

Abstract
The prevailing view is that enhancement of dopamine (DA) transmission in the mesolimbic system, consisting of DA neurons in the ventral tegmental area (VTA) that project to the nucleus accumbens (NAc), underlies the reward properties of ethanol (EtOH) and nicotine (NIC). We have shown previously that EtOH and NIC modulation of DA release in the NAc is mediated by α6-containing nicotinic acetylcholine receptors (α6*-nAChRs), that α6*-nAChRs mediate low-dose EtOH effects on VTA GABA neurons and EtOH preference, and that α6*-nAChRs may be a molecular target for low-dose EtOH. However, the most sensitive target for reward-relevant EtOH modulation of mesolimbic DA transmission and the involvement of α6*-nAChRs in the mesolimbic DA reward system remains to be elucidated. The aim of this study was to evaluate EtOH effects on GABAergic modulation of VTA GABA neurons and VTA GABAergic input to cholinergic interneurons (CINs) in the NAc. Low-dose EtOH enhanced GABAergic input to VTA GABA neurons that was blocked by knockdown of α6*-nAChRs. Knockdown was achieved either by α6-miRNA injected into the VTA of VGAT-Cre/GAD67-GFP mice or by superfusion of the α-conotoxin MII[H9A;L15A] (MII). Superfusion of MII blocked EtOH inhibition of mIPSCs in NAc CINs. Concomitantly, EtOH enhanced CIN firing rate, which was blocked by knockdown of α6*-nAChRs with α6-miRNA injected into the VTA of VGAT-Cre/GAD67-GFP mice. The firing rate of CINs was not enhanced by EtOH in EtOH-dependent mice, and low-frequency stimulation (LFS; 1 Hz, 240 pulses) caused inhibitory long-term depression at this synapse (VTA-NAc CIN-iLTD) which was blocked by knockdown of α6*-nAChR and MII. Ethanol inhibition of CIN-mediated evoked DA release in the NAc was blocked by MII. Taken together, these findings suggest that α6*-nAChRs in the VTA-NAc pathway are sensitive to low-dose EtOH and play a role in plasticity associated with chronic EtOH.
Keywords: Alcohol, Nicotine (NIC), Cholinergic interneurons (CINs), Nicotinic acetylcholine receptors (nAChRs), Alpha6 nAChRs (α6*-nAChRs)
Introduction
Alcohol use disorder (AUD) and tobacco use disorder (TUD) are leading causes of disease and premature death in the USA, with both substances often co-abused. Approximately 80% of persons with AUD are tobacco smokers [1, 2]. Nearly 17 million Americans have an AUD, but only approximately 1.3 million adults receive treatment [3], leaving over 90% untreated. The estimated economic costs of AUD to the USA in 2015 were $249 billion, with almost three-quarters related to binge drinking [3, 4]. The estimated direct medical care costs associated with tobacco smoking in 2014 was $170 billion [5, 6]. The economic, societal, familial, and personal costs associated with AUD and TUD are staggering. Understanding how alcohol and tobacco affect the brain on a molecular level is imperative in order to develop effective, long-lasting treatments.
The Mesolimbic Dopamine Reward System
The compulsion to consume ethanol (EtOH) and nicotine (NIC) stems from both their positive reinforcing properties, including anxiolytic and euphoric effects, and negative reinforcing properties, including aversive withdrawal symptoms that result from abstinence [7]. Symptoms of dependence accumulate with repeated drug taking and withdrawal [8]. A large body of research has supported the idea that the rewarding effects of EtOH, NIC, and other psychoactive drugs are dependent on mesolimbic dopamine (DA)-mediated neuromodulation [9–13]. Current dogma maintains that DA neuron activation and release in the mesolimbic DA system originating in the midbrain ventral tegmental area (VTA) and projecting to the nucleus accumbens (NAc) of the ventral striatum is a scalar index of EtOH and NIC reward [14]. The emerging view is that the dysregulated homeostasis that accompanies the development of drug addiction may result from experience-dependent neuroadaptations that hijack normal synaptic transmission in this system [15–18].
