

Abstract
Inherited genetic variations in the melanocortin-1 receptor (MC1R) responsible for human red hair color (RHC-variants) are associated with impaired DNA damage repair and increased melanoma risk. MC1R signaling is critically dependent on palmitoylation, primarily mediated by the protein acyltransferase ZDHHC13. A better understanding of how ZDHHC13 is physiologically activated could help identify approaches to prevent melanomagenesis in red-heads. Here we report that AMPK phosphorylates ZDHHC13 at S208 to strengthen the interaction between ZDHHC13 and MC1R-RHC, leading to enhanced MC1R palmitoylation in red-heads. Consequently, phosphorylation of ZDHHC13 by AMPK increased MC1R-RHC downstream signaling. AMPK activation and MC1R palmitoylation repressed UVB-induced transformation of human melanocytes in vitro and delayed melanomagenesis in vivo in C57BL/6J-MC1R-RHC mice. The importance of AMPK to MC1R signaling was validated in human melanomas where AMPK upregulation correlated with expression of factors downstream from MC1R signaling and with prolonged patient survival. These findings suggest AMPK activation as a promising strategy to reduce melanoma risk, especially for individuals with red hair.
Significance:
Phosphorylation of ZDHHC13 by AMPK at S208 promotes MC1R activation and suppresses melanocyte transformation, indicating activation of AMPK as a potential approach to prevent melanoma in people with red hair.
Keywords: MC1R, AMPK, palmitoylation, red hair, melanomagenesis
Introduction
Skin cancer is the most common cancer in the United States, with about 5 million cases every year (1). Melanoma is less common than other types of skin cancer, including basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), but it causes the majority of skin cancer deaths (1). Melanoma risk is associated with environmental factors, such as exposure to ultraviolet irradiation (UVR), and genetic background. Notably, the central regulator of pigmentation, the melanocortin-1 receptor (MC1R), is involved in melanoma development (2). MC1R is a G protein-coupled receptor (GPCR) and has a pivotal role in regulating hair and skin pigmentation (2). UVR stimulates the secretion of α-melanocyte-stimulating hormone (α-MSH), and the binding between MC1R and α-MSH activates cAMP signaling, enhances melanin synthesis, and promotes DNA repair (2).
The MC1R gene is highly polymorphic with more than 200 variants being described (3). Some of these variants are strongly correlated with phenotypes, including red hair, fair skin, freckling, inability to tan, and elevated risk of melanoma, which are defined as red-hair-color (RHC) variants (4). RHC variants are relatively common in people with ancient European ancestry, and it is estimated that more than half of the northern European population has the RHC variant (5). RHC variants have also been classified into strong (R) and weak (r) RHC alleles. The most common R variants are R151C, R160W, and D294H, as they are associated with more than 60% of all red hair phenotypes (6). These RHC variants impair MC1R signaling, stimulate pheomelanin production and make red-heads more susceptible to skin cancer (7,8). Individuals carrying one RHC variant have a nearly 40% increased risk of melanoma, while two or more RHC variants double the risk compared to wild-type MC1R (9).
Considering the central role of MC1R in pigmentation, RHC variants may increase melanoma risk partly through skin pigmentation as pheomelanin increases melanomagenesis through UVR-independent oxidative damage (7). However, dark-pigmented subjects with RHC variants also present an elevated risk of melanoma development (10). Studies show that MC1R plays additional non-pigmentary roles in melanomagenesis. For example, MC1R promotes efficient repair of the UVB-induced DNA damage products cyclobutane pyrimidine dimer (CPD) and 6–4 photoproduct (6–4PP) (11). In addition, MC1R regulates UVB-induced G1-like cell cycle arrest and subsequent onset of premature senescence, which when bypassed leads to the malignant transformation of melanocytes (12). Hence, we are interested in revealing the potential intervention strategies to directly activate MC1R signaling and reduce the melanoma incidence associated with RHC variants.
S-Palmitoylation is a reversible post-translational modification of GPCRs, in which a palmitic acid covalently attaches to a cysteine residue of the C-terminal tail or the intracellular loops and profoundly affects the structure, stability, subcellular localization, or protein-protein interactions. Zinc Finger DHHC-Type Palmitoyltransferase 13 (ZDHHC13) is the major palmitoyl acyltransferase (PAT) responsible for MC1R palmitoylation, which has been proved to be essential for activating MC1R signaling (8). Overexpression of ZDHHC13 or inhibition of palmitoyl-protein thioesterases (PPTs) enhances MC1R signaling and prevents RHC variants-associated increased risk of melanoma (8,13). However, treatment with PPT inhibitors may lead to a generic increase in palmitoylation of multiple signaling molecules and might not therefore be desirable as an intervention in a large-scale healthy population with MC1R variants. However, whether we can directly activate ZDHHC13 to enhance MC1R palmitoylation remains unknown.
Here we show that 5’-adenosine monophosphate (AMP)-activated protein kinase (AMPK) directly phosphorylates ZDHHC13 at S208 to strengthen its interaction with MC1R RHC variants and promote MC1R palmitoylation. AMPKα1 expression correlates with MC1R signaling and survival in human melanomas, and AMPK activation can rescue RHC variant-induced defects in MC1R signaling in vitro and in vivo to suppress UVR-induced melanomagenesis.
Material and methods
Immunoblots and immunoassays
Western blot, IP and the ABE palmitoylation assay were performed as described previously (8,12,13). Briefly, cells were lysed in buffer (50 mM Tris pH 7.4, 1% Triton X-100, 0.5 mM EDTA, 0.5 mM EGTA, 150 mM NaCl, 10% Glycerol) with Protease and Phosphatase Inhibitor Cocktail (Pierce #78446). The lysate was centrifuged at 15,000g (15 min, 4 °C), and the collected supernatant was precleared by 20 μl Protein G Agarose Beads (Thermo Scientific #20397). Samples were incubated with primary antibodies or anti-Flag/HA beads overnight at 4 °C. Anti-ZDHHC13 S208 phosphorylated rabbit polyclonal antibodies (HD16MA0305/0306) were generated by Sino Biological US Inc. Antibody used: Streptavidin-HRP (1:2000) (Thermo Scientific #21130), ZDHHC13 (1:1000) (Abcam #ab28759, RRID:AB_1603048), MC1R (1:1000) (Abcam #ab125031), MC1R (1:500) (Santa Cruz #sc-6875), AMPKα1/2 (1:1000) (Santa Crus #c-74461), β-Actin-HRP (1:20000) (Sigma #A5441), Flag-HRP (1:2000) (Sigma #A8592), HA-HRP (1:1000) (Sigma #H6533), anti-mouse secondary (1:2000) (Sigma #A4416), anti-rabbit secondary (1:2000) (Sigma #A-4914), AMPKα (1:1000) (Cell Signaling #5831), phospho-AMPK substrate motif (1:1000) (Cell Signaling #5759), phospho-T172-AMPKα(1:1000) (Cell Signaling #2535). All immunoblots shown are representatives of three independent experiments. Other related products: cAMP Direct Immunoassay Kit (Abcam #ab65355), Flag agarose beads (Sigma #A2220), HA agarose beads (Sigma #A2095).
In vitro kinase assay
Bacterially purified GST-ZDHHC13 protein was incubated with active recombinant full-length human AMPK (A1/B1/G2) kinase (Promega # V4012) in kinase buffer (pH 7.5, 25 mM MOPS, 1 mM EGTA, 0.1 mM Na3VO4, 15 mM MgCl2) supplemented with 300 μM AMP and 100 μM ATP (or ATP mix with γ−32P-ATP) for 60 min at 30 °C. Subsequent western blot and autoradiography were performed to detect the phosphorylation signal.
Cell culture and UV irradiation
HPMs were isolated from normal discarded foreskins as described previously (8). Isolated HPMs were cultured in Medium 254 with HMGS (Thermo Scientific #S0025). Cells were washed twice by PBS and exposed to UVB in the UVP Crosslinker CL-3000M (Analytik Jena). The emittance of UVB was confirmed by the UV photometer (UVP LLC). Cell lines, B16-F10 (CRL-6475-ATCC) and HEK293T (CRL-3216-ATCC), are purchased from and authenticated using short tandem repeat (STR) fingerprinting by ATCC, and tested mycoplasma negative using LookOut Mycoplasma Detection Kit (Sigma-Aldrich #MP0035). Cells were used for five to six passages from thawing after cryopreservation.
