

Abstract
VNPP433–3β (compound 2, (3β-(1H-imidazole-1-yl)-17-(1H-benzimidazole-1-yl)-androsta-5,16-diene), a multitarget anticancer agent has emerged as our lead next generation galeterone analogs (NGGA). Compound 2 is currently in development as potential new therapeutic for prostate and pancreatic cancers. The preliminary toxicity study reveals that the compound 2 was better tolerated by the normal male CD-1 mice than the male Nude mice. The maximum tolerated dose (MTD) in the Nude mice was estimated to be between 25 < 50 mg/kg. After oral dosing of compound 2 to male and female rats, the plasma concentration versus time curves were very consistent between animals and the AUClast increased with dose. Many plasmas concentration versus time curves profiles were nearly flat over 24 hr., suggesting extended absorption from the GI tract. Consequently, reliable values for half-life and AUCinf were not determined. Calculated oral bioavailability (using oral AUClast and excluding the outlier IV animal) ranged from 32–47%. This should be considered a minimum value since the contribution to true AUC beyond 24 hr. is clearly not zero. Clearly, these toxicology and pharmacokinetics parameters pave the way for understanding the anticancer pharmacological actions and provide a meaningful basis for further preclinical development and eventual clinical development.
Keywords: VNPP433–3β (2), anti-prostate cancer agent, toxicology, pharmacokinetics, oral bioavailability
Graphical Abstract

1. Introduction:
New treatment modalities are urgently needed to prolong the time to development of castration-resistant prostate cancer (CRPC), attack CRPC and metastatic disease (mCRPC) and to improve the quality of life of men suffering from advanced prostate cancer (PC). The progression to CRPC, metastatic disease and resistant to therapy correlates with gain-of-function of androgen receptor (AR) [both full-length (fAR) and splice variants, especially, AR-V7 and ARv567es] [1–8] and hyper-activation of eukaryotic translation initiation factor 4E (eIF4E), the nexus where the well-established PI3K/Akt/mTOR and RAS/MAPK oncogenic signaling pathways converge [9–13].
We recently developed clinical candidate Galeterone (Gal, 1, Fig. 1) which disrupts AR signaling via multiple mechanisms of action [reviewed in [14] ]. Gal progressed into pivotal Phase III clinical studies in patients with mCRPC [14–16]]. However, the disappointing termination of ARMOR3-SV trial and the required 2550 mg/day high therapeutic dose of Gal (because of its low oral bioavailability and short half-live) underscores the need to further systematic refinements to enable development of next generation galeterone analogs (NGGAs) with enhanced efficacies, improved pharmaceutical properties and high therapeutic indices at low dose-administration expected to result in safer, more effective PC therapy, in addition to lowering costs of drug production.
Fig. 1.
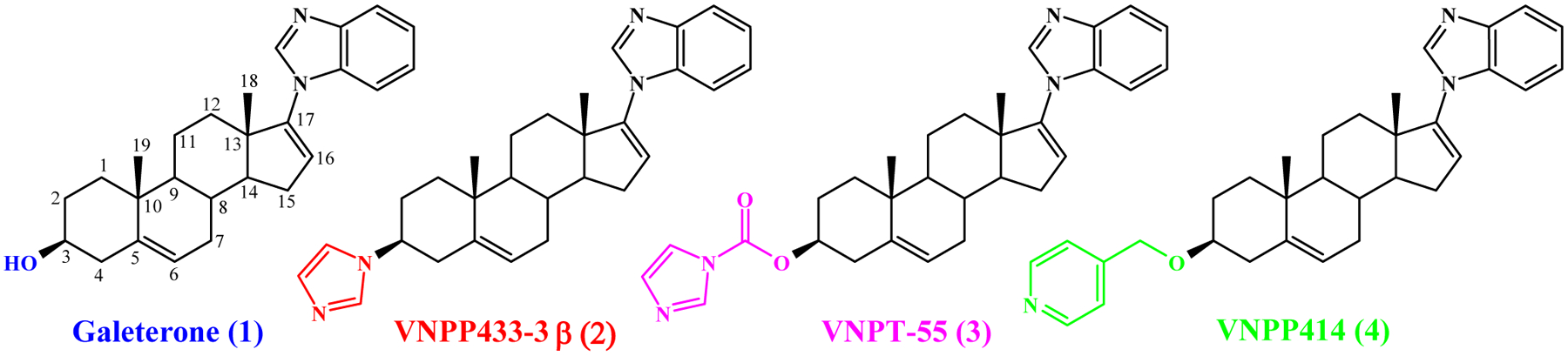
Chemical structures of Galeterone (1), VNPP433-3β (2), VNPT-155 (3) and VNPP414 (4)
During studies to develop more potent androgen receptor degraders (ARDs) to modulate AR signaling in PC models [17] , we modified structure of Gal at C3, C16 and C17 positions. Among the C3 modifications we designed the imidazole substituted compound (3β-(1H-imidazole-1-yl)-17-(1H-benzimidazole-1-yl)-androsta-5,16-diene (VNPP433–3β, 2, Fig. 1)) as an alternate moiety for the 3β-hydroxyl group of Gal to maintain hydrophilic interactions with active site of the biological targets.
We envisioned that this modification would avoid metabolic modification by steroidogenic enzyme, 3β-hydroxysteroid dehydrogenase (3βHSD). In our initial synthetic efforts to synthesize VNPP433–3β (2), we serendipitously discovered Gal 3β-imidazole carbamate (3, Fig. 1) [17]. We discovered that Gal and the novel more efficacious ARDs also effectively target oncogenic eukaryotic protein translation, via modulation of Mnk-eIF4E axis [18]. These agents also strongly suppress expression of oncogenic peIF4E via degradation of Mnk1 and 2 and as such are also referred to as Mnk1/2 degraders (MNKDs) (reviewed in [19]).
In recent studies, we successfully synthesized and evaluated VNPP433–3β and 3β-(pyridine-4-ylmethoxy)-17-(1H-benzimidazol-1-yl)androsta-5,16-diene (VNPP414, 4, Fig. 1) [20]. These two compounds exhibited improved in vitro anticancer activities over Gal [20], which led to additional studies that enabled improved procedures for their gram-scale syntheses [21]. Due to the chemical nature of the C-3 moieties (absence of Δ5, 3β-hydroxyl structure) of these two lead compounds (Fig. 1), we predicted that they are likely to be metabolically more stable than Gal. It is important to state here that in addition to our earlier report of the pharmacokinetics of Gal following subcutaneous (SC) administration to male SCID mice [22], there is now extensive literature on the pharmacokinetics and metabolism of Gal [23] and structurally related prostate cancer drug, abiraterone (Abi) [24, 25] in mice. These two compounds are metabolized mainly by two steroidogenic enzymes, 3βHSD and steroid-5α-reductase (SRD5A).
