

Abstract
Objective:
Photosensitivity is one of the most common manifestations of systemic lupus erythematosus (SLE), yet its pathogenesis is not well understood. Normal-appearing epidermis of patients with SLE exhibits increased UVB-driven cell death that persists in cell culture. Here we investigate the role of epigenetic modification and Hippo signaling in enhanced UVB-induced apoptosis seen in SLE keratinocytes.
Methods:
DNA methylation was analyzed using bisulfite sequencing from cultured SLE keratinocytes compared to healthy control (n=6, each). Protein expression was validated in cultured keratinocytes using immunoblotting and immunofluorescence. An immortalized keratinocyte line overexpressing WWC1 was generated via lentiviral vector. WWC1-driven changes were inhibited using a LATS1/2 inhibitor (TRULI) and siRNA. YAP-TEAD interaction was inhibited via overexpression of TEADi protein. Apoptosis was assessed using cl-caspase 3/7 and TUNEL staining.
Results:
Hippo signaling is the top differentially methylated pathway in SLE versus control keratinocytes. SLE keratinocytes show significant hypomethylation (Δβ = −0.153) and thus overexpression of the Hippo regulator WWC1 (p=0.002, n=6). WWC1 overexpression increases LATS1/2 kinase activation, leading to YAP cytoplasmic retention and altered pro-apoptotic transcription in SLE keratinocytes. Accordingly, UVB-mediated apoptosis in keratinocytes can be enhanced by WWC1 overexpression or YAP-TEAD inhibition, mimicking SLE keratinocytes. Importantly, chemical and siRNA-mediated LATS1/2 inhibition effectively eliminates enhanced UVB-apoptosis in SLE keratinocytes.
Conclusion:
Our work unravels a novel driver of photosensitivity in SLE: overactive Hippo signaling in SLE keratinocytes restricts YAP transcriptional activity, leading to shifts that promote UVB-apoptosis.
Keywords: lupus, autoimmunity, hippo signaling, photosensitivity
Graphical Abstract

INTRODUCTION
Development of rashes in response to ultraviolet (UV) light exposure, termed photosensitivity, is present in 57–73% of patients with systemic lupus erythematosus (SLE)(1). Photosensitivity causes significant morbidity due to skin symptoms, emotional distress, and functional impairment (1), and it can even trigger life-threatening systemic disease flares (2, 3). Despite this considerable health burden, the pathogenesis of photosensitivity is largely undescribed, hampering development of effective therapies.
Abnormalities in normal-appearing, non-lesional skin of patients with SLE may contribute to inflammation and enhanced keratinocyte apoptosis following UV light exposure(4). Recent studies of non-lesional SLE keratinocytes have shown that heightened cell death following UV exposure persists in cell culture (4), suggesting epigenetic changes may promote this phenotype. However, the role of epigenetics in SLE keratinocyte susceptibility to cell death has never been studied.
Hippo signaling regulates cell proliferation and apoptosis. When activated, Hippo signaling results in phosphorylation and cytosolic retention of the transcriptional coactivator YAP. This prevents YAP nuclear translocation and binding to TEAD family transcription factors, through which YAP drives expression of pro-proliferative genes (5). Investigations of Hippo signaling have primarily focused on cancer, including UV-induced skin cancers (6, 7). In skin, YAP is also required for epidermal cell proliferation, and its deletion promotes cell death (8). Upstream regulation of Hippo signaling is an area of intense study given the broad role of YAP in oncogenesis and thus the great potential for therapeutic targeting. The role of Hippo signaling in inflammatory skin diseases such as SLE/CLE has not previously been investigated.
In this study, we identified Hippo signaling as the most significantly dysregulated pathway in lupus keratinocytes by differential methylation analysis. We show that that the upstream Hippo regulator WWC1 is overexpressed in SLE keratinocytes, restricting YAP nuclear access. This diminishes YAP’s anti-apoptotic effects, leading to enhanced UV-mediated apoptosis in SLE keratinocytes. Overexpression of WWC1 in immortalized keratinocytes results in a phenocopy of SLE keratinocyte sensitivity to UV. Further, inhibition of YAP phosphorylation via multiple approaches attenuates SLE keratinocyte UV-mediated apoptosis. Altered Hippo signaling thus contributes to enhanced UV-mediated apoptosis in the epidermis of patients with SLE and may represent a tractable target in treating photosensitivity and preventing disease flares.
METHODS
Patient population
6 patients with SLE per revised ACR criteria (25) were recruited for the methylation analysis (Supplementary Table 1). Controls were matched for age, sex and self -reported race/ethnicity. Pre- and post- UVB and scRNA-seq studies, we used 10 SLE patients (defined above) and 6 healthy controls from our Taubman Institute Innovative Research Program Personalized Medicine through Integration of Immune Phenotypes in Autoimmune Skin Disease (TIIP-PerMIPA) cohort. 14 patients with psoriasis from the TIIP-PerMIPA cohort were used as disease control in the scRNA-seq analysis. Demographics for SLE patients and controls are in Supplementary Table 2. According to the declaration of Helsinki, all patients and controls gave written, informed consent prior to inclusion in the study.
DNA extraction and bisulfite conversion
DNA was extracted from primary KC at passage 1 using the DNeasy Blood and Tissue Kit (Qiagen, MD, USA) according to manufacturer’s instructions. DNA concentration was measured using a Qubit dsDNA Broad Range Assay Kit (Thermo Fisher Scientific, MA, USA). DNA was bisulfite converted for DNA methylation studies using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA) using the provided protocol and thermocycler settings recommended by Zymo for Illumina DNA methylation array samples included in the product literature.
DNA methylation profiling
Genome-wide DNA methylation analysis was performed on extracted KC DNA using the Infinium MethylationEPIC BeadChip Kit (Illumina, San Diego, CA). This array interrogates over 850,000 methylation sites (primarily CpG dinucleotides) allowing for a comprehensive genome-wide coverage. All array handling, sample hybridization, and array scanning was performed at the University of Michigan Advanced Genomics Core.
Methylation data processing
DNA methylation analysis was performed using the GenomeStudio (v2011.1) Methylation Module (v1.9.0; Illumina, San Diego, CA) as previously described (24). Red and green channel values were normalized to internal, non-CpG control probes and array background subtracted using unhybridized in-array control probes. Methylation beta values (β) were calculated on a scale of 0% to 100% methylation using normalized, background-corrected signal values. A probe-wise DiffScore was calculated using the Illumina Custom Model to report average differential methylation between SLE patients and healthy controls with adjustment for array-wide false discovery rate. To remove probes influenced by technical effects, we filtered out any probe with a single nucleotide polymorphism within 10bp of the 3’ end of the array probe. Probes with detection P-values < 0.05 were also removed. A significant difference in average DNA methylation (Δβ) for this study was any probe with a |Δβ| ≥ 15% and a |DiffScore| ≥ 22 which is equivalent to a false discovery rate (FDR) corrected P value < 0.01. Genes associated with differentially methylated CpG sites were used as input for gene set enrichment using Ingenuity Pathway Analysis (Qiagen, Germantown, MD, USA). Methylation data are publicly available in GEO with the accession number GSE198972.
Cell Culture
Primary human keratinocytes were established from SLE vs. control as previously described (26). Keratinocytes were cultured in keratinocyte-specific medium (Epilife with keratinocyte growth supplement) and carefully passaged at 60% confluency to avoid differentiation. Media was replaced every 2–3 days and keratinocyte culture purity was confirmed by morphology. Experiments were performed at primary keratinocyte passage 2–4. Immortalized keratinocytes, (N/TERTs)(27) were obtained with permission of Dr. James G. Rheinwald. N/TERT TEADi cells, which express GFP-tagged TEAD-interacting domains that bind to TEAD and antagonize YAP1 activity by blocking YAP1 binding to TEAD were kindly obtained from Dr. Ramiro Iglesias-Bartolome(18).
RNA-Sequencing
Libraries for RNA-seq were generated from polyadenylated RNA and sequenced at six libraries per lane on the Illumina Genome(28) Analyzer Iix. We used Tophat2(29) to align RNA-seq reads to the human genome, using annotations of GENCODE as gene model(29). HTSeq was used to quantify gene expression levels(29); normalization and differential expression analysis were performed by DESeq2(31).
Single cell RNA-sequencing
Samples with >80% viability was run on the 10X Genomics system (10X Single Cell Immune Profiling). Single-cell 3’ libraries were generated using the 10x V2 protocols as described, and sequencing was done using the Illumina NovaSeq6000 platform. For analysis, we used Cell-Ranger pipeline supported by 10X Genomics to conduct alignment and barcode and unique molecular identifier (UMI) read counting. T-distributed stochastic neighbor embedding (tSNE) approach was used for dimension reduction, and the cell subpopulations were clustered and SCDE, a statistical package for analyzing single-cell RNAseq data, was used for differential expression analysis and pathway(32) and gene set over-dispersion analysis(33)[Li, 2019 #2171].
