

Summary
Similar to their pivotal roles in nervous system development, neurons have emerged as critical regulators of cancer initiation, maintenance and progression. Focusing on nervous system tumors, we describe the normal relationships between neurons and other cell types relevant to normal nerve function, and discuss how disruptions of these interactions promote tumor evolution, focusing on electrical (gap junctions) and chemical (synaptic) coupling, as well as the establishment of new paracrine relationships. We also review how neuron-tumor communication contributes to some of the complications of cancer, including neuropathy, chemobrain, seizures, and pain. Lastly, we consider the implications of cancer neuroscience in establishing risk for tumor penetrance and in the design of future anti-tumoral treatments.
Keywords: brain tumors, nerves, synapse, microglia, T cells, glioma, neuronal activity, neurotransmitter
eTOC Blurb
In this review, Anastasaki and colleagues describe the normal relationships between neurons and other cell types relevant to normal nervous system function, and discuss how disruptions of these interactions promote tumor formation, growth, and complications in cancer of the nervous system.
Introduction
Cancers initiate, evolve, and progress within a local environment rich in non-neoplastic cells, which largely reflect the normal cellular constituency of their surrounding tissue milieu. The non-neoplastic cells in these tumor microenvironments contain immune system cells (T and B lymphocytes, monocytes, mast cells), fibroblasts, and vascular elements, which communicate with the cancer cells to regulate overall tumor fitness, as well as contribute to the ability of cancers to evade conventional and targeted therapies1–3. While considerable research has focused on cancer-associated fibroblasts4,5, immune cells6–9, and endothelial cells10,11, until recently, comparatively less emphasis has been placed on the role of nerve cells (neurons) in the pathogenesis of cancer12.
The idea that neurons might participate in cancer pathogenesis originated with Hans Scherer in the 1930s, who first described invasive brain cancer cells encircling neuronal cell bodies and dendrites13,14. This characteristic cell grouping, known as “perineuronal satellitosis”, mirrors the clustering of macroglia (astrocytes, oligodendrocytes) around neurons in the healthy nervous system, as originally reported in 1899 by Santiago Ramon y Cajal, raising the intriguing possibility that a symbiotic relationship exists between glial and neuronal elements in both health and disease15. Importantly, this association is not unique to the nervous system neoplasias, but is also observed in cancers outside of the brain, where perineural invasion of tumor cells is a common feature associated with poor prognosis in pancreatic ductal adenocarcinoma, gastric carcinoma, colorectal cancer, prostate cancer, head and neck cancer, biliary tract tumor, and cervical cancer16–23.
With the recognition that neurons commonly integrate into most solid tumors and that neuron-glial relationships exist even in the absence of disease, it becomes increasingly important to consider neuronal contributions to cancer as an extension of their homeostatic and adaptive roles in the development and maintenance of the healthy body. In this review, we focus on nervous system tumors and discuss how these cancers usurp normal neuronal interactions to facilitate tumor initiation, maintenance, and progression.
Neurons interact with other cell types during nervous system development and homeostasis
Neurons are first born during embryonic brain development24–26, when they begin to instruct the proliferation, differentiation, and specification of the central nervous system (CNS)27 through interactions with oligodendrocyte lineage cells, astrocytes, microglia, and T lymphocytes (Figure 1). One of the major mechanisms by which neurons control CNS development is through their electrical activity. In this regard, neuronal activity is critical for neural induction, neural stem cell and precursor cell proliferation, migration and differentiation, synaptogenesis, oligodendrogenesis, and myelination28. As such, productive associations with oligodendrocyte precursor cells (OPCs) are critical for proper myelination and function of neurons29–32, while crosstalk with astrocytes helps dictate neuronal synapse formation, function, and elimination33–39. Similarly, neuron-secreted neurotransmitters depolarize neural progenitors to inhibit DNA synthesis during development, as well as induce neurogenesis in the adult brain27,40,41. Moreover, neurons in the peripheral nervous system (PNS) interact with macroglia (Schwann cells) to control their proliferation and survival, as well as influence their own myelination42,43.
Figure 1. Neurons interact with numerous cell types during nervous system development and maintenance.
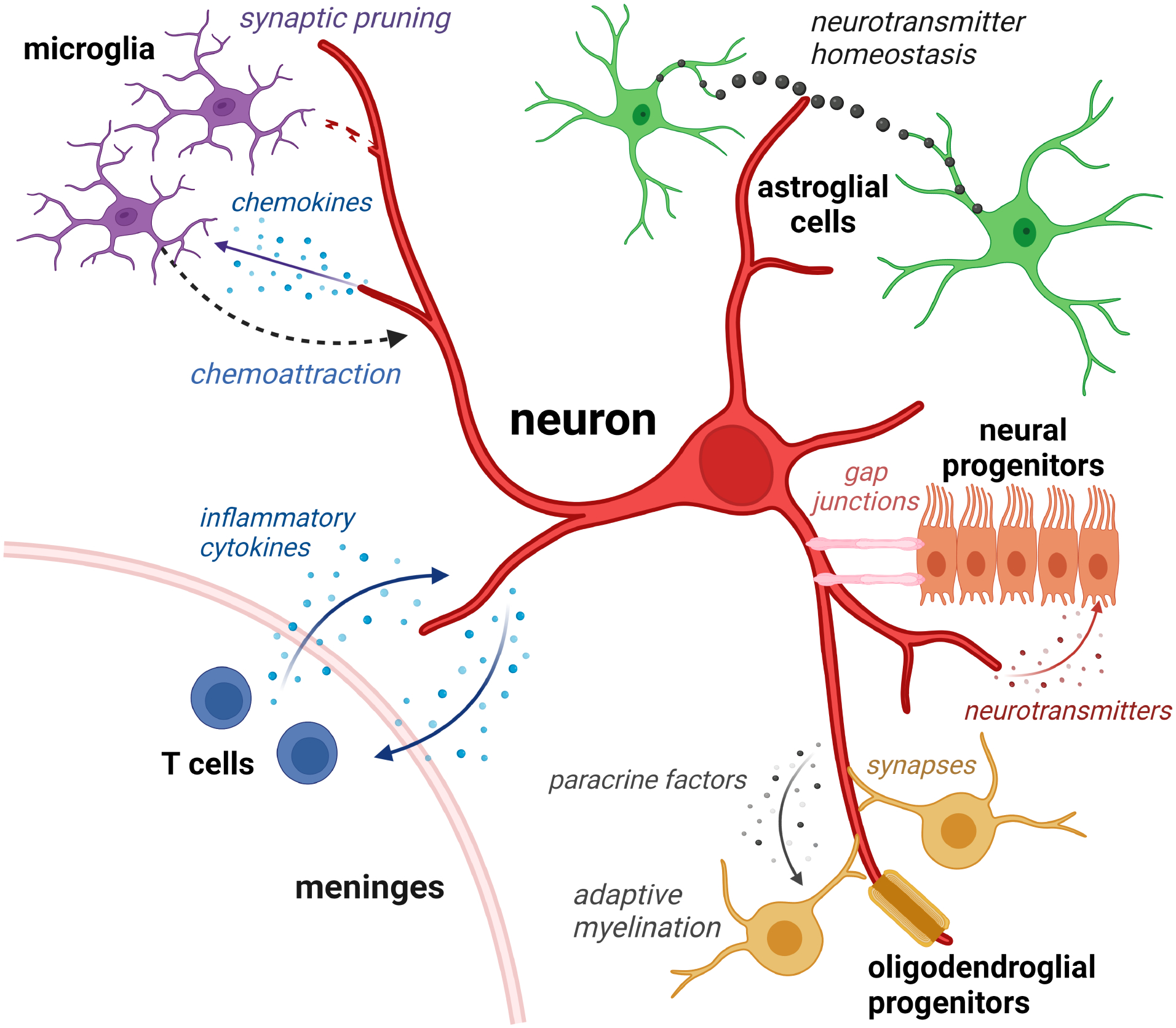
Neurons in the central nervous system interact with microglia through chemokine attraction (chemoattraction) to enable synaptic pruning and induce neuroplasticity, but can also communicate with T cells in the meningeal spaces to modify neuronal function and behavior. In addition, neurons form cooperative relationships with astroglial cells (neurotransmitters), neural progenitors (gap junctions), and oligodendrocyte precursors (OPCs, direct synapses, neurotransmitters) to regulate neuroglial function, neurogenesis, and adaptive myelination. Similar interactions also occur in the peripheral nervous system between immune system cells, Schwann cells, and neurons.
Neurons can additionally interact with immune system cells, such as resident brain macrophages (microglia)44–46. Neuronal activity regulates microglia phagocytosis to selectively eliminate synapses during development (synaptic pruning)47,48, a process that strengthens the remaining neuronal circuits. Neurons also communicate with T lymphocytes, which in turn, can change the function of other cells in the healthy CNS49 to influence learning and behavior50–55. In an analogous manner, neurons can induce T cell motility in the peripheral nervous system. Aging sciatic nerve neurons express elevated levels of the chemokine CXCL13, which acts as a chemoattractant for T cells to promote age-dependent neurodegeneration in response to injury56.
Generally, neurons communicate with their cellular neighbors by transmitting electrical activity through one of three mechanisms: (1) the establishment of gap junctions, (2) the formation of bona fide synapses, and (3) the release of neurotransmitters and paracrine factors.
Gap junctions are intercellular channels containing transmembrane proteins (connexins) that permit the direct transfer of ions and small molecules between cells. During cortical neurogenesis, these specialized conduits can couple ventricular zone stem cells (tanycytes, radial glial cells) with embryonic neural progenitors to form functional circuits that exhibit synchronous depolarization57,58. In this manner, tanycytes directly communicate with one another through connexin-43-containing gap junctions to create an electrical syncytium. Similarly, radial glial cells can use gap junctions for the propagation of calcium currents to control cortical neuron production. Gap junctions can also create neuronal circuits with postnatal neural progenitors59, as well as with mature astrocytes, to mediate synaptic plasticity and learning60,61.
In addition to gap junctions, neurons can form bona fide synapses on OPCs62, where a presynaptic neuron creates a synapse with a postsynaptic structure on OPCs to allow for signal transduction. Similar to canonical neuron-to-neuron synapses, these intercellular junctions facilitate the rapid transfer of information via presynaptic neurotransmitter release and post-synaptic neurotransmitter receptor-mediated signal transduction63. Although the exact mechanism governing the generation of neuron-OPC synapses remains to be fully elucidated, experience (e.g., learning, light exposure) and neuronal activity stimulate OPC proliferation and differentiation, which, in turn, regulate adaptive myelination and motor function30,64–67.
Synaptic transmission between two neurons typically involves neurotransmitter release from a presynaptic neuron, resulting in neurotransmitter receptor activation and downstream signaling within a post-synaptic neuron. These chemical synapses are classified according to the specific neurotransmitter released, and can result in either inhibitory (e.g., GABA-mediated) or excitatory (e.g., glutamate-mediated) effects on post-synaptic neuron function.
In addition to traditional inter-neuronal inhibition or excitation, neurotransmitter secretion by neurons can also regulate neural progenitor cell proliferation, migration, and differentiation, independent of the formation of bona fide synapses40,68. For example, non-synaptic glutamate and GABA release causes ventricular zone neural stem cell depolarization through ionotropic glutamate and GABA receptors expressed on the neural progenitor cells69 to increase their proliferation during forebrain development70,71. Other neurotransmitters can similarly regulate postnatal neurogenesis72–76. For example, depletion of dopamine, which is present during early neuronal development and in adult subventricular zones, or loss of dopamine (D2 and D4) receptor function, can result in reduced proliferation of neural progenitor cells75,76. Similarly, acetylcholine reduction can decrease neurogenesis in the hippocampus, while increased acetylcholine-mediated muscarinic receptor signaling can increase neural stem cell proliferation77.
Besides neurotransmitter secretion, neurons can establish other paracrine relationships with non-neuronal lineage cells. Release of neurotrophins, such as nerve growth factor and brain-derived neurotrophic factor (BDNF), is regulated by neuronal activity78,79, which have profound effects on the proliferation, migration, maturation, survival and myelination capacity of oligodendrocytes and their precursors80–82. For example, neuronal activity can influence microglia and T cell biology. In this manner, neuronal activity-dependent secretion of chemokines, such as Cx3cl183, or neurotransmitters, such as glutamate, dopamine, and GABA84–86, can attract microglia to modulate their activation and phagocytic function. In addition, T lymphocytes located in the meningeal spaces and choroid plexus produce inflammatory mediators (IL-4, IFN-γ) that regulate neuronal function and excitability relevant to normal mouse learning and behavior51,53,87.
