

Abstract
Although utilization of fluorine compounds has a long history, synthesis of chiral fluorinated amino acid derivatives with structural diversity and high stereoselectivity is still very appealing and challenging. Here, we report a biomimetic study of enantioselective [1,3]-proton shift of β,β-difluoro-α-imine amides catalyzed by chiral quinine derivatives. A wide range of corresponding β,β-difluoro-α-amino amides were achieved in good yields with high enantioselectivities. The optically pure β,β-difluoro-α-amino acid derivatives were further obtained, which have high application values in the synthesis of fluoro peptides, fluoro amino alcohols and other valuable fluorine-containing molecules.
Subject terms: Organocatalysis, Asymmetric catalysis
Organocatalysed 1,3-proton shifts can offer efficient access to chiral nonracemic fluorinated products. Here chiral amino amides are obtained in high enantiomeric ratio via enantioselective 1,3- proton shift of β,β-difluoro-αimino amides.
Introduction
Organic fluorides are a class of unique fluorine compounds, which feature with strong electronegativity, similar size to hydrogen, great influence on pKa and lipophilicity, and behavior of hydrogen-bond receptors1–7. Although fluorine-containing compounds have special physicochemical and biochemical properties, naturally occurring organofluorine compounds are extremely rare8. Fluorine earns other extensive applications in modern chemistry. For example, at least 25% of drugs and 50% agrochemicals contain fluorine9–11. Chiral-fluorinated amino acids are a class of fluorine-containing building blocks, which are wildely utilized in biorthogonal chemistry, drug modification, and asymmetric catalysis12–16. However, the development of chiral fluorine-containing amino acids still has numerous limitations17 as below. (i) The preparation of fluorine-containing peptides from natural amino acids is extremely rare, and the application of natural chemical connection is still in its infancy;18–21 (ii) compared with aromatic fluorination, the structural diversity of alkyl-fluorinated amino acids needs further study;22,23 (iii) the self-disproportionation of enantiomers is also an important factor to prevent the formation of optically pure fluorinated amino acids24,25.
Difluoromethylene has the highest dipole moment in the fluoromethane series and also exerts as bioisostere of ketone and ether, which may change the protein structure26–29. Chiral β,β-difluoro-α-amino acids (dFAAs) are vital building blocks for assembling bioactive molecules30,31. As shown in Fig. 1, the β,β-difluoro compound III acts as a better substrate for human CCRF-CEM folypoly-γ-glutamate synthetase;32 3,3-difluoro-3,4-dideoxy-KRN7000 analog II is identified as potent immunostimulator;33 the β,β-difluorophenylalanyl puromycin I reacts faster than puromycin at neutral or acidic condition34. However, the synthetic methods of chiral dFAAs are not mature, especially in catalytic asymmetric induction. There are only three main routes to prepare chiral dFAAs (Fig. 2). The group of Ayi reported the enzymatic hydrolysis of β,β-difluoro-α-amino esters via kinetic resolution35. Liu and his colleagues disclosed an effective method for controlling enantioselectivity by introducing chiral auxiliaries36–40. Uneyama uncovered an amazing asymmetric hydrogenation of α-fluoroiminoesters via catalytic amount of chiral BINAP ligand and palladium41–43. However, in the current situation, it is urgent to develop a general, efficient, and highly enantioselective dFAAS method under mild conditions.
Fig. 1. Representative β,β-difluoro-α-amino acid derivatives.
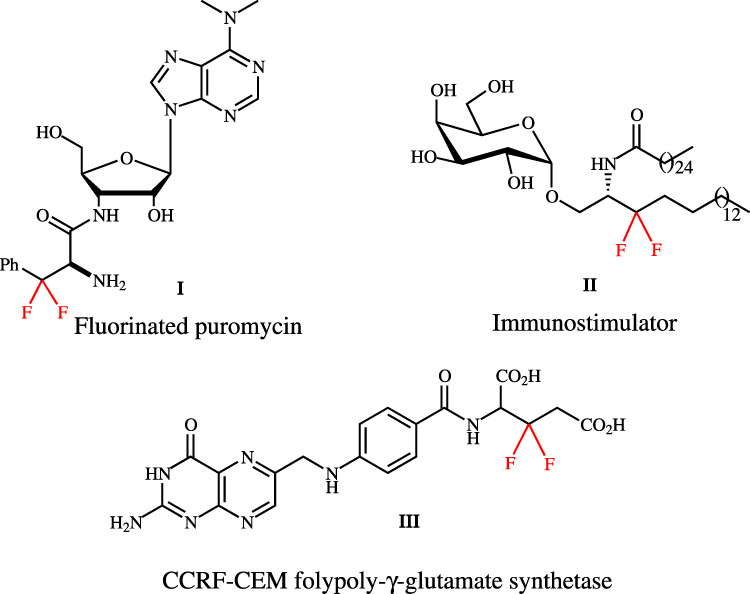
I: β,β-difluorophenylalanyl puromycin; II: 3,3-difluoro-3,4-dideoxy-KRN7000 analog; III:CCRF-CEM folypoly-γ-glutamate synthetase.