Immunofluorescence
Immunofluorescence was performed on frozen sections obtained from skin biopsies from healthy control or non-sun exposed, non-lesional SLE or non-lesional psoriasis patient skin. Thawed sections were fixed in −20 °C acetone, washed, and blocked with 5% BSA for 1 hr at RT. Slides were placed in primary antibody overnight at 4°C, washed and placed in secondary antibody for 1.5 hrs at RT. Antibodies used: YAP 1:200 (Cell signaling, Catalog no. 4912), p-YAP 1:200 (Cell signaling, catalog no. 13008). Images were acquired using Zeiss Axioskop 2 microscope and analyzed by SPOT software. Images are representative of n=4 control and n=4 SLE. Each dot represents average of three images for each patient. The integrated density was calculated of a region of interest of the entire epidermis was taken and divided by the number of nuclei using ImageJ.
Identification of Hippo-associated genes and calculation of the Hippo score
From the Ingenuity Pathway Analysis software (33) (www.ingenuity.com), the Hippo signaling pathway contained 21 molecules having EntrezGene IDs (Supplementary Table 3). Three of those genes were not expressed in the lesional cutaneous lupus erythematosus (CLE) microarrays (AMOT, MER and SCRIB). A total of 18 genes was thus used for calculating an “Hippo score”, using the algorithm described by Feng et al(34).
UVB irradiation, FAM-FLICA caspase 3/7and TUNEL assays
Keratinocyte cultures at 80% confluence were irradiated with 50mJ/cm2 UVB (310nm) via a UV-2 irradiator (Tyler Industries, Alberta, Canada). Cells were grown for 6 hrs and stained using FAM-FLICA caspase 3/7 (37) or TUNEL (Sigma). Percent cl-caspase 3/7 cells was quantified using ImageJ (# cells with active caspase 3/7 and DAPI (/(# cells DAPI). Images presented are representative triplicate images of three separate experiments.
Inhibition of YAP phosphorylation using Small-molecule LATS inhibitor TRULI
The compound N-(3-benzylthiazol-2(3H)-ylidene)-1H-pyrrolo[2,3-b]pyridine- 3-carboxamide (CAS number 1424635–83-5) and herein termed TRULI, was obtained from CSN Pharm (Cat No CSN 26140). Primary SLE or control keratinocyte at 60% confluence were treated with 10 uM TRULI for 24 hours following by UVB (50mJ/cm2) and FAM-FLICA caspase 3/7, as above. LATS1/2 inhibition was confirmed using immunoblot of p-YAP and YAP in total cell lysate.
Evaluation of UVB mediated Hippo response in primary SLE vs. control keratinocytes
RNA was isolated (RNA MiniPrep (Qiagen)) and cDNA was made. RT-PCR for SNAI1, MYC, AREG, WWC1 (normalized to RPLP0) were performed using QuantStudio 12K. PCR primers used: SNAI forward, 5’-ACTATGCCGCGCTCTTTCCT-3’, SNAI reverse 5’-GCTGCTGGAAGGTAAACTCTGG-3’, MYC forward 5’-ATTTGGGGACACTTCCCCGC-3’, MYC reverse 5’-CACCGAGTCGTAGTCGAGGT-3’. AREG forward, 5’-TGTCGCTCTTGATACTCGGCT-3’, AREG reverse 5’-TGGTTCACGCTTCCCAGAGTAG-3’, WWC1 forward 5’-GTTCGAAACTCCCTGGAGCG-3’, WWC1 reverse 5’-GACTTGACCGAGGAAGGCTG-3’.
Generation of WWC1 OE and Ctr-GFP cell lines
Overexpression of WWC1 in N/TERTs was made via pLVX lentiviral vector (from TaKaRa) expressing wild-type WWC1 (pLVX-GFP-WWC1) or an empty vector GFP control (pLVX-GFP). 293T cells were co-transfected with the pLVX-GFP-WWC1 (the construct of this plasmid was generated by sub-cloning of WWC1 from pBabe-KIBRA vector (Addgene 40887) to pLVX-GFP vector) or pLVX-GFP-WWC1 and packaging plasmids pxPAX2 and pMD2 by Lipofectamine 2000 to produce the lentivirus (modified from (36)), and the supernatant containing virus was used to infect keratinocytes.
Western Blot
Total protein was isolated from cultured cells using Pierce RIPA buffer (ThermoFisher #89900) and run on gel (Bio-Rad # 456–1004S). The membrane was blocked and incubated in primary antibody (WWC1, Cell Signaling, Catalog #: 8774S), p-YAP, Cell signaling, #: 13308, YAP, Cell signaling #: 14074, pLATS1 Cell signaling, #: 9157, LATS, Cell signaling, #: 3477, GFP, Cell signaling, #: 2956, beta actin, sigma #A5441), then secondary antibody (anti-rabbit IgG AP-linked Antibody, Cell signaling #7054), washed 5 times, substrate added (Fisher Scientific # 45–000-947) and imaged on Omega lum C (Aplegen). Ratio of phosphorylated/total protein was quantified using Image J.
Accel siRNA knock down and qRT-PCR
SiRNA knockdown was performed as previously described(37) according to manufacturer’s instructions.
Statistics
For in vitro experiments, calculations were made using GraphPad Prism v.7. For most studies, comparisons of the means between experimental variables were made via unpaired two-sided Student’s t-test for normally distributed variables and via Mann-Whitney for non-normally distributed variables. Paired testing was used for siRNA studies. For both microarray and RNA-seq, false discovery rate was used to control for multiple testing.
Study approval
This study was approved by the University of Michigan IRBMED, which includes affiliated and unaffiliated members of the public. Lupus patients were not specifically involved in the design of this study.
RESULTS
The Hippo pathway is differentially methylated in SLE keratinocytes
To investigate the role of epigenetic changes in non-lesional SLE keratinocytes that may regulate persistent differences in apoptosis predisposition in culture, we analyzed genome-wide DNA methylation from keratinocytes isolated from 6 patients with SLE. Samples were analyzed in parallel with age-, sex-, and self-identified race/ethnicity-matched healthy controls (HCs) from identical sites (upper outer thigh). 2,461 differentially methylated CpG sites were identified, including 1,618 (66%) hypomethylated and 843 (34%) hypermethylated CpG sites. These CpG sites overlapped with 1,443 genes, 924 (64%) being hypomethylated and 519 (36%) being hypermethylated in keratinocytes compared to keratinocytes from the matched healthy control cohort (Fig.1a). Supplementary Table 4 shows the top 10 hypomethylated and top 10 hypermethylated sites in SLE keratinocytes compared to control. A list of all differentially methylated sites between patients and healthy controls is shown in Supplementary Table 5.
Figure 1. Hippo pathway is differentially methylated in SLE keratinocytes (KC).