We have reported that compound 2 exhibited improved pharmacokinetic profile over Gal in mice and as expected, 2 (at 7.53-fold lower equimolar dose than Gal) markedly suppressed (84% vs. Gal, 47%; p < 0.01) the growth of castration-resistant PC (CRPC) CWR22Rv1 xenograft tumors, with no apparent host toxicity and was more efficacious than enzalutamide [26]. Because of the impressive and promising anti-PC activities, compound 2 was selected to receive targeted resources from National Institutes of Health’s National Cancer Institute (NCI) through a new program called ‘Stepping Stones’ to advance compound 2 towards clinical trials (https://dtp.cancer.gov/discovery_development/stepping_stones/default.htm). Recently, we reported the large-scale synthesis of Gal and VNPP433-3β (2), with support from the ‘Stepping Stones’ Program [27]. We have also reported that VNPP433-3β (2) inhibits PC stem cells by downregulating epithelial-mesenchymal transition and stem cell marked [28], and on its molecular mechanisms of action in AR-overexpressing CRPC in vitro and in vivo models [29].
In this manuscript, we report for the first time on the acute toxicity in male CD-1 normal male mice, acute and chronic toxicological profile of VNPP433-3β (2) in male Nude mice, the model used for the antitumor efficacy assessments, and the oral and intravenous pharmacokinetics and bioavailability in male and female rats.
2. Experimental section
2.1. Chemicals and reagents:
(3β-(1H-imidazole-1-yl)-17-(1H-benzimidazole-1-yl)-androsta-5,16-diene (VNPP433-3β, 2) was synthesized in our laboratory as previously reported [27, 30]. Abiraterone was purchased from Sigma-Aldrich, St. Louis, MO, USA.
2.2. Animal Studies
The studies with animals were conducted at three locations, including University of Maryland School of Medicine, Baltimore, USA, GVK Biosciences Pvt. Ltd. (now called Aragen Life Sciences Private Limited (Aragen), Hyderabad, India, and University of Pittsburgh, Pittsburgh, PA, USA
2.2.1. Animal handling at University of Maryland School of Medicine, Baltimore:
All animal studies were performed according to the guidelines approved by the Animal Care Committee of the University of Maryland, School of Medicine (UMSOM), Baltimore. Male Nude mice 5–6 weeks of age were obtained from the National Cancer Institute (Fredrick, MD) and were fed autoclaved pellets and sterile water ad libitum. Mice were allowed to acclimate to the UMSOM Animal Facility for at least 5 days before use.
2.2.2. Animal handling at GVK Bio:
The study was conducted at GVK BIO., Hyderabad, India, in accordance with Study Protocol No.: 045–16- IPH. This study was performed with approval from the Institutional Animal Ethics Committee (IAEC) in accordance with the requirement of Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), India. The study was not performed as per Good Laboratory Practices (GLP) regulations and was not audited by Quality Assurance; however, all appropriate study documentation including study plan and raw data are maintained in the study file. Study phases, generated data and the report were verified for accuracy by the study group. Male CD-1 mice 9–10 weeks old were used for the acute toxicology dosing study. Mice were allowed to acclimate to the GVK BIO Animal Facility for at least 5 days before use.
2.2.3. Animal handling at University of Pittsburgh:
Specific pathogen-free, adult Sprague Dawley male and female rats with catheterized jugular veins were purchased from Charles River (Wilmington, MA, USA). Rats were allowed to acclimate to the UPMC Hillman Cancer Center Animal Facility for at least 5 days before being used. To minimize infection, rats were maintained in micro-isolator cages in a separate room and handled in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996) and on a protocol approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Ventilation and airflow were set to 12 changes per hour. Room temperatures were regulated at 22 ± 1 °C, and rooms were kept on automatic 12 h light/dark cycles. Rats received Prolab ISOPRO RMH 3000 Irradiated Lab Diet (PMI Nutrition International, St. Louis, MO, USA) and water ad libitum. Rats were 8–12 weeks old at the time of dosing and were stratified into 3 groups of 3 animals each per sex. Rats received a single dose of compound 2 either 1 mg/kg IV, 10 mg/kg PO or 50 mg/kg PO (with curved blunt tip oral gavage needle) at a dosing volume of 10 ml/kg body weight. Drug was formulated as a solution in freshly prepared chremophor:ethanol:saline (1:1:8), pH 4.0 (with acetic acid). At 5, 15, 30 min, and 1, 2, 4, 6, and 24 h post-dose, EDTA blood was collected and processed to obtain plasma by centrifugation at 12,000 × g for 4 min.
2.3. Bioanalysis and Pharmacokinetics
Bioanalysis was performed with a generic assay approach as reported previously [31]. In brief, the assay utilized 50 μL of plasma and 8 potential internal standards (IS), of which abiraterone was selected for use. Preparation consisted of acetonitrile protein precipitation (150 μL) and aqueous dilution (125 μL supernatant and 500 μL water) in a 96 well-plate format. Chromatographic separation was achieved with a Kinetex C18 reverse phase (2.6 μm, 2 mm × 50 mm) column and a gradient of 0.1 % formic acid in acetonitrile and water over an 8 min run time. Mass spectrometric detection was performed on an AB SCIEX4000QTRAP with electrospray, positive-mode ionization. One microliter of supernatant was injected into the LC-MS/MS system. The approximate retention time of 2 was 2.71 min and for the internal standard (abiraterone) was 3.09 min, respectively. The MRM m/z transitions monitored were 439.2>371.2 for 2 and 350.0>156.2 for abiraterone. In both analytical runs, calibrators, and QC’s met FDA criteria. PK parameters were estimated using standard noncompartmental methods with Phoenix WinNonlin (Certara, Princeton, NJ).