Recombinant DNA
MC1R and ZDHHC13 cDNAs were subcloned into pQCXIP (RRID: Addgene_15714) at the NotI/EcoRI sites as described previously (8,12). Flag-AMPKα1 (RC218572), Flag-AMPKα2 (RC210226) were purchased from Origene. Mutations were generated by QuickChange II Site-Directed Mutagenesis kit (Agilent). Human pLKO.1 lentiviral MC1R shRNA target set and Human pLKO.1 lentiviral AMPKα1 shRNA target set were purchased from Open Biosystems. The most efficient knockdown of shMC1R (RHS3979–201743320) and shAMPKα1 (RHS3979–201733131) were used in experiment. pLKO.1 lentiviral shRNAs were co-transfected with psPAX2 (RRID: Addgene_12260) and pMD2.G (RRID: Addgene_12259) in HEK293T cells using Lipofectamine 3000 (Thermo Scientific). Lentiviruses were collected after 48 h and used to infect cells for 24 h in the presence of 8 μg/mL polybrene. The infected cells were selected by 2 μg/mL puromycine. pQCXIP retroviral vectors were co-transfected with VSV-G and pUMVC (RRID: Addgene_8449) using Lipofectamine 3000. Retroviruses were collected after 48 h and cells were infected with retroviruses in the presence of 8 μg/mL polybrene. After 24 h, cells were selected by 2 μg/mL puromycine.
Melanocytes malignant transformation assay
Melanocytes malignant transformation detected by clonogenic survival and soft agar methods were performed as described previously (8,13,14). Briefly, for clonogenic survival assay, engineered hTERT/p53DD/CDK4(R24C)/BRAFV600E melanocytes were pre-treated with 1 μM α-MSH and irradiated by 20 J/m2 UVB, and seeded into 6-well plate at 1,000 cells/well, crystal violet was used to stain colonies 15 days after UVB irradiation. For soft agar assay, after α-MSH and UVB treatment, 10,000 engineered melanocytes were seeded into DMEM with 0.5% low-melting-point agarose and 10% FBS, layered onto DMEM with 0.8% agarose and 10% FBS. After 30 days, the agar was solubilized and the cells were quantified by CyQUANT™ Cell Proliferation Assay (Thermo Scientific), or the colonies >50 μm were counted under a light microscope.
Mouse experiment
All animal studies were approved by Institutional Animal Care and Use Committee (IACUC) of Boston University School of Medicine and Cleveland Clinic. Transgenic MC1R RHC-variant mice were generated by Boston University School of Medicine Transgenic and Genome Engineering Core (8) and Tyr-Cre-BRAFCA mice (B6.Cg-Braftm1Mmcm Tg(Tyr-cre/ERT2)Bos/BosJ) (Stock No: 017837) were purchased from The Jackson Laboratory. All mice were housed in pathogen-free facility of Animal Science Center (ASC) of Boston University Medical Campus and Animal core of Lerner Research Institute at Cleveland Clinic with a time cycle of 12 h of light and 12 h of dark and allowed free access to sterilized diet and water. The MC1RR151C mice show not only a paler coat color than WT, but also a mosaic coat color, which could arise as a consequence of the promoter used in transgene, sites of integration, or mouse strain background. All mice are monitored daily for signs of health status and distress and if they show any signs of distress, they will be euthanized. For the melanoma free survival analysis, mice were administered tamoxifen in corn oil by intraperitoneal injection at 75 mg per kg body weight for 5 consecutive days to active BRAFV600E. UVB treatment procedure was performed as described previously (8), the mice were irradiated one day per week for 500 J/m2 UVB treatment in a custom-made lucite chamber (Plastic Design Corporation) for 12 minutes. For the tumorigenesis assay, 3×106 engineered hTERT/p53DD/CDK4(R24C)/BRAFV600E melanocytes were mixed with matrigel by 1:1 and subcutaneously injected into the flanks of 8–10 weeks old female NCr nude mice (Taconic Biosciences). Tumor sizes were measured by caliper and did not exceed 20 mm at the largest diameter. Mice were randomly assigned to groups. Conditions that would lead to euthanasia include, but are not limited to, inability to ambulate, weight loss exceeding 20%, inability to eat or drink, severe dyspnea, obvious signs of discomfort, or other unexpected indications of distress upon consultation with a veterinarian. Sample size was determined by previous model-specific experience. The investigators were blinded to allocation during research.
Immunohistochemical staining and melanin staining
Immunohistochemical staining of mouse melanoma samples was performed as described previously (8,14,15). Briefly, mouse melanoma samples were fixed by 10% formalin at 4°C overnight. Then the samples were paraffin-embedded and cut into 5 μm thick sections. The tissue sections were heated in 10 mM sodium citrate buffer (pH 6.0) in the boiling water bath for 20 min for antigen retrieval and blocked by TBS with 0.1% Tween-20 and 5% normal goat serum. Then the tissue sections were incubated with anti-S-100 (1:200) (Dako North America, Inc.) at 4°C overnight, and followed by subsequent incubation of secondary antibody and processed with DAB substrate kit (Thermo Scientific). Lastly coverslips were mounted onto tissue sections by Permount. Melanin staining for mouse ears was performed using Abcam Fontana-Masson Stain Kit (ab150669) followed by the manufacturer’s protocol.
Bioinformatics analysis
Pearson’s correlation between different mRNA expressions in TCGA SKCM were calculated by GEPIA2 (Gene Expression Profiling Interactive Analysis, RRID:SCR_018294) (16). Survival curves were calculated by UCSC Xena platform using Kaplan-Meier method and log-rank test (17).
Quantification and statistical analysis
Unpaired (Two Sample) t-Test was performed for all quantitative data between indicated two groups, the statistical significance was labeled as *p<0.05, **p<0.01, ***p<0.001. The quantitative data were presented as the Mean ± SD or Mean ± SEM as labeled of at least three independent experiments.
Data availability
Mass spec data are available via ProteomeXchange with identifier PXD038934. All other data are available within the article and supplementary data files. Raw data are available upon reasonable request from the corresponding author.
Results
AMPK phosphorylates ZDHHC13 at Serine 208
ZDHHC13 is the major PAT of MC1R and serves as a critical regulator for MC1R function upon UVB irradiation (8) (Supplementary Fig. S1A). To better understand the regulatory mechanism of ZDHHC13 phosphorylation and how ZDHHC13 is controlled under different physiological conditions, we performed mass spectrometry to identify ZDHHC13 phosphorylation sites. The result revealed a novel phosphorylation site of ZDHHC13 at S208 (Supplementary Fig. S1B). By analyzing the protein sequence of ZDHHC13, we identified that S208 is highly conserved across various species (Fig. 1A), and is located in a potential AMPK substrate consensus motif located within the Ankyrin (ANK) domain of ZDHHC13 (Fig. 1B; Supplementary Fig. S1C). These results indicate that AMPK might phosphorylate ZDHHC13 at S208. To support this hypothesis, we identified by co-immununoprecipitation that endogenous AMPKα interacts with ZDHHC13 in Human Primary Melanocytes (HPMs) (Fig. 1C). When ectopically expressed, ZDHHC13 specifically binds to AMPKα1 but not AMPKα2 (Fig. 1D–E), indicating that the AMPKα1-containing AMPK complex is potentially the major isoform that regulates ZDHHC13 phosphorylation. To further confirm the analysis, we performed an in vitro kinase assay with bacterially purified GST-ZDHHC13 and AMPK heterotrimeric complex. The result revealed that full-length WT ZDHHC13, but not an S208A mutant, could be efficiently phosphorylated by purified AMPK complex (Fig. 1F). To better observe the endogenous level of ZDHHC13 S208 phosphorylation, we generated a pS208-ZDHHC13 phosphorylation-specific antibodies that recognized a ZDHHC13 peptide containing phosphor-S208, but not a non-modified peptide (Supplementary Fig. S1D–F). Consistent with AMPK phosphorylating S208, the S208A mutant largely impaired AMPKα1-induced ZDHHC13 phosphorylation by both using the AMPK substrate motif antibody and the pS208-ZDHHC13 phosphorylation-specific antibody (Fig. 1G). A769662 is a direct AMPK activator that mimics the effects of AMP that at 100 μM activates AMPK in cell culture (18), and as phosphorylation at T172 of the AMPKα subunit activates AMPK (19), AMPK pT172 is commonly used as an AMPK activation marker. Our results showed that A769662 activated AMPK and induced ZDHHC13 phosphorylation in HPMs (Supplementary Fig. S1G). Using the specific pS208-ZDHHC13 antibody, we found that activation of AMPK by the specific AMPK agonist A769662 induced ZDHHC13 pS208 phosphorylation in HPMs (Fig. 1H–I).