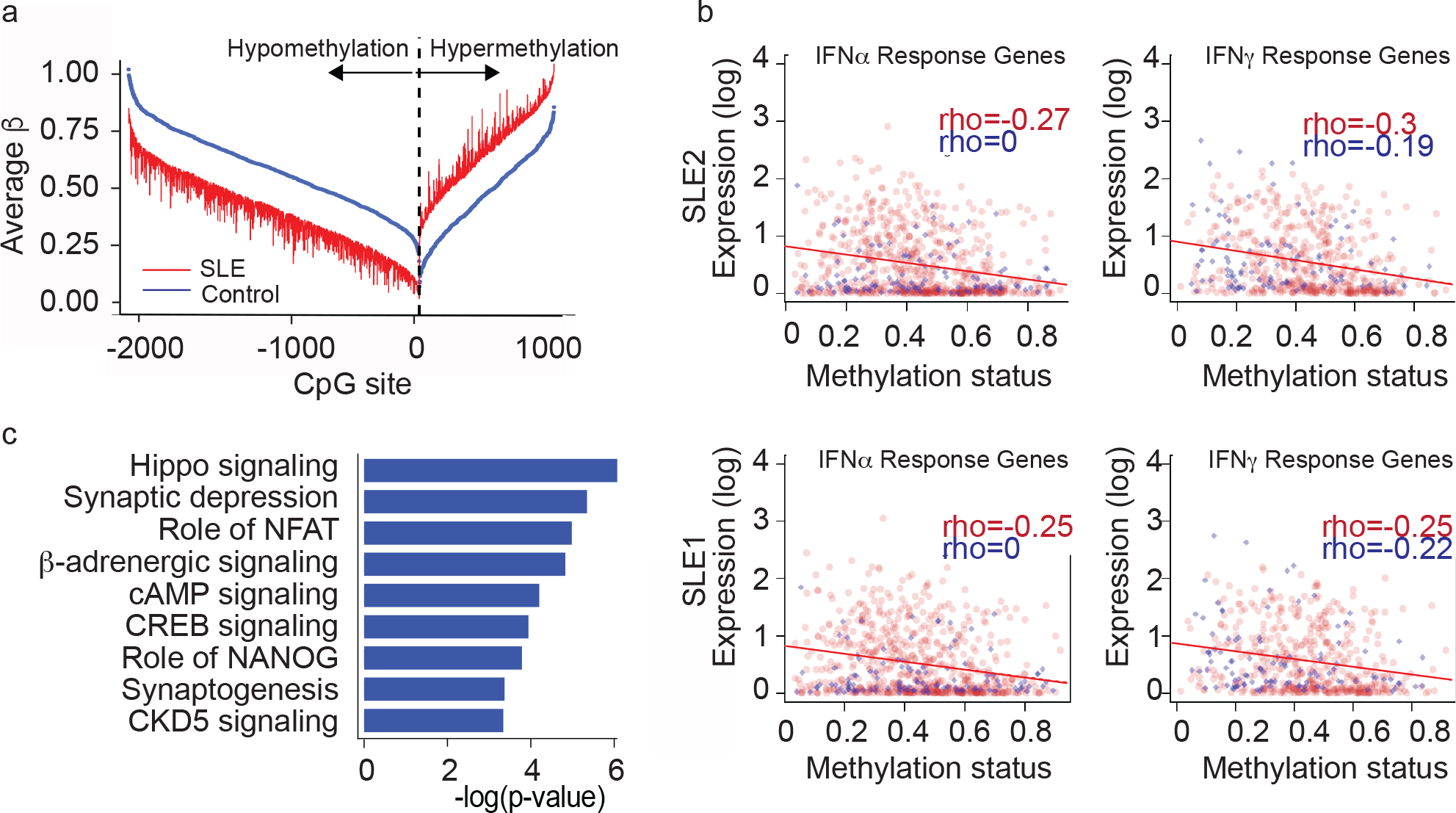
a. Illumina methylation of non-lesional, SLE vs. HC KCs (age, sex, race matched; n=6 each)
b. Correlations of genes induced by IFN and methylation signatureso
c. Ingenuity pathway analysis of methylation changes
To determine functional relevance of our methylation data, we examined whether SLE methylation changes correlated with stimulated gene transcription (i.e. would hypomethylated genes exhibit increased expression and hypermethylated genes exhibit decreased expression). We thus treated two of the same SLE keratinocyte lines used in our methylation analysis with 1000 U/mL IFN-α or 5 ng/mL IFN-γ and examined gene expression via RNA-sequencing. As shown in Fig. 1b, expected negative correlations between genes induced by either IFN-α or IFN-γ and methylation signatures were noted, suggesting that the methylation changes we identified result in functional gene expression differences in vivo after treatment with cytokines known to be increased in SLE skin(4,9–10). These data suggest that SLE KC undergo methylation changes that persist in culture and regulate relevant gene expression pathways.
We next performed pathway analysis using Ingenuity to determine the affected signaling mechanisms regulated by methylation changes. Surprisingly, a robust hypomethylation signature in IFN-regulated genes, which has been reported in other cell types in SLE (11, 12), was not observed in SLE keratinocytes, suggesting that direct IFN stimulation may be a more relevant regulator of IFN gene expression in SLE KCs. Instead, we identified Hippo signaling as the most significantly differentially methylated pathway in SLE vs. control keratinocytes (Fig. 1c, Supplementary Table 6). Several members of the Hippo pathway had differentially methylated CpG sites in SLE keratinocytes compared to HC, including WWC1 (Supplementary Table 6). WWC scaffolding proteins are critical regulators of Hippo signaling in that only increased expression is required to trigger Hippo signaling through recruitment of kinases to phosphorylate and activate LATS1/2. LATS1/2 kinases phosphorylate YAP, preventing its nuclear translocation and promotion of proliferative gene expression via TEAD transcription factors (13). The reduced methylation of WWC1 led us to hypothesize that increased WWC1 expression in SLE keratinocytes would promote activation of LATS1/2 and subsequent YAP phosphorylation and cytoplasmic retention. As YAP and TEAD activity are essential for maintenance of skin renewal(14), blockade of YAP/TEAD interaction could sensitize keratinocyte to UVB-apoptosis through a shift towards apoptotic vs. proliferative gene expression.
scRNA seq of normal appearing skin shows differential activation of Hippo signaling in SLE
To identify ex vivo evidence for activation of Hippo signaling in SLE skin, we performed scRNA-seq on non-lesional skin from an additional 10 patients (all with active CLE) and 6 healthy controls. The final dataset comprised 39,086 cells, with an average of 2,252 genes and 10,970 transcripts per cell. Cell type clustering was performed as previously described (15). Visualization using Uniform Manifold Approximation and Projection (UMAP) of characteristic keratinocytes markers identified 7 sub-clusters: basal, spinous, supraspinous, granular, follicular, basal inflammatory and spinous inflammatory (Fig. 2a), each comprising cells from lesional, non-lesional and healthy control keratinocytes (Fig. 2b).
Figure 2. scRNA seq of ‘normal appearing’ SLE skin and both CLE subtypes show differential activation of Hippo signaling.
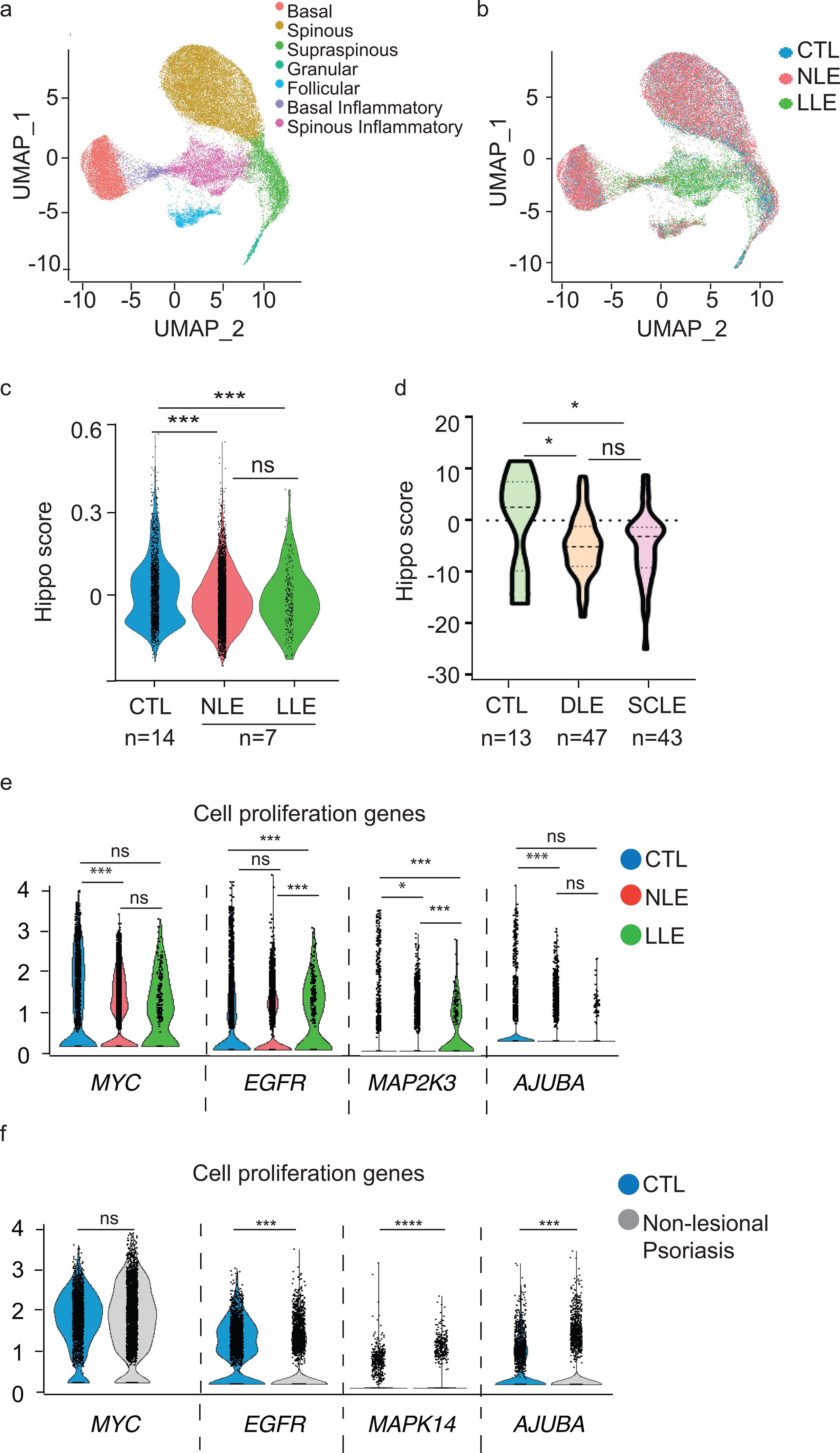
a. UMAP plot of 39,086 keratinocytes colored by sub-cluster from all samples: CTL, healthy control skin, n=6. NLE, non-lesional lupus skin. LLE, lesional lupus skin, n=10 each.