3. Results and Discussions:
3.1. Acute and Chronic Toxicity Evaluation in Male Nude Mice
Prior to investigating the in vivo anti-tumor efficacy of compound 2, we evaluated the maximum tolerable doses that could be used in the study. First, we performed an acute dosing study, where male nude mice were injected intraperitoneally once with 25, 50 or 100 mg/kg body weight of 2 formulated in 40% β-cyclodextrin in saline and observed for two weeks. Considering the efficacy of 2 in vitro and the fact that it is several folds more efficacious than gal, we decided to use 100 mg/kg body weight as the highest dose. Body weights were taken twice a week and animals examined for any adverse effects (inactivity, skin changes, or bleeding). One of the three mice in the 100 mg/kg group died on the second day after injections. The rest of the animal in groups 25 and 50 mg/kg did not exhibit any signs of toxicity. From the overall average body weights and physical appearance of mice there were no apparent toxicities from the treatment (Fig. 2A).
Fig. 2:
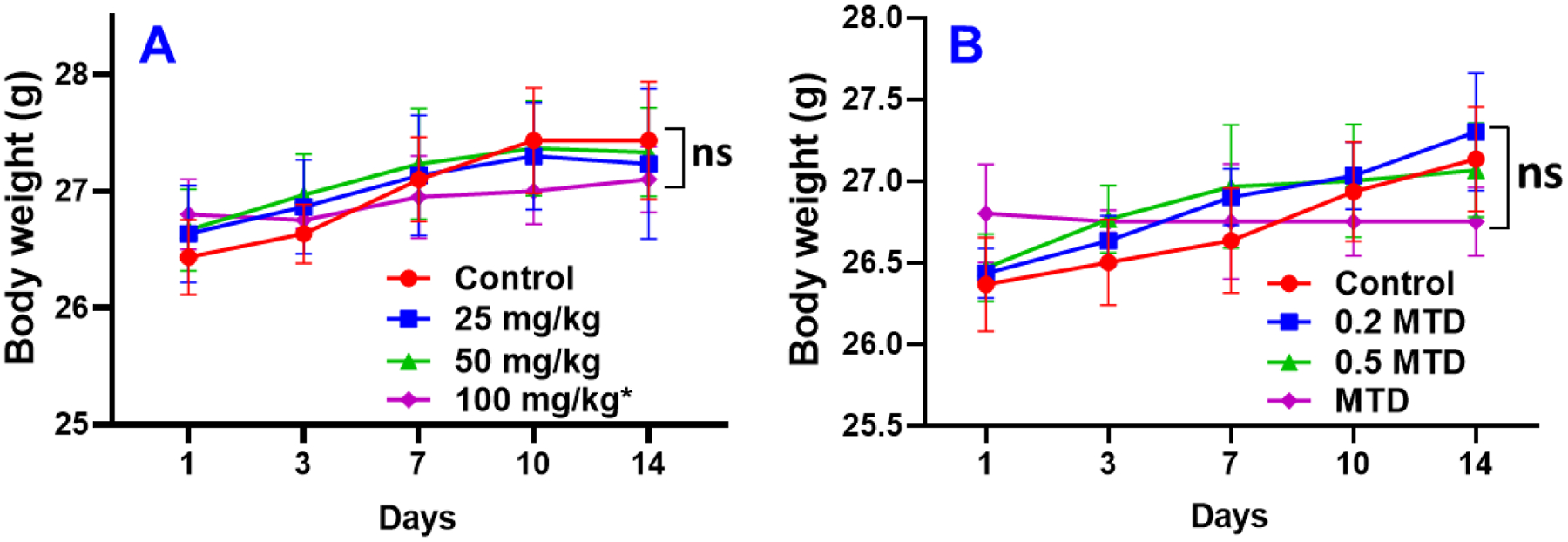
Mean body weights of nude mice (A) following treated with one bolus of compound 2 and (B) during chronic dosing of compound 2. Note: * It should be noted that one mouse died in the 100 mg/kg group and the mean body weight in this group was of two mice. ns = not significant.
Results from the acute dosing indicated that 100 mg/kg body weight of 2 was tolerated well by the two mice in that group. Hence, we decided to use 100 mg/kg as the maximum tolerable dose (MTD) for the next phase in determining the viable dose for in vivo studies. However, two astute reviewers brough to our attention that because one mouse died in the 100 mg/kg group, our assumption of 100 mg/kg MTD was incorrect. Based on the correct definition of MTD, the MTD of compound 2 in our acute dosing study is 50 mg/kg, and we have revised Fig. 2B accordingly. Thus, we performed a chronic dosing administering 0.2, 0.5 and MTD, five days a week for two weeks. Body weights were taken as before, twice a week and any adverse conditions noted (Fig. 2B). After 14 days, experiment was terminated, and animals sacrificed. Two mice died in the 0.5 MTD group on day 7. All other groups looked healthy, including the lone mice in group MTD. Based on the acute and chronic dosing results, we estimate that the MTD for 2 in nude mice is between 25 and <50 mg/kg body weight. However, more rigorous toxicology studies are required to characterize compound 2 adverse effects.
3.2. Acute Toxicity Evaluation in Male CD-1 Mice
Because the nude mice are immunocompromised, we conducted another MTD study in normal immunocompetent male CD-1 mice. Compound 2 was formulated in 20% β-cyclodextrin in saline, resulting in milky white thick suspensions. One vehicle control group and three dose groups (3 mice per group) were in the study i.e., low (100 mg/kg), medium (300 mg/kg), and high (1000 mg/kg) dose groups via oral gavage (PO). Animals were dosed 100 mg/kg and observed for 72 hrs. Body weights were taken on day 0, 1, 2, and 3 and any adverse conditions noted (Fig. 3). This dose was well tolerated, and no mortality was observed. Mice in the medium group were dosed 300 mg/kg and observed for 72 hrs. Thereafter, the high dose was opted, and animals were dosed with 1000 mg/kg and observed till mortality. All mice in the low and medium dose groups were normal after dosing. After 72 hrs., the animals were necropsied, and no signs of toxicity were observed. This study was stopped at day 3 based on the approved rat acute toxicity study of GVK BIO, a renown Certified Research Organization (CRO).
