another unrelated individual had all the other typical manifestations of VDDR1A without seizures [14]. All the affected individuals (P1, P2, and P3) in the present study had biochemical profiles similar to the findings of VDDR1A [7] (Supplementary Table S1). Three affected individuals are described in the present study with variants in CYP27B1, one of the individuals (P1) showed the complete and severe spectrum of disease with enamel hypoplasia and hypocalcemic seizures with early age of onset (4 mo of age). Interestingly, the novel variant identified in P1 was nonsynonymous as compared to truncating variant found in the other two individuals (P2 and P3). The difference in disease severity could be possibly due to phenotypic variability [14].
Vitamin-D-dependent rickets type 1B, VDDR1B (MIM # 600081) is an autosomal recessive disease caused by biallelic variants in CYP2R1 [15]. Only 6 variants in 31 patients have been described in literature hitherto [16]. Through the present study, the authors add two more novel variants in the literature, c.595C>T and c.1330G>C, which are identified in compound heterozygous state in CYP2R1 in P4. Recently, Bakhamis et al. suggested a semi-dominant inheritance pattern of this disease affecting individuals equally with both biallelic and single-heterozygous variants [16]. The only clinical feature different in both groups of individuals was hypocalcemic manifestations. The typical biochemical findings of VDDR1B (except for PTH and 25-OH vitamin D levels) corroborate with the parameters of the affected subject (P4) in the present study (Supplementary Table S1) [7].
The third entity of the VDDR group of disorders, vitamin-D-dependent rickets type 2A, VDDR2A (MIM # 277440), is an autosomal recessive disease caused by biallelic variants in the vitamin D receptor gene, VDR [17]. VDR constitutes two functional domains, namely the N-terminal dual zinc finger DNA binding domain (DBD), and the C-terminal ligand-binding domain (LBD) [17]. Genotype–phenotype correlation in VDDR2A suggests that all the inactivating biallelic variants found in the DBD result in severe clinical presentation (with alopecia) due to complete loss of function whereas a milder phenotype is observed in individuals with variants in the LBD due to partial loss of function of VDR [18, 19]. However, there are exceptions to this rule [20]. The affected individual (P5) in the present cohort also had a severe spectrum (with alopecia) of disease presentation; however, the variant was located in LBD. Functional analysis performed for this variant revealed that Arg391Cys leads to poor VDRE binding in vivo and reduced transactivation thus leading to a severe phenotype [20]. The biochemical findings (except for PTH and 25-OH vitamin D levels) observed in the affected boy (P5) in the present study corroborate with the laboratory profile of VDDR2A (Supplementary Table S1) [7].
X-linked dominant hypophosphatemic rickets, XLDHR (MIM # 307800) belongs to the FGFR3-dependent rickets category, and it is caused by pathogenic variants in PHEX (phosphate-regulating endopeptidase homolog X-linked). A recent study has stated that individuals harboring pathogenic variants in the N-terminal region have an earlier onset of disease as compared to subjects with variants in C-terminus [21]. Nevertheless, the affected individuals (P6, P7, and P8) in the present cohort had similar ages of onset of disease (average: 2 y) irrespective of the location of variants (2 variants in C terminus and 1 variant in N terminus). The authors report three known truncating variants, c.1482+5G>C, c.1586_1586+1del and c.58C>T in PHEX reported earlier to cause XLDHR [22–24]. Both the canonical and noncanonical splice variants cause the skipping of exons 13 and 14, respectively. The phenotype observed in P6 harboring the canonical splice variant was comparatively more severe (Supplementary Material S1) than in the other two individuals (P7 and P8). The biochemical findings of affected individuals (P7 and P8) with variants in PHEX are mentioned in Supplementary Table S1.
Hypophosphatemic rickets with hypercalciuria, HHRH (MIM # 241530) is an autosomal recessive disease caused due to biallelic variants in SLC34A3 (solute carrier family 34, member 3) [25]. A review of all the previously reported patients shows that affected individuals with rickets have markedly low levels of serum phosphate as compared to subjects with only renal phenotype. Additionally, most individuals with homozygous variants have rickets as compared to subjects with compound heterozygous variants whose phenotype is variable [26]. The affected individual (P9) in the present cohort harbored a novel variant, c.1336-11_1336-1del (in SLC34A3) in the homozygous state with all the characteristic features of HHRH including clinical and biochemical profiles (Table 1 and Supplementary Table S1) [7]. Subjects with a single-heterozygous variant (carriers) have also been reported earlier with mild symptoms [26].