b. UMAP plot of keratinocytes colored by disease state. CTL, healthy control skin, n=6. NLE, non-lesional lupus skin. LLE, lesional lupus skin, n=10 each.
c. Violin plots of Hippo module score from scRNA seq analysis of KC cluster
d. Violin plots of Hippo module score from microarray analysis of CLE lesions. DLE = discoid lupus erythematosus; n=47, SCLE =subacute cutaneous lupus erythematosus; n=43
e. Violin plots showing the expression level of representative cell proliferation genes in healthy control (n=6), nonlesional and lesional lupus (n=10) in basal keratinocyte subcluster; *=p<0.05, ***=p<0.001
f. Violin plots showing the expression level of representative cell proliferation genes in the basal keratinocyte sub-cluster in healthy control (n=6) and nonlesional psoriasis (n=14); ***=p<0.001, ****=p<0.0001
To investigate whether Hippo responses were differentially expressed in SLE keratinocytes, we calculated a Hippo score derived from downstream cell proliferation genes of the Hippo pathway that are expressed when YAP binds TEAD and generated feature plots displaying module scores for basal keratinocyte sub-type and disease state. This Hippo module score was decreased in non-lesional and lesional SLE KCs implicating activation of hippo signaling and retention of YAP in the cytoplasm in the epidermis of SLE patients (Fig 2c). We also validated a repression of the Hippo score of both DLE and SCLE skin lesions in our previously published microarray data of 90 CLE skin lesions from SLE and CLE-only patients (16, 17) (Fig 2d), suggesting that in sum, alterations in the Hippo pathway persist from non-lesional to lesional skin and may thus play a role in disease pathogenesis.
To examine gene expression on a more granular level, we plotted relative expression of YAP/TEAD regulated cell-proliferation markers, MYC, EGFR, MAP2K3, and AJUBA in the basal KC clusters. As shown in Fig 2e, these genes were decreased in NLE and LLE compared to control (Fig. 2e). Similar findings of YAP/TEAD driven genes from the microarray data are shown in Supplementary Figure 1.
To evaluate whether these changes were specific to SLE or were reflective of skin inflammation in general, we examined scRNA-seq analysis from basal keratinocytes from non-lesional skin from 10 psoriasis patients and 6 matched control skin samples. Opposite to what we see in lupus, ‘normal-appearing’, non-lesional skin of psoriasis patients exhibited increased cell proliferation gene expression of MYC, EGFR, MAPK, and AJUBA compared to controls, consistent with the proliferative epidermal phenotype in psoriasis. Taken together, these data suggest a role of activated Hippo signaling leading to a reduction in YAP/TEAD proliferative signals in SLE that is not seen in other inflammatory skin diseases such as psoriasis.
Hippo changes translate to significant differences in protein expression
We then examined protein expression of Hippo pathway members from cultured non-lesional SLE vs. control keratinocytes at the same cell passage and confluency. Corroborating our methylation analysis, we observed increased WWC1 expression in SLE compared to control (Fig 3a–b, p=0.002, n=6 each). Total YAP protein levels were also increased in SLE (Fig 3a,c), and consistent with the expected function of increased WWC1 expression, we also observed an increase in phosphorylated YAP (p-YAP)/total YAP levels in 6 unique SLE and 6 healthy control samples (p= 0.014) (Fig 3a,d). As WWC1 recruits upstream kinases to activate LATS1/2 kinase by phosphorylation, we also examined the phosphorylation of LATS1/2. As expected, we note increased phosphorylated LATS1/2 (pLATS1/2) to total LATS protein expression (p = 0.029) in SLE keratinocytes (Fig 3a, e). We also confirmed increased phosphorylation of YAP in vivo via immunofluorescent staining of healthy control and SLE non-lesional biopsies (Fig.3f). We again used non-lesional psoriasis skin as a disease comparator (Fig 3–i). While there was no significant difference of total YAP expression between SLE and HC in vivo (Fig. 3g), there was an impressive increase in the ratio of p-YAP/total YAP (p<0.0001) in SLE vs. control (Fig. 3h,i). There was no significant difference in either YAP or p-YAP/YAP in non-lesional psoriasis compared to control (Fig. 3g,i). These data suggest that the observed increase in WWC1 expression results in a chronic increase in LATS1/2 activation and YAP phosphorylation in SLE keratinocytes in vitro and in vivo.
Figure 3. Hippo signaling methylation changes translate to significant difference in protein expression in situ.
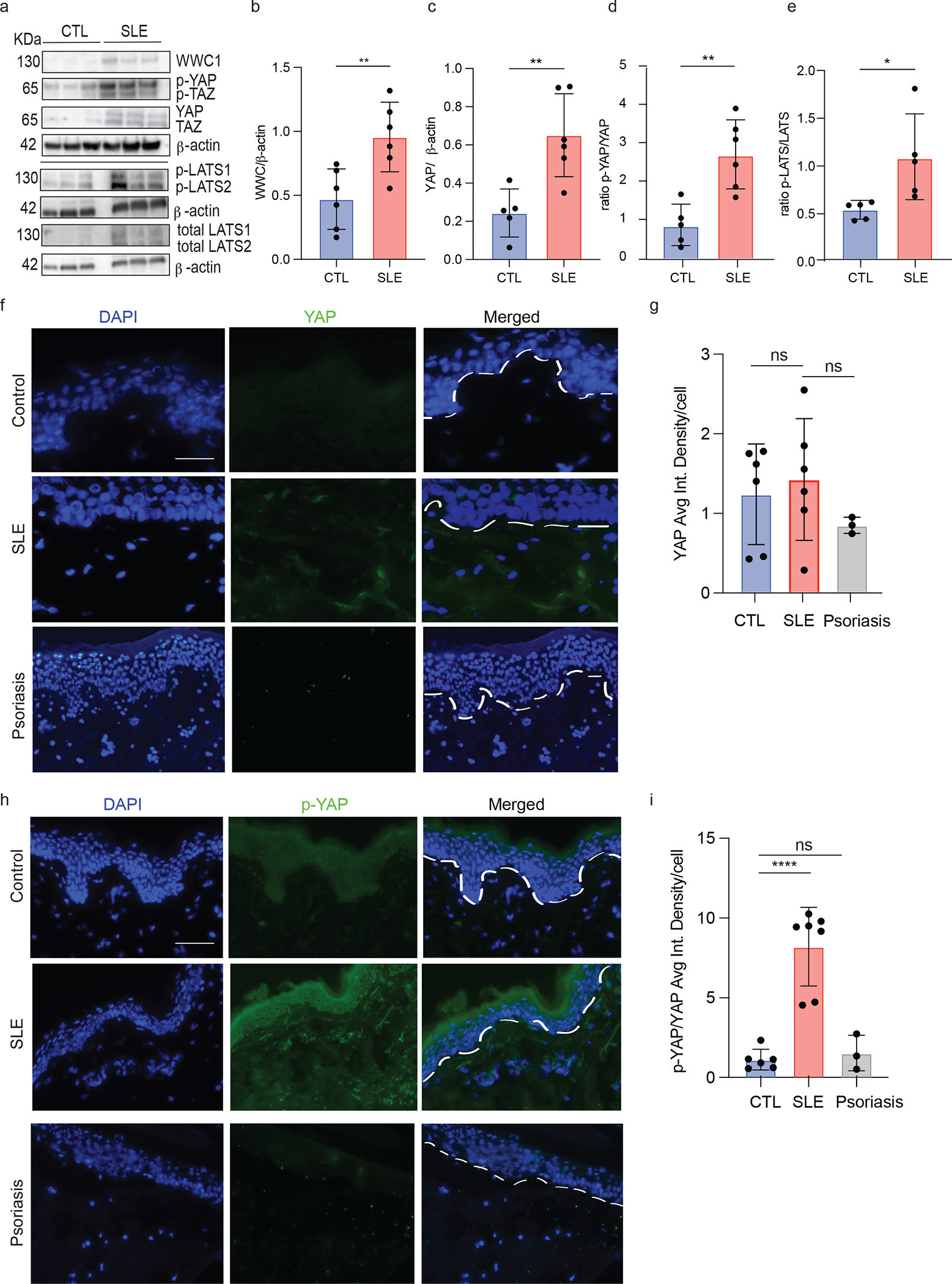
a. WWC1, p-YAP, pTAZ, pLATS1/2, total LATS1/2 and beta actin levels by Western blot. Blots were divided and probed with respective primary antibody
b. Bar graph of ratio of WWC1 to beta actin in 6 SLE and 6 HC KC primary cell lines
c. Bar graph of ratio of YAP/beta actin in 6 CTL and 6 SLE KC primary cell lines
d. Bar graph of ratio of p-YAP/YAP in 6 SLE and 6 CTL primary cell lines
e. Bar graph of ratio of pLATS/LATS in 6 SLE and 6 CTL primary cell lines
f. Immunofluorescence of YAP from frozen skin biopsies in 6 CTL and non-lesional frozen skin biopsies from 6 SLE patients and 3 psoriasis patients. Scale bar= 100 μM
g. Bar graph of averaged integrated density per cell of YAP in 6 CTL and non-lesional frozen skin biopsies from 6 SLE patients and 3 psoriasis patients.