Fig. 3:
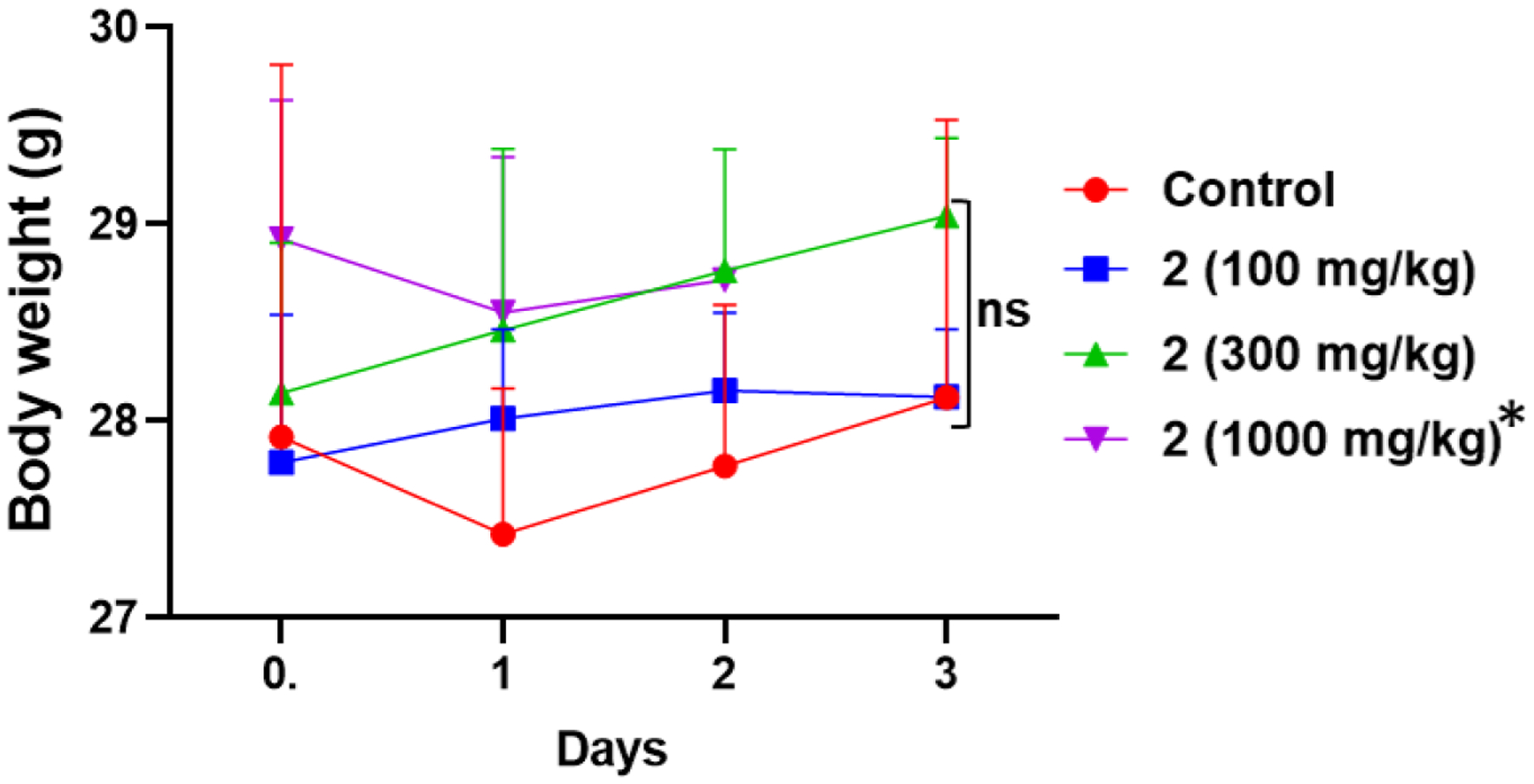
Mean body weights of CD-1 mice on day 0, 1, 2 and 3 following treatments (oral gavage, PO) with three doses of compound 2. * Note: For the group dosed with 1000 mg.kg of compound 2, two mice died on days 2 and the third mice died on day 3.
All three animals in the 1000 mg/kg group were normal after dosing. However, after 24 hrs. animals exhibited some clinical signs like piloerection, apathy (inactive) and hypothermia. Changes in gait were observed as well. Two mice were in a moribund stage and found dead 36 hrs. post dose, the third mice died 72 hrs. post dose. Dead animals were necropsied and accumulation of compound 2 in stomach and intestine was observed. Gross necropsy of mice in all three groups postmortem revealed no evident organ toxicity, however the clinical signs at 1000 mg/kg dose seemed toxic to animals. Thus, the NOAEL (No Observed Adverse Effect Level) was greater than 300 mg/kg via oral gavage. To further study 2 organ toxicity/safety profile, repeated dose toxicity studies will be conducted in the future.
3.3. Plasma concentration pharmacokinetic parameter in male and female rats
For IV dosing, the concentration versus time profiles of VNPP433-3β in male and female rats is presented in Fig. 4 and the PK parameters are presented in Table 1. No overt toxic effects were observed during conduct of this PK study. The third male rat dosed at 1 mg/kg IV had a strikingly deviant profile relative to the other 2 rats in that group. The rise in plasma levels is slower than that observed in rats dosed PO, and therefore likely represent extravascular dosing. Summary statistics therefore excluded this rat. After IV dosing, C0 values were similar for both sexes, confirming similar initial volumes of distribution. Half-lives were in the same order of magnitude between the sexes, with large variability in the males. AUC0-inf was moderately captured with AUC0-last. Clearance values were generally in the 5–7 mL/min/kg range with the male clearance being higher, agreeing with the shorter half-life. Volume of distribution was ~6 L/kg, suggesting extensive tissue distribution.
Fig. 4.