Neuronal regulation of nervous system tumor formation and growth
Taking advantage of already established interactions important for healthy nervous system development and maintenance, neurons also regulate the formation and growth of central and peripheral nervous system tumors. Neuronal activity governs tumor formation and progression through multiple mechanisms, including (1) the establishment of paracrine factor dependencies involving growth factors, cytokines and neurotrophins (Figure 2), (2) non-synaptic neuron-tumor cell electrical coupling via microtubes (Figure 3), and (3) the formation of bona fide glutamatergic synapses (Figure 3). In addition, neuronal control of cancer cell growth can be strengthened through the aberrant expression of ion channels by the cancer cells themselves.
Figure 2. Neurons interact with tumor cells through the elaboration of paracrine factors.
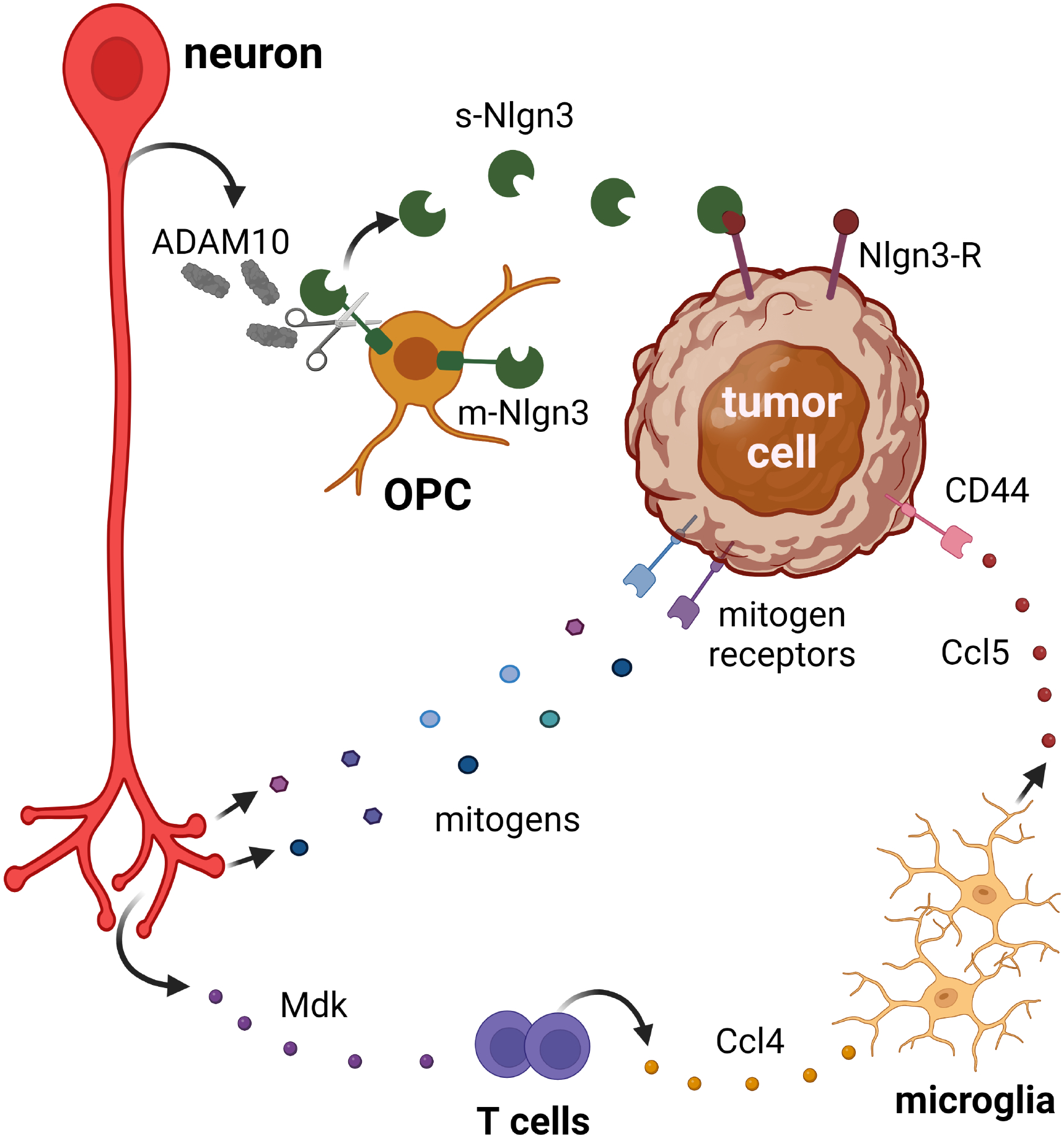
Neurons can increase tumor cell growth through activity-regulated cleavage (ADAM10-mediated) of membrane-bound Nlgn3 (m-Nlgn3) to generate a bioavailable soluble Nlgn3 molecule (s-Nlgn3) that increases tumor cell growth. Neurons can also control tumor cell growth either directly through secretion of other mitogens that bind mitogen receptors on tumor cells, or indirectly through immune cells (T cells and microglia) via the elaboration of paracrine factors (midkine, Mdk; Ccl4; Ccl5).
Figure 3. Neurons directly and indirectly interact with tumor cells.
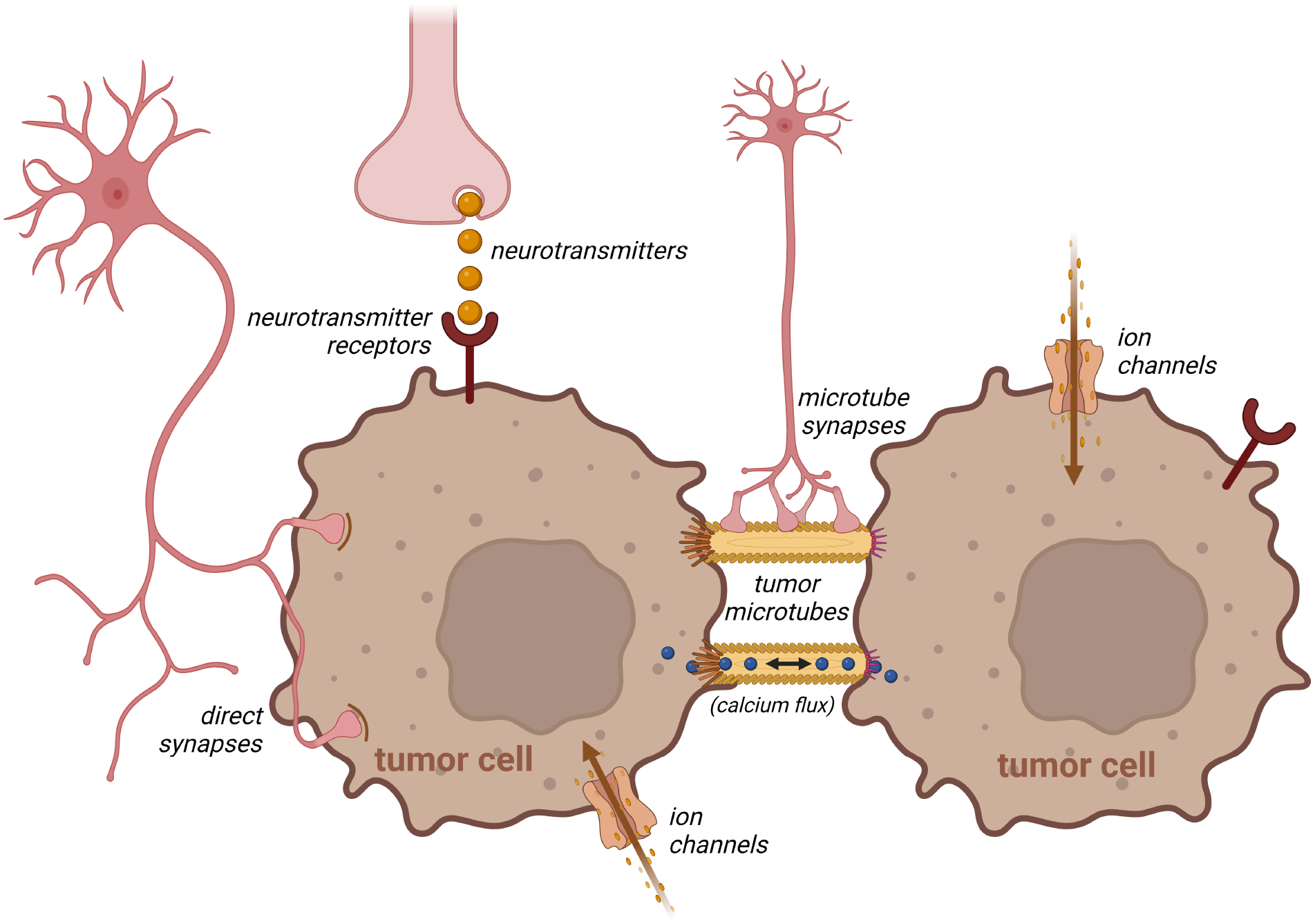
Neurons can form bona fide synapses or respond to local neurotransmitters to regulate tumor cell growth, which can be propagated between adjacent tumor cells through tumor microtubes, thus creating interconnected electrically coupled syncytia. Aberrant expression of ion channels on cancer cells can additionally modulate tumor expansion, and neurons can also directly synapse onto tumor microtubes.
Neuron-tumor cell paracrine relationships regulate tumor initiation
Neuronal activity can directly drive the development (initiation) of both low- and high-grade central nervous system tumors (gliomas). Leveraging murine models of the cancer predisposition syndrome88, Neurofibromatosis type 1 (NF1), Nf1-mutant mice that form low-grade gliomas of the optic nerve and chiasm were employed to define the role of neuronal activity in tumorigenesis. Since the axons of the optic nerve originate in the retina (retinal ganglion cells) and transmit light-induced photoreceptor signals to the brain, optogenetic stimulation of optic nerve activity increases optic glioma cell proliferation, while light deprivation (dark rearing) prevents tumor formation89. The molecular etiology for this activity-dependent regulation of gliomagenesis reflects the impact of Nf1 mutation on retinal ganglion cell (RGC) neuronal activity. Nf1 mutation in RGCs causes increased production of a proteolytic enzyme, ADAM10, as a consequence of increased neuronal activity. ADAM10 then cleaves a membrane bound protein, neuroligin-3 (Nlgn3), expressed on OPCs90 to generate a soluble bioactive protein capable of increasing tumor cell proliferation. Consistent with their roles in tumor initiation, both genetic Nlgn3 loss and pharmacologic ADAM10 inhibition abrogate Nf1-OPG formation.
Analogously, using an autochthonous murine model of adult malignant glioma originating from oligodendroglial progenitors, odorant stimulation and subsequent olfactory receptor neuronal activation results in the preferential development of tumors within the olfactory bulb, where the majority of olfactory signals are processed91. In this model, high-grade glioma formation results from activity-dependent olfactory receptor neuron insulin growth factor-1 production, which, in turn, induces the principal olfactory output neurons (mitral and tufted cells) to drive gliomagenesis.
Neuron-tumor cell paracrine relationships regulate tumor progression
In addition to their capacity to stimulate tumor development (initiation), neuron activity-dependent paracrine factors can also regulate tumor progression (continued growth after tumor induction). Using a xenograft model of high-grade glioma, optogenetic induction of neuronal activity increases tumor growth through the elaboration of NLGN3 from OPCs92, which results from the cleavage of NLGN3 by neuron-produced ADAM10. Similar to Nf1 low-grade optic gliomas, ADAM10 inhibitors reduce high grade-glioma growth in vivo90, serving as the preclinical foundation for a recent clinical trial (NCT04295759). In addition to NLGN3, neurotrophins (e.g., BDNF, NT3) have been shown to increase the growth of both low-grade and high-grade glioma cells through the engagement of their cognate receptors expressed on cancer cells78,89,92,93. Further supporting a role for growth factor signaling in glioma biology, pediatric low-grade gliomas (pilocytic astrocytomas) can arise from mutations in the BDNF receptor (NTRK2) or fibroblast growth factor receptor (FGFR1)71,94. In an analogous fashion, progression of oral mucosa carcinomas in nutrient-poor microenvironments depends upon tumor-associated nociceptive neuron secretion of nerve growth factor (NGF)-triggered calcitonin gene-related peptide (CGRP)95.