h. Immunofluorescence of p-YAP from frozen skin biopsies in 6 CTL and non-lesional frozen skin biopsies from 6 SLE patients and 3 psoriasis patients. Scale bar= 100 μM
i. Bar graph of average integrated density per cell of p-YAP in 6 CTL and non-lesional frozen skin biopsies from 6 SLE patients and 3 psoriasis patients.
f,h. Each dot represents the average of 3 images from each patient. Scale bar 100 μM. ns= not significant, * p<0.05, **p<0.01 unpaired t-test.
WWC1 overexpression and interruption of YAP-TEAD interaction results in exaggerated UVB- driven apoptosis
SLE keratinocytes have increased apoptosis to UV stimulation, and this is thought to drive photosensitivity in SLE and CLE (4). To investigate the role of Hippo signaling in UV-mediated apoptosis, we used an immortalized human keratinocyte cell line (N/TERT) overexpressing GFP-tagged WWC1 (WWC1OE) or GFP control (Ctr-GFP) (Fig. 4 a,b). Consistent with the activation of hippo signaling via WWC1 overexpression, GFP-WWC1 cells exhibit increased upregulation of the apoptosis marker Snail Family Transcriptional Repressor 1 (SNAI1) and cell death (measured by TUNEL and cl-caspase 3/7) compared to Ctr-GFP cells (Fig. 4 c–f). Following treatment with UVB, cell death (ratio 2.4 UVB/control) and SNAI1 transcription (ratio 3.4 UVB/control) was increased in CTL-GFP lines as expected. In WWC1OE lines, a higher baseline in apoptosis was noted but the ratio of increase remained about the same: a 2.2 fold increase in cell death and 2.8 increase in SNAI1 expression (Fig. 4c–e). These data support a role for WWC1 overexpression in promoting cell death. As WWC1 overexpression modulated cell morphology in our KC line, we wanted to confirm the role of YAP-TEAD in regulation of UVB-mediated apoptosis in an inducible model. We thus used an N/TERT line expressing a tetracycline-inducible GFP-tagged protein that inhibits YAP-TEAD binding (TEADi), mimicking the effect of increased YAP phosphorylation and cytoplasmic retention(18). Validation of induced GFP-TEADi expression is shown in Fig 4h. N/TERT TEADi cells demonstrated a small but significant increase in apoptosis at baseline compared to control, and this was further robustly increased after 6 hours of UVB exposure (0.34 fold in WT and 0.83 fold in TEADi) (Fig. 4.i,j). These data suggest that increased WWC1 and mimicking its physiologic effect by inhibition of TEAD-YAP interactions primes cells for increased apoptosis after UVB exposure.
Figure 4. WWC1 overexpression and interruption of YAP-TEAD interactions results in exaggerated apoptosis.
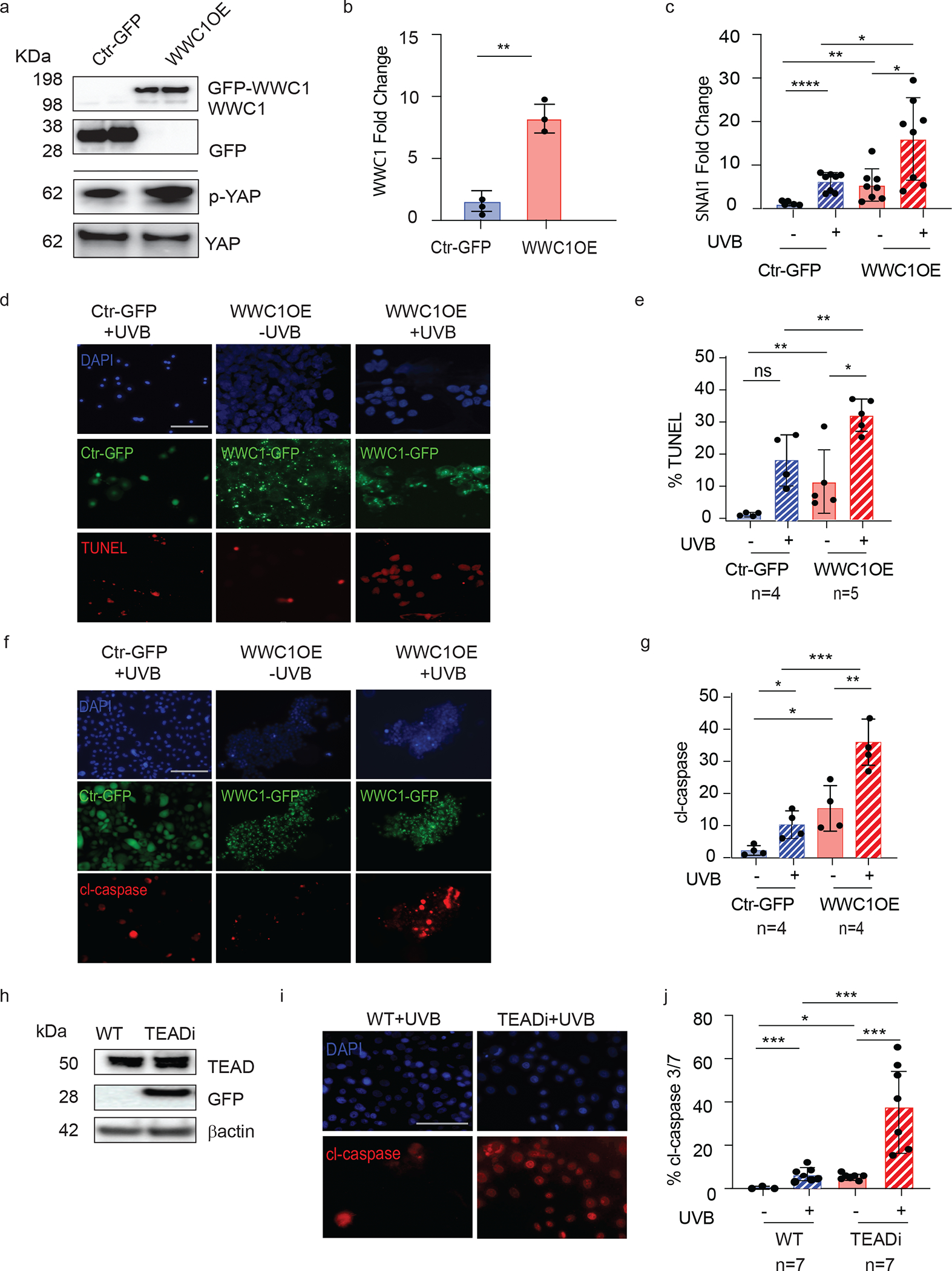
a. Western blot of WWC1 overexpression and GFP in N/TERT2G keratinocyte line, probed with WWC1 and GFP primary antibody as indicated by box. Separate gel was run for p-YAP and YAP in Ctr-GFP and WWC1OE cell lines.
b. Expression of WWC1 in Ctr-GFP and WWC1OE cell lines by RT qPCR (n=3)
c. Expression of apoptosis gene SNAI1 in WWC1OE cell lines vs. Ctr-GFP by RT qPCR (n=8 for Ctr-GFP and n=8 for WWC1OE)
d. Representative TUNEL images of WWC1OE vs. Ctr-GFP treated with or without UVB, n=4. Scale bar = 100 μM
e. Quantification of TUNEL in WWC1OE vs Ctr-GFP (n=5, each),
f. Representative cl-caspase 3/7 images of WWC1OE vs. Ctr-GFP treated with or without UVB, n=4. Scale bar = 100 μM.
g. Quantification of cl-caspase 3/7 in WWC1OE vs Ctr-GFP in f (n=4, each)
h. Western blot of TEAD, GFP and beta-actin in WT and N/TERT TEADi-GFP cell lines
i. Representative images of cl-caspase 3/7 staining in WT N/TERT and TEADi (n=7, each) with and without UVB. Scale bar = 100 μM
j. Quantification of %cl-caspase 3/7 in WT vs. TEADi N/TERTs in i, * p<0.05, **p<0.01. ***p<0.001 unpaired t-test.