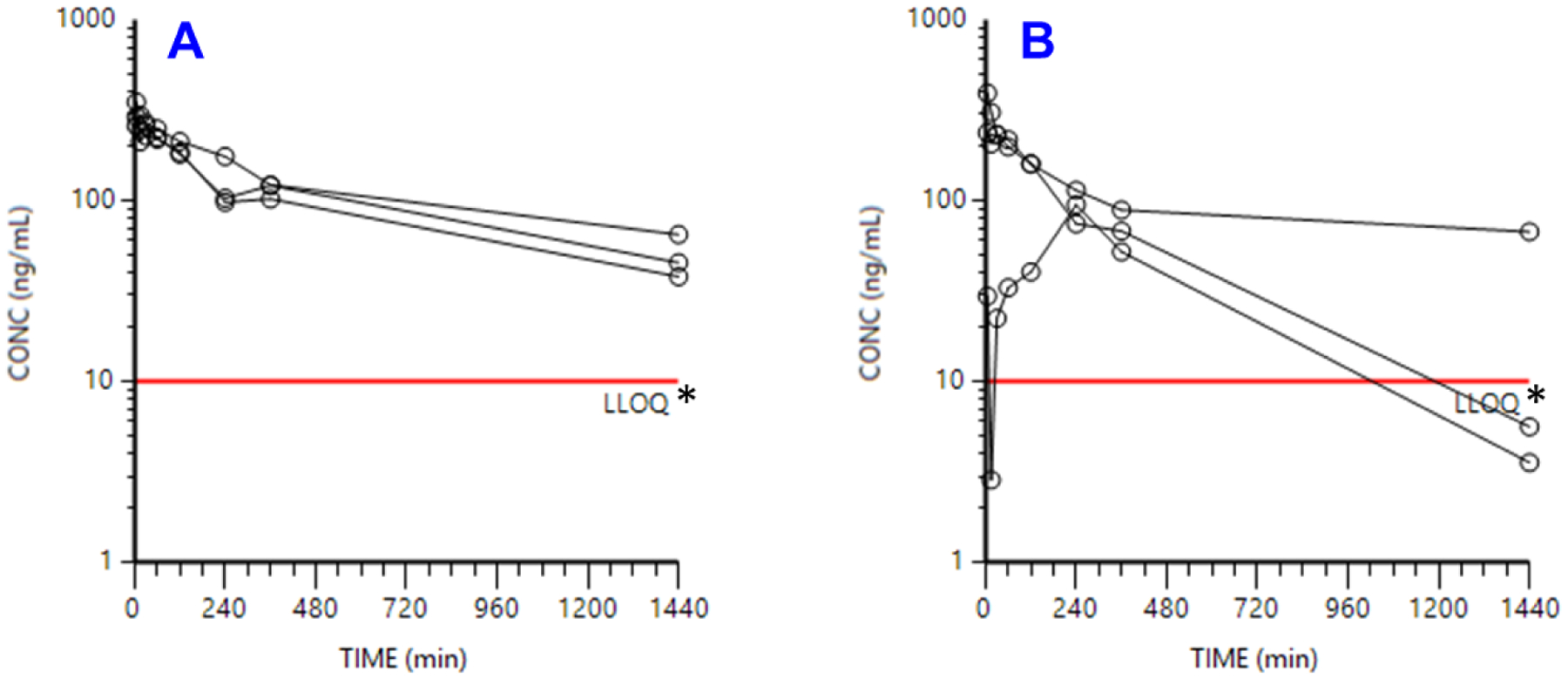
Concentration vs. time profiles of compound 2 dosed intravenously to (A) female and (B) male rats at 1 mg/kg. Note: * LLOQ = lower limit of quantification.
Table 1.
PK parameters of compound 2 dosed intravenously to male and female rats at 1 mg/kg.
| SEX | Dose (mg/kg) |
C0 (ng/mL) |
Cmax (ng/mL) |
Cmax/D ng/mL / mg/kg) |
Tmax (min) |
T1/2 (h) |
AUClast (min•μg/mL) |
AUCinf (min•μg/mL) |
CL (mL/min/kg) |
Vz (L/kg) |
Vss (L/kg) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| F | 1 | 259 | 296 | 296 | 15 | 14.1 | 168 | 248 | 4.04 | 4.93 | 4.96 |
| 342 | 291 | 291 | 5.0 | 15.0 | 140 | 199 | 5.03 | 6.51 | 5.99 | ||
| 418 | 350 | 350 | 5.0 | 13.8 | 126 | 171 | 5.85 | 6.98 | 6.31 | ||
| GeoMean | 333 | 311 | 311 | 7.21 | 14.3 | 144 | 203 | 4.92 | 6.08 | 5.73 | |
| GeoSD | 1.27 | 1.11 | 1.11 | 1.89 | 1.0 | 1.16 | 1.20 | 1.20 | 1.20 | 1.13 | |
| M | 1 | 254 | 237 | 237 | 5.0 | 2.96 | 46.6 | 64.0 | 15.6 | 4.00 | 4.14 |
| 443 | 392 | 392 | 5.0 | 31.4 | 139 | 321 | 3.11 | 8.46 | 8.14 | ||
| 29.7* | 94.8* | 94.8* | 240* | 2.32* | 20.2* | 30.6* | 32.7* | 6.55* | 11.0* | ||
| GeoMean | 336 | 305 | 305 | 5.00 | 9.64 | 80.4 | 143 | 6.97 | 5.82 | 5.81 | |
| GeoSD | 1.48 | 1.43 | 1.43 | 1.00 | 5.3 | 2.16 | 3.13 | 3.13 | 1.70 | 1.61 | |
Note:
Excluded from summary statistics. C0: extrapolated plasma concentration at time 0; Cmax: maximum observed plasma concentration; Tmax: time to maximum concentration; T1/2: elimination half-life; AUClast: area under the concentration-time curve from dosing (time 0) to the time of the last measured concentration; AUC (0−∞): area under the concentration-time curve from time of dosing extrapolated to infinity; CL: clearance; Vz: apparent volume of distribution during the terminal phase; Vss: steady state volume of distribution
The concentration versus time profiles of 2 in male and female rats following oral dosing at 10 mg/kg or 50 mg/kg is presented in Fig. 5 and the PK parameters are presented in Table 2. After oral dosing, PK was quite consistent as suggested by low geometric SD values. All profiles suggest continued absorption, likely through the last time point of 24 h. Not enough data is available to definitively conclude it, but there may be absorption rate limited elimination. In agreement with slightly higher clearance in males, indeed, the male PO profiles peak at 6 h, while the female PO profiles peak later. Bioavailability estimates were based on PO AUC0-last values, given that the extrapolated percentage of AUC0-inf was so large (17–82% and beyond). Even this underestimate of oral absorption suggested robust absorption of at least 32–34% (at 10 mg/kg) to 47% (at 50 mg/kg), with exact values likely much higher. Absorption was remarkably similar between sexes.
Fig. 5.