Hypophosphatemia is a common feature of many other diseases which are not considered to be a classical form of HR. Fanconi–Bickel syndrome, FBS (MIM # 227810) an autosomal recessive disease is one such entity that mimics hypophosphatemic rickets as well as osteogenesis imperfecta (OI) [27]. FBS is caused due to biallelic variants in SLC2A2 (solute carrier family 2 member 2) or GLUT2 (glucose transporter 2 protein) known to mediate bidirectional glucose transport [28]. Affected individuals with variants in this gene are also susceptible to noninsulin-dependent diabetes mellitus (NIDDM) [29]. The variant, c.589 G>C (in SLC2A2) observed in the present study (in P10) is a novel alternate variant for the first-ever reported variant in SLC2A2 in the homozygous state which causes a different missense change at the same amino acid codon [28]. The reported variant, c.589G>A p.Val197Leu (in SLC2A2) has been in discussion since its first description as it was also reported in heterozygous state in African American women with diabetes mellitus (DM). Genotype–phenotype correlation by a recent study highlights that biallelic/nonfunctional variants show a complete spectrum of FBS including hepatonephromegaly owing to glycogen accumulation, renal tubular dysfunction, and hypophosphatemic rickets whereas subjects with single-heterozygote variants are susceptible to NIDDM owing to impaired sugar transport in the kidney [29]. Clinical features and biochemical findings (Supplementary Table S1) observed in the patient described in the present study (P10) are concordant with the complete phenotypic spectrum of FBS.
Diagnosis of rickets requires a multifaceted approach which begins with the collection of detailed family history, clinical evaluation, radiographic and biochemical investigations, and use of next-generation sequencing (NGS) to receive a definitive molecular diagnosis. Often, the clinical profile of monogenic rickets is similar to classical nutritional deficiency rickets, which leads to a delay in diagnosis and failure of treatment. However, in the present era, a low index of suspicion of genetic rickets and early deployment of genetic tests are necessary to achieve a rapid and accurate diagnosis. Recently, Marik et al. reported a cohort of 63 individuals affected with rickets and proposed a gene panel for the diagnosis of hypophosphatemic rickets [30]. Molecular diagnosis plays a major role in early intervention and helps in redirecting the treatment plan which would lead to significant improvement in clinical, biochemical, and radiological features.
Conclusion
In this study, the authors have presented profiles of six different genetic forms of rickets including vitamin-D-dependent (VDDR1A, VDDR2A, and VDDR2B) rickets, hypophosphatemic rickets (XLDHR and HHRH), and a disease entity with rickets as one of the clinical features, Fanconi–Bickel syndrome (FBS). With the addition of six novel variants in four known genes (CYP27B1, CYP2R1, SLC34A3, and SLC2A2), the present study provides an update to the mutation spectrum.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank all the families for their participation in the study.
Authors' Contributions
PJ: Writing - original draft, investigation, data curation, writing - review & editing; GSB and PU: Validation, investigation, writing - review & editing; ZW, SVH, KD, NK, and SI: Methodology, investigation, validation, writing - review & editing; RDS, KH: Validation, investigation, writing - review & editing; AD and HS: Funding acquisition, conceptualization, supervision, writing - review & editing, methodology; KMG: Conceptualization, methodology, supervision, funding acquisition, writing - review & editing. KMG will act as the guarantor for this paper.
Funding
Open access funding provided by Manipal Academy of Higher Education, Manipal. This study was supported by the following: Department of Science and Technology, Government of India funded the project entitled ‘Application of Autozygosity Mapping and Exome Sequencing to Identify Genetic Basis of Disorders of Skeletal Development’ (SB/SO/HS/005/2014) to Katta M. Girisha and Department of Biotechnology, Government of India funded the project entitled ‘Development of Genomic Technologies for Predictive Genetic Health and Forensic Profiling’ (Grant No. BTl/AAQ/01/CDFD-Flagship/2019) to Ashwin Dalal. This study was also supported partially by DBT/Wellcome Trust India Alliance Fellowship (India Alliance) under the project entitled ‘Center for Rare Disease Diagnosis, Research and Training’ (Grant ID: GR-0011; Reference number: IA/ CRC/20/1/600002) awarded to Katta M. Girisha.