Fig. 2. Strategies for asymmetric synthesis of β,β-difluoro-α-amino acid derivatives.

Strategies, a enzymatic hydrolysis; b asymmetric hydrogenation; c chiral auxiliaries. Valuable molecules: difluoro peptides, difluoro amino alcohols, and difluoro alkaloid.
In the 1990s, Soloshonok et al. first studied the biomimetic [1,3]-proton shift, which opened up a new door for the synthesis of chiral β-fluoroalkyl β-amino acids44–46. Then, Jørgensen47, Deng48–50, Shi51,52, and others53–55 further extended this strategy to construct chiral amino acids and chiral trifluoromethylamines. Building upon our continued interests in organic fluorides56–61 and the limitations for the rapid synthesis of chiral fluoro amino acids, we report here an approach to achieve dFAAS via biomimetic enantioselective [1,3]-proton-shift reactions. Notably, the yielded chiral dFAAs are useful synthons for producing various valuable molecules, such as difluoro peptides, difluoro amino alcohols, and difluoro alkaloid62.
Results and discussion
Reaction optimization
We first studied the reaction of N-benzyl-2-((2-chlorobenzyl)imino)-3,3-difluoro-5-phenylpentanamide 1a as model substrate and toluene as solvent. The results are shown in Table 1. When cinchonan-6′, diol A was exploited, the expected 2a was acquired in 34% yield and 81% ee (entry 1). Almost racemic 2a was obtained by replacing the catalyst with quinine B (entry 2), while the yield and enantioselectivity were improved by using catalysts C and D (entries 3, 4). Further tests on catalysts E and F showed no significant change in yield or ee (entries 5, 6). To our delight, when G was tested, 2a was successfully formed with 87% yield and 95% ee and pseudoenantiomer H was also tested for delivering the enantiomer of 2a with 80% yield and 95% ee (entries 7 and 8). Solvent evaluation experiments reveal that toluene is the best choice (entries 9–12). Furthermore, reducing the loading of catalyst G to 5 mol% (entry 13) without decreasing the yield or ee implies that this highly enantioselective biomimetic [1,3]-proton shift can be achieved under suitable conditions. Even if the time is extended to 24 h and the loading is further reduced to 1 mol% (entry 14), the requirement of useless yield can not be met.
Table 1.
Optimization of asymmetric isomerization of 1aa.
 | ||||
|---|---|---|---|---|
| Entry | NHC | Solvent | Yield [%]b | ee [%]c |
| 1 | A | Toluene | 34 | 81 |
| 2 | B | Toluene | 18 | 2 |
| 3 | C | Toluene | 79 | 93 |
| 4 | D | Toluene | 76 | 95 |
| 5 | E | Toluene | 83 | 90 |
| 6 | F | Toluene | 43 | 95 |
| 7 | G | Toluene | 87 | 95 |
| 8 | H | Toluene | 80 | 95f |
| 9 | G | DCM | 81 | 93 |
| 10 | G | CHCl3 | 85 | 94 |
| 11 | G | CH3CN | 51 | 84 |
| 12 | G | THF | 49 | 77 |
| 13d | G | Toluene | 86 | 95 |
| 14e | G | Toluene | 27 | 96 |
aConditions: 1a (0.05 mmol), catalyst (10 mol%), solvent (0.5 mL), room temperature, 2 h.
bIsolated yield after flash column chromatography.
cEnantiomeric excess (ee) determined via chiral-phase HPLC analysis.
dCat. G (5 mol%) was used; 4 h.
eCat. G (1 mol%) was used; 24 h.
fEnantiomer was obtained.
Substrate scope
Under the optimized catalytic conditions, we turned our attention to explore the generality of biomimetic [1,3]-proton-shift reaction. As illustrated in Fig. 3, R1 group was examined first. In the process of generating the [1,3]-proton-shift products 2a and 2b, it was found that the yield and enantioselectivity are independent of the adjacent CH2CH2 groups of the substituents (1a and 1b). Alkene (1c) is also compatible under this condition. Furthermore, the CH2 adjoint groups containing cyclohexyl, 1,3-dioxolan-2-yl, isopropyl, phenyl, methyl and alkene were well tolerated and provided good yields and high enantioselectivities (2d–2i). β,β-difluoro-2-aminobutyric amide (2j) was obtained with good yield and excellent ee. In the case of 2 l that is a hydrolyzed derivative and bearing ester group, 72% ee and 62% yield were observed. Then, we turned to amide group R2, where isobutyl can be installed smoothly (2 m). It is gratifying that dipeptides (2n and 2o) bearing leukine have been successfully provided for peptide synthesis and the diastereoisomers indicated that the source of chirality is induced by the catalyst rather than substrate itself. Pleasingly, the imines with or without substituted phenyl group are well tolerated under these conditions (2p–2r). β,β-difluoro natural amino acid derivatives (e.g., β,β-difluoro glutamine, β,β-difluoro leucine, and β,β-difluoro phenylalanine) were all obtained in good yields and high ee’s.
Fig. 3. Scope of β,β-difluorinated imines[a,b,c].