Figure 1.

AMPK phosphorylates ZDHHC13 at Serine 208. A, Sequence alignments of ZDHHC13 among different species containing conserved AMPK substrate motif. B, Alignments of identified AMPK substrates motif. C, IB analysis of whole-cell-lysate (WCL) and anti-AMPKα immunoprecipitates from HPMs. D-E, IB analysis of WCL, anti-Flag or anti-HA immunoprecipitates from HEK293T cells transfected with HA-ZDHHC13 and the indicated Flag-AMPK constructs. F, in vitro kinase assay showing bacterially purified ZDHHC13 could be phosphorylated by AMPK. ZDHHC13 protein was incubated with AMPK in kinase buffer supplemented with 100 μM γ−32P-ATP for 60 min at 30 °C. G, IB analysis of WCL, or anti-HA immunoprecipitates from HEK293T cells transfected with the indicated HA-ZDHHC13 constructs and Flag-AMPKα1. H, IB analysis of HPMs treated with 100 μM A769662 for the indicated time. I, IB analysis of HPMs treated with A769662 for 2 h and the indicated concentration.
Similar upregulation of ZDHHC13 pS208 phosphorylation was also observed in HPMs stimulated by the AMPK activator metformin (Supplementary Fig. S1H–I) and 2-DG that activates AMPK by blocking glycolysis to increase the intracellular AMP:ATP ratio (Supplementary Fig. S1J). Collectively, these evidence suggests that AMPK is a physiological upstream kinase that phosphorylates ZDHHC13 at S208.
AMPK promotes ZDHHC13-mediated MC1R palmitoylation
Palmitoylation of GPCRs is critical for its activation (20). As a GPCR, MC1R is palmitoylated at C315 in the C-terminus (8). As ZDHHC13 is the major PAT of MC1R, we next investigated whether AMPK-induced phosphorylation of ZDHHC13 affects MC1R palmitoylation. HPMs were infected with control shRNA or AMPKα1 shRNA, then pre-treated with α-MSH for 30 min and subjected to 100J/m2 UVB irradiation. By using the acyl-biotin exchange (ABE) assay, we showed that knockdown of AMPKα1 dramatically reduced MC1R palmitoylation (Fig. 2A; Supplementary Fig. S2A). Phosphorylation of Acetyl-CoA Carboxylase (ACC), a well characterized AMPK substrate, at S79, was used as a control for knockdown of AMPK activity. Consistently, treatment of Compound C, a widely used selective AMPK inhibitor, also significantly reduced the level of MC1R palmitoylation (Fig. 2B). To further address the role of AMPK activation in MC1R palmitoylation, we activated AMPK in HPMs with A769662, metformin or 2-DG. Using A769662 in the presence of α-MSH and UVB stimulation, the MC1R R151C variant exhibited an increased palmitoylation when AMPK was activated (Fig. 2C). A769662 also induced a moderate increase in palmitoylation of WT MC1R in HPMs (Fig. 2C), but as expected, no palmitoylation was detected in the MC1R C315S mutant. Similarly, metformin, or 2-DG, did not show strong effects on WT MC1R but both significantly increased the palmitoylation of the MC1R R151C variant (Supplementary Fig. S2B–C). Although WT ZDHHC13 led to an increase of the palmitoylation level of the MC1R R151C variant, this effect was abrogated by the ZDHHC13 S208A mutant (Fig. 2D); remarkably, S208 phospho-mimic mutant, ZDHHC13 S208D, further strengthened the palmitoylation level of the MC1R R151C variant compared to WT ZDHHC13 (Supplementary Fig. S2D), indicative of the crucial role of AMPK-mediated ZDHHC13 S208 phosphorylation in MC1R palmitoylation.
Figure 2.
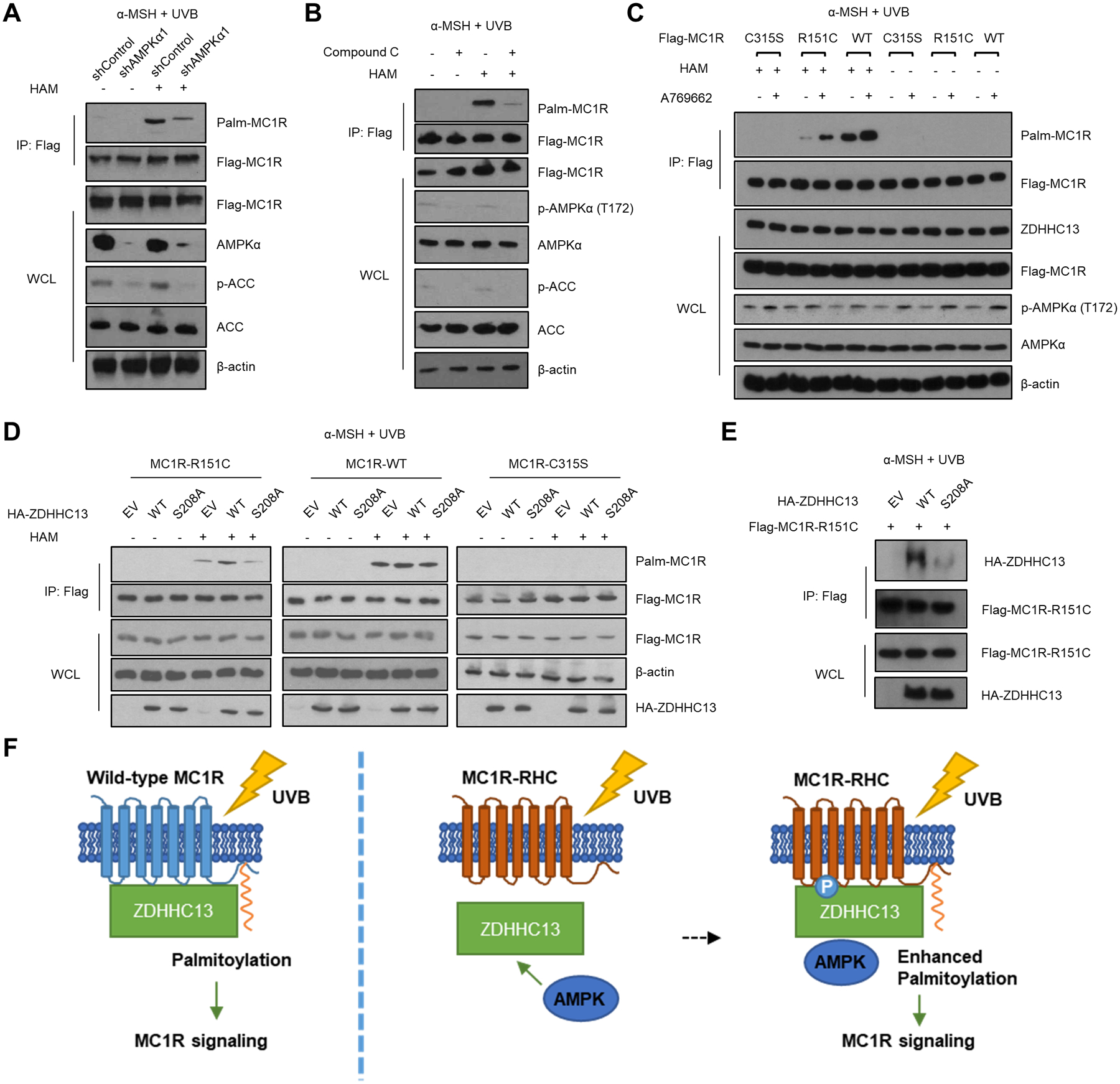
AMPK promotes ZDHHC13-mediated MC1R palmitoylation. A, HPMs with stable Flag-MC1R expression were infected shAMPKα1 or shControl. Then the cells were pre-treated with 1 μM α-MSH for 30 min followed by 100 J/m2 UVB irradiation. 3 h after UVB exposure, the treated cells were subjected for IP, ABE and IB analysis. B, HPMs with stable Flag-MC1R expression were treated with or without 10 μM AMPK inhibitor Compound C together with 1 μM α-MSH. Then the cells were irradiated with 100 J/m2 UVB 30 min after the drug treatment. 3 h after UVB exposure, the treated cells were subjected for IP, ABE and IB analysis. C, MC1R-depleted HPMs were infected with the indicated Flag-MC1R-encoding constructs. Then the cells were pre-treated with 1 μM α-MSH and 100 μM A769662 for 30 min followed by 100 J/m2 UVB irradiation. Finally, the cells were harvested for IP, ABE and IB analysis 3 h after UVB exposure. D, MC1R-depleted HPMs were infected with the indicated Flag-MC1R-encoding and HA-ZDHHC13 constructs. Then the cells were pre-treated with 1 μM α-MSH for 30 min followed by 100 J/m2 UVB irradiation. 3 h after UVB exposure, the treated cells were subjected for IP, ABE and IB analysis. E, MC1R-depleted HPMs were infected with Flag-MC1R-R151C and the indicated HA-ZDHHC13 constructs. Then the cells were pre-treated with 1 μM α-MSH for 30 min followed by 100 J/m2 UVB irradiation. 3 h after UVB exposure, the treated cells were subjected for IP and IB analysis. F, Schematic illustration of proposed model. AMPK-mediated phosphorylation of ZDHHC13 at S208 strengthen the binding between ZDHHC13 and MC1R RHC-variant.