Declarations
Ethics Approval
Ethical clearance (IEC:921/2018) was obtained for this study from the Institutional Ethics Committee, Kasturba Medical College and Hospital, Manipal.
Consent to Participate
Written informed consent was obtained from the parents/legal guardian/individual participants included in the study.
Conflict of Interest
None.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Misra M, Pacaud D, Petryk A, Collett-Solberg PF, Kappy M; Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Pediatrics. 2008;122:398–417. [DOI] [PubMed]
- 2.Wharton B, Bishop N. Rickets Lancet. 2003;362:1389–1400. doi: 10.1016/S0140-6736(03)14636-3. [DOI] [PubMed] [Google Scholar]
- 3.Forrest KYZ, Stuhldreher WL. Prevalence and correlates of vitamin D deficiency in US adults. Nutr Res. 2011;31:48–54. doi: 10.1016/j.nutres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Gupta P, Dabas A, Seth A, et al. Indian Academy of Pediatrics Revised (2021) guidelines on prevention and treatment of vitamin D deficiency and rickets. Indian Pediatr. 2022;59:142–158. doi: 10.1007/s13312-022-2448-y. [DOI] [PubMed] [Google Scholar]
- 5.Fukumoto S, Ozono K, Michigami T, et al. Pathogenesis and diagnostic criteria for rickets and osteomalacia - proposal by an expert panel supported by Ministry of Health, Labour and Welfare, Japan, The Japanese Society for Bone and Mineral Research and The Japan Endocrine Society. Endocr J. 2015;62:665–671. doi: 10.1507/endocrj.EJ15-0289. [DOI] [PubMed] [Google Scholar]
- 6.Beck-Nielsen SS, Brock-Jacobsen B, Gram J, Brixen K, Jensen TK. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. Eur J Endocrinol. 2009;160:491–497. doi: 10.1530/EJE-08-0818. [DOI] [PubMed] [Google Scholar]
- 7.Acar S, Demir K, Shi Y. Genetic causes of rickets. J Clin Res Pediatr Endocrinol. 2017;9(Suppl 2):88–105. doi: 10.4274/jcrpe.2017.S008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roizen JD, Li D, O’Lear L, et al. CYP3A4 mutation causes vitamin D–dependent rickets type 3. J Clin Invest. 2018;128:1913–1918. doi: 10.1172/JCI98680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michałus I, Rusińska A. Rare, genetically conditioned forms of rickets: differential diagnosis and advances in diagnostics and treatment. Clin Genet. 2018;94:103–114. doi: 10.1111/cge.13229. [DOI] [PubMed] [Google Scholar]
- 10.Kaur P, do Rosario MC, Hebbar M, et al. Clinical and genetic spectrum of 104 Indian families with central nervous system white matter abnormalities. Clin Genet. 2021;100:542–50. [DOI] [PMC free article] [PubMed]
- 11.Richards S, Aziz N, Bale S, et al; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23. [DOI] [PMC free article] [PubMed]
- 12.Wang JT, Lin CJ, Burridge SM, et al. Genetics of vitamin D 1alpha-hydroxylase deficiency in 17 families. Am J Hum Genet. 1998;63:1694–1702. doi: 10.1086/302156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaygusuz SB, Alavanda C, Kirkgoz T, et al. Does genotype-phenotype correlation exist in vitamin D-dependent rickets type IA: report of 13 new cases and review of the literature. Calcif Tissue Int. 2021;108:576–586. doi: 10.1007/s00223-020-00784-2. [DOI] [PubMed] [Google Scholar]
- 14.Durmaz E, Zou M, Al-Rijjal RA, et al. Clinical and genetic analysis of patients with vitamin D-dependent rickets type 1A. Clin Endocrinol (Oxf) 2012;77:363–369. doi: 10.1111/j.1365-2265.2012.04394.x. [DOI] [PubMed] [Google Scholar]