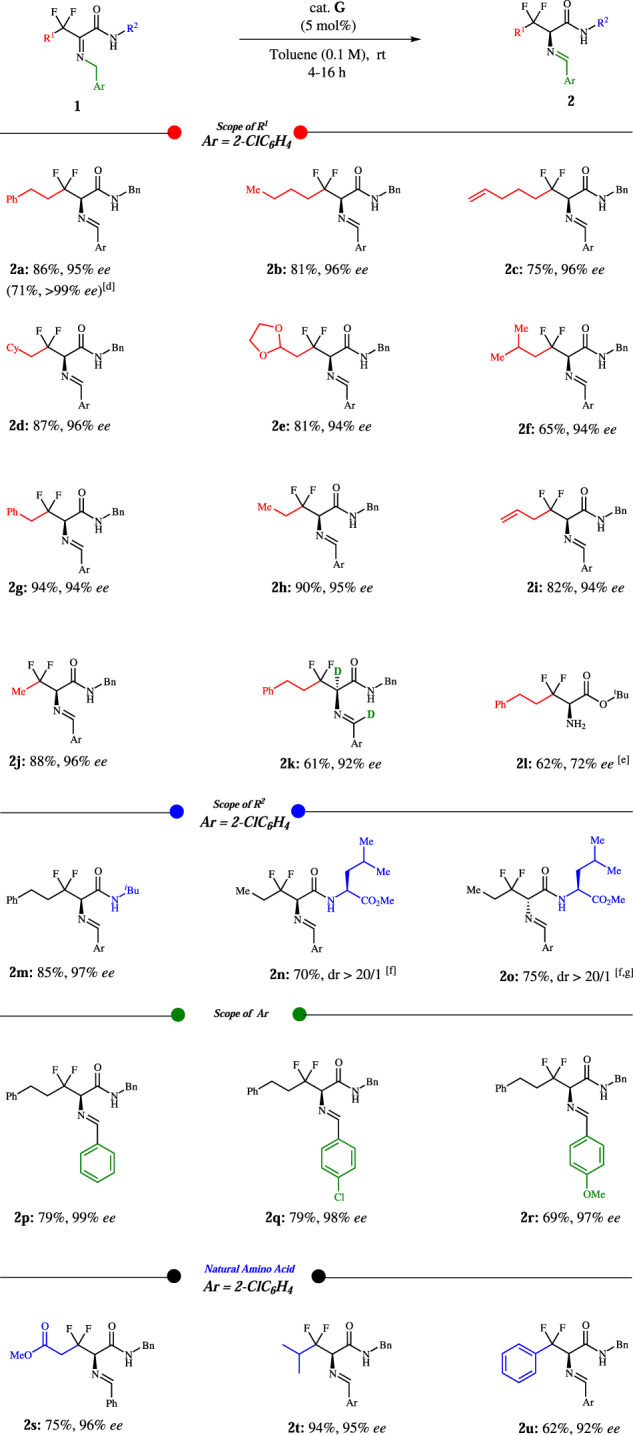
a Conditions: 1a (0.1 mmol), cat. G (5 mol%), toluene (1.0 mL), room temperature, 4–16 h. b Isolated yield after flash-column chromatography. c Enantiomeric excess (ee) determined via chiral-phase HPLC analysis. d Recrystallization from hexane/ethyl acetate (20/1). e After hydrolysis. f dr was determined via crude 1H NMR. g Cat. H (5 mol%) was used.
Mechanistic studies and postulated mechanism
Next, we started to investigate the reaction mechanism. We want to understand whether the [1,3]-proton-shift process is intermolecular or intramolecular. Benzyl-deuterated 1a-d2 was prepared and the reaction carried out under standard conditions. The results show that 2k has no erosion on deuterated ratio (Fig. 4a). As a contrast, when deuterium oxide was added to the reaction system, no deuterium product was found (Fig. 4b).
Fig. 4. Mechanistic studies.
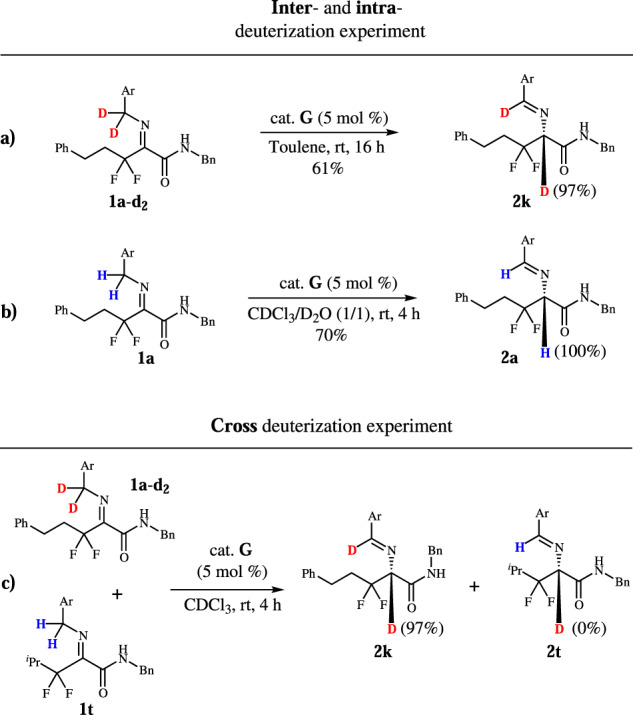
Deuterization and cross deuterization experiment.