Neurons can also create supportive microenvironments for brain tumor progression through communication with immune system cells. In this regard, neurons produce many cytokines and chemokines that attract and control T cell and monocyte function96. As such, following rabies infection, neurons produce CXCL1097, while bacterial infection induces neuronal cytokine and chemokine production98 to recruit T lymphocytes. Additionally, neurons are the major source of CX3CL1 (fractalkine), a potent chemoattractant for resident brain microglia99. In the setting of pancreatic cancer, increased vagus nerve cholinergic signaling reprograms the immune microenvironment, resulting in decreased CD8+ T cell infiltration, altered T helper cell ratios, and increased tumor growth100. Conversely, severing the vagus nerve (vagotomy) reverses these effects on T cells and improves mouse survival. Additionally, in Nf1-optic glioma mice, where low-grade glioma progression is dependent on T cell and microglia interaction101, interrupting immune cell function during tumor evolution inhibits optic glioma progression102. In these tumors, Nf1-mutant RGCs (neurons) secrete midkine, which stimulates T cells to produce Ccl4103,104. Ccl4 then induces the elaboration of Ccl5 from microglia to increase tumor cell growth104.
Lastly, comparing Nf1-mutant mouse strains with different propensities to develop central (optic gliomas) and peripheral (neurofibromas) nervous system tumors, neurons from mice with tumor-causing Nf1 gene mutations are inherently hyperexcitable105. This basal hyperexcitability is mediated, in part, by the Hyperpolarization Activated Cyclic Nucleotide Gated Potassium Channel (HCN), such that agonism (lamotrigine) or antagonism (ZD7288) of HCN channel function modulates neuronal mitogen elaboration in both central and peripheral nervous system neurons. In the setting of Nf1 optic gliomas, HCN channel function inhibition reduces midkine production in RGCs relevant to optic glioma growth. Analogously, in the peripheral nervous system, sensory neurons which are in association with peripheral nerve sheath tumors (neurofibromas) make collagen (Col1a2) in an activity-dependent manner105. In contrast, mice with a germline Nf1 mutation found in NF1 patients who do not develop either gliomas or neurofibromas lack neuronal hyperexcitability and do not form brain or peripheral nerve tumors, owing to a failure to induce increased neuronal midkine and Col1a2 expression, respectfully. Important for future potential therapeutics, treatment of Nf1-mutant mice harboring optic gliomas or neurofibromas with lamotrigine to restore HCN channel function and dampen neuronal hyperactivity attenuates tumor progression in vivo.
With respect to Nf1 optic glioma formation and progression, the finding that Nf1 optic glioma initiation is controlled by visual experience (light-induced retinal ganglion cell activity), while basal neuronal hyperexcitability regulates brain tumor progression through midkine-mediated immune microenvironment support, suggests that neurons have the capacity to control different phases of tumorigenesis in an activity-dependent manner. The fact that visual experience controls the ADAM10/NLGN3 axis, but not midkine production, and HCN1 modulation only affects midkine expression, raises the intriguing idea that neuronal excitability can be fine-tuned to alter the tumor microenvironment throughout the life cycle of cancer, as well as potentially in response to treatment.
Non-synaptic potassium-evoked currents amplified in a gap-junction-coupled network
Non-synaptic potassium currents originating from firing neurons in the vicinity of cancer cells can develop as a consequence of neuronal potassium leaking into the extracellular space. Inward rectifying channels expressed on cancer cells then uptake this leaked potassium, causing calcium influx into the glioma cells, which can be propagated through a network of glioma cells via gap junctions (Figure 3). These specialized gap junctions, named tumor microtubes, also known as tunneling nanotubes or cytonemes, facilitate long-range communication between cells, and additionally allow for the transfer of mitochondria, proteins, and infectious particles106–109. Tumor microtubes are similar to membrane tubes formed by healthy tissues110, resembling long cellular protrusions106. Tumor microtubes comprise thicker tubes that arborize into thinner ones, mitochondria (indicative of local ATP production and vesicle trafficking), and actin filaments - all features reminiscent of axonal and dendritic outgrowths28,106. Moreover, glioma microtubes contain connexin-43, a gap junction protein involved in regulating the synchronicity of calcium current propagation and the propagation of spontaneous excitatory postsynaptic currents106.
In gliomas, microtubes also act as post-synaptic contacts for neurons, enabling rapid coupling between nerves and neuron-stimulated glioma cells. These junctions transmit calcium waves to other glioma cells to form a functional network106. This syncytial electrical coupling not only increases the growth of glioma cells, but also regulates their motility and invasiveness111. As such, tumors connected by a network of tumor microtubes are largely protected from the cytotoxic effects of radiation106 and chemotherapy112,113, while unconnected tumor cells are susceptible to treatment114: Genetic silencing of the connexin-43 gap junction protein found in tumor microtubes decreases the radioprotective effect of the tumor microtube network. Clinically, the presence of microtubes is related to tumor aggressiveness, as glioblastomas and astrocytomas form expansive tumor microtube networks, while oligodendrogliomas, which are less invasive, do not form such junctions106.
Bona fide neuron-glioma synapses
Electron microscopy revealed the presence of bona fide neuronal synapses on human high-grade glioma cells xenografted in mice115,116 (Figure 3). These synapses are also seen in experimental mouse models of low-grade optic pathway glioma89, as well as in murine glioblastoma, fresh operative human glioma specimens in situ, human glioma cells co-cultured with neurons in vitro116 and patient-derived xenograft models111. Similar to neuron-neuron synapses, neuron-glioma synapses exhibit the hallmark features of glutamatergic synapses and contain presynaptic vesicles, a synaptic cleft, a presynaptic active zone with docked vesicles, and a postsynaptic density area116. Further characterization of these neuron-glioma synapses revealed three main morphological types: (a) single synaptic contacts onto glioma microtubes, (b) multi-synaptic contacts with both glioma microtubes and other neurons, and (c) “pseudo-tripartite” perisynaptic connections.
The bona fide glutamatergic synapses are formed between neurons and glioma cells through ionotropic glutamate (AMPA) receptors to induce glioma cell membrane depolarization and calcium influx. These synapses primarily form on tumor microtubes, while some exist on glioma cell bodies (somas)116. As only a small proportion of the tumor cells are connected to neurons, these synapses generate calcium currents and stimulate the entire glioma network through the induction of new tumor microtubes between cancer cells111 or existing tumor microtube networks106. This enhanced neuronal activity and glutamatergic signaling facilitates tumor invasion111,117,118 and proliferation119. Importantly, regardless of the underlying mechanism, membrane depolarization itself can drive glioma cell growth115. Conversely, genetic silencing of the AMPA GluR1 subunit on tumor cells inhibits glioma proliferation120, while AMPA receptor blockade suppresses cancer cell migration and induces apoptosis121 through the Akt signaling pathway122.
In addition to glutamate, other neurotransmitters, including acetylcholine123 and dopamine75, have been implicated in brain cancer progression, reminiscent of their pro-tumorigenic role in peripheral solid tumors124,125. Using a high-content neurochemical compound screen, antagonists to dopamine receptor D4 (DRD4) signaling, as well as to serotonergic and cholinergic neurotransmission, were found to selectively inhibit malignant glioma cell growth and increase the differentiation of non-neoplastic neural stem cells. As such, blockade of DRD4 on cancer cells results in an accumulation of autophagic vacuoles, cell cycle arrest, and apoptosis. Inhibition of tumor progression by targeting dopamine G protein coupled receptors (GPCRs) nicely parallels the observation that pharmacologic interruption of GPCR-cyclic AMP signaling attenuates malignant brain tumor growth74.
In contrast to the bona fide synapses in primary gliomas, metastatic tumors to the brain, such as breast-to-brain metastases, associate with neurons in a perisynaptic manner without establishing true synaptic connections126. In these tumors, excitatory (glutamatergic) neurons transmit glutamate through “pseudo-tripartite” synapses, akin to those formed by two neurons and astrocytes in the normal brain127. These pseudo-tripartite synapses increase glutamatergic signaling through NMDA receptors on the tumor cells, and promote metastatic cancer colonization and spread. However, the biological function of pseudo-tripartite synaptic structures in the setting of primary gliomas is not clear. Similarly, whether actual synapses are formed between peripheral neurons and peripheral nervous system tumors remains to be fully elucidated.
Cancer cell ion channels
Often overexpressed in cancer cells, ion channels convert neuron-derived extracellular cues into intracellular molecular cascades128,129 that coordinate cell excitability and cell proliferation130 and migration131. Specifically, these ion channels include anion (chloride-conducting) and cation (potassium-, sodium-, and calcium-conducting) channels132–134, as well as non-selective transient receptor potential (TRP) channels135,136. The presence of such cation (calcium) microtubes permits cancer cell network connectivity and autonomous rhythmic activity within a subset of glioblastoma cells, which collectively acts to increase overall tumor growth137. While some of the etiologic mechanisms remain incompletely characterized, the CLIC1 chloride channel, overexpressed by many cancers, regulates brain tumor cell cycle progression138. Similarly, increased potassium channel expression in brain tumors139,140 supports cancer stem cell viability141, while the PIEZO mechanosensitive cation ion channel, overexpressed in glioma, is associated with poor patient prognosis142. In addition, overexpression of the TRPV1 channel in gliomas regulates tumor cell survival through endoplasmic reticulum stress pathway activation143, while overexpressed TRPM7 channel controls glioma cell migration, invasion, and proliferation144. These findings prompted the report of an 18-ion channel gene signature, which was found to be predictive of overall survival in patients with glioma145.
Complications of cancer
Tumors of the nervous system not only depend upon neurons for regulation of cancer formation and progression, but their intimate relationship with neurons also influences normal nerve cell function both at baseline and in the setting of tumor treatment. These effects include tumor-induced neuronal hyperexcitability and chemotherapy-induced neuronal dysfunction (Figure 4).
Figure 4. Tumors interact with non-neoplastic cells in the tumor microenvironment to influence their local milieu and create neuronal dysfunction.
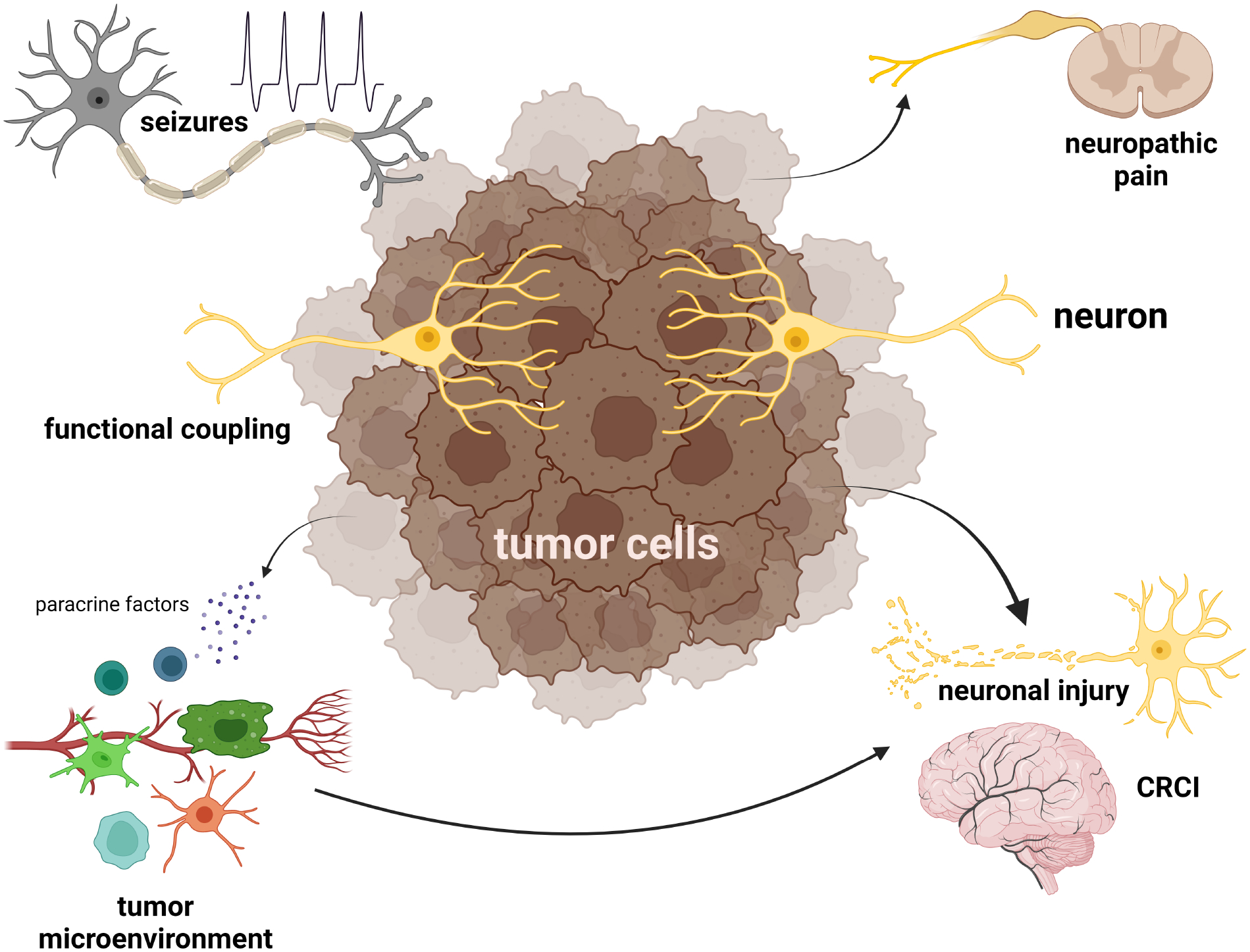
Gliomas are functionally coupled with neurons in the brain to impair normal brain function, induce seizures, or cause pain. Additionally, nervous system tumors secrete paracrine factors that modify the tumor microenvironment to increase tumor growth or promote resistance to anti-neoplastic therapies, but also interrupt the normal relationships between glial cells and neurons relative to chemotherapy-related cognitive impairment (CRCI) and neuronal injury.