- 15.Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci USA. 2004;101:7711–7715. doi: 10.1073/pnas.0402490101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bakhamis S, Imtiaz F, Ramzan K, et al. 25-Hydroxylase vitamin D deficiency in 27 Saudi Arabian subjects: a clinical and molecular report on CYP2R1 mutations. Endocr Connect. 2021;10:767–775. doi: 10.1530/EC-21-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes MR, Malloy PJ, Kieback DG, et al. Point mutations in the human vitamin D receptor gene associated with hypocalcemic rickets. Science. 1988;242:1702–1705. doi: 10.1126/science.2849209. [DOI] [PubMed] [Google Scholar]
- 18.Marx SJ, Bliziotes MM, Nanes M. Analysis of the relation between alopecia and resistance to 1,25-dihydroxyvitamin D. Clin Endocrinol (Oxf) 1986;25:373–381. doi: 10.1111/j.1365-2265.1986.tb01703.x. [DOI] [PubMed] [Google Scholar]
- 19.Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D-resistant rickets. Endocr Rev. 1999;20:156–188. doi: 10.1210/edrv.20.2.0359. [DOI] [PubMed] [Google Scholar]
- 20.Whitfield GK, Selznick SH, Haussler CA, et al. Vitamin D receptors from patients with resistance to 1,25-dihydroxyvitamin D3: point mutations confer reduced transactivation in response to ligand and impaired interaction with the retinoid X receptor heterodimeric partner. Mol Endocrinol. 1996;10:1617–1631. doi: 10.1210/mend.10.12.8961271. [DOI] [PubMed] [Google Scholar]
- 21.Zhang C, Zhao Z, Sun Y, et al. Clinical and genetic analysis in a large Chinese cohort of patients with X-linked hypophosphatemia. Bone. 2019;121:212–220. doi: 10.1016/j.bone.2019.01.021. [DOI] [PubMed] [Google Scholar]
- 22.Francis F, Strom TM, Hennig S, et al. Genomic organization of the human PEX gene mutated in X-linked dominant hypophosphatemic rickets. Genome Res. 1997;7:573–585. doi: 10.1101/gr.7.6.573. [DOI] [PubMed] [Google Scholar]
- 23.Tyynismaa H, Kaitila I, Näntö-Salonen K, Ala-Houhala M, Alitalo T. Identification of fifteen novel PHEX gene mutations in Finnish patients with hypophosphatemic rickets. Hum Mutat. 2000;15:383–384. doi: 10.1002/(SICI)1098-1004(200004)15:4<383::AID-HUMU18>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 24.Gaucher C, Walrant-Debray O, Nguyen TM, Esterle L, Garabédian M, Jehan F. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum Genet. 2009;125:401–411. doi: 10.1007/s00439-009-0631-z. [DOI] [PubMed] [Google Scholar]
- 25.Bergwitz C, Roslin NM, Tieder M, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. 2006;78:179–192. doi: 10.1086/499409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhadada SK, Sridhar S, Dhiman V, et al. Hypophosphatemic rickets with hypercalciuria: a novel homozygous mutation in SLC34A3 and literature review. AACE Clin Case Rep. 2020;6:e105–e112. doi: 10.4158/ACCR-2019-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hadipour F, Sarkheil P, Noruzinia M, Hadipour Z, Baghdadi T, Shafeghati Y. Fanconi-Bickel syndrome versus osteogenesis imperfeeta: an Iranian case with a novel mutation in glucose transporter 2 gene, and review of literature. Indian J Hum Genet. 2013;19:84–86. doi: 10.4103/0971-6866.112906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santer R, Schneppenheim R, Dombrowski A, Götze H, Steinmann B, Schaub J. Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome. Nat Genet. 1997;17:324–326. doi: 10.1038/ng1197-324. [DOI] [PubMed] [Google Scholar]
- 29.Grünert SC, Schumann A, Baronio F, et al. Evidence for a genotype–phenotype correlation in patients with pathogenic GLUT2 (SLC2A2) variants. Genes (Basel) 2021;12:1785. doi: 10.3390/genes12111785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marik B, Bagga A, Sinha A, Khandelwal P, Hari P, Sharma A. Genetic and clinical profile of patients with hypophosphatemic rickets. Eur J Med Genet. 2022;65:104540. doi: 10.1016/j.ejmg.2022.104540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.