Consistent with a role of S208 phosphorylation on ZDHHC13-mediated MC1R palmitoylation, knockdown of AMPKα1, or inhibition of AMPK by Compound C, both moderately reduced the interaction between MC1R and ZDHHC13 (Supplementary Fig. S2E–F). By contrast, activation of AMPK by A769662, metformin or 2-DG, all increased the interaction between ZDHHC13 and the MC1R R151C variant (Supplementary Fig. S2G–I), whereas little effect was observed using WT MC1R. Furthermore, S208A-ZDHHC13 failed to efficiently interact with R151C variant (Fig. 2E). The conserved S208 site is located in the Ankyrin (ANK) domain of ZDHHC13 (Fig. 1A), which often mediates protein-protein interaction (21). Taken together, these data are consistent with ZDHHC13 S208 phosphorylation enhancing MC1R/ZDHHC13 interaction and being critical for MC1R palmitoylation (Fig. 2F).
Phosphorylation of ZDHHC13 at S208 by AMPK enhances MC1R function
To test whether AMPK-induced phosphorylation of ZDHHC13 at S208 is essential for MC1R function, we first measured the levels of cAMP, a direct downstream signal activated by MC1R, in HPMs engineered to express WT, R151C variant, or C315S mutant forms of MC1R after UVB irradiation and α-MSH stimulation (1 μM). Whereas ectopic expression of WT ZDHHC13 significantly increased cAMP levels in HPMs bearing the RHC variant, the S208A mutation (Fig. 3A), or inhibition of AMPK by Compound C (Supplementary Fig. S3A), disrupted the effects of ZDHHC13 on cAMP production. In addition, the ZDHHC13 S208D mutant further increased cAMP levels in HPMs bearing the RHC variant compared to WT ZDHHC13 (Supplementary Fig. S3B). WT ZDHHC13 slightly increased the high basal level of cAMP in WT MC1R HPMs, while S208A ZDHHC13 showed marginal effects (Fig. 3A). The non-palmitoylatable MC1R C315S mutant was used as a negative control. By using the similar experimental setting, we also determined the mRNA expression of another two MC1R downstream effectors, the microphthalmia-associated transcription factor (MITF) which is a critical coordinator of melanocyte and melanoma biology, and its target DCT (Dopachrome Tautomerase or Tyrosine-Related Protein 2) (Supplementary Fig. S3C–D). As expected, the ZDHHC13 S208A mutant failed to enhance MITF or DCT expression in HPMs with the R151C variant (Supplementary Fig. S3C–D), while with WT MC1R the S208A mutation blocked the moderate stimulation of MITF and DCT mRNA expression mediated by WT ZDHHC13. These results suggest that phosphorylation of S208 at ZDHHC13 is important for stimulating signaling downstream from MC1R in melanocytes expressing an MC1R RHC-variant.
Figure 3.

Phosphorylation of ZDHHC13 at S208 by AMPK enhances MC1R function. A, MC1R-depleted HPMs were infected with the indicated Flag-MC1R and HA-ZDHHC13 constructs and then pre-treated with 1 μM α-MSH for 30 min followed by 100 J/m2 UVB irradiation. The cells were collected after 3 h for cAMP immunoassay. Three independent experiments were measured and calculated as mean ± SD, n=3. B, MC1R-depleted HPMs were infected with the indicated Flag-MC1R and HA-ZDHHC13 constructs and then pre-treated with 1 μM α-MSH for 30 min followed by 100 J/m2 UVB irradiation. Genomic DNA samples were collected at the indicated time points and CPD or 6–4PP were detected by ELISA. Three independent experiments were measured and calculated as mean ± SD, n=3. C-D, MC1R-depleted HPMs were infected with the indicated Flag-MC1R and HA-ZDHHC13 constructs and then pre-treated with 1 μM α-MSH for 30 min followed by 25 J/m2 UVB irradiation. Then the cells were subjected to SA-β-gal staining assay 7 days after UVB treatment. The quantification of SA-β-gal positive percentage and representative pictures were shown. Three independent experiments were measured and calculated as mean ± SD, n=3. E, MC1R-depleted HPMs were infected with the indicated Flag-MC1R-encoding constructs. Then the cells were pre-treated with 1 μM α-MSH and 100 μM A769662 for 30 min followed by 100 J/m2 UVB irradiation. The cells were collected after 3 h for cAMP immunoassay. Three independent experiments were measured and calculated as mean ± SD, n=3. F, MC1R-depleted HPMs were infected with the indicated Flag-MC1R-encoding constructs. Then the cells were pre-treated with 1 μM α-MSH and 100 μM A769662 for 30 min followed by 100 J/m2 UVB irradiation. Genomic DNA samples were collected at the indicated time points and CPD or 6–4PP were detected by ELISA. Three independent experiments were measured and calculated as mean ± SD, n=3. G, MC1R-depleted HPMs were infected with the indicated Flag-MC1R-encoding constructs. Then the cells were pre-treated with 1 μM α-MSH and 100 μM A769662 for 30 min followed by 25 J/m2 UVB irradiation. Then the cells were subjected to SA-β-gal staining assay 7 days after UVB treatment. The quantification of SA-β-gal positive percentage and representative pictures were shown. Three independent experiments were measured and calculated as mean ± SD, n=3.
To examine the role of ZDHHC13 phosphorylation in α-MSH/MC1R-regulated DNA repair, cyclobutane pyrimidine dimer (CPD), and 6–4 photoproducts (6–4 PPs) were examined following UVB irradiation of HPMs reconstituted with different forms of MC1R (Fig. 3B; Supplementary Fig. S3E). Although WT ZDHHC13 promoted photoproduct clearance in HPMs expressing the R151C variant, the S208A mutant form of ZDHHC13 failed to increase lesion repair (Fig. 3B) whereas little effect was observed of the S208A mutant on repair in the presence of WT MC1R or the non-palmitoylatable C315S mutant (Supplementary Fig. S3E). Consistent with previous work (8,13), ectopic expression of WT ZDHHC13 protein augments low dose UVB-induced premature senescence in HPMs (Fig. 3C–D) and B16 melanoma cells (Supplementary Fig. S3F) expressing an MC1R RHC-variant. However, the increase in senescence was largely dependent on phosphorylation of S208 of ZDHHC13; when S208 was mutated to alanine, ZDHHC13 exhibited only a minor effect on UVB-induced premature senescence (Fig. 3C–D; Supplementary Fig. S3F). These results suggested that phosphorylation on S208 is essential for ZDHHC13 to enhance MC1R RHC-variant function.