Since brain tumors arise within an existing functional network critical for normal brain function, it is not surprising that they can become integrated into these same circuits. As such, gliomas can locally disrupt the synchronization of neural communication, which is important for processing motor and sensory information. Additionally, the infiltration of tumor cells with the ability to engage in electric or chemical synapse transmission could create new integrated circuits or degrade the amount and quality of information transmitted in these functional networks. Using intraoperative electrocorticography and magnetoencephalography in subjects with malignant glioma, the tumor-infiltrated cortex was found to engage in coordinated neural responses, which impaired normal language processing146,147. Similarly, motor and language function is inhibited by electrical stimulation of glioma-infiltrated cortex148, and resection of tumor-infiltrated brain regions with high degrees of functional connectivity causes permanent neurological damage147.
Although electrochemical neuron-glioma communication is traditionally thought to occur between presynaptic neurons and postsynaptic glioma cells, this interaction is in fact bidirectional. The tight integration of glioma cells into functional neuronal networks also affects normal neuronal activity through the induction of hyperexcitable states (seizures). In this regard, seizures occur in 40–80% of patients with glioma149–151. Increased neuronal excitability and seizures could result from the release of glutamate from glioma cells, or a reduction in GABAergic inhibition152–154. In this regard, inhibition of glutamate release from tumor cells reduces the frequency of seizures in glioma-bearing mice153. Additionally, glioma cells can transfer genetic material to neighboring neurons via extracellular vesicles, which can also increase neuronal activity and ultimately stimulate tumor growth155. To gain insights into other possible mechanisms for the reciprocal crosstalk between neurons and glioma cells, an in vivo high-throughput screening study revealed that some PIK3CA gene mutations selectively initiate neuronal excitability through differential glioma cell secretion of glypican-3 (GPC3)156. These GPC3-driven tumors have greater excitatory and inhibitory synapse formation, resulting in seizure induction and further enhancement of glioma growth. Conversely, genetic silencing of GPC3 in glioma cells eliminates the early onset neuronal hyperexcitability and extends mouse survival.
The intimate relationship established between neurons and oligodendrocytes is important for adaptive myelination in the healthy brain29,30 and can be disrupted following anti-cancer treatment. One of the unintended consequences of chemotherapy is the development of chemotherapy-related cognitive impairment (CRCI; “chemobrain”)157, which results from impaired neuron activity-dependent myelination66. In a mouse model of methotrexate (MTX) chemotherapy158, MTX reduced neuronal BDNF expression and impaired TrkB signaling in OPCs to disrupt activity-dependent myelination. This neurotoxicity involved microglia, which have also been implicated in other chemotherapy-induced cognitive impairments66,159,160.
Lastly, chemotherapy-induced peripheral neuronal damage (neuropathy) is similarly influenced by microglia161,162, T cells161,163,164 and inflammatory cytokine release165,166. In the setting of cancer, perineural invasion and neuropathic pain involves dysregulated neurotrophin (NGF) signaling167,168 and/or dysfunctional neuroimmune interactions169,170.
Implications and Future Directions
Since neurons and tumors establish bi-directional dependencies that reflect the normal connections between neurons and their local cellular milieu, it is likely that tumors create their own microenvironment by usurping existing developmental and homeostatic relationships12,33,171,172. Defining the molecular bases for each of these interactions will be important as we consider future therapies that interrupt neuron-cancer communication. These interventions could include repurposing drugs that inhibit neuronal hyperexcitability105,173, targeting tumor-specific synapses/receptors174, or employing ADAM10 inhibitors to interrupt paracrine circuits89,90.
In addition, the wealth of evidence arguing that neurons are key drivers of tumor formation and growth supports a reconceptualization of the hallmarks of cancer175. Incorporating studies from numerous laboratories in the cancer neuroscience field176, we now include cell (neuron and tumor cell) excitability, as well as the relationships between cell excitability and other key tumor features (e.g., tumor invasion, immune cell function) as major hallmarks, using brain tumors (gliomas) as an illustrative example (Figure 5).
Figure 5. Integrated hallmarks of cancer.
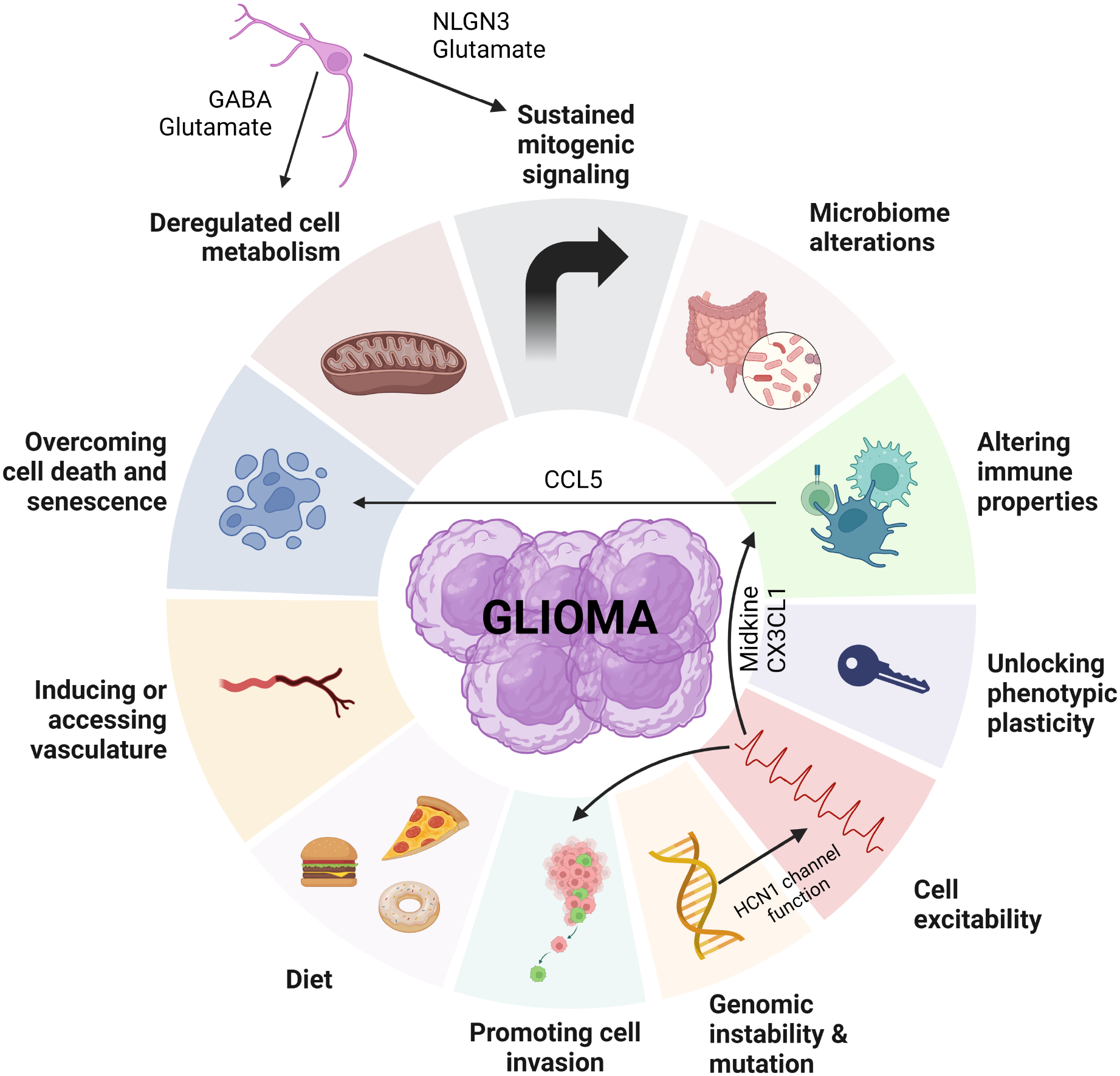
Modification of the hallmarks of cancer, now incorporating the relationships between cell (neuron and tumor cell) excitability and other properties, such as genetic mutation, cell invasion, cell metabolism, immune properties, mitogenic signaling, and cell death/senescence, as relevant to brain tumor (glioma) pathobiology.
Moreover, the idea that neurons are central regulators of tumorigenesis89,105 raises the provocative concept that neurons might create set points for tumor risk (Figure 6). As such, it is possible that specific cancer-associated genetic alterations (perhaps even single nucleotide variations) alter neuronal hyperexcitability through the modulation of ion channel or neurotransmitter function at the genomic or transcriptional level. For instance, postnatal loss of one of the genes implicated in the tuberous sclerosis cancer predisposition syndrome (Tsc1) increases the excitability of striatonigral neurons177 due to a reduction in inhibitory transmission178, as well as reduces the intrinsic excitability of dopaminergic neurons179. Likewise, p53 mutation, as seen in patients with Li-Fraumeni syndrome, reduces the firing frequency and the number of excitatory synapses formed in layer 5 pyramidal neurons of the mouse primary somatosensory cortex180. As described above, different NF1 patient germline NF1 mutations have varying effects on neuronal hyperexcitability, which in turn, differentially dictate tumor formation and progression in mice105. In addition to the specific gene mutation, environmental factors, tissue injury, systemic diseases, and perinatal infections may also modify cancer risk by interrupting interactions between neurons and other cell types. In this regard, we have found that asthma, which is associated with reduced risk of glioma in children with NF1, modifies neuroimmune interactions102 critical for establishing a microenvironment supportive of brain tumor growth103. Further work on these and related risk factors may identify new intersections between neurons and tumor cells relevant to future precision medicine strategies.
Figure 6. Risk factors operate at the level of the neuron to modulate cancer risk.