When 1a-d2 cross-reacted with 1t, no deuterium was found in product 2t (Fig. 4c). These experiments reveal that the [1,3]-proton-shift process is intramolecular. The parallel kinetic isotope effects (KIE) were measured with kH/kD of 4.0, indicating that the rate-determining step (RDS) is the process of hydrogen leaving from benzyl group (Fig. 5).
Fig. 5. KIE from parallel experiments.

The parallel experiments of 1a and 1a-d2.
Based on the condition optimization, substrate scope, and the initial mechanistic experiments, a plausible mechanism for the biomimetic enantioselective [1,3]-proton-shift reaction was proposed (Fig. 6). First, the possible intramolecular hydrogen-bond interaction (blue) of the amide NH to imine of the substrate is essential, and without it, the ee decreased dramatically (e.g., 2 l bearing an ester group resulted in 72% ee). Then, the catalyst with free OH of phenol is needful (e.g., cat. B with OMe only induced 2% ee) for assembling another hydrogen bond between it and the N of the amide (green). The bulk isopentyloxy of the catalyst enforced deprotonation of the inner H of benzyl (red) and this process was a RDS (TS-I). Finally, the asymmetric protonation of 2-azaallyl anion from Si face (TS-II) to deliver the target product with R-configuration.
Fig. 6. Proposed transition model.

The possible mechanistic of the enantioselective [1,3]-proton-shift reaction.
Synthetic transformations and applications
In addition, our protocol is potentially suitable for large-scale preparation. As illustrated in Fig. 7, the use of 2 mol% catalyst G is sufficient to produce 2i (0.91 g) in 80% yield and 95% ee. Compound 3 can be achieved via hydrolysis and N-Phth protection. Next, the conversion of amide to ester group can generate the key intermediate 463. Finally, 3,3-difluoro-3,4-dideoxy-KRN7000 analog II can be made smoothly from 4a based on formal synthesis33. Meanwhile, the corresponding Fmoc-protected amino acid 5 can be prepared from 4b for further medicinal study. To further prove the practicability of dFAAs, difluoro leukine was assembled into linear difluoro-oxytocin (28% yield for overall steps), which could be folded smoothly by using glutathione oxidation (63% yield) (Fig. 8). Additionally, we demonstrated that the stability of folded difluoro-oxytocin (3 mM GSH, 100 mM PBS, pH 7.0, rt) is higher than WT oxytocin64–67(Fig. 9).
Fig. 7. Gram-scale synthesis and transformations.

The gram-scale synthesis of 1i. The transformation of 2i to II and 2t to 5.
Fig. 8. Solid-phase peptide synthesis of difluorinated oxytocin.

a About 20% piperidine/DMF, 10 min, double; b 1.5 equiv. fluorinated leucine, 1.5 equiv. HOBt, 1.5 equiv. DIC, minimum DMF, rt, 16 h; c 10% piperidine/DMF, 3 mins, double; d 3 equiv. amino acid, 2.9 equiv. HBTU, 6 equiv. DIPEA, minimum DMF, rt, 1.5 h; e 10% piperidine/DMF, 3 mins, double; f TFA cocktail, global deprotection, rt, 3 h; g GSH/GSSG folding, rt, over-night. Fmoc = fluorenylmethoxycarbonyl, HOBt = hydroxybenzotriazole, DIC = N,N’-diisopropylcarbodiimide, HBTU = N,N,N’,N’-tetramethyl-O-(1H-benzotriazol-1-yl)-uronium hexafluorophosphate, DIPEA = N,N-diisopropylethylamine, GSSG = glutathione disulfide.
Fig. 9. Folded difluoro-oxytocin stable than folded WT oxytocin.

s4: fold WT-oxytocin, s5: linear WT oxytocin; s4′: fold difluoro-oxytocin, s5′: linear difluoro-oxytocin, s1–s3/s6–s9 scrambling, s1′–s3′/s6′–s9′ scrambling.
In summary, a biomimetic enantioselective [1,3]-proton shift of difluoroimines has been developed. This new protocol allows the rapid assembly of enantiomerically enriched dFAAs from readily available starting materials under mild conditions. Preliminary mechanism studies show that the proton transfer is intramolecular and the deprotonation step is RDS. More details on enantiocontrol are being conducted in our laboratory and will be reported in due course.
Methods
Synthesis of 2
To a flame-dried Schlenk reaction tube equipped with a magnetic stir bar, was added the 1 (0.1 mmol) and cat. G (1.9 mg, 0.005 mmol). The Schlenk tube was closed with a septum, and toluene (1.0 mL) was added. The mixture was then stirred at 25 °C and monitored by TLC until 1 was consumed. The mixture was concentrated under reduced pressure and purified by column chromatography on silica gel (hexane/EtOAc = 20:1–10:1) to afford the desired product 2. Full experimental details can be found in the Supplementary Methods.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
Generous financial support for this work is provided by the Bayer Investigator fellow, the National Natural Science Foundation of China (21672121, 21871160, and 22071130), and the fellowship of Tsinghua-Peking Centre for Life Sciences (CLS). We warmly thank Prof. Christian Hackenberger for providing chemicals for peptide synthesis and assistance on the peptide characterizations. We thank Prof. Lei Liu for insightful comments and discussions of fluorine-containing peptides. We thank Jiekang Tian, Rudolf Volkmer, and Ines Kretzschmar for technical support.