To further determine the role of AMPK activation in MC1R function, we treated cells with A769662 and measured cAMP levels, expression of MITF and DCT, UVB-induced photoproducts, and premature senescence. AMPK activation by A769662 significantly stimulated cAMP production (Fig. 3E), and MITF and DCT expression (Supplementary Fig. S3G) in HPMs expressing the MC1R R151C variant, and showed minimum effects on HPMs expressing WT or C315S mutant form of MC1R. A769662 also dramatically enhanced DNA damage repair and cleared UVB-induced CPD and 6–4PP photoproducts more efficiently in HPMs with the MC1R R151C variant (Fig. 3F) while little effect was observed in cells expressing MC1R WT or the C315S mutant (Supplementary Fig. S3H–I). In further support of the role of AMPK activation in MC1R function, A769662 rescued the defect of the MC1R R151C variant in bypassing UVB-induced premature senescence in both HPMs (Fig. 3G) and B16 melanoma cells (Supplementary Fig. S3J).
AMPK-mediated phosphorylation of ZDHHC13 at S208 attenuates MC1R-RHC-induced melanocyte malignant transformation
MC1R is a known tumor suppressor that protects melanocytes from UVB-induced cellular transformation, and ectopic expression of BRAFV600E cooperates with MC1R deficiency to drive malignant transformation in engineered human immortal melanocytes (hTERT/p53DD/CDK4(R24C)) (8,12,22). In this assay, hTERT/p53DD/CDK4(R24C) melanocytes require TPA and a cAMP agonist for survival, which is a hallmark of non-transformed melanocytes (22). Loss of, or deficient, MC1R cooperates with BRAFV600E to transform hTERT/p53DD/CDK4(R24C) melanocytes and stimulates cell growth in the absence of TPA and cAMP agonist (12). As such hTERT/p53DD/CDK4(R24C) melanocytes represent an appropriate model to study melanocyte transformation. To ask if phosphorylation of ZDHHC13 at S208 would affect UVB-induced melanomagenesis, we further engineered hTERT/p53DD/CDK4(R24C) immortalized human primary melanocytes to express WT, R151C, and C315S forms of MC1R with the additional expression of BRAFV600E and pretreatment with a low dose of UVB (20 J/m2). ZDHHC13 affected the palmitoylation level of the engineered melanocytes similarly compared to HPMs: an increase of the palmitoylation level of the MC1R R151C variant was induced by WT ZDHHC13, but not by the ZDHHC13 S208A mutant (Supplementary Fig. S4A). Clonogenic survival (Fig. 4A) and anchorage-independent soft agar growth assays (Fig. 4B; Supplementary Fig. S4B) indicated that the ZDHHC13 S208A mutant failed to diminish MC1R RHC-variant induced malignant transformation. To examine the effects of S208 phosphorylation of ZDHHC13 on malignant transformation in vivo, hTERT/p53DD/CDK4(R24C)/BRAFV600E/MC1RR151C melanocytes were subcutaneously inoculated into each flank of nude mice. Examination of xenograft tumor volumes (Fig. 4C) and weight (Fig. 4D) indicated that phosphorylation of ZDHHC13 at S208 is required to suppress MC1R RHC-variant induced malignant transformation in vivo.
Figure 4.
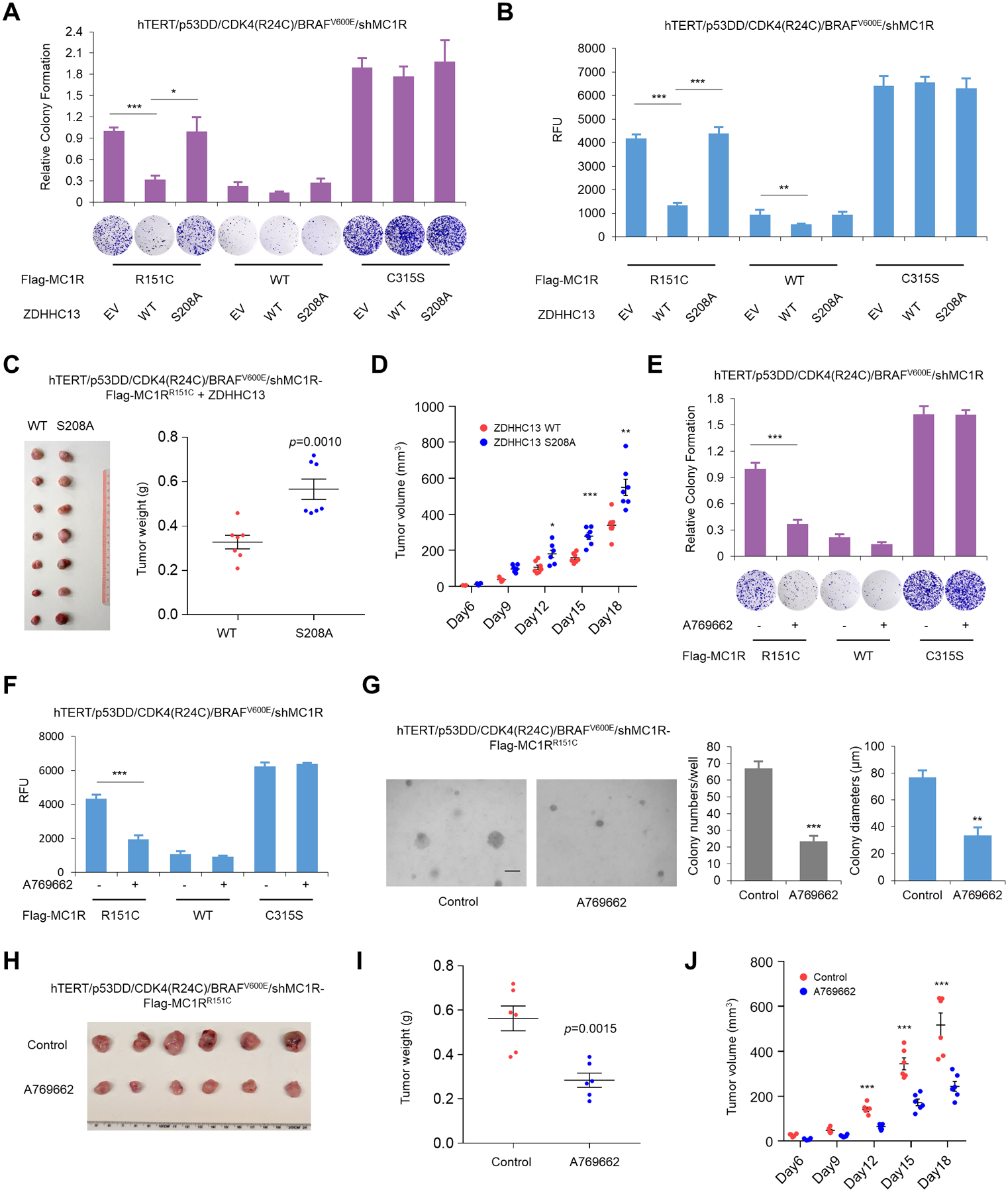
AMPK-mediated phosphorylation of ZDHHC13 at S208 attenuates MC1R-RHC-induced melanocyte malignant transformation. A, MC1R-depleted hTERT/p53DD/CDK4(R24C)/BRAFV600E melanocytes were infected with the indicated Flag-MC1R and HA-ZDHHC13 constructs. Then the cells were pre-treated with 1 μM α-MSH for 30 min before 20 J/m2 UVB irradiation. The Cells were subjected to clonogenic survival assay (crystal violet staining) 15 days after UVB treatment. Three independent experiments were measured and calculated as mean ± SD, n=3. B, MC1R-depleted hTERT/p53DD/CDK4(R24C)/BRAFV600E melanocytes were infected with the indicated Flag-MC1R and HA-ZDHHC13 constructs. Then the cells were pre-treated with 1 μM α-MSH for 30 min before 20 J/m2 UVB irradiation. Cells were finally seeded 10,000 cells per well in 0.5% low-melting-point agarose in DMEM + 10% FBS, layered onto 0.8% agarose in DMEM + 10% FBS. After 30 days, agar was solubilized and the cells were collected for quantification by CyQuant assay. Three independent experiments were measured and calculated as mean ± SD, n=3. C-D, MC1R-depleted hTERT/p53DD/CDK4(R24C)/BRAFV600E melanocytes were infected with Flag-MC1R-R151C and the indicated HA-ZDHHC13 constructs. Then the cells were pre-treated with 1 μM α-MSH for 30 min before 20 J/m2 UVB irradiation. After 24 h, 3×106 cells were inoculated subcutaneously into each flank of nude mice. Tumor growth and weight were measured. Error bars represent ±SEM. E, MC1R-depleted hTERT/p53DD/CDK4(R24C)/BRAFV600E melanocytes were infected with the indicated Flag-MC1R constructs. Then the cells were treated with 1 μM α-MSH and 100 μM A769662 for 30 min before 20 J/m2 UVB irradiation. The Cells were subjected to clonogenic survival assay (crystal violet staining) 15 days after UVB treatment. Three independent experiments were measured and calculated as mean ± SD, n=3. F-G, MC1R-depleted hTERT/p53DD/CDK4(R24C)/BRAFV600E melanocytes were infected with the indicated Flag-MC1R constructs. Then the cells were treated with 1 μM α-MSH and 100 μM A769662 for 30 min before 20 J/m2 UVB irradiation. Cells were finally seeded 10,000 cells per well in 0.5% low-melting-point agarose in DMEM + 10% FBS, layered onto 0.8% agarose in DMEM + 10% FBS. After 30 days, agar was solubilized and the cells were collected for quantification by CyQuant assay. Alternatively, colonies were also counted under a light microscope with the number of colonies (diameter > 50μm) per well and the average diameters (10 random fields). Three independent experiments were measured and calculated as mean ± SD, n=3. H-J, MC1R-depleted hTERT/p53DD/CDK4(R24C)/BRAFV600E melanocytes were infected with Flag-MC1R-R151C and the indicated HA-ZDHHC13 constructs. Then the cells were treated with 1 μM α-MSH and 100 μM A769662 for 30 min before 20 J/m2 UVB irradiation. After 24 h, 3×106 cells were inoculated subcutaneously into each flank of nude mice. Tumor growth and weight were measured. Error bars represent ±SEM.