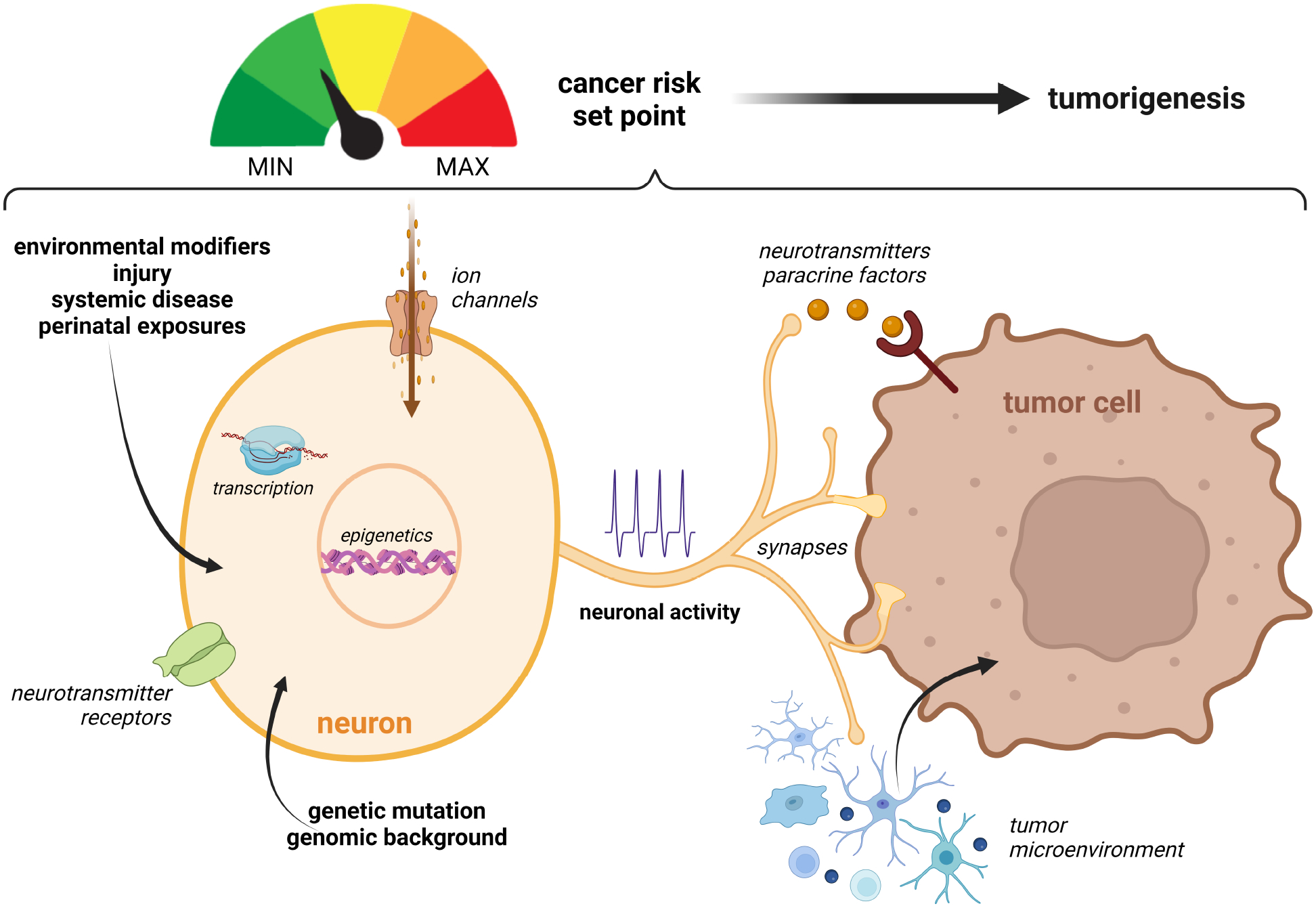
Operating at the transcriptional or epigenetic level, genetic mutations and genomic background can alter neuronal excitability in numerous ways, including ion channel and neurotransmitter receptor function, the formation of neuron-tumor synapses, and the elaboration of paracrine factors that act either directly on the cancer cells or indirectly through non-neoplastic cells in the tumor microenvironment. Environmental factors, nervous system injury, systemic diseases, and perinatal exposures (infection) can additionally operate to disrupt interactions between neurons and cancer cells, such that the combination of these factors establish a ground state of neuronal excitability that makes cancer development more or less likely to occur.
Acknowledgements
Funding.
This work was funded by grants from the Giorgio Foundation, Neurofibromatosis Acceleration Therapeutics Program, and National Institutes of Health (R35NS097211 and R01CA258384 to DHG; 1-R50-CA233164-01 to CA).
The figures were generated in BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
Lead Contact Statement
Enquiries for further information should be directed to and will be fulfilled by the lead contact, David H. Gutmann, gutmannd@wustl.edu.
References
- 1.Taeb S, Ashrafizadeh M, Zarrabi A, Rezapoor S, Musa AE, Farhood B, and Najafi M (2022). Role of Tumor Microenvironment in Cancer Stem Cells Resistance to Radiotherapy. Curr Cancer Drug Targets 22, 18–30. 10.2174/1568009622666211224154952. [DOI] [PubMed] [Google Scholar]
- 2.Barker HE, Paget JT, Khan AA, and Harrington KJ (2015). The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer 15, 409–425. 10.1038/nrc3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinshaw DC, and Shevde LA (2019). The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res 79, 4557–4566. 10.1158/0008-5472.CAN-18-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y, McAndrews KM, and Kalluri R (2021). Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol 18, 792–804. 10.1038/s41571-021-00546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. (2020). A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 20, 174–186. 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayik D, and Lathia JD (2021). Cancer stem cell-immune cell crosstalk in tumour progression. Nat Rev Cancer 21, 526–536. 10.1038/s41568-021-00366-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garner H, and de Visser KE (2020). Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat Rev Immunol 20, 483–497. 10.1038/s41577-019-0271-z. [DOI] [PubMed] [Google Scholar]
- 8.van der Leun AM, Thommen DS, and Schumacher TN (2020). CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer 20, 218–232. 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fridman WH, Pages F, Sautes-Fridman C, and Galon J (2012). The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12, 298–306. 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 10.Huinen ZR, Huijbers EJM, van Beijnum JR, Nowak-Sliwinska P, and Griffioen AW (2021). Anti-angiogenic agents - overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol 18, 527–540. 10.1038/s41571-021-00496-y. [DOI] [PubMed] [Google Scholar]
- 11.Hjelmeland AB, Lathia JD, Sathornsumetee S, and Rich JN (2011). Twisted tango: brain tumor neurovascular interactions. Nat Neurosci 14, 1375–1381. 10.1038/nn.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monje M, Borniger JC, D’Silva NJ, Deneen B, Dirks PB, Fattahi F, Frenette PS, Garzia L, Gutmann DH, Hanahan D, et al. (2020). Roadmap for the Emerging Field of Cancer Neuroscience. Cell 181, 219–222. 10.1016/j.cell.2020.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillespie S, and Monje M (2018). An active role for neurons in glioma progression: making sense of Scherer’s structures. Neuro Oncol 20, 1292–1299. 10.1093/neuonc/noy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scherer HJ (1938). Structural development in gliomas. Am J Cancer Res, 333–351. [Google Scholar]
- 15.Brownson RH (1956). Perineuronal satellite cells in the motor cortex of aging brains. J Neuropathol Exp Neurol 15, 190–195. 10.1097/00005072-195604000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Chen SH, Zhang BY, Zhou B, Zhu CZ, Sun LQ, and Feng YJ (2019). Perineural invasion of cancer: a complex crosstalk between cells and molecules in the perineural niche. Am J Cancer Res 9, 1–21. [PMC free article] [PubMed] [Google Scholar]
- 17.Yang YH, Liu JB, Gui Y, Lei LL, and Zhang SJ (2017). Relationship between autophagy and perineural invasion, clinicopathological features, and prognosis in pancreatic cancer. World J Gastroenterol 23, 7232–7241. 10.3748/wjg.v23.i40.7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aurello P, Berardi G, Tierno SM, Rampioni Vinciguerra GL, Socciarelli F, Laracca GG, Giulitti D, Pilozzi E, and Ramacciato G (2017). Influence of perineural invasion in predicting overall survival and disease-free survival in patients With locally advanced gastric cancer. Am J Surg 213, 748–753. 10.1016/j.amjsurg.2016.05.022. [DOI] [PubMed] [Google Scholar]
- 19.Quintana JM, Gonzalez N, Lazaro S, Bare M, Fernandez-de-Larrea N, Redondo M, Briones E, Escobar A, Sarasqueta C, Garcia-Gutierrez S, et al. (2018). Predictors of 1- and 2-year mortality in patients with rectal cancer. Colorectal Dis 20, 676–687. 10.1111/codi.14250. [DOI] [PubMed] [Google Scholar]
- 20.Tollefson MK, Karnes RJ, Kwon ED, Lohse CM, Rangel LJ, Mynderse LA, Cheville JC, and Sebo TJ (2014). Prostate cancer Ki-67 (MIB-1) expression, perineural invasion, and gleason score as biopsy-based predictors of prostate cancer mortality: the Mayo model. Mayo Clin Proc 89, 308–318. 10.1016/j.mayocp.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Albergotti WG, Schwarzbach HL, Abberbock S, Ferris RL, Johnson JT, Duvvuri U, and Kim S (2017). Defining the Prevalence and Prognostic Value of Perineural Invasion and Angiolymphatic Invasion in Human Papillomavirus-Positive Oropharyngeal Carcinoma. JAMA Otolaryngol Head Neck Surg 143, 1236–1243. 10.1001/jamaoto.2017.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murakami Y, Uemura K, Sudo T, Hashimoto Y, Kondo N, Nakagawa N, Muto T, Sasaki H, Urabe K, and Sueda T (2013). Perineural invasion in extrahepatic cholangiocarcinoma: prognostic impact and treatment strategies. J Gastrointest Surg 17, 1429–1439. 10.1007/s11605-013-2251-0. [DOI] [PubMed] [Google Scholar]
- 23.Zhu Y, Zhang G, Yang Y, Cui L, Jia S, Shi Y, Song S, and Xu S (2018). Perineural invasion in early-stage cervical cancer and its relevance following surgery. Oncol Lett 15, 6555–6561. 10.3892/ol.2018.8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Batsakis JG (1985). Nerves and neurotropic carcinomas. Ann Otol Rhinol Laryngol 94, 426–427. [PubMed] [Google Scholar]
- 25.Liebig C, Ayala G, Wilks JA, Berger DH, and Albo D (2009). Perineural invasion in cancer: a review of the literature. Cancer 115, 3379–3391. 10.1002/cncr.24396. [DOI] [PubMed] [Google Scholar]
- 26.Kostovic I, Sedmak G, and Judas M (2019). Neural histology and neurogenesis of the human fetal and infant brain. Neuroimage 188, 743–773. 10.1016/j.neuroimage.2018.12.043. [DOI] [PubMed] [Google Scholar]
- 27.Deisseroth K, Singla S, Toda H, Monje M, Palmer TD, and Malenka RC (2004). Excitation-neurogenesis coupling in adult neural stem/progenitor cells. Neuron 42, 535–552. 10.1016/s0896-6273(04)00266-1. [DOI] [PubMed] [Google Scholar]
- 28.Jung E, Osswald M, Blaes J, Wiestler B, Sahm F, Schmenger T, Solecki G, Deumelandt K, Kurz FT, Xie R, et al. (2017). Tweety-Homolog 1 Drives Brain Colonization of Gliomas. J Neurosci 37, 6837–6850. 10.1523/JNEUROSCI.3532-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monje M, and Karadottir RT (2021). The bright and the dark side of myelin plasticity: Neuron-glial interactions in health and disease. Semin Cell Dev Biol 116, 10–15. 10.1016/j.semcdb.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, et al. (2014). Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344, 1252304. 10.1126/science.1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hines JH, Ravanelli AM, Schwindt R, Scott EK, and Appel B (2015). Neuronal activity biases axon selection for myelination in vivo. Nat Neurosci 18, 683–689. 10.1038/nn.3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mensch S, Baraban M, Almeida R, Czopka T, Ausborn J, El Manira A, and Lyons DA (2015). Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat Neurosci 18, 628–630. 10.1038/nn.3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Venkatesh H, and Monje M (2017). Neuronal Activity in Ontogeny and Oncology. Trends Cancer 3, 89–112. 10.1016/j.trecan.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jung E, Alfonso J, Osswald M, Monyer H, Wick W, and Winkler F (2019). Emerging intersections between neuroscience and glioma biology. Nat Neurosci 22, 1951–1960. 10.1038/s41593-019-0540-y. [DOI] [PubMed] [Google Scholar]
- 35.Xie Y, Kuan AT, Wang W, Herbert ZT, Mosto O, Olukoya O, Adam M, Vu S, Kim M, Tran D, et al. (2022). Astrocyte-neuron crosstalk through Hedgehog signaling mediates cortical synapse development. Cell Rep 38, 110416. 10.1016/j.celrep.2022.110416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stogsdill JA, and Eroglu C (2017). The interplay between neurons and glia in synapse development and plasticity. Curr Opin Neurobiol 42, 1–8. 10.1016/j.conb.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jakovcevski I, Filipovic R, Mo Z, Rakic S, and Zecevic N (2009). Oligodendrocyte development and the onset of myelination in the human fetal brain. Front Neuroanat 3, 5. 10.3389/neuro.05.005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen NJ, and Eroglu C (2017). Cell Biology of Astrocyte-Synapse Interactions. Neuron 96, 697–708. 10.1016/j.neuron.2017.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chung WS, Allen NJ, and Eroglu C (2015). Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb Perspect Biol 7, a020370. 10.1101/cshperspect.a020370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LoTurco JJ, Owens DF, Heath MJ, Davis MB, and Kriegstein AR (1995). GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15, 1287–1298. 10.1016/0896-6273(95)90008-x. [DOI] [PubMed] [Google Scholar]
- 41.Luk KC, and Sadikot AF (2004). Glutamate and regulation of proliferation in the developing mammalian telencephalon. Dev Neurosci 26, 218–228. 10.1159/000082139. [DOI] [PubMed] [Google Scholar]
- 42.Maurel P, and Salzer JL (2000). Axonal regulation of Schwann cell proliferation and survival and the initial events of myelination requires PI 3-kinase activity. J Neurosci 20, 4635–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morrissey TK, Levi AD, Nuijens A, Sliwkowski MX, and Bunge RP (1995). Axon-induced mitogenesis of human Schwann cells involves heregulin and p185erbB2. Proc Natl Acad Sci U S A 92, 1431–1435. 10.1073/pnas.92.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheridan GK, and Murphy KJ (2013). Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol 3, 130181. 10.1098/rsob.130181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marinelli S, Basilico B, Marrone MC, and Ragozzino D (2019). Microglia-neuron crosstalk: Signaling mechanism and control of synaptic transmission. Semin Cell Dev Biol 94, 138–151. 10.1016/j.semcdb.2019.05.017. [DOI] [PubMed] [Google Scholar]