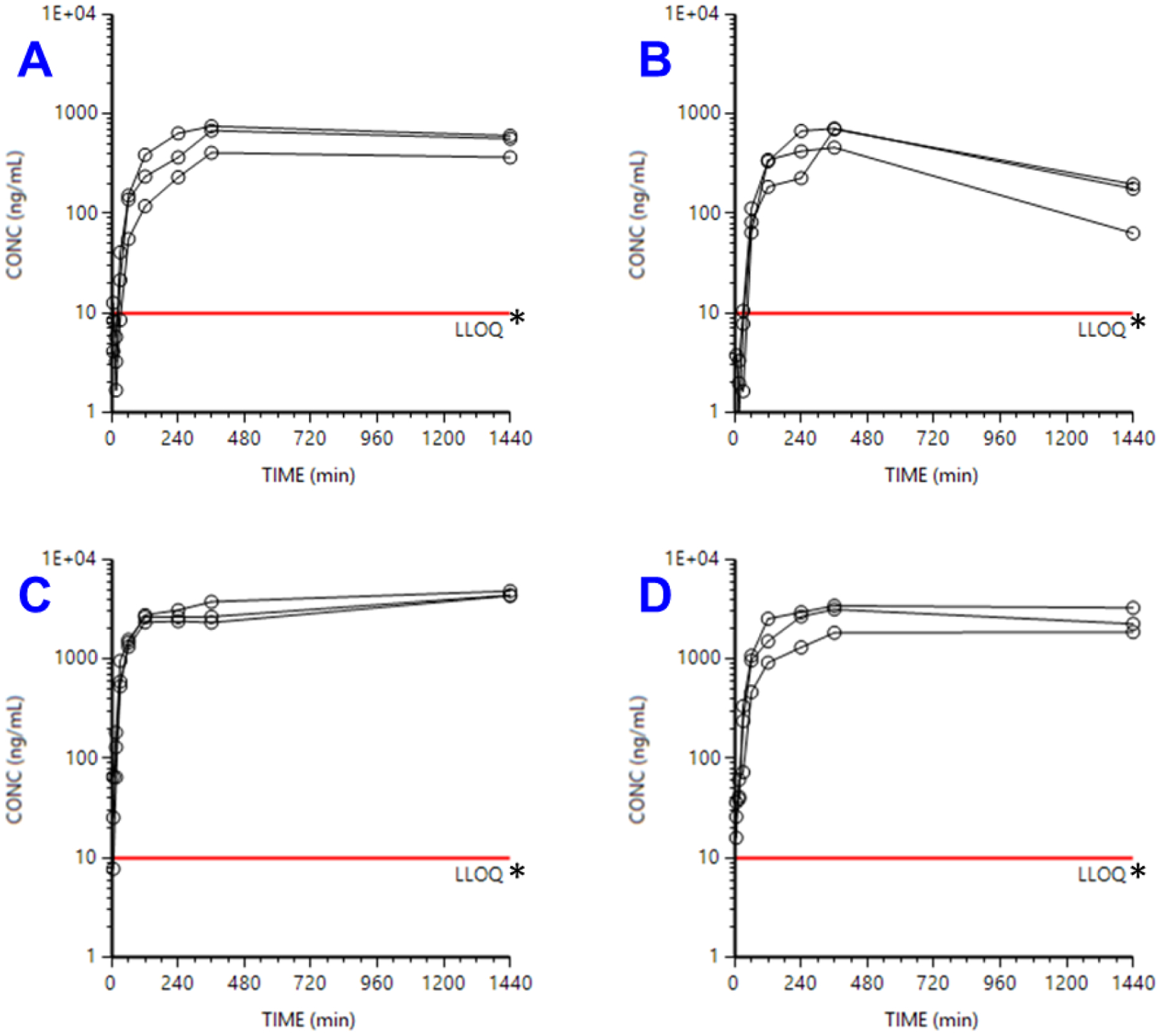
Concentration vs. time profiles of compound 2 dosed orally to female and male rats at 10 mg/kg (A and B) or 50 mg/kg (C and D). Note: * LLOQ = lower limit of quantification.
Table 2.
PK parameters of VNPP433-3β dosed orally to male and female rats at 10 or 50 mg/kg.
| SEX | DOSE_1 (mg/kg) |
Cmax (ng/mL) |
Cmax/D (ng/mL / mg/kg) |
Tmax (min) |
T1/2 (h) |
AUClast (min•μg/mL) |
AUCinf (min•μg/mL) |
AUC% Extrap (%) |
CL/F (mL/min/kg) |
Vz/F (L/kg) |
|---|---|---|---|---|---|---|---|---|---|---|
| F | 10 | 755 | 75.5 | 360 | 57 | 896 | 3870 | 76.9 | 2.58 | 12.7 |
| 408 | 40.8 | 360 | 120 | 484 | 4230 | 88.6 | 2.37 | 24.1 | ||
| 681 | 68.1 | 360 | 67 | 785 | 4090 | 80.8 | 2.45 | 14.3 | ||
| GeoMean | 594 | 59.4 | 360 | 76.7 | 698 | 4060 | 81.9 | 2.46 | 16.3 | |
| GeoSD | 1.39 | 1.39 | 1.00 | 1.5 | 1.38 | 1.05 | 1.07 | 1.05 | 1.41 | |
| F | 50 | 4820 | 96.4 | 1400 | * | 5560 | ||||
| 4320 | 86.4 | 1400 | * | 4300 | ||||||
| 4380 | 87.6 | 1400 | * | 4610 | ||||||
| GeoMean | 4500 | 90.0 | 1440 | 4800 | ||||||
| GeoSD | 1.06 | 1.06 | 1.00 | 1.14 | ||||||
| M | 10 | 702 | 70.2 | 360 | 9.8 | 519 | 686 | 24.4 | 14.6 | 12.4 |
| 460 | 46.0 | 360 | 6.3 | 329 | 364 | 9.55 | 27.5 | 15.0 | ||
| 715 | 71.5 | 360 | 9.0 | 577 | 715 | 19.3 | 14.0 | 10.9 | ||
| GeoMean | 613 | 61.3 | 360 | 8.22 | 462 | 563 | 16.5 | 17.8 | 12.6 | |
| GeoSD | 1.28 | 1.28 | 1.00 | 1.3 | 1.35 | 1.46 | 1.63 | 1.46 | 1.18 | |
| M | 50 | 1870 | 37.4 | 1400 | * | 2380 | ||||
| 3430 | 68.6 | 360 | 260 | 4460 | 78400 | 94.3 | 0.638 | 14.4 | ||
| 3160 | 63.2 | 360 | 37 | 3600 | 10900 | 66.9 | 4.60 | 14.8 | ||
| GeoMean | 2730 | 54.5 | 571 | 98.6 | 3370 | 29200 | 79.5 | 1.71 | 14.6 | |
| GeoSD | 1.39 | 1.39 | 2.23 | 4.0 | 1.38 | 4.04 | 1.27 | 4.04 | 1.02 | |
Note:
Negative half-life due to the last concentration still being higher than the preceding concentration, indicating more prolonged absorption of drug, resulting in incomplete capture of data with the sampling scheme used. C0: extrapolated plasma concentration at time 0; Cmax: maximum observed plasma concentration; Tmax: time to maximum concentration; T1/2: elimination half-life; AUClast: area under the concentration-time curve from dosing (time 0) to the time of the last measured concentration; AUC (0−∞): area under the concentration-time curve from time of dosing extrapolated to infinity; CL: clearance; Vz: apparent volume of distribution during the terminal phase; Vss: steady state volume of distribution.