We next treated hTERT/p53DD/CDK4(R24C)/BRAFV600E/shMC1R melanocytes that expressed WT, R151C variant, and C315S mutant MC1R with the AMPK activator A769662. ABE results showed that A769662 induced a moderate increase in palmitoylation of WT MC1R, but significantly increased palmitoylation of MC1R R151C variant in engineered melanocytes (Supplementary Fig. S4C). The clonogenic survival (Fig. 4E) and anchorage-independent soft agar growth assays (Fig. 4F–G; Supplementary Fig. S4D) showed that activation of AMPK by A769662 significantly suppressed MC1R RHC-variant-induced malignant transformation. Moreover, mouse xenograft studies using hTERT/p53DD/CDK4(R24C)/BRAFV600E/MC1RR151C melanocytes indicated the activation of AMPK by A769662 markedly inhibited MC1R R151C-variant-augmented malignant transformation (Fig. 4H–J). Together these data are consistent with AMPK-induced ZDHHC13 phosphorylation suppressing MC1R RHC-variant induced melanocytic malignant transformation.
Activation of AMPK inhibits UVB-induced melanomagenesis in redheads in vivo
Next, we further tested the effects of AMPK activation on MC1R in vivo by using previous generated MC1RR151C (8,13) and WT MC1R mice (Supplementary Fig. S5A). Mice ears, where epidermal melanocytes locate (23), were topically treated with 100 μL A769662 (100 μM) daily for 14 days, before processing by Fontana-Masson staining and biochemical melanin assays. Enhanced pigmentation was observed in the ears of A769662 treated MC1RR151C mice (Fig. 5A–B). Notably, pigmentation enhancement was not detected in MC1RWT mice (Fig. 5A–B). Downstream of MC1R signaling, Mitf, and Dct mRNA expression were also measured in mice ears treated with A769662 (Fig. 5C–D). The qRT-PCR result showed that AMPK activation by A769662 significantly increased the mRNA levels of Mitf and Dct in MC1RR151C mouse ears (Fig. 5C–D). No significant change was detected in MC1RWT mice. To investigate the potential role of AMPK activation in melanoma initiation, the Tyr-CreERT2-BRAFCA melanoma mouse model was used. Here we crossed B6.Cg-Braftm1MmcmTg(Tyr-cre/ERT2)13Bos/BosJ (24) with the MC1RR151C mice to generate Tyr-Cre-BRAFCA/ MC1RR151C mice. Tamoxifen was administrated to induce the activity of Tyr-Cre-ERT2 fusion to activate BRAFV600E expression. Mice were given a dose of 500 J/m2 UVB irradiation each week for four weeks and prior to UVB irradiation, 10 mg/kg A769662 or vehicle control were injected intraperitoneally into mice. Mice were then observed for melanoma incidence for 90 days (Supplementary Fig. S5B). Melanomas were observed in both UVB-exposed Tyr-Cre-BRAFCA/MC1RR151C mouse groups. No melanoma was detected in a Tyr-Cre mouse control group (Fig. 5E). In the treatment group, melanoma was first diagnosed 52 days after the final UV irradiation, while melanoma was first diagnosed 33 days after UVB treatment in the vehicle control group, indicating that activation of AMPK by A769662 delayed the onset of melanoma (Fig. 5E). 90 days after the final UVB irradiation, melanoma was diagnosed in 70% (7/10) and 30% (3/10) of mice with vehicle control and A769662 treatment, respectively (Fig. 5E). These data also suggest that A769662 significantly reduced melanoma incidence in UVB-induced Tyr-Cre-BRAFCA/ MC1RR151C mice (log-rank test, p=0.0452) (Fig. 5E). All diagnosed melanomas displayed similar morphological and histologic features (Fig. 5F). Therefore, these results are consistent with AMPK activation playing a critical role in melanoma prevention for redheads in vivo.
Figure 5.

Activation of AMPK inhibits UVB-induced melanomagenesis in redheads in vivo. A, Fontana-Masson Staining of indicated mouse ear that were topically treated with 100 μL A769662 for 14 days. B, Melanin quantification of indicated mouse ear samples. Three independent experiments were measured and calculated as mean ± SD, n=3. C-D, qRT-PCR were performed to measure the expression of Mitf and Dct in indicated mice ears. Three independent experiments were measured and calculated as mean ± SD, n=3. E, Melanoma-free survival of Tyr-Cre-BRAFV600E-MC1RR151C, n=10, Tyr-Cre-BRAFV600E-MC1RR151C + A769662, n=10. In the UVB radiation period, 10 mg/kg A769662 was injected intraperitoneally into mice before the UVB treatment. F, H&E staining of histological sections and immunohistochemistry staining of S100 of representative cutaneous melanomas. G-H, Pearson’s correlation between AMPKα1 and MITF or DCT in TCGA SKCM, plots show the correlations from RNA-seq data in TCGA melanoma calculated by GEPIA (Gene Expression Profiling Interactive Analysis).
To study the role of AMPK in human melanoma samples, we used GEPIA2 (Gene Expression Profiling Interactive Analysis) (16) and RNA sequencing (RNA-seq) data from the human TCGA melanoma cohort (Fig. 5G–H; Supplementary Fig. S5C–D). AMPKα1, encoded by PRKAA1 gene, is expressed in both melanoma and non-melanoma skin tissues, while AMPKα2 is expressed at a relatively low level (Supplementary Fig. S5C). We found that the mRNA levels of AMPKα1, not AMPKα2, positively correlated with MITF and DCT expression, which are downstream from MC1R signaling (Fig. 5G–H; Supplementary Fig. S5D). Further TCGA clinical data analysis using the Xena platform (17) showed that high mRNA levels of AMPKα1, not AMPKα2, are associated with a survival benefit in melanoma patients (Supplementary Fig. S5E–F). These results suggest that higher expression levels of AMPKα1 are correlated with more robust activation of MC1R signaling in human melanomas.