- 46.Lenz KM, and Nelson LH (2018). Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front Immunol 9, 698. 10.3389/fimmu.2018.00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilton DK, Dissing-Olesen L, and Stevens B (2019). Neuron-Glia Signaling in Synapse Elimination. Annu Rev Neurosci 42, 107–127. 10.1146/annurev-neuro-070918-050306. [DOI] [PubMed] [Google Scholar]
- 48.Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, Walker AJ, Heller MD, Umemori H, Chen C, and Stevens B (2018). CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron 100, 120–134 e126. 10.1016/j.neuron.2018.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellwardt E, Walsh JT, Kipnis J, and Zipp F (2016). Understanding the Role of T Cells in CNS Homeostasis. Trends Immunol 37, 154–165. 10.1016/j.it.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 50.Tanabe S, and Yamashita T (2018). The role of immune cells in brain development and neurodevelopmental diseases. Int Immunol 30, 437–444. 10.1093/intimm/dxy041. [DOI] [PubMed] [Google Scholar]
- 51.Herz J, Fu Z, Kim K, Dykstra T, Wall M, Li H, Salvador AF, Zou B, Yan N, Blackburn SM, et al. (2021). GABAergic neuronal IL-4R mediates T cell effect on memory. Neuron 109, 3609–3618 e3609. 10.1016/j.neuron.2021.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alves de Lima K, Rustenhoven J, Da Mesquita S, Wall M, Salvador AF, Smirnov I, Martelossi Cebinelli G, Mamuladze T, Baker W, Papadopoulos Z, et al. (2020). Meningeal gammadelta T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat Immunol 21, 1421–1429. 10.1038/s41590-020-0776-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, Overall CC, Gadani SP, Turner SD, Weng Z, et al. (2016). Unexpected role of interferon-gamma in regulating neuronal connectivity and social behaviour. Nature 535, 425–429. 10.1038/nature18626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kipnis J, Gadani S, and Derecki NC (2012). Pro-cognitive properties of T cells. Nat Rev Immunol 12, 663–669. 10.1038/nri3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lewitus GM, Zhu J, Xiong H, Hallworth R, and Kipnis J (2007). CD4(+)CD25(−) effector T-cells inhibit hippocampal long-term potentiation in vitro. Eur J Neurosci 26, 1399–1406. 10.1111/j.1460-9568.2007.05788.x. [DOI] [PubMed] [Google Scholar]
- 56.Zhou L, Kong G, Palmisano I, Cencioni MT, Danzi M, De Virgiliis F, Chadwick JS, Crawford G, Yu Z, De Winter F, et al. (2022). Reversible CD8 T cell-neuron cross-talk causes aging-dependent neuronal regenerative decline. Science 376, eabd5926. 10.1126/science.abd5926. [DOI] [PubMed] [Google Scholar]
- 57.Szilvasy-Szabo A, Varga E, Beliczai Z, Lechan RM, and Fekete C (2017). Localization of connexin 43 gap junctions and hemichannels in tanycytes of adult mice. Brain Res 1673, 64–71. 10.1016/j.brainres.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 58.Weissman TA, Riquelme PA, Ivic L, Flint AC, and Kriegstein AR (2004). Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron 43, 647–661. 10.1016/j.neuron.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 59.Yu YC, He S, Chen S, Fu Y, Brown KN, Yao XH, Ma J, Gao KP, Sosinsky GE, Huang K, and Shi SH (2012). Preferential electrical coupling regulates neocortical lineage-dependent microcircuit assembly. Nature 486, 113–117. 10.1038/nature10958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hosli L, Binini N, Ferrari KD, Thieren L, Looser ZJ, Zuend M, Zanker HS, Berry S, Holub M, Mobius W, et al. (2022). Decoupling astrocytes in adult mice impairs synaptic plasticity and spatial learning. Cell Rep 38, 110484. 10.1016/j.celrep.2022.110484. [DOI] [PubMed] [Google Scholar]
- 61.Mayorquin LC, Rodriguez AV, Sutachan JJ, and Albarracin SL (2018). Connexin-Mediated Functional and Metabolic Coupling Between Astrocytes and Neurons. Front Mol Neurosci 11, 118. 10.3389/fnmol.2018.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bergles DE, Roberts JD, Somogyi P, and Jahr CE (2000). Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405, 187–191. 10.1038/35012083. [DOI] [PubMed] [Google Scholar]
- 63.Sudhof TC (2021). The cell biology of synapse formation. J Cell Biol 220. 10.1083/jcb.202103052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McKenzie IA, Ohayon D, Li H, de Faria JP, Emery B, Tohyama K, and Richardson WD (2014). Motor skill learning requires active central myelination. Science 346, 318–322. 10.1126/science.1254960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiao L, Ohayon D, McKenzie IA, Sinclair-Wilson A, Wright JL, Fudge AD, Emery B, Li H, and Richardson WD (2016). Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat Neurosci 19, 1210–1217. 10.1038/nn.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Geraghty AC, Gibson EM, Ghanem RA, Greene JJ, Ocampo A, Goldstein AK, Ni L, Yang T, Marton RM, Pasca SP, et al. (2019). Loss of Adaptive Myelination Contributes to Methotrexate Chemotherapy-Related Cognitive Impairment. Neuron 103, 250–265 e258. 10.1016/j.neuron.2019.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swire M, Kotelevtsev Y, Webb DJ, Lyons DA, and Ffrench-Constant C (2019). Endothelin signalling mediates experience-dependent myelination in the CNS. Elife 8. 10.7554/eLife.49493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ohtaka-Maruyama C, Okamoto M, Endo K, Oshima M, Kaneko N, Yura K, Okado H, Miyata T, and Maeda N (2018). Synaptic transmission from subplate neurons controls radial migration of neocortical neurons. Science 360, 313–317. 10.1126/science.aar2866. [DOI] [PubMed] [Google Scholar]
- 69.Wadhwa S, Nag TC, Jindal A, Kushwaha R, Mahapatra AK, and Sarkar C (2003). Expression of the neurotrophin receptors Trk A and Trk B in adult human astrocytoma and glioblastoma. J Biosci 28, 181–188. 10.1007/BF02706217. [DOI] [PubMed] [Google Scholar]
- 70.Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, et al. (2014). The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46, 444–450. 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DA, et al. (2013). Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45, 927–932. 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al. (2013). The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet 45, 1141–1149. 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walker AJ, Card T, Bates TE, and Muir K (2011). Tricyclic antidepressants and the incidence of certain cancers: a study using the GPRD. Br J Cancer 104, 193–197. 10.1038/sj.bjc.6605996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shchors K, Massaras A, and Hanahan D (2015). Dual Targeting of the Autophagic Regulatory Circuitry in Gliomas with Repurposed Drugs Elicits Cell-Lethal Autophagy and Therapeutic Benefit. Cancer Cell 28, 456–471. 10.1016/j.ccell.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 75.Dolma S, Selvadurai HJ, Lan X, Lee L, Kushida M, Voisin V, Whetstone H, So M, Aviv T, Park N, et al. (2016). Inhibition of Dopamine Receptor D4 Impedes Autophagic Flux, Proliferation, and Survival of Glioblastoma Stem Cells. Cancer Cell 29, 859–873. 10.1016/j.ccell.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li J, Zhu S, Kozono D, Ng K, Futalan D, Shen Y, Akers JC, Steed T, Kushwaha D, Schlabach M, et al. (2014). Genome-wide shRNA screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotarget 5, 882–893. 10.18632/oncotarget.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mohapel P, Leanza G, Kokaia M, and Lindvall O (2005). Forebrain acetylcholine regulates adult hippocampal neurogenesis and learning. Neurobiol Aging 26, 939–946. 10.1016/j.neurobiolaging.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 78.Hong EJ, McCord AE, and Greenberg ME (2008). A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron 60, 610–624. 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lindholm D, Castren E, Berzaghi M, Blochl A, and Thoenen H (1994). Activity-dependent and hormonal regulation of neurotrophin mRNA levels in the brain--implications for neuronal plasticity. J Neurobiol 25, 1362–1372. 10.1002/neu.480251105. [DOI] [PubMed] [Google Scholar]
- 80.McTigue DM, Horner PJ, Stokes BT, and Gage FH (1998). Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J Neurosci 18, 5354–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vondran MW, Clinton-Luke P, Honeywell JZ, and Dreyfus CF (2010). BDNF+/− mice exhibit deficits in oligodendrocyte lineage cells of the basal forebrain. Glia 58, 848–856. 10.1002/glia.20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nicholson M, Wood RJ, Fletcher JL, van den Buuse M, Murray SS, and Xiao J (2018). BDNF haploinsufficiency exerts a transient and regionally different influence upon oligodendroglial lineage cells during postnatal development. Mol Cell Neurosci 90, 12–21. 10.1016/j.mcn.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 83.Madrigal JL, Caso JR, Garcia-Bueno B, Gutierrez IL, and Leza JC (2017). Noradrenaline induces CX3CL1 production and release by neurons. Neuropharmacology 114, 146–155. 10.1016/j.neuropharm.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 84.Nimmerjahn A, Kirchhoff F, and Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 85.Farber K, Pannasch U, and Kettenmann H (2005). Dopamine and noradrenaline control distinct functions in rodent microglial cells. Mol Cell Neurosci 29, 128–138. 10.1016/j.mcn.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 86.Logiacco F, Xia P, Georgiev SV, Franconi C, Chang YJ, Ugursu B, Sporbert A, Kuhn R, Kettenmann H, and Semtner M (2021). Microglia sense neuronal activity via GABA in the early postnatal hippocampus. Cell Rep 37, 110128. 10.1016/j.celrep.2021.110128. [DOI] [PubMed] [Google Scholar]
- 87.Kim IJ, Beck HN, Lein PJ, and Higgins D (2002). Interferon gamma induces retrograde dendritic retraction and inhibits synapse formation. J Neurosci 22, 4530–4539. 20026431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guillamo JS, Creange A, Kalifa C, Grill J, Rodriguez D, Doz F, Barbarot S, Zerah M, Sanson M, Bastuji-Garin S, et al. (2003). Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain 126, 152–160. 10.1093/brain/awg016. [DOI] [PubMed] [Google Scholar]
- 89.Pan Y, Hysinger JD, Barron T, Schindler NF, Cobb O, Guo X, Yalcin B, Anastasaki C, Mulinyawe SB, Ponnuswami A, et al. (2021). NF1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature 594, 277–282. 10.1038/s41586-021-03580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM, Ni J, Duveau DY, Morris PJ, Zhao JJ, et al. (2017). Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 549, 533–537. 10.1038/nature24014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen P, Wang W, Liu R, Lyu J, Zhang L, Li B, Qiu B, Tian A, Jiang W, Ying H, et al. (2022). Olfactory sensory experience regulates gliomagenesis via neuronal IGF1. Nature. 10.1038/s41586-022-04719-9. [DOI] [PubMed] [Google Scholar]