Author contributions
Q. P. conducted the main chemical part experiments; B. Y. conducted peptide part experiments; F. L., M. L., B. Z. and D. G. prepared several starting materials. D. B. and J. W. conceptualized and directed the project, and drafted the paper with assistance from coauthors. All authors contributed to discussions.
Data availability
For 1H NMR, 13C NMR, and 19F NMR spectra see Supplementary Figs. 1–75 and highperformance liquid-chromatography spectra see Supplementary Figs. 76–118.The X-ray crystallographic coordinates for Fmoc-2l (Supplementary Data 1) reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 2102965. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Communications Chemistry thanks Xinfang Xu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Qiupeng Peng, Bingjia Yan.
Contributor Information
Donald Bierer, Email: donald.bierer1@bayer.com.
Jian Wang, Email: wangjian2012@tsinghua.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-021-00586-z.
References
- 1.O’Hagan D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2008;37:308–319. doi: 10.1039/B711844A. [DOI] [PubMed] [Google Scholar]
- 2.Fujiwara T, O’Hagan D. Successful fluorine-containing herbicide agrochemicals. J. Fluor. Chem. 2014;167:16–29. doi: 10.1016/j.jfluchem.2014.06.014. [DOI] [Google Scholar]
- 3.Zhu Y, et al. Modern approaches for asymmetric construction of carbon–fluorine quaternary stereogenic centers: synthetic challenges and pharmaceutical needs. Chem. Rev. 2018;118:3887–3964. doi: 10.1021/acs.chemrev.7b00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma J-A, Cahard D. Asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions. Chem. Rev. 2004;104:6119–6146. doi: 10.1021/cr030143e. [DOI] [PubMed] [Google Scholar]
- 5.Uneyama K, Katagiri T, Amii H. α-Trifluoromethylated carbanion synthons. Acc. Chem. Res. 2008;41:817–829. doi: 10.1021/ar7002573. [DOI] [PubMed] [Google Scholar]
- 6.Campbell MG, Ritter T. Modern carbon–fluorine bond forming reactions for aryl fluoride synthesis. Chem. Rev. 2015;115:612–633. doi: 10.1021/cr500366b. [DOI] [PubMed] [Google Scholar]
- 7.Champagne PA, Desroches J, Hamel J-D, Vandamme M, Paquin J-F. Monofluorination of organic compounds: 10 years of innovation. Chem. Rev. 2015;115:9073–9174. doi: 10.1021/cr500706a. [DOI] [PubMed] [Google Scholar]
- 8.Walker MC, Chang MC. Natural and engineered biosynthesis of fluorinated natural products. Chem. Soc. Rev. 2014;43:6527–6536. doi: 10.1039/C4CS00027G. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Y, et al. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II–III clinical trials of major pharmaceutical companies: new structural trends and therapeutic areas. Chem. Rev. 2016;116:422–518. doi: 10.1021/acs.chemrev.5b00392. [DOI] [PubMed] [Google Scholar]
- 10.Ogawa, Y., Tokunaga, E., Kobayashi, O., Hirai, K. & Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. iScience23, 101467 (2020). [DOI] [PMC free article] [PubMed]
- 11.Yerien DE, Bonesi S, Postigo A. Fluorination methods in drug discovery. Org. Biomol. Chem. 2016;14:8398–8427. doi: 10.1039/C6OB00764C. [DOI] [PubMed] [Google Scholar]
- 12.Mei H, et al. Fluorine‐containing drugs approved by the FDA in 2018. Chem. Eur. J. 2019;25:11797–11819. doi: 10.1002/chem.201901840. [DOI] [PubMed] [Google Scholar]
- 13.Han J, Sorochinsky AE, Ono T, Soloshonok VA. Biomimetic transamination - a metal-free alternative to the reductive amination. Application for generalized preparation of fluorine-containing amines and amino acids. Curr. Org. Synth. 2011;8:281–294. doi: 10.2174/157017911794697277. [DOI] [Google Scholar]
- 14.Mei, H. et al. Applications of fluorine-containing amino acids for drug design. Eur. J. Med. Chem. 186, 111826 (2020). [DOI] [PubMed]
- 15.Gao F, et al. Stereoselective synthetic strategies of stereogenic carbon centers featuring a difluoromethyl group. Org. Chem. Front. 2021;8:2799–2819. doi: 10.1039/D1QO00032B. [DOI] [Google Scholar]