Discussion
AMPK is a conserved serine/threonine kinase and a classical sensor of energy homeostasis (19,25). AMPK is a heterotrimeric kinase complex consists of a catalytic subunit α (α1 and α1), a scaffolding subunit β (β1 and β2), and a regulatory subunit γ (γ1, γ2 and γ3). During the activation of AMPK, AMP binds to the γ-subunit and exposes the catalytic domain of the α-subunit (26). A cascade of AMPK-triggered phosphorylation events eventually inhibits ATP-consuming anabolic processes and stimulates ATP-producing catabolic processes. AMPK was first described for its role in lipid metabolism, and the role of AMPK in regulating metabolism is well known. The increase of ADP or AMP level will activate AMPK and AMPK downstream signaling. For example, AMPK activation leads to the phosphorylation and inactivation of acetyl CoA carboxylase (ACC), thus inhibiting fatty acid synthesis (27). AMPK is also known to suppress energy consuming glycolysis and promote oxidative respiration to restore energy production (28). Interestingly, loss of Zdhhc13 in mouse cortex and cerebellum increased glycolysis in vivo (29) and impaired mitochondrial function and reduced oxygen consumption were observed in hepatocytes from Zdhhc13-deficient mice and ZDHHC13 knockdown liver cells (30). Thus, AMPK may phosphorylate ZDHHC13 at S208 to regulate energy homeostasis physiologically.
It is well established that AMPK acts as a tumor suppressor because activation of AMPK restrains cell growth and division; however, AMPK may support the cancer cells to survive under many environmental stresses such as hypoxia and glucose deprivation, which often occur when cancer cells outgrow blood supply (31). Therefore, the role of AMPK in cancer prevention and treatment is context-dependent. An AMPK activator, OSU-53, significantly suppressed breast cancer growth (32). In contrast, AMPK supported the growth of H-RAS-transformed embryonic fibroblasts under hypoxia conditions (33). AMPK was found activated in human prostate cancers and increases prostate cancer cell proliferation (34).
In addition, AMPK activation may play important roles in melanoma. Metformin can suppress melanoma cell proliferation by activating autophagy and apoptosis, although the mechanism is unknown (35,36). BRAF is phosphorylated at S729 by AMPK, which promotes an interaction between BRAF and a 14–3-3 protein, leading to a suppression of MEK-ERK signaling (18). However, additional phosphorylation of S729 on oncogenic BRAFV600E mutant did not affect malignant transformation of NIH 3T3 cells (37,38). Furthermore, AMPK activation failed to affect the interaction between BRAFV600E and 14–3-3 protein in MEFs and did not interrupt MEK-ERK signaling (18). In our model, MC1R RHC-variant cooperates with BRAFV600E to drive malignant transformation of hTERT/p53DD/CDK4(R24C) melanocytes under UVB irradiation (8,13); for the first time, we identified ZDHHC13 as a novel substrate of AMPK, and demonstrated that AMPK activation disrupts RHC variant-induced melanocyte transformation in the context of BRAFV600E through ZDHHC13 phosphorylation at S208.
More palmitoylated proteins have been identified recently, many of which are shown to affect cancer metabolism. For example, ZDHHC4 and ZDHHC5 stimulate fatty acid intake via palmitoylating CD36 (39). Both ZDHHC7 and ZDHHC21 palmitoylate ERα at C451 and activate ERα to support the survival of breast cancer cells (40). ZDHHC7 also palmitoylates GLUT4 at C223 and increases glucose uptake in 3T3 cells (41,42). PD-L1, the critical checkpoint blockade therapy target, is palmitoylated by ZDHHC3 and palmitoylation stabilizes PD-L1 by blocking its ubiquitination (43). AKT is palmitoylated at C344 and palmitoylation-deficient AKT blocks AKT-dependent preadipocyte differentiation (44). Interestingly, metformin could suppress AKT palmitoylation, but only when macrophages were activated by LPS (45). Our studies previously identified ZDHHC13 as a major regulator of MC1R signaling in melanocytes (8,13). However, how activities of ZDHHCs are regulated remains largely unknown. Here, we provided a detailed mechanism to explain how AMPK phosphorylates ZDHHC13 at S208 and enhances its interaction with MC1R. The conserved S208 site is in the ANK domain which regulates protein-protein interaction (21). Our findings that AMPK-induced ZDHHC13 phosphorylation is indispensable for rescuing the defect of MC1R RHC-variant provides a novel intervention strategy for redheads to prevent UVB-induced melanomagenesis.
AMPK selects its substrate based on AMPK consensus motif. Optimally, AMPK prefers hydrophobic residues at −5 and +4 positions, and basic residues at −3 position of the actual phosphorylation site (46). Consistently, our identified ZDHHC13 S208 contains a Leucine (L) at −5 position and a Valine (V) at +4 position. Although ZDHHC13 S208 lacks a basic residue at −3 position, there are also known AMPK substrates that do not contain this AMPK consensus motif, such as JAK1 S518 containing a Serine (S) (47), HAT1 S190 containing a Glutamic acid (E) (28), and ARMC10 S45 containing a Serine (S) at −3 position (48). In addition, our site-specific pS208-ZDHHC13 antibody and in vitro kinase assay validated that AMPK indeed phosphorylates ZDHHC13 at S208.
Red-haired people are known for increased risk of melanoma. It is estimated that redheads make up one to two percent of the world’s population. For decades, scientists have aimed to modulate MC1R function for clinical benefit in redheads. The cyclic AMP agonist forskolin was shown to increase pigmentation and prevent melanomagenesis, but it is difficult for forskolin to penetrate the epidermis when topically applied (49). The identification of ZDHHC13/APT2 as essential regulators of MC1R function made depalmitoylation inhibitors a potential approach for redheads to prevent melanoma (8,13). However, depalmitoylation inhibitors may have side effects and treatment of inhibitors to healthy individuals may not be desirable. Our results define the critical role of AMPK-ZDHHC13-MC1R axis in melanocytes and suggest AMPK activation could be a novel target for melanoma prevention for redheads. There are many natural ways to activate AMPK effectively, including exercise and caloric restriction (50). Red-haired individuals may consider changing their lifestyle to maintain AMPK activation when exposed to UVR by increasing exercise and taking low calorie food. For non-redheads without any RHC-variants, it could be beneficial to avoid low AMPK status, which mainly arises as a consequence of lack of exercise, overabundance of nutrients, and metabolic disorders, to maintain normal MC1R activity to prevent UVB-induced DNA damage.
In summary, our study reveals the essential role of ZDHHC13 S208 phosphorylation induced by AMPK in melanomagenesis. We show that AMPK activation rescues MC1R RHC-variant-induced defect of MC1R signaling in vitro and in vivo. Our study advances our current understanding of how ZDHHC13 and MC1R signaling are maintained and provides a potential strategy to impede melanomagenesis for redheads.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grant K99/R00 CA234097 (S. Chen) and the Outrun the Sun Melanoma Research Scholar Program (S. Chen). C. R. Goding was supported by Ludwig Cancer Research. J. Arbesman is advisory board for Castle Biosciences and receives research funding to the institution from Castle Biosciences. No other potential conflicts of interest were disclosed.
Footnotes
Authors’ Disclosures
JA is advisory board for Castle Biosciences. The authors declare no other competing interests.