Inhibition of LATS1/2 abrogates exaggerated UVB-apoptosis in SLE keratinocytes.
SLE KCs exhibit increased apoptosis after UVB compared to healthy control KCs (4). We next evaluated if we could reverse the increased apoptotic profile of SLE keratinocytes by inhibiting Hippo signaling. We used two approaches in primary SLE keratinocytes: inhibition of LATS1/2 gene expression with siRNA and a commercially available LATS1/2 kinase inhibitor, TRULI. Validation of siRNA knockdown of LATS1 and LATS2 protein and gene expression is shown in two healthy control KC lines in Fig 5a–c. Inhibition of LATS1/2 gene expression via siRNA decreased the apoptosis in both HC and SLE KCs but had a larger effect size in the SLE patients’ cells (ratio of effect in SLE KCs is 67% reduction in apoptosis vs. 37% reduction in HC KCs) (Figure 5d,e). Using a second, complimentary approach, pretreatment of SLE keratinocytes with TRULI, an inhibitor of LATS1/2 kinase activity, resulted in decreased apoptosis at baseline (measured by cleaved caspase 3) (p=0.0034, n=3) and a notable decrease in apoptosis following UVB exposure (p=0.003) (ratio of effect in SLE KCs is 88% reduction in apoptosis vs. 52% reduction in HC KCs) (Fig. 5f, g). In SLE samples, where p-YAP levels are elevated at baseline, a reduction in p-YAP with and without UV exposure was noted after treatment with TRULI (Fig 5h, i), confirming the kinase inhibition effect on LATS1/2. In healthy controls, the effect of TRULI on p-YAP was less pronounced, especially after UV exposure, This difference may potentially be secondary to reduced p-YAP at baseline or secondary to more variable levels of yap after UV treatment. We cannot rule out an effect of other kinases on yap phosphorylation as well. These data suggest that inhibition of Hippo signaling through targeting of LATS1/2 ameliorates UVB-mediated cell death and has a large effect in SLE keratinocytes.
Figure 5. Inhibition of LATS1/2 represses exaggerated UVB apoptosis in SLE keratinocytes.
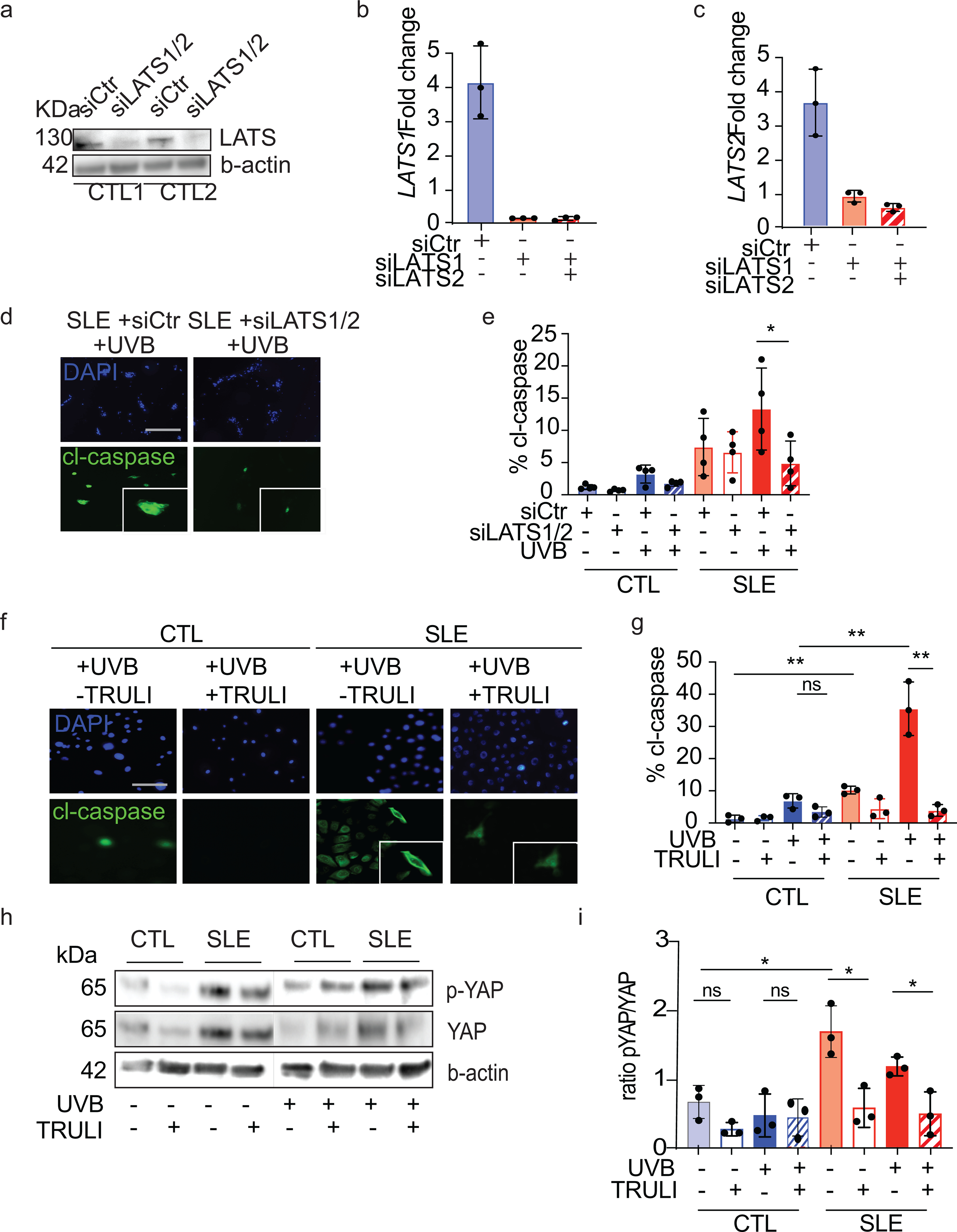
a. Western blot showing siRNA knockdown of LATS1/2 (siLATS1/2) or nontarget control (siCtr) in two different primary healthy control KC lines.
b. Expression changes of LATS1 in siRNA treated control primary KCs by qPCR, n=3, each.
c. Expression changes of LATS2 in siRNA treated control primary KCs by qPCR, n=3, each.
d. Representative images of UVB apoptotic response measured by cl-caspase 3/7 in siRNA treated primary KCs, n=4. Scale bar 50 μM.
e. Quantification of % active cl-caspase 3/7 of triplicate samples from SLE keratinocytes (n=4) and healthy control (n=4). Un-paired t test. *=p<0.05
f. Representative images of UVB apoptotic response measured by cl-caspase 3/7 in CTL (n=3) and SLE keratinocytes (n=3) following 24 hr pre-treatment with 10 μM LATS1/2 kinase inhibitor, TRULI. **=p<0.01
g. Quantification of cl-caspase 3/7 in primary CTL and SLE KCs treated with and without UVB and TRULI, n=3. Un-paired t test. Scale bar 100 μM
h. Representative Western blot of p-YAP and YAP after 24 hr treatment +/− TRULI with and without UV stimulation.
i. Quantification of p-YAP/YAP ratios for Western blot in h for three different healthy control and 3 different SLE primary KC lines. *=p<0.05.
SLE keratinocytes show a skewed pro-apoptotic gene profile that is reversible with TRULI
To determine whether we could detect direct evidence of TEAD-mediated pro-apoptotic signaling (genes that are upregulated when YAP does not bind TEAD) in SLE non-lesional keratinocytes, we performed RNA-sequencing of SLE (n=6) vs HC basal keratinocytes (n=7) at baseline and after UVB exposure. In line with our hypothesis, SLE keratinocytes exhibit a decrease in YAP/TEAD-regulated cell proliferation genes BMP4, TWIST1/2, MYC (19, 20) and an increase in pro-apoptotic genes (DDL4, LGR5, SNAI1)(20–22) following UV exposure compared to healthy control KCs (Fig 6a). Importantly, treatment with LATS1/2 siRNA reversed the UVB-mediated upregulation of the pro-apoptotic gene SNAI (Fig 6b). Similar findings were noted when SLE but not HC keratinocytes were treated with TRULI, including normalization of pro-apoptotic genes SNAI and DDL4 (p=0.0319 and p=0.01, respectively (Fig. 6c). These data suggest that the increased apoptotic phenotype in SLE keratinocytes is regulated by YAP phosphorylation and that the predisposition for increased apoptosis after UVB exposure is reversible through LATS1/2 inhibition.