- 16.Gao F, et al. Catalytic asymmetric construction of tertiary carbon centers featuring an α-difluoromethyl group with CF2H-CH2-NH2 as the “Building Block”. Org. Lett. 2021;23:2584–2589. doi: 10.1021/acs.orglett.1c00497. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X-X, et al. Recent advances in catalytic enantioselective synthesis of fluorinated α- and β-amino acids. Adv. Synth. Catal. 2020;362:4763–4793. doi: 10.1002/adsc.202000966. [DOI] [Google Scholar]
- 18.Ma J-A. Recent developments in the catalytic asymmetric synthesis of α- and β-amino acids. Angew. Chem. Int. Ed. 2003;42:4290–4299. doi: 10.1002/anie.200301600. [DOI] [PubMed] [Google Scholar]
- 19.Weiner B, Szymański W, Janssen DB, Minnaard AJ, Feringa BL. Recent advances in the catalytic asymmetric synthesis of β-amino acids. Chem. Soc. Rev. 2010;39:1656–1691. doi: 10.1039/b919599h. [DOI] [PubMed] [Google Scholar]
- 20.Metz AE, Kozlowski MC. Recent advances in asymmetric catalytic methods for the formation of acyclic α,α-disubstituted α-amino acids. J. Org. Chem. 2015;80:1–7. doi: 10.1021/jo502408z. [DOI] [PubMed] [Google Scholar]
- 21.Noda H, Shibasaki M. Recent advances in the catalytic asymmetric synthesis of β2- and β2,2-amino acids. Eur. J. Org. Chem. 2020;2020:2350–2361. doi: 10.1002/ejoc.201901596. [DOI] [Google Scholar]
- 22.Mei H, et al. Tailor-made amino acids and fluorinated motifs as prominent traits in modern pharmaceuticals. Chem. Eur. J. 2020;26:11349–11390. doi: 10.1002/chem.202000617. [DOI] [PubMed] [Google Scholar]
- 23.Hu, X.-S., Yu, J.-S. & Zhou, J. Catalytic selective mono- and difluoroalkylation using fluorinated silyl enol ethers. Chem. Commun. 55, 13638–13648 (2019). [DOI] [PubMed]
- 24.Han J, Kitagawa O, Wzorek A, Klika KD, Soloshonok VA. The self-disproportionation of enantiomers (SDE): a menace or an opportunity? Chem. Sci. 2018;9:1718–1739. doi: 10.1039/C7SC05138G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soloshonok VA. Remarkable amplification of the self-disproportionation of enantiomers on achiral-phase chromatography columns. Angew. Chem. Int. Ed. 2006;45:766–769. doi: 10.1002/anie.200503373. [DOI] [PubMed] [Google Scholar]
- 26.Meanwell NA. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 2018;61:5822–5880. doi: 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- 27.Levin MD, Ovian JM, Read JA, Sigman MS, Jacobsen EN. Catalytic enantioselective synthesis of difluorinated alkyl bromides. J. Am. Chem. Soc. 2020;142:14831–14837. doi: 10.1021/jacs.0c07043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geri JB, Wade Wolfe MM, Szymczak NK. The difluoromethyl group as a masked nucleophile: a Lewis acid/base approach. J. Am. Chem. Soc. 2018;140:9404–9408. doi: 10.1021/jacs.8b06093. [DOI] [PubMed] [Google Scholar]
- 29.Li F, Qiu C, Yin G, Wang C, Li Z. Metal- and base-free synthesis of functionalized α,α-difluoroimines via electrophilic fluorination of N-substituted enamines. Org. Biomol. Chem. 2018;16:6895–6899. doi: 10.1039/C8OB01941J. [DOI] [PubMed] [Google Scholar]
- 30.Moschner J, et al. Approaches to obtaining fluorinated α-amino acids. Chem. Rev. 2019;119:10718–10801. doi: 10.1021/acs.chemrev.9b00024. [DOI] [PubMed] [Google Scholar]
- 31.O’Hagan D, Wang Y, Skibinski M, Slawin AM. Influence of the difluoromethylene group (CF2) on the conformation and properties of selected organic compounds. Pure Appl. Chem. 2012;84:1587–1595. doi: 10.1351/PAC-CON-11-09-26. [DOI] [Google Scholar]
- 32.Hart BP, et al. Synthesis and biological activity of folic acid and methotrexate analogues containing l-threo-(2S,4S)-4-fluoroglutamic acid and dl-3,3-difluoroglutamic acid. J. Med. Chem. 1996;39:56–65. doi: 10.1021/jm950515e. [DOI] [PubMed] [Google Scholar]
- 33.Hunault J, et al. 3-Fluoro- and 3,3-Difluoro-3,4-dideoxy-KRN7000 analogues as new potent immunostimulator agents: total synthesis and biological evaluation in human invariant natural killer T cells and mice. J. Med. Chem. 2012;55:1227–1241. doi: 10.1021/jm201368m. [DOI] [PubMed] [Google Scholar]
- 34.Okuda K, Seila AC, Strobel SA. Uncovering the enzymatic pKa of the ribosomal peptidyl transferase reaction utilizing a fluorinated puromycin derivative. Biochemistry. 2005;44:6675–6684. doi: 10.1021/bi047419c. [DOI] [PubMed] [Google Scholar]