- 92.Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS, et al. (2015). Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 161, 803–816. 10.1016/j.cell.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lawn S, Krishna N, Pisklakova A, Qu X, Fenstermacher DA, Fournier M, Vrionis FD, Tran N, Chan JA, Kenchappa RS, and Forsyth PA (2015). Neurotrophin signaling via TrkB and TrkC receptors promotes the growth of brain tumor-initiating cells. J Biol Chem 290, 3814–3824. 10.1074/jbc.M114.599373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fisher MJ, Jones DTW, Li Y, Guo X, Sonawane PS, Waanders AJ, Phillips JJ, Weiss WA, Resnick AC, Gosline S, et al. (2021). Integrated molecular and clinical analysis of low-grade gliomas in children with neurofibromatosis type 1 (NF1). Acta Neuropathol 141, 605–617. 10.1007/s00401-021-02276-5. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Y, Lin C, Liu Z, Sun Y, Chen M, Guo Y, Liu W, Zhang C, Chen W, Sun J, et al. (2022). Cancer cells co-opt nociceptive nerves to thrive in nutrient-poor environments and upon nutrient-starvation therapies. Cell Metab 34, 1999–2017 e1910. 10.1016/j.cmet.2022.10.012. [DOI] [PubMed] [Google Scholar]
- 96.de Haas AH, van Weering HR, de Jong EK, Boddeke HW, and Biber KP (2007). Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol Neurobiol 36, 137–151. 10.1007/s12035-007-0036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chai Q, She R, Huang Y, and Fu ZF (2015). Expression of neuronal CXCL10 induced by rabies virus infection initiates infiltration of inflammatory cells, production of chemokines and cytokines, and enhancement of blood-brain barrier permeability. J Virol 89, 870–876. 10.1128/JVI.02154-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wochal P, Rathinam VA, Dunne A, Carlson T, Kuang W, Seidl KJ, Hall JP, Lin LL, Collins M, Schattgen SA, et al. (2014). TRIL is involved in cytokine production in the brain following Escherichia coli infection. J Immunol 193, 1911–1919. 10.4049/jimmunol.1302392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arnoux I, and Audinat E (2015). Fractalkine Signaling and Microglia Functions in the Developing Brain. Neural Plast 2015, 689404. 10.1155/2015/689404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang MW, Tao LY, Jiang YS, Yang JY, Huo YM, Liu DJ, Li J, Fu XL, He R, Lin C, et al. (2020). Perineural Invasion Reprograms the Immune Microenvironment through Cholinergic Signaling in Pancreatic Ductal Adenocarcinoma. Cancer Res 80, 1991–2003. 10.1158/0008-5472.CAN-19-2689. [DOI] [PubMed] [Google Scholar]
- 101.Guo X, Pan Y, and Gutmann DH (2019). Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell-specific chemokine recruitment of T cells and microglia. Neuro Oncol 21, 1250–1262. 10.1093/neuonc/noz080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.de Andrade Costa A, Chatterjee J, Cobb O, Cordell E, Chao A, Schaeffer S, Goldstein A, Dahiya S, and Gutmann DH (2022). Immune deconvolution and temporal mapping identifies stromal targets and developmental intervals for abrogating murine low-grade optic glioma formation. Neurooncol Adv 4, vdab194. 10.1093/noajnl/vdab194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chatterjee J, Sanapala S, Cobb O, Bewley A, Goldstein AK, Cordell E, Ge X, Garbow JR, Holtzman MJ, and Gutmann DH (2021). Asthma reduces glioma formation by T cell decorin-mediated inhibition of microglia. Nat Commun 12, 7122. 10.1038/s41467-021-27455-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guo X, Pan Y, Xiong M, Sanapala S, Anastasaki C, Cobb O, Dahiya S, and Gutmann DH (2020). Midkine activation of CD8(+) T cells establishes a neuron-immune-cancer axis responsible for low-grade glioma growth. Nat Commun 11, 2177. 10.1038/s41467-020-15770-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anastasaki C, Mo J, Chen J-K, Chatterjee J, Pan Y, Scheaffer SM, Cobb O, Monje M, Le LQ, and Gutmann DH (2022). Neuronal hyperexcitability drives central and peripheral nervous system tumor progression in models of Neurofibromatosis-1. Nature Communications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, Weil S, Horstmann H, Wiestler B, Syed M, et al. (2015). Brain tumour cells interconnect to a functional and resistant network. Nature 528, 93–98. 10.1038/nature16071. [DOI] [PubMed] [Google Scholar]
- 107.Rustom A, Saffrich R, Markovic I, Walther P, and Gerdes HH (2004). Nanotubular highways for intercellular organelle transport. Science 303, 1007–1010. 10.1126/science.1093133. [DOI] [PubMed] [Google Scholar]
- 108.Lou E, Fujisawa S, Morozov A, Barlas A, Romin Y, Dogan Y, Gholami S, Moreira AL, Manova-Todorova K, and Moore MA (2012). Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS One 7, e33093. 10.1371/journal.pone.0033093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Joseph JV, Magaut CR, Storevik S, Geraldo LH, Mathivet T, Latif MA, Rudewicz J, Guyon J, Gambaretti M, Haukas F, et al. (2022). TGF-beta promotes microtube formation in glioblastoma through thrombospondin 1. Neuro Oncol 24, 541–553. 10.1093/neuonc/noab212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Valdebenito S, Malik S, Luu R, Loudig O, Mitchell M, Okafo G, Bhat K, Prideaux B, and Eugenin EA (2021). Tunneling nanotubes, TNT, communicate glioblastoma with surrounding non-tumor astrocytes to adapt them to hypoxic and metabolic tumor conditions. Sci Rep 11, 14556. 10.1038/s41598-021-93775-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Venkataramani V, Yang Y, Schubert MC, Reyhan E, Tetzlaff SK, Wissmann N, Botz M, Soyka SJ, Beretta CA, Pramatarov RL, et al. (2022). Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 185, 2899–2917 e2831. 10.1016/j.cell.2022.06.054. [DOI] [PubMed] [Google Scholar]
- 112.Weil S, Osswald M, Solecki G, Grosch J, Jung E, Lemke D, Ratliff M, Hanggi D, Wick W, and Winkler F (2017). Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol 19, 1316–1326. 10.1093/neuonc/nox070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, et al. (2013). Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 11, 94. 10.1186/1479-5876-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Osswald M, Solecki G, Wick W, and Winkler F (2016). A malignant cellular network in gliomas: potential clinical implications. Neuro Oncol 18, 479–485. 10.1093/neuonc/now014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M, Tam LT, Espenel C, Ponnuswami A, Ni L, et al. (2019). Electrical and synaptic integration of glioma into neural circuits. Nature 573, 539–545. 10.1038/s41586-019-1563-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T, Korber C, Kardorff M, Ratliff M, Xie R, et al. (2019). Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 573, 532–538. 10.1038/s41586-019-1564-x. [DOI] [PubMed] [Google Scholar]
- 117.Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, and Sontheimer H (2007). Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res 67, 9463–9471. 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Piao Y, Lu L, and de Groot J (2009). AMPA receptors promote perivascular glioma invasion via beta1 integrin-dependent adhesion to the extracellular matrix. Neuro Oncol 11, 260–273. 10.1215/15228517-2008-094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Takano T, Lin JH, Arcuino G, Gao Q, Yang J, and Nedergaard M (2001). Glutamate release promotes growth of malignant gliomas. Nat Med 7, 1010–1015. 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- 120.de Groot JF, Piao Y, Lu L, Fuller GN, and Yung WK (2008). Knockdown of GluR1 expression by RNA interference inhibits glioma proliferation. J Neurooncol 88, 121–133. 10.1007/s11060-008-9552-2. [DOI] [PubMed] [Google Scholar]
- 121.Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H, Miwa A, Kurihara H, Nakazato Y, Tamura M, et al. (2002). Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med 8, 971–978. 10.1038/nm746. [DOI] [PubMed] [Google Scholar]
- 122.Ishiuchi S, Yoshida Y, Sugawara K, Aihara M, Ohtani T, Watanabe T, Saito N, Tsuzuki K, Okado H, Miwa A, et al. (2007). Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J Neurosci 27, 7987–8001. 10.1523/JNEUROSCI.2180-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Guizzetti M, Costa P, Peters J, and Costa LG (1996). Acetylcholine as a mitogen: muscarinic receptor-mediated proliferation of rat astrocytes and human astrocytoma cells. Eur J Pharmacol 297, 265–273. 10.1016/0014-2999(95)00746-6. [DOI] [PubMed] [Google Scholar]
- 124.Wang X, Wang ZB, Luo C, Mao XY, Li X, Yin JY, Zhang W, Zhou HH, and Liu ZQ (2019). The Prospective Value of Dopamine Receptors on Bio-Behavior of Tumor. J Cancer 10, 1622–1632. 10.7150/jca.27780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Song P, Sekhon HS, Fu XW, Maier M, Jia Y, Duan J, Proskosil BJ, Gravett C, Lindstrom J, Mark GP, et al. (2008). Activated cholinergic signaling provides a target in squamous cell lung carcinoma. Cancer Res 68, 4693–4700. 10.1158/0008-5472.CAN-08-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W, McCabe BD, Galvan JA, Robinson HPC, Zlobec I, et al. (2019). Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 573, 526–531. 10.1038/s41586-019-1576-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Perea G, Navarrete M, and Araque A (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32, 421–431. 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 128.Fan JJ, and Huang X (2022). Ion Channels in Cancer: Orchestrators of Electrical Signaling and Cellular Crosstalk. Rev Physiol Biochem Pharmacol 183, 103–133. 10.1007/112_2020_48. [DOI] [PubMed] [Google Scholar]
- 129.Prevarskaya N, Skryma R, and Shuba Y (2018). Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol Rev 98, 559–621. 10.1152/physrev.00044.2016. [DOI] [PubMed] [Google Scholar]
- 130.Lang F, Foller M, Lang KS, Lang PA, Ritter M, Gulbins E, Vereninov A, and Huber SM (2005). Ion channels in cell proliferation and apoptotic cell death. J Membr Biol 205, 147–157. 10.1007/s00232-005-0780-5. [DOI] [PubMed] [Google Scholar]
- 131.Schwab A, Fabian A, Hanley PJ, and Stock C (2012). Role of ion channels and transporters in cell migration. Physiol Rev 92, 1865–1913. 10.1152/physrev.00018.2011. [DOI] [PubMed] [Google Scholar]
- 132.Saberbaghi T, Wong R, Rutka JT, Wang GL, Feng ZP, and Sun HS (2019). Role of Cl(−) channels in primary brain tumour. Cell Calcium 81, 1–11. 10.1016/j.ceca.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 133.Liu J, Qu C, Han C, Chen MM, An LJ, and Zou W (2019). Potassium channels and their role in glioma: A mini review. Mol Membr Biol 35, 76–85. 10.1080/09687688.2020.1729428. [DOI] [PubMed] [Google Scholar]
- 134.Takayasu T, Kurisu K, Esquenazi Y, and Ballester LY (2020). Ion Channels and Their Role in the Pathophysiology of Gliomas. Mol Cancer Ther 19, 1959–1969. 10.1158/1535-7163.MCT-19-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lefranc F (2021). Transient Receptor Potential (TRP) Ion Channels Involved in MalignantGlioma Cell Death and Therapeutic Perspectives. Front Cell Dev Biol 9, 618961. 10.3389/fcell.2021.618961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chinigo G, Castel H, Chever O, and Gkika D (2021). TRP Channels in Brain Tumors. Front Cell Dev Biol 9, 617801. 10.3389/fcell.2021.617801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hausmann D, Hoffmann DC, Venkataramani V, Jung E, Horschitz S, Tetzlaff SK, Jabali A, Hai L, Kessler T, Azorin DD, et al. (2022). Autonomous rhythmic activity in glioma networks drives brain tumour growth. Nature. 10.1038/s41586-022-05520-4. [DOI] [PubMed] [Google Scholar]