4. Conclusion
Herein, we report for the first time on the acute toxicity in male CD-1 normal male mice, acute and chronic toxicological profile of compound 2 in male Nude mice, and the pharmacokinetics in male and female rats which have human-like physiology. The PK parameters for compound 2 in rats observed in this study are like those previously reported by us in mice [26]. Given the highly potent anticancer efficacy of compound 2 [26, 28, 32], the toxicity and PK profiles reported here provides confidence that the compound can be further developed as potential new anticancer drug.
Highlights:
A generic assay LC-MS/MS approach was utilized to detect the concentration of VNPP433-3β (2) in rat plasma.
Pharmacokinetics of VNPP433-3β (2), a potent anti-cancer agent, in male and female rats, has been accomplished.
The toxicity profile of VNPP433-3β (2) in CD-1 and Nude mice has been address.
The maximum tolerated dose of VNPP433-3β (2) in Nude mice has been determined.
ACKNOWLEDGEMENTS
We thank all the agencies for the financial support of this study. VCON is an Inaugural Distinguished University Professor at University of Maryland. Baltimore (UMB).
Funding
This work was supported in part by a grant from the National Institutes of Health (NIH) and the National Cancer Institute (NCI) (R01CA224696) awarded to VCON and NCI ‘Stepping-Stones’ Program (Contract HHSN261201600022I, control number N02CM-2016-00022 (NCI)), grant R50CA211241 (NCI) awarded to JHB. This project used the UPMC Hillman Cancer Center Cancer Pharmacokinetics and Pharmacodynamics Facility (CPPF) and was supported in part by award P30CA47904 (NCI).
List of Abbreviations/Acronyms and Their Definitions
- Abi
Abiraterone
- AR
Androgen Receptor
- ARD
Androgen Receptor Degraders
- AUC
Area Under the Curve
- CL
Clearance
- CRPC
Castration-Resistant Prostate Cancer
- EDTA
Ethylenediaminetetraacetic Acid
- eIF4E
Eukaryotic Translation Initiation Factor 4E
- fAR
Full-length Androgen Receptor
- Gal
Galeterone
- GI
Gastrointestinal
- IV
Intravenous dosing
- LC/MS/MS
Liquid Chromatography/Tandem-Mass Spectrometry
- LLOQ
Lower Limit of Quantification
- mCRPC
Metastatic Castration-Resistant Prostate Cancer
- Mnk1/2
MAP Kinase-Interacting Serine/Threonine-Protein Kinase 1 and 2
- MNKD
Mnk1/2 Degraders
- MS
Mass Spectrometry
- MTD
Maximum Tolerated Dose
- NGGA
Next Generation Galeterone analogs
- NOAEL
No Observed Adverse Effect Level
- PC
Prostate Cancer
- p-eIF4E
Phospho- Eukaryotic Translation Initiation Factor 4E
- PK
Pharmacokinetics
- PO
Oral Dosing
- SC
Subcutaneous
- SRD5A
Steroid-5α-Reductase
- T1/2
Elimination half-life
- Tmax
Time to Maximum Concentration
- Vss
Steady state volume of distribution
- Vz
Apparent volume of distribution during the terminal phase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
Vincent C. O. Njar is the lead inventor of VNPP433-3β, the patents and technologies thereof are owned by the University of Maryland, Baltimore. Puranik Purushottamachar is a co-inventor of VNPP433-3β. The other authors declare no potential conflict of interest.
References
- [1].Guo Z, Qiu Y, A new trick of an old molecule: androgen receptor splice variants taking the stage?!, Int J Biol Sci 7(6) (2011) 815–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AM, Edwards J, Qiu Y, A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth, Cancer Res 69(6) (2009) 2305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kahn B, Collazo J, Kyprianou N, Androgen receptor as a driver of therapeutic resistance in advanced prostate cancer, Int J Biol Sci 10(6) (2014) 588–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kong D, Sethi S, Li Y, Chen W, Sakr WA, Heath E, Sarkar FH, Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of EMT and expression of stem cell marker genes, Prostate 75(2) (2015) 161–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, Nelson PS, Montgomery RB, Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants, Clin Cancer Res 17(18) (2011) 5913–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schrader AJ, Schrader MG, Cronauer MV, Words of wisdom. Re: androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines, Eur Urol 64(1) (2013) 169–70. [DOI] [PubMed] [Google Scholar]
- [7].Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, Nelson PS, Plymate SR, Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant, J Clin Invest 120(8) (2010) 2715–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, Vakar-Lopez F, Vessella RL, Plymate SR, Androgen receptor variants occur frequently in castration resistant prostate cancer metastases, PLoS One 6(11) (2011) e27970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M, Gaboury LA, Pandolfi PP, Saad F, Sonenberg N, eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression, Proc Natl Acad Sci U S A 107(32) (2010) 14134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Graff JR, Konicek BW, Lynch RL, Dumstorf CA, Dowless MS, McNulty AM, Parsons SH, Brail LH, Colligan BM, Koop JW, Hurst BM, Deddens JA, Neubauer BL, Stancato LF, Carter HW, Douglass LE, Carter JH, eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival, Cancer Res 69(9) (2009) 3866–73. [DOI] [PubMed] [Google Scholar]
- [11].Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, Ferrari M, Egevad L, Rayford W, Bergerheim U, Ekman P, DeMarzo AM, Tibshirani R, Botstein D, Brown PO, Brooks JD, Pollack JR, Gene expression profiling identifies clinically relevant subtypes of prostate cancer, Proc Natl Acad Sci U S A 101(3) (2004) 811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, Dhanasekaran SM, Kalyana-Sundaram S, Wei JT, Rubin MA, Pienta KJ, Shah RB, Chinnaiyan AM, Integrative molecular concept modeling of prostate cancer progression, Nat Genet 39(1) (2007) 41–51. [DOI] [PubMed] [Google Scholar]