- 35.Ayi AI, Guedj R, Septe B. Enzymatic hydrolysis of methyl 3, 3-difluoro-2-amino esters. Synthesis of D-and L-3, 3-difluoro-2-amino acids and their derivatives. J. Fluor. Chem. 1995;73:165–169. doi: 10.1016/0022-1139(94)03224-N. [DOI] [Google Scholar]
- 36.Li K, Leriche C, Liu H-W. Synthesis of β-difluorine-containing amino acids. Bioorg. Med. Chem. Lett. 1998;8:1097–1100. doi: 10.1016/S0960-894X(98)00168-1. [DOI] [PubMed] [Google Scholar]
- 37.Schlosser M, Brügger N, Schmidt W, Amrhein N. β, β-Difluoro analogs of α-oxo-β-phenylpropionic acid and phenylalanine. Tetrahedron. 2004;60:7731–7742. doi: 10.1016/j.tet.2004.06.086. [DOI] [Google Scholar]
- 38.Wang X-J, Zhang F, Liu J-T. Asymmetric synthesis of β, β-difluoroamino acids via cross-coupling and Strecker reactions. Tetrahedron. 2008;64:1731–1735. doi: 10.1016/j.tet.2007.12.002. [DOI] [Google Scholar]
- 39.Katagiri T, Handa M, Matsukawa Y, Kumar JD, Uneyama K. Efficient synthesis of an optically pure β-bromo-β, β-difluoroalanine derivative, a general precursor for β, β-difluoroamino acids. Tetrahedron Asymmetry. 2001;12:1303–1311. doi: 10.1016/S0957-4166(01)00237-3. [DOI] [Google Scholar]
- 40.Honraedt A, Van Der Lee A, Campagne J-M, Leclerc E. α,α-Difluoro-α-(trimethylsilyl)acetamides as versatile reagents for the preparation of difluorinated aldol and Mannich adducts. Adv. Synth. Catal. 2017;359:2815–2823. doi: 10.1002/adsc.201700371. [DOI] [Google Scholar]
- 41.Suzuki A, Mae M, Amii H, Uneyama K. Catalytic route to the synthesis of optically active β, β-difluoroglutamic acid and β, β-difluoroproline derivatives. J. Org. Chem. 2004;69:5132–5134. doi: 10.1021/jo049789c. [DOI] [PubMed] [Google Scholar]
- 42.Doebelin C, He Y, Kamenecka TM. Multigram-scale synthesis of enantiopure 3,3-difluoroproline. Tetrahedron Lett. 2016;57:5658–5660. doi: 10.1016/j.tetlet.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abe H, Amii H, Uneyama K. Pd-catalyzed asymmetric hydrogenation of α-fluorinated iminoesters in fluorinated alcohol: a new and catalytic enantioselective synthesis of fluoro α-amino acid derivatives. Org. Lett. 2001;3:313–315. doi: 10.1021/ol0002471. [DOI] [PubMed] [Google Scholar]
- 44.Soloshonok VA, Kirilenko AG, Galushko SV, Kukhar VP. Catalytic asymmetric synthesis of β-fluoroalkyl-β-amino acids via biomimetic [1, 3]-proton shift reaction. Tetrahedron Lett. 1994;35:5063–5064. doi: 10.1016/S0040-4039(00)73320-X. [DOI] [Google Scholar]
- 45.Soloshonok VA, Kukhar VP. Biomimetic base-catalyzed [1, 3]-proton shift reaction. A practical synthesis of β-fluoroalkyl-β-amino acids. Tetrahedron. 1996;52:6953–6964. doi: 10.1016/0040-4020(96)00300-6. [DOI] [Google Scholar]
- 46.Michaut V, Metz F, Paris J-M, Plaquevent J-C. Enantioselective organocatalytic route to trifluoromethyl-β-amino acids using chiral bases. J. Fluor. Chem. 2007;128:500–506. doi: 10.1016/j.jfluchem.2006.12.013. [DOI] [Google Scholar]
- 47.Knudsen, K. R., Bachmann, S. & Jørgensen, K. A. Catalytic enantioselective transaminiation of α-keto esters: an organic approach to enzymatic reactions. Chem. Commun., 10.1039/B308395K (2003). [DOI] [PubMed]
- 48.Zhou X, Wu Y, Deng L. Cinchonium betaines as efficient catalysts for asymmetric proton transfer catalysis: the development of a practical enantioselective isomerization of trifluoromethyl imines. J. Am. Chem. Soc. 2016;138:12297–12302. doi: 10.1021/jacs.6b08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu Y, Hu L, Li Z, Deng L. Catalytic asymmetric umpolung reactions of imines. Nature. 2015;523:445–450. doi: 10.1038/nature14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu Y, Deng L. Asymmetric synthesis of trifluoromethylated amines via catalytic enantioselective isomerization of imines. J. Am. Chem. Soc. 2012;134:14334–14337. doi: 10.1021/ja306771n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao X, Xie Y, Su C, Liu M, Shi Y. Organocatalytic asymmetric biomimetic transamination: from α-Keto esters to optically active α-amino acid derivatives. J. Am. Chem. Soc. 2011;133:12914–12917. doi: 10.1021/ja203138q. [DOI] [PubMed] [Google Scholar]