Figure 6. SLE shows skewed pro-apoptotic gene profile that is reversible with TRULI.
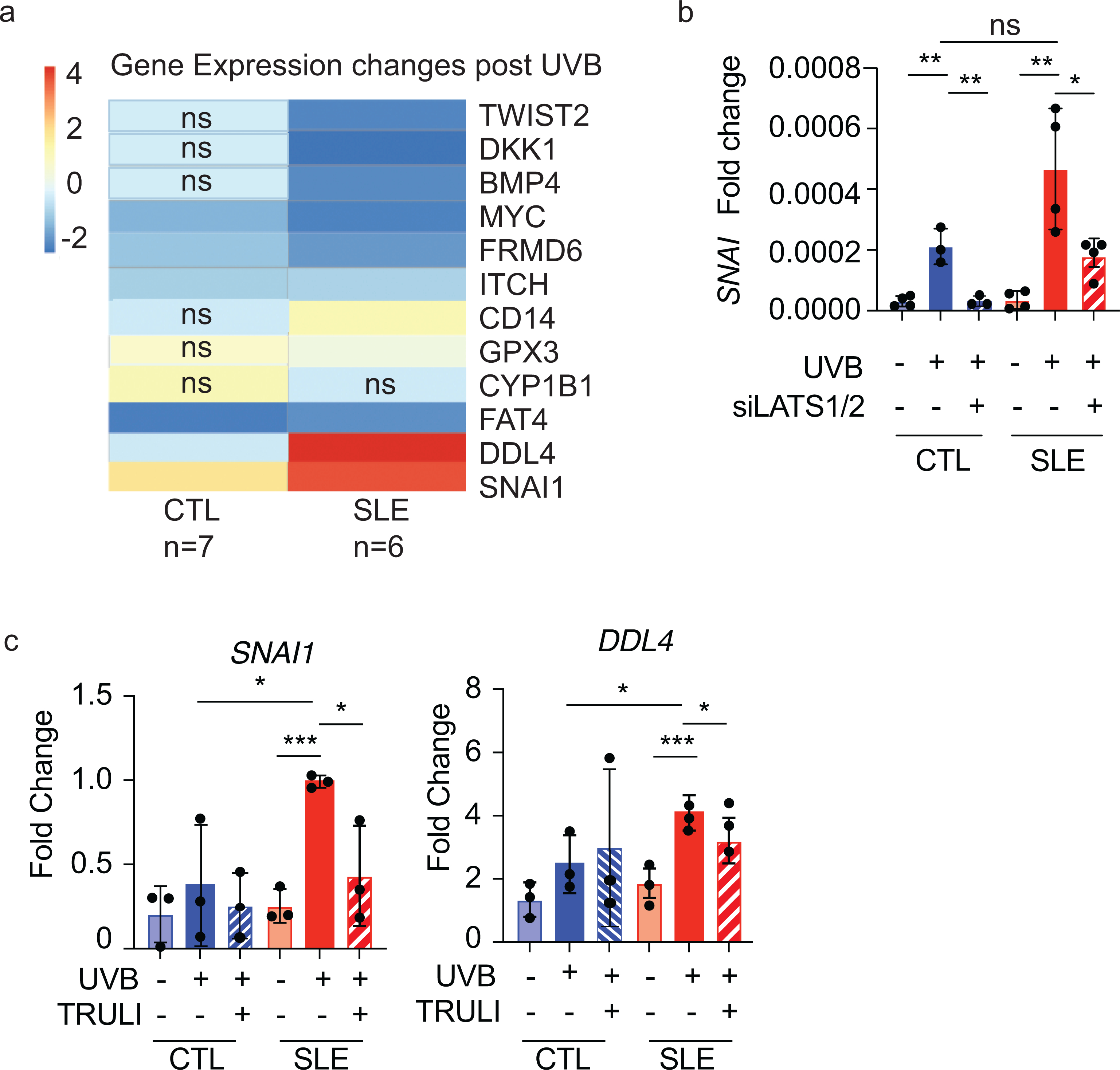
a. Change in gene expression following UVB stimulation in CTL (n=7) and SLE (n=6) cultured KCs. Expression changes between comparisons are significant with p<0.05 unless annotated.
b. Mean gene expression (+/− SD) of Hippo-regulated pro-apoptosis gene SNAI1 in CTL (n=3) and SLE (n=4) treated with UVB and LATS1/2 siRNA.
c. Gene expression of Hippo regulated pro-apoptosis genes SNAI1 and DDL4 in SLE (n=3) and CTL (n=3) KCs treated with and without UVB and TRULI. Dots represent triplicate biological replicates for 3 unique SLE and 3 unique CTL samples.
DISCUSSION
Our data demonstrate that epigenetic changes in the Hippo pathway of SLE keratinocytes have functional relevance for driving an enhanced UVB-mediated apoptotic phenotype, which may contribute to photosensitivity pathogenesis. We demonstrate that WWC1 is hypomethylated and overexpressed in SLE keratinocytes and this leads to activation of LATS1/2, increased p-YAP, and transcriptional changes consistent with inhibition of YAP/TEAD interactions. These changes shift transcription toward pro-apoptotic genes that are amplified after UV exposure. We demonstrate inhibition of LATS1/2 diminishes the exaggerated UV-mediated apoptosis in SLE keratinocytes and could be considered as a potential therapeutic option for photosensitivity in CLE and SLE.
Hippo signaling is regulated by protein-protein interactions and the formation of large membrane-associated multiprotein complexes which include the key scaffolding protein WWC1. WWC1 recruitment of MOB kinase activator 1 A and B (MOB1A/B) and Hippo homolog kinases MST1/2 both activate LATS1/2 and phosphorylation of YAP and its paralog TAZ. p-YAP/p-TAZ are confined to the cytoplasm via association with the 14–3-3 family and may be degraded. Liberation of YAP/TAZ via dephosphorylation by PTPN14 or decreased expression of WWC1 leads to nuclear translocation of YAP/TAZ and binding to TEAD family members (TEAD 1–4) to drive expression of pro-proliferative genes. In this study, we chose to focus on the changes in YAP (which are likely paralleled by TAZ); however, the role of these other Hippo signaling mediators in SLE should be studied in future work.
Lending further relevance to Hippo signaling in autoimmune skin disease, epidermal expression of vestigial like family member 3 (VGLL3) was shown to be sufficient to drive a SLE-like phenotype in mice, with characteristic interface dermatitis and apoptotic keratinocytes (22). It is known that VGLL proteins bind to TEAD proteins at the same domain as YAP in other organs (23). When YAP is phosphorylated and retained in the cytoplasm, VGLL3 may bind TEAD and influence downstream gene expression. Thus, VGLL3 overexpression, which theoretically would inhibit YAP binding to TEAD and thus mimic our findings in SLE, also results in results SLE-like disease in mice (22). Further studies are needed to determine the connection between YAP, VGLL3 and TEAD in SLE skin.
The role of hippo signaling in autoimmune phenotypes is an exciting new avenue for research. We have identified activation of YAP phosphorylation and subsequent repression of pro-proliferative hippo-regulated gene expression in both non-lesional and lesional SLE skin and contrast our findings in non-lesional SLE skin with non-lesional psoriasis, where p-YAP is not increased. Previously, total YAP expression has been identified to be increased in psoriasis lesions, where it contributes to a hyper-proliferative phenotype (38), presumably because the YAP is not phosphorylated and thus is free to bind TEADs and promote cellular proliferation. Thus, the behavior of different autoimmune skin diseases may be regulated by the expression and activation of hippo signaling. Other autoimmune tissues may also exhibit a role for hippo signaling. For example, regulation of hippo signaling mediators via Vgll3 may contribute to interferon production in rheumatoid arthritis fibroblast-like synoviocytes(39). Further research into hippo signaling regulation of autoimmune disease phenotypes is warranted.