- [13].Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, Shah RB, Chandran U, Monzon FA, Becich MJ, Wei JT, Pienta KJ, Ghosh D, Rubin MA, Chinnaiyan AM, Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression, Cancer Cell 8(5) (2005) 393–406. [DOI] [PubMed] [Google Scholar]
- [14].Njar VC, Brodie AM, Discovery and development of Galeterone (TOK-001 or VN/124–1) for the treatment of all stages of prostate cancer, J Med Chem 58(5) (2015) 2077–87. [DOI] [PubMed] [Google Scholar]
- [15].McKay RR, Mamlouk K, Montgomery B, Taplin ME, Treatment With Galeterone in an Elderly Man With Castration-Resistant Prostate Cancer: A Case Report, Clin Genitourin Cancer 13(4) (2015) e325–e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Montgomery B, Eisenberger MA, Rettig MB, Chu F, Pili R, Stephenson JJ, Vogelzang NJ, Koletsky AJ, Nordquist LT, Edenfield WJ, Mamlouk K, Ferrante KJ, Taplin ME, Androgen Receptor Modulation Optimized for Response (ARMOR) Phase I and II Studies: Galeterone for the Treatment of Castration-Resistant Prostate Cancer, Clin Cancer Res 22(6) (2016) 1356–63. [DOI] [PubMed] [Google Scholar]
- [17].Purushottamachar P, Godbole AM, Gediya LK, Martin MS, Vasaitis TS, Kwegyir-Afful AK, Ramalingam S, Ates-Alagoz Z, Njar VC, Systematic structure modifications of multitarget prostate cancer drug candidate galeterone to produce novel androgen receptor down-regulating agents as an approach to treatment of advanced prostate cancer, J Med Chem 56(12) (2013) 4880–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kwegyir-Afful AK, Bruno RD, Purushottamachar P, Murigi FN, Njar VC, Galeterone and VNPT55 disrupt Mnk-eIF4E to inhibit prostate cancer cell migration and invasion, FEBS J 283(21) (2016) 3898–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].R S Ramamurthy VP, Kwegyir-Afful AK, Hussain A, Njar VCO, Therapeutic targeting of protein translation as a new paradigm for prostate cancer, Current Opinion in Oncology 29(3) (2017) 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Purushottamachar PK-A, K. A; Martin MS; Ramamurthy VP; Ramalingam S; Njar VCO, Identification of Novel Steroidal Androgen Receptor Degrading Agents Inspired by Galeterone 3β-Imidazole Carbamate, ACS Med. Chem.Lett 7 (2016) 708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Purushottamachar PM, F. N; Njar VCO, Improved Procedures for Gram-Scale Synthesis of Galeterone 3β-Imidazole and Galeterone 3β-Pyridine Methoxylate, Potent Androgen Receptor/Mnk Degrading Agents, Org. Process Res. Dev 20 (2016) 1654–1661. [Google Scholar]
- [22].Handratta VD, Vasaitis TS, Njar VC, Gediya LK, Kataria R, Chopra P, Newman D Jr., Farquhar R, Guo Z, Qiu Y, Brodie AM, Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model, J Med Chem 48(8) (2005) 2972–84. [DOI] [PubMed] [Google Scholar]
- [23].Alyamani M, Li Z, Berk M, Li J, Tang J, Upadhyay S, Auchus RJ, Sharifi N, Steroidogenic Metabolism of Galeterone Reveals a Diversity of Biochemical Activities, Cell Chem Biol 24(7) (2017) 825–832 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li Z, Alyamani M, Li J, Rogacki K, Abazeed M, Upadhyay SK, Balk SP, Taplin ME, Auchus RJ, Sharifi N, Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy, Nature 533(7604) (2016) 547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ, Sharifi N, Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer, Nature 523(7560) (2015) 347–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kwegyir-Afful AK, Ramalingam S, Ramamurthy VP, Purushottamachar P, Murigi FN, Vasaitis TS, Huang W, Kane MA, Zhang Y, Ambulos N, Tiwari S, Srivastava P, Nnane IP, Hussain A, Qiu Y, Weber DJ, Njar VCO, Galeterone and The Next Generation Galeterone Analogs, VNPP414 and VNPP433-3beta Exert Potent Therapeutic Effects in Castration-/Drug-Resistant Prostate Cancer Preclinical Models In Vitro and In Vivo, Cancers (Basel) 11(11) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Purushottamachar P, Thomas E, Thankan RS, Rudchenko V, Huang G, Njar VCO, Large-scale synthesis of galeterone and lead next generation galeterone analog VNPP433–3beta, Steroids 185 (2022) 109062. [DOI] [PubMed] [Google Scholar]
- [28].Thomas E, Thankan RS, Purushottamachar P, Huang W, Kane MA, Zhang Y, Ambulos N, Weber DJ, Njar VCO, Transcriptome profiling reveals that VNPP433-3beta, the lead next-generation galeterone analog inhibits prostate cancer stem cells by downregulating epithelial-mesenchymal transition and stem cell markers, Mol Carcinog 61 (2022) 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Thomas E, Thankan RS, Purushottamachar P, Huang W, Kane MA, Zhang Y, Ambulos NP, Weber DJ, Njar VCO, Novel AR/AR-V7 and Mnk1/2 Degrader, VNPP433–3beta: Molecular Mechanisms of Action and Efficacy in AR-Overexpressing Castration Resistant Prostate Cancer In Vitro and In Vivo Models, Cells 11(17) (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Purushottamachar P, Murigi Francis N., Njar Vincent C. O., Improved Procedures for Gram-Scale Synthesis of Galeterone 3β-Imidazole and Galeterone 3β-Pyridine Methoxylate, Potent Androgen Receptor/Mnk Degrading Agents, Organic Process Research & Development 20(9) (2016) 1647–1653. [Google Scholar]
- [31].Parise RA, Covey JM, Hollingshead MG, Srivastava AK, Synold TW, Beumer JH, Development and validation of an LC-MS/MS generic assay platform for small molecule drug bioanalysis, J Pharm Biomed Anal 203 (2021) 114185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Thomas E TR, Purushottamachar P, Njar VCO, Mechanistic insights on the effects of the lead next generation galeterone analog, VNPP433-3β in castration resistant prostate cancer., Molecular Cancer Therapeutics 20(12 Suppl.) (2021) Abstract nr LBA027. [Google Scholar]