- 52.Xie Y, Pan H, Liu M, Xiao X, Shi Y. Progress in asymmetric biomimetic transamination of carbonyl compounds. Chem. Soc. Rev. 2015;44:1740–1748. doi: 10.1039/C4CS00507D. [DOI] [PubMed] [Google Scholar]
- 53.Chen P, et al. Phosphine-catalyzed asymmetric umpolung addition of trifluoromethyl ketimines to Morita–Baylis–Hillman Carbonates. Angew. Chem. Int. Ed. 2016;55:13316–13320. doi: 10.1002/anie.201607918. [DOI] [PubMed] [Google Scholar]
- 54.Feng B, et al. Umpolung of imines enables catalytic asymmetric regio-reversed [3+2] cycloadditions of iminoesters with nitroolefins. Angew. Chem. Int. Ed. 2018;57:5888–5892. doi: 10.1002/anie.201802492. [DOI] [PubMed] [Google Scholar]
- 55.Yoshida Y, Hiroshige T, Omori K, Mino T, Sakamoto M. Chemo- and regioselective asymmetric synthesis of cyclic enamides through the catalytic umpolung organocascade reaction of α-imino amides. J. Org. Chem. 2019;84:7362–7371. doi: 10.1021/acs.joc.9b01036. [DOI] [PubMed] [Google Scholar]
- 56.Peng Q, et al. N-Heterocyclic carbene-catalyzed enantioselective hetero-[10+2] annulation. Commun. Chem. 2020;3:177. doi: 10.1038/s42004-020-00425-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng Q, Zhang B, Xie Y, Wang J. Carbene-catalyzed [4+2] annulation of 2H-azirine-2-carboxaldehydes with ketones via azolium Aza-dienolate intermediate. Org. Lett. 2018;20:7641–7644. doi: 10.1021/acs.orglett.8b03378. [DOI] [PubMed] [Google Scholar]
- 58.Wu J, Lang M, Wang J. Photoredox-catalyzed cross-coupling of enamides for the assembly of β-difluoroimine synthons. Org. Lett. 2017;19:5653–5656. doi: 10.1021/acs.orglett.7b02809. [DOI] [PubMed] [Google Scholar]
- 59.Wang X, Wu Z, Wang J. α-Fluoroallenoate synthesis via N-heterocyclic carbene-catalyzed fluorination reaction of alkynals. Org. Lett. 2016;18:576–579. doi: 10.1021/acs.orglett.5b03615. [DOI] [PubMed] [Google Scholar]
- 60.Li F, Wu Z, Wang J. Oxidative enantioselective α-fluorination of aliphatic aldehydes enabled by N-heterocyclic carbene catalysis. Angew. Chem. Int. Ed. 2015;54:656–659. doi: 10.1002/anie.201409473. [DOI] [PubMed] [Google Scholar]
- 61.Zhang B, Peng Q, Guo D, Wang J. NHC-catalyzed radical trifluoromethylation enabled by Togni reagent. Org. Lett. 2020;22:443–447. doi: 10.1021/acs.orglett.9b04203. [DOI] [PubMed] [Google Scholar]
- 62.Callejo R, et al. Fluorinated musk fragrances: the CF2 group as a conformational bias influencing the odour of civetone and (R)‐muscone. Chem. Eur. J. 2016;22:8137–8151. doi: 10.1002/chem.201600519. [DOI] [PubMed] [Google Scholar]
- 63.Shendage DM, Fröhlich R, Haufe G. Highly efficient stereoconservative amidation and deamidation of α-amino acids. Org. Lett. 2004;6:3675–3678. doi: 10.1021/ol048771l. [DOI] [PubMed] [Google Scholar]
- 64.Collins J, et al. In Situ conjugation of dithiophenol maleimide polymers and oxytocin for stable and reversible polymer–peptide conjugates. Bioconjugate. Chem. 2015;26:633–638. doi: 10.1021/bc5006202. [DOI] [PubMed] [Google Scholar]
- 65.Qu Q, et al. Synthesis of disulfide surrogate peptides incorporating large-span surrogate bridges through a native-chemical-ligation-assisted diaminodiacid strategy. Angew. Chem. Int. Ed. 2020;59:6037–6045. doi: 10.1002/anie.201915358. [DOI] [PubMed] [Google Scholar]
- 66.Anderson ME. Glutathione: an overview of biosynthesis and modulation. Chem. Biol. Interact. 1998;111:1–14. doi: 10.1016/S0009-2797(97)00146-4. [DOI] [PubMed] [Google Scholar]
- 67.Hunter L, Butler S, Ludbrook SB. Solid phase synthesis of peptides containing backbone-fluorinated amino acids. Org. Biomol. Chem. 2012;10:8911–8918. doi: 10.1039/c2ob26596f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
For 1H NMR, 13C NMR, and 19F NMR spectra see Supplementary Figs. 1–75 and highperformance liquid-chromatography spectra see Supplementary Figs. 76–118.The X-ray crystallographic coordinates for Fmoc-2l (Supplementary Data 1) reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 2102965. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.