References
- 1.Guy GP Jr., Thomas CC, Thompson T, Watson M, Massetti GM, Richardson LC, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982–2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6 [PMC free article] [PubMed] [Google Scholar]
- 2.Guida S, Guida G, Goding CR. MC1R Functions, Expression, and Implications for Targeted Therapy. J Invest Dermatol 2022;142:293–302 e1 [DOI] [PubMed] [Google Scholar]
- 3.García‐Borrón JC, Sánchez‐Laorden BL, Jiménez‐Cervantes CJPcr. Melanocortin‐1 receptor structure and functional regulation. 2005;18:393–410 [DOI] [PubMed] [Google Scholar]
- 4.Schioth HB, Phillips SR, Rudzish R, Birch-Machin MA, Wikberg JE, Rees JL. Loss of function mutations of the human melanocortin 1 receptor are common and are associated with red hair. Biochem Biophys Res Commun 1999;260:488–91 [DOI] [PubMed] [Google Scholar]
- 5.Tagliabue E, Fargnoli MC, Gandini S, Maisonneuve P, Liu F, Kayser M, et al. MC1R gene variants and non-melanoma skin cancer: a pooled-analysis from the M-SKIP project. Br J Cancer 2015;113:354–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong TH, Rees JL. The relation between melanocortin 1 receptor (MC1R) variation and the generation of phenotypic diversity in the cutaneous response to ultraviolet radiation. Peptides 2005;26:1965–71 [DOI] [PubMed] [Google Scholar]
- 7.Mitra D, Luo X, Morgan A, Wang J, Hoang MP, Lo J, et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 2012;491:449–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen S, Zhu B, Yin C, Liu W, Han C, Chen B, et al. Palmitoylation-dependent activation of MC1R prevents melanomagenesis. Nature 2017;549:399–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wendt J, Rauscher S, Burgstaller-Muehlbacher S, Fae I, Fischer G, Pehamberger H, et al. Human determinants and the role of melanocortin-1 receptor variants in melanoma risk independent of UV radiation exposure. 2016;152:776–82 [DOI] [PubMed] [Google Scholar]
- 10.Han J, Kraft P, Colditz GA, Wong J, Hunter DJ. Melanocortin 1 receptor variants and skin cancer risk. Int J Cancer 2006;119:1976–84 [DOI] [PubMed] [Google Scholar]
- 11.Dong L, Wen J, Pier E, Zhang X, Zhang B, Dong F, et al. Melanocyte-stimulating hormone directly enhances UV-Induced DNA repair in keratinocytes by a xeroderma pigmentosum group A-dependent mechanism. Cancer Res 2010;70:3547–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao J, Wan L, Hacker E, Dai X, Lenna S, Jimenez-Cervantes C, et al. MC1R is a potent regulator of PTEN after UV exposure in melanocytes. Mol Cell 2013;51:409–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen S, Han C, Miao X, Li X, Yin C, Zou J, et al. Targeting MC1R depalmitoylation to prevent melanomagenesis in redheads. Nat Commun 2019;10:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu B, Chen S, Wang H, Yin C, Han C, Peng C, et al. The protective role of DOT1L in UV-induced melanomagenesis. Nat Commun 2018;9:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu B, Tang L, Chen S, Yin C, Peng S, Li X, et al. Targeting the upstream transcriptional activator of PD-L1 as an alternative strategy in melanoma therapy. Oncogene 2018 [DOI] [PubMed] [Google Scholar]
- 16.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 2017;45:W98–W102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 2020;38:675–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen CH, Yuan P, Perez-Lorenzo R, Zhang Y, Lee SX, Ou Y, et al. Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol Cell 2013;52:161–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012;13:251–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qanbar R, Bouvier M. Role of palmitoylation/depalmitoylation reactions in G-protein-coupled receptor function. Pharmacol Ther 2003;97:1–33 [DOI] [PubMed] [Google Scholar]
- 21.Foord R, Taylor IA, Sedgwick SG, Smerdon SJ. X-ray structural analysis of the yeast cell cycle regulator Swi6 reveals variations of the ankyrin fold and has implications for Swi6 function. Nat Struct Biol 1999;6:157–65 [DOI] [PubMed] [Google Scholar]
- 22.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005;436:117–22 [DOI] [PubMed] [Google Scholar]
- 23.Nordlund JJ, Collins CE, Rheins LA. Prostaglandin-E2 and Prostaglandin-D2 but Not Msh Stimulate the Proliferation of Pigment-Cells in the Pinnal Epidermis of the Dba/2 Mouse. Journal of Investigative Dermatology 1986;86:433–7 [DOI] [PubMed] [Google Scholar]
- 24.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr., et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 2009;41:544–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature cell biology 2011;13:1016–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology 2011;13:132–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett 1987;223:217–22 [DOI] [PubMed] [Google Scholar]
- 28.Marin TL, Gongol B, Zhang F, Martin M, Johnson DA, Xiao H, et al. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci Signal 2017;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Napoli E, Song G, Liu S, Espejo A, Perez CJ, Benavides F, et al. Zdhhc13-dependent Drp1 S-palmitoylation impacts brain bioenergetics, anxiety, coordination and motor skills. Sci Rep 2017;7:12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen LF, Chen YJ, Liu KM, Haddad ANS, Song IW, Roan HY, et al. Role of S-Palmitoylation by ZDHHC13 in Mitochondrial function and Metabolism in Liver. Sci Rep 2017;7:2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu CC, Peng D, Cai Z, Lin HK. AMPK signaling and its targeting in cancer progression and treatment. Semin Cancer Biol 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee KH, Hsu EC, Guh JH, Yang HC, Wang D, Kulp SK, et al. Targeting energy metabolic and oncogenic signaling pathways in triple-negative breast cancer by a novel adenosine monophosphate-activated protein kinase (AMPK) activator. J Biol Chem 2011;286:39247–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, et al. 5’-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol 2006;26:5336–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park HU, Suy S, Danner M, Dailey V, Zhang Y, Li H, et al. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol Cancer Ther 2009;8:733–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomic T, Botton T, Cerezo M, Robert G, Luciano F, Puissant A, et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis 2011;2:e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janjetovic K, Harhaji-Trajkovic L, Misirkic-Marjanovic M, Vucicevic L, Stevanovic D, Zogovic N, et al. In vitro and in vivo anti-melanoma action of metformin. Eur J Pharmacol 2011;668:373–82 [DOI] [PubMed] [Google Scholar]
- 37.Brummer T, Martin P, Herzog S, Misawa Y, Daly RJ, Reth M. Functional analysis of the regulatory requirements of B-Raf and the B-Raf(V600E) oncoprotein. Oncogene 2006;25:6262–76 [DOI] [PubMed] [Google Scholar]
- 38.Ritt DA, Monson DM, Specht SI, Morrison DK. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol 2010;30:806–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao L, Zhang C, Luo X, Wang P, Zhou W, Zhong S, et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J Hepatol 2018;69:705–17 [DOI] [PubMed] [Google Scholar]
- 40.Pedram A, Razandi M, Deschenes RJ, Levin ER. DHHC-7 and −21 are palmitoylacyltransferases for sex steroid receptors. Mol Biol Cell 2012;23:188–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren W, Sun Y, Du K. Glut4 palmitoylation at Cys223 plays a critical role in Glut4 membrane trafficking. Biochem Biophys Res Commun 2015;460:709–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du K, Murakami S, Sun Y, Kilpatrick CL, Luscher B. DHHC7 Palmitoylates Glucose Transporter 4 (Glut4) and Regulates Glut4 Membrane Translocation. J Biol Chem 2017;292:2979–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng 2019;3:306–17 [DOI] [PubMed] [Google Scholar]
- 44.Blaustein M, Piegari E, Martinez Calejman C, Vila A, Amante A, Manese MV, et al. Akt Is S-Palmitoylated: A New Layer of Regulation for Akt. Front Cell Dev Biol 2021;9:626404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiong W, Sun KY, Zhu Y, Zhang X, Zhou YH, Zou X. Metformin alleviates inflammation through suppressing FASN-dependent palmitoylation of Akt. Cell Death Dis 2021;12:934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaffer BE, Levin RS, Hertz NT, Maures TJ, Schoof ML, Hollstein PE, et al. Identification of AMPK Phosphorylation Sites Reveals a Network of Proteins Involved in Cell Invasion and Facilitates Large-Scale Substrate Prediction. Cell Metab 2015;22:907–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rutherford C, Speirs C, Williams JJ, Ewart MA, Mancini SJ, Hawley SA, et al. Phosphorylation of Janus kinase 1 (JAK1) by AMP-activated protein kinase (AMPK) links energy sensing to anti-inflammatory signaling. Sci Signal 2016;9:ra109. [DOI] [PubMed] [Google Scholar]
- 48.Chen Z, Lei C, Wang C, Li N, Srivastava M, Tang M, et al. Global phosphoproteomic analysis reveals ARMC10 as an AMPK substrate that regulates mitochondrial dynamics. Nat Commun 2019;10:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.D’Orazio JA, Nobuhisa T, Cui R, Arya M, Spry M, Wakamatsu K, et al. Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature 2006;443:340–4 [DOI] [PubMed] [Google Scholar]
- 50.Canto C, Auwerx J. Calorie restriction: is AMPK a key sensor and effector? Physiology (Bethesda) 2011;26:214–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spec data are available via ProteomeXchange with identifier PXD038934. All other data are available within the article and supplementary data files. Raw data are available upon reasonable request from the corresponding author.