The limitations of this study include limited knowledge of the upstream drivers of the methylation changes in SLE skin. Our non-lesional biopsies were taken from the upper thigh, which is not chronically sun exposed. This suggests that circulating factors could influence the epigenome that regulates Hippo activation. Alternatively, epigenetic changes can start in utero, so whether the skin is primed from birth or becomes so later in life should be investigated. In addition, we cannot rule out an effect of hydroxychloroquine on methylation changes as all patients recruited to the study were taking this medication. Understanding these changes will inform the biology of SLE/CLE and help identify therapeutic targets
Collectively, our work implicates a novel and mechanistic paradigm through which increased WWC1 restricts YAP transcriptional activity and leads to enhanced apoptosis in SLE keratinocytes after UVB exposure. Future consideration of therapeutic modulation of this pathway in SLE should be considered.
Supplementary Material
Supplementary Table 1. Patient demographics used in methylation analysis
Supplementary Table 2. Patient demographics of all patients
Supplementary Table 3. Genes used in calculating ‘Hippo score’
Supplementary Table 4. Top 10 hypomethylated and top 10 hypermethylated sites in SLE keratinocytes compared to control
Supplementary Table 5. All differentially methylated sites between patients and healthy control
ACKNOWLEDGEMENTS
National Institutes of Health grant R01-AR071384 (JMK)
National Institutes of Health grant R03-AR066337 (JMK)
National Institutes of Health grant K24-AR076975 (JMK)
National Institutes of Health grant R01-AR060802 (JEG)
National Institutes of Health grant F31-AR077988 (SNE)
National Institutes of Health grant T32-AR007197 (GAH)
National Institutes of Health grant K08-AR078251 (ACB)
National Institutes of Health grant P30-AR075043 (JEG, JMK)
National Institute of Allergy and Infectious Diseases T32AI007413 (JWSM)
A. Alfred Taubman Medical Research Institute via Innovative Programs (JEG, JMK)
Parfet Emerging Scholar Award (JMK)
Lupus Research Alliance (JMK)
Footnotes
Competing Interest Statement: JMK has received Grant support from Q32 Bio, Celgene/BMS, Ventus Therapeutics, Rome Therapeutics. and Janssen. JEG has received Grant support from Celgene/BMS, Janssen, Eli Lilly and Almirall. JMK has served on advisory boards for AstraZeneca, Eli Lilly, GlaxoSmithKline, Gilead, Bristol Myers Squibb, Avion Pharmaceuticals, Lupus Therapeutics, Provention Bio, Aurinia Pharmaceuticals, Ventus Therapeutics, and Boehringer Ingelheim. JEG has served on advisory boards for AstraZeneca, Sanofi, Eli Lilly, Boehringer Ingelheim, Novartis, Janssen, Almirall, BMS. All other authors have nothing to disclose.
REFERENCES
- 1.Foering K, et al. Prevalence of self-report photosensitivity in cutaneous lupus erythematosus. J Am Acad Dermatol. 2012;66(2):220–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pirner K, et al. Significance of ultraviolet light in the pathogenesis of systemic lupus erythematosus: case report and discussion of the literature. J Rheumatol. 1992;51(1):20–4. [PubMed] [Google Scholar]
- 3.Ansel JC, et al. Effects of UV radiation on autoimmune strains of mice: increased mortality and accelerated autoimmunity in BXSB male mice. J Invest Dermatol. 1985;85(3):181–6. [DOI] [PubMed] [Google Scholar]
- 4.Sarkar MK, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis. 2018;77(11):1653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan Y, et al. YAP1/TAZ-TEAD transcriptional networks maintain skin homeostasis by regulating cell proliferation and limiting KLF4 activity. Nat Commun. 2020;11(1):1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pellegrini C, et al. Understanding the Molecular Genetics of Basal Cell Carcinoma. Int J Mol Sci. 2017;18(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vincent-Mistiaen Z, et al. YAP drives cutaneous squamous cell carcinoma formation and progression. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elbediwy A, et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development. 2016;143(10):1674–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson ES, et al. The role of cytokines in the pathogenesis of cutaneous lupus erythematosus. Cytokine. 2015. Jun;73(2):326–34. [DOI] [PubMed] [Google Scholar]
- 10.Berthier CC, et al. Molecular Profiling of Cutaneous Lupus Lesions Identifies Subgroups Distinct from Clinical Phenotypes. J Clin Med. 2019. Aug 17;8(8):1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coit P, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naïve CD4+ T cells from lupus patients. J Autoimmun. 2013;43:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coit P, et al. Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils. J Autoimmun. 2015;58:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Höffken VA-O, et al. WWC Proteins: Important Regulators of Hippo Signaling in Cancer. Cancers (Basel). 2021. Jan 15;13(2):306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uttagomol J, et al. Evidence for the Desmosomal Cadherin Desmoglein-3 in Regulating YAP and Phospho-YAP in Keratinocyte Responses to Mechanical Forces. Int J Mol Sci. 2019;20(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Billi AC, et al. Non-lesional and Lesional Lupus Skin Share Inflammatory Phenotypes that Drive Activation of CD16+ Dendritic Cells. Sci Transl. Med. 2021. In press. [Google Scholar]
- 16.Berthier CC, et al. Molecular Profiling of Cutaneous Lupus Lesions Identifies Subgroups Distinct from Clinical Phenotypes. J Clin Med. 2019;8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abernathy-Close L, et al. B Cell Signatures Distinguish Cutaneous Lupus Erythematosus Subtypes and the Presence of Systemic Disease Activity. Front Immunol. 2021;12:775353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan Y, et al. YAP1/TAZ-TEAD transcriptional networks maintain skin homeostasis by regulating cell proliferation and limiting KLF4 activity. Nat Commun. 2020. Mar 19;11(1):1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debaugnies M, et al. YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep. 2018. Jul;19(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barry ER, et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature, 2013. Jan 3;493(7430):106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Der E, et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol. 2019. Jul;20(7):915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Billi AC, et al. The female-biased factor VGLL3 drives cutaneous and systemic autoimmunity. JCI Insight. 2019;4(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin KC, et al. Regulation of the Hippo Pathway Transcription Factor TEAD. Trends Biochem Sci. 2017;42(11):862–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller S, et al. Hypomethylation of STAT1 and HLA-DRB1 is associated with type-I interferon-dependent HLA-DRB1 expression in lupus CD8+ T cells. Ann Rheum Dis. 2019;78(4):519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum.1997.Sep;40(9):1725. [DOI] [PubMed] [Google Scholar]
- 26.Stannard JN, et al. Lupus Skin Is Primed for IL-6 Inflammatory Responses through a Keratinocyte-Mediated Autocrine Type I Interferon Loop. J Invest Dermatol. 2017;137(1):115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dickson MA, et al. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. 2000. Feb;20(4):1436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012. Mar 1;7(3):562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harrow J, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012. Sep;22(9):1760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fau Love Mi et al. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kharchenko PV, et al. Bayesian approach to single-cell differential expression analysis. Nat Methods. 2014. Jul;11(7)740–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan JA-O, et al. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat Methods. 2016. Mar 13(3):241–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Green C, et al. A comparison of the efficacy and relapse rates of narrowband UVB (TL-01) monotherapy vs. etretinate (re-TL-01) vs. etretinate-PUVA (re-PUVA) in the treatment of psoriasis patients. Br J Dermatol. 1992;127(1):5–9. [DOI] [PubMed] [Google Scholar]
- 34.Feng X, et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54(9):2951–62. [DOI] [PubMed] [Google Scholar]
- 35.Dustin ML, et al. Adhesion of T lymphoblasts to epidermal keratinocytes is regulated by interferon gamma and is mediated by intercellular adhesion molecule 1 (ICAM-1). J Exp Med. 1988. Apr 1;167(4):1323–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu B, et al. Multiple roles for the non-coding RNA SRA in regulation of adipogenesis and insulin sensitivity. PLoS One. 2010;5(12):e14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsoi LC, et al. Hypersensitive IFN Responses in Lupus Keratinocytes Reveal Key Mechanistic Determinants in Cutaneous Lupus. J Immunol. 2019;202(7):2121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jia J et al. Yes-associated protein promotes the abnormal proliferation of psoriatic keratinocytes via an amphiregulin dependent pathway. Sci Rep. 2018. Oct 15;8(1):14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du Y, et al. Regulation of type I interferon signature by VGLL3 in the fibroblast-like synoviocytes of rheumatoid arthritis patients via targeting the Hippo pathway . Arthritis Res Ther. 2022. Aug 8;24(1):188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Patient demographics used in methylation analysis
Supplementary Table 2. Patient demographics of all patients
Supplementary Table 3. Genes used in calculating ‘Hippo score’
Supplementary Table 4. Top 10 hypomethylated and top 10 hypermethylated sites in SLE keratinocytes compared to control
Supplementary Table 5. All differentially methylated sites between patients and healthy control