- 138.Francisco MA, Wanggou S, Fan JJ, Dong W, Chen X, Momin A, Abeysundara N, Min HK, Chan J, McAdam R, et al. (2020). Chloride intracellular channel 1 cooperates with potassium channel EAG2 to promote medulloblastoma growth. J Exp Med 217. 10.1084/jem.20190971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Huang X, He Y, Dubuc AM, Hashizume R, Zhang W, Reimand J, Yang H, Wang TA, Stehbens SJ, Younger S, et al. (2015). EAG2 potassium channel with evolutionarily conserved function as a brain tumor target. Nat Neurosci 18, 1236–1246. 10.1038/nn.4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huang X, Dubuc AM, Hashizume R, Berg J, He Y, Wang J, Chiang C, Cooper MK, Northcott PA, Taylor MD, et al. (2012). Voltage-gated potassium channel EAG2 controls mitotic entry and tumor growth in medulloblastoma via regulating cell volume dynamics. Genes Dev 26, 1780–1796. 10.1101/gad.193789.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang HY, Wang W, Liu YW, Li MY, Liang TY, Li JY, Hu HM, Lu Y, Yao C, Ye YY, et al. (2017). Role of KCNB1 in the prognosis of gliomas and autophagy modulation. Sci Rep 7, 14. 10.1038/s41598-017-00045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chen X, Wanggou S, Bodalia A, Zhu M, Dong W, Fan JJ, Yin WC, Min HK, Hu M, Draghici D, et al. (2018). A Feedforward Mechanism Mediated by Mechanosensitive Ion Channel PIEZO1 and Tissue Mechanics Promotes Glioma Aggression. Neuron 100, 799–815 e797. 10.1016/j.neuron.2018.09.046. [DOI] [PubMed] [Google Scholar]
- 143.Stock K, Kumar J, Synowitz M, Petrosino S, Imperatore R, Smith ES, Wend P, Purfurst B, Nuber UA, Gurok U, et al. (2012). Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat Med 18, 1232–1238. 10.1038/nm.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ji D, Fleig A, Horgen FD, Feng ZP, and Sun HS (2022). Modulators of TRPM7 and its potential as a drug target for brain tumours. Cell Calcium 101, 102521. 10.1016/j.ceca.2021.102521. [DOI] [PubMed] [Google Scholar]
- 145.Wang R, Gurguis CI, Gu W, Ko EA, Lim I, Bang H, Zhou T, and Ko JH (2015). Ion channel gene expression predicts survival in glioma patients. Sci Rep 5, 11593. 10.1038/srep11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Aabedi AA, Lipkin B, Kaur J, Kakaizada S, Valdivia C, Reihl S, Young JS, Lee AT, Krishna S, Berger MS, et al. (2021). Functional alterations in cortical processing of speech in glioma-infiltrated cortex. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2108959118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lee AT, Faltermeier C, Morshed RA, Young JS, Kakaizada S, Valdivia C, Findlay AM, Tarapore PE, Nagarajan SS, Hervey-Jumper SL, and Berger MS (2020). The impact of high functional connectivity network hub resection on language task performance in adult low- and high-grade glioma. J Neurosurg 134, 1102–1112. 10.3171/2020.1.JNS192267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sanai N, Mirzadeh Z, and Berger MS (2008). Functional outcome after language mapping for glioma resection. N Engl J Med 358, 18–27. 10.1056/NEJMoa067819. [DOI] [PubMed] [Google Scholar]
- 149.Berendsen S, Varkila M, Kroonen J, Seute T, Snijders TJ, Kauw F, Spliet WG, Willems M, Poulet C, Broekman ML, et al. (2016). Prognostic relevance of epilepsy at presentation in glioblastoma patients. Neuro Oncol 18, 700–706. 10.1093/neuonc/nov238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.van Breemen MS, Wilms EB, and Vecht CJ (2007). Epilepsy in patients with brain tumours: epidemiology, mechanisms, and management. Lancet Neurol 6, 421–430. 10.1016/S1474-4422(07)70103-5. [DOI] [PubMed] [Google Scholar]
- 151.Klein M, Engelberts NH, van der Ploeg HM, Kasteleijn-Nolst Trenite DG, Aaronson NK, Taphoorn MJ, Baaijen H, Vandertop WP, Muller M, Postma TJ, and Heimans JJ (2003). Epilepsy in low-grade gliomas: the impact on cognitive function and quality of life. Ann Neurol 54, 514–520. 10.1002/ana.10712. [DOI] [PubMed] [Google Scholar]
- 152.Campbell SL, Robel S, Cuddapah VA, Robert S, Buckingham SC, Kahle KT, and Sontheimer H (2015). GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 63, 23–36. 10.1002/glia.22730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Buckingham SC, Campbell SL, Haas BR, Montana V, Robel S, Ogunrinu T, and Sontheimer H (2011). Glutamate release by primary brain tumors induces epileptic activity. Nat Med 17, 1269–1274. 10.1038/nm.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Campbell SL, Buckingham SC, and Sontheimer H (2012). Human glioma cells induce hyperexcitability in cortical networks. Epilepsia 53, 1360–1370. 10.1111/j.1528-1167.2012.03557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gao X, Zhang Z, Mashimo T, Shen B, Nyagilo J, Wang H, Wang Y, Liu Z, Mulgaonkar A, Hu XL, et al. (2020). Gliomas Interact with Non-glioma Brain Cells via Extracellular Vesicles. Cell Rep 30, 2489–2500 e2485. 10.1016/j.celrep.2020.01.089. [DOI] [PubMed] [Google Scholar]
- 156.Yu K, Lin CJ, Hatcher A, Lozzi B, Kong K, Huang-Hobbs E, Cheng YT, Beechar VB, Zhu W, Zhang Y, et al. (2020). PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis. Nature 578, 166–171. 10.1038/s41586-020-1952-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Koppelmans V, Breteler MM, Boogerd W, Seynaeve C, Gundy C, and Schagen SB (2012). Neuropsychological performance in survivors of breast cancer more than 20 years after adjuvant chemotherapy. J Clin Oncol 30, 1080–1086. 10.1200/JCO.2011.37.0189. [DOI] [PubMed] [Google Scholar]
- 158.Gibson EM, Nagaraja S, Ocampo A, Tam LT, Wood LS, Pallegar PN, Greene JJ, Geraghty AC, Goldstein AK, Ni L, et al. (2019). Methotrexate Chemotherapy Induces Persistent Tri-glial Dysregulation that Underlies Chemotherapy-Related Cognitive Impairment. Cell 176, 43–55 e13. 10.1016/j.cell.2018.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Allen BD, Apodaca LA, Syage AR, Markarian M, Baddour AAD, Minasyan H, Alikhani L, Lu C, West BL, Giedzinski E, et al. (2019). Attenuation of neuroinflammation reverses Adriamycin-induced cognitive impairments. Acta Neuropathol Commun 7, 186. 10.1186/s40478-019-0838-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shi DD, Huang YH, Lai CSW, Dong CM, Ho LC, Wu EX, Li Q, Wang XM, Chung SK, Sham PC, and Zhang ZJ (2019). Chemotherapy-Induced Cognitive Impairment Is Associated with Cytokine Dysregulation and Disruptions in Neuroplasticity. Mol Neurobiol 56, 2234–2243. 10.1007/s12035-018-1224-4. [DOI] [PubMed] [Google Scholar]
- 161.Wang Z, Jiang C, He Q, Matsuda M, Han Q, Wang K, Bang S, Ding H, Ko MC, and Ji RR (2020). Anti-PD-1 treatment impairs opioid antinociception in rodents and nonhuman primates. Sci Transl Med 12. 10.1126/scitranslmed.aaw6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tozaki-Saitoh H, Masuda J, Kawada R, Kojima C, Yoneda S, Masuda T, Inoue K, and Tsuda M (2019). Transcription factor MafB contributes to the activation of spinal microglia underlying neuropathic pain development. Glia 67, 729–740. 10.1002/glia.23570. [DOI] [PubMed] [Google Scholar]
- 163.Kuhn JA, Vainchtein ID, Braz J, Hamel K, Bernstein M, Craik V, Dahlgren MW, Ortiz-Carpena J, Molofsky AB, Molofsky AV, and Basbaum AI (2021). Regulatory T-cells inhibit microglia-induced pain hypersensitivity in female mice. Elife 10. 10.7554/eLife.69056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Durante M, Squillace S, Lauro F, Giancotti LA, Coppi E, Cherchi F, Di Cesare Mannelli L, Ghelardini C, Kolar G, Wahlman C, et al. (2021). Adenosine A3 agonists reverse neuropathic pain via T cell-mediated production of IL-10. J Clin Invest 131. 10.1172/JCI139299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Luo X, Huh Y, Bang S, He Q, Zhang L, Matsuda M, and Ji RR (2019). Macrophage Toll-like Receptor 9 Contributes to Chemotherapy-Induced Neuropathic Pain in Male Mice. J Neurosci 39, 6848–6864. 10.1523/JNEUROSCI.3257-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Navia-Pelaez JM, Choi SH, Dos Santos Aggum Capettini L, Xia Y, Gonen A, Agatisa-Boyle C, Delay L, Goncalves Dos Santos G, Catroli GF, Kim J, et al. (2021). Normalization of cholesterol metabolism in spinal microglia alleviates neuropathic pain. J Exp Med 218. 10.1084/jem.20202059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhu Z, Friess H, diMola FF, Zimmermann A, Graber HU, Korc M, and Buchler MW (1999). Nerve growth factor expression correlates with perineural invasion and pain in human pancreatic cancer. J Clin Oncol 17, 2419–2428. 10.1200/JCO.1999.17.8.2419. [DOI] [PubMed] [Google Scholar]
- 168.da Silva JT, Evangelista BG, Venega RAG, Seminowicz DA, and Chacur M (2019). Anti-NGF treatment can reduce chronic neuropathic pain by changing peripheral mediators and brain activity in rats. Behav Pharmacol 30, 79–88. 10.1097/FBP.0000000000000422. [DOI] [PubMed] [Google Scholar]
- 169.Lowy DB, Makker PGS, and Moalem-Taylor G (2021). Cutaneous Neuroimmune Interactions in Peripheral Neuropathic Pain States. Front Immunol 12, 660203. 10.3389/fimmu.2021.660203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fumagalli G, Monza L, Cavaletti G, Rigolio R, and Meregalli C (2020). Neuroinflammatory Process Involved in Different Preclinical Models of Chemotherapy-Induced Peripheral Neuropathy. Front Immunol 11, 626687. 10.3389/fimmu.2020.626687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Boilly B, Faulkner S, Jobling P, and Hondermarck H (2017). Nerve Dependence: From Regeneration to Cancer. Cancer Cell 31, 342–354. 10.1016/j.ccell.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 172.Zahalka AH, and Frenette PS (2020). Nerves in cancer. Nat Rev Cancer 20, 143–157. 10.1038/s41568-019-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Grossman SA, Ye X, Chamberlain M, Mikkelsen T, Batchelor T, Desideri S, Piantadosi S, Fisher J, and Fine HA (2009). Talampanel with standard radiation and temozolomide in patients with newly diagnosed glioblastoma: a multicenter phase II trial. J Clin Oncol 27, 4155–4161. 10.1200/JCO.2008.21.6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.De Meulenaere V, Bonte E, Verhoeven J, Kalala Okito JP, Pieters L, Vral A, De Wever O, Leybaert L, Goethals I, Vanhove C, et al. (2019). Adjuvant therapeutic potential of tonabersat in the standard treatment of glioblastoma: A preclinical F98 glioblastoma rat model study. PLoS One 14, e0224130. 10.1371/journal.pone.0224130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hanahan D (2022). Hallmarks of Cancer: New Dimensions. Cancer Discov 12, 31–46. 10.1158/2159-8290.CD-21-1059. [DOI] [PubMed] [Google Scholar]
- 176.Amit M (2022). Cancer Neuroscience: Evidence for a New Hallmark of Cancer. Adv Biol (Weinh) 6, e2200192. 10.1002/adbi.202200192. [DOI] [PubMed] [Google Scholar]
- 177.Benthall KN, Ong SL, and Bateup HS (2018). Corticostriatal Transmission Is Selectively Enhanced in Striatonigral Neurons with Postnatal Loss of Tsc1. Cell Rep 23, 3197–3208. 10.1016/j.celrep.2018.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, and Sabatini BL (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522. 10.1016/j.neuron.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kosillo P, Doig NM, Ahmed KM, Agopyan-Miu A, Wong CD, Conyers L, Threlfell S, Magill PJ, and Bateup HS (2019). Tsc1-mTORC1 signaling controls striatal dopamine release and cognitive flexibility. Nat Commun 10, 5426. 10.1038/s41467-019-13396-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kuang H, Liu T, Jiao C, Wang J, Wu S, Wu J, Peng S, Davidson AM, Zeng SX, Lu H, and Mostany R (2022). Genetic Deficiency of p53 Leads to Structural, Functional, and Synaptic Deficits in Primary Somatosensory Cortical Neurons of Adult Mice. Front Mol Neurosci 15, 871974. 10.3389/fnmol.2022.871974. [DOI] [PMC free article] [PubMed] [Google Scholar]