Nicotinic Receptors in the Mesolimbic Dopamine System
Neurons within the VTA express a wide variety of nicotinic acetylcholine (ACh) receptors (nAChRs) [19], with heteromeric α4β2- and homomeric α7-nAChRs being the most widely expressed subtypes in the mammalian brain. In the VTA and substantia nigra, heteromeric α6 -containing nAChRs α6*-nAChRs (* denotes α6 subunits combined with other nAChR subunits) are expressed on DAergic neurons [20], but this expression is mainly on striatal DAergic terminals [21], which are known to project to the striatum [14]. In the VTA, α6*-nAChRs are also found on GABAergic terminals that project onto DA neurons [21]. Application of nicotine desensitizes these nAChRs on GABAergic neurons, while glutamatergic synapses are not desensitized [22]. This leads to increased activation of VTA DAergic neurons with NIC through disinhibition via GABAergic synapses and direct activation through glutamatergic synapses [21]. Heteromeric α6*-nAChRs have been proposed as a substrate for EtOH. Administration of α-conotoxin MII (α-Ctx MII), a potent antagonist of α6*-nAChRs, locally into the VTA reduces EtOH-induced NAc DA release [23], reduces EtOH-induced locomotor activity [24], blocks recognition of EtOH-associated cues [25], and blocks voluntary EtOH drinking in rodents [23]. Likewise, expressing hypersensitive α6*-nAChRs (α6L9’S subunits) alters EtOH-induced behaviors [26, 27]. Although α4β2- and α7-nAChRs are the most widely expressed neuronal subtypes in the mammalian brain, there is considerable expression of heteromeric α6*-nAChRs in the VTA. In fact, α6*-nAChR subunit mRNA levels are 16-fold higher than other nAChR subunits in the VTA [21], suggesting α6*-nAChRs are major molecular regulators of addiction. Accumulating evidence suggests nAChRs serve as common substrates that regulate the motivational properties of both NIC and EtOH. Nicotine, for example, stimulates EtOH drinking in both EtOH-dependent humans and rodents [28–33]. Conversely, the α4β2-nAChR partial agonist varenicline, marketed as the smoking cessation agent Chantix, decreases EtOH intake in EtOH-dependent humans [34–36] and rodents [37, 38]. On a molecular level, EtOH has been documented to play a direct role in the allosteric modulation, stabilization, desensitization, and internalization of nAChRs as well as the up- and downregulation of these functions [39].
We have previously shown that α6*-nAChRs are expressed on GABA terminals projecting to VTA GABA neurons and mediate low-dose EtOH effects [40], that high-dose EtOH inhibits DA release in the NAc via α6*-nAChRs [41], that VTA GABA neurons express α6*-nAChRs [42], that functional α6*-nAChRs can be expressed in cell lines [43], that α6*-nAChRs are a highly sensitive target for low-dose EtOH on GABAAR-mediated inhibition to VTA GABA neurons [40], that α6*-nAChRs are involved in spontaneous DA release [42], and that α6*-nAChRs mediate EtOH reward [40]. However, the specific location in the brain where α6*-nAChRs mediate EtOH effects on DA release in the mesolimbic system and EtOH reward cannot be determined from these studies. This requires selective activation and inactivation of the specific pathway in the brain that expresses α6*-nAChRs, which was the main objective of this study. The dogma is that α6*-nAChRs are expressed on DA terminals [44]. However, the VTA has high levels of α6*-nAChR expression, and α6*-nAChRs are expressed primarily in the VTA and NAc [21]. Most importantly, we and others have shown that at least one population of VTA GABA neurons expresses α6*-nAChRs and projects to the NAc. However, the implications of striatal cholinergic interneuron (CIN)-mediated DA release are becoming increasingly appreciated. A number of recent optogenetic studies demonstrate that selective activation of VTA GABA neurons inhibits DA neurons, drives conditioned place aversion [45], and disrupts reward consumption [46]. Studies also demonstrate that projections of GABA neurons to the NAc inhibit CINs to enhance associative learning [47]. We have recently reported that VTA GABA neurons project to CINs and inhibit them via an atypical GABA receptor, which is sensitive to EtOH [48], providing compelling evidence for the importance of VTA-NAc GABAergic pathways in regulating EtOH effects on DA transmission in the reward pathway. Given that some VTA GABA neurons express α6*-nAChRs [40], and our evidence demonstrating that α6*-nAChRs mediate EtOH enhancement of CIN-mediated DA release, the core hypothesis of this study was that α6*-nAChR-containing VTA GABA neuron input to CINs in the NAc mediates EtOH effects on CINs and ultimately DA release. We hypothesized that α6*-nAChRs will mediate EtOH effects on VTA GABAergic synaptic inhibition of CINs and CIN-mediated DA release and that adaptive effects accrue to chronic EtOH exposure, including plasticity of VTA-NAc GABAergic synaptic transmission to CINs.
Methods
Animal Subjects
VGAT-Cre mice (Jackson Labs stock Slc32a1tm2(cre)Lowl/J; Strain #016962) were crossed with mice from our colony of glutamate decarboxylase-67 (GAD67) green fluorescent protein (GFP) mice [GAD-67-GFP knock-in on a CD-1 (white albino) mice [49] (VGAT-Cre/GAD67-GFP)], which enabled visualization and electrophysiology of VTA and NAc neurons. ChAT-ChR2-eYFP mice (Jackson Labs stock B6.Cg-Tg(Chat-COP4*H134R/EYFP,Slc18a3)6Gfng/J; Strain #014546) were also used which enabled visualization, electrophysiology of CINs, and activation of CIN-mediated DA release in the NAc. Mostly male mice were used for this project with a total of 160 male and female mice used. For each methodology employed, animals were treated in strict accordance with the Brigham Young University (BYU) Animal Research Committee (IACUC) guidelines, which incorporate and exceed current NIH guidelines. The BYU IACUC reviewed and approved the procedures detailed herein. Once weaned at PND 21, all mice were housed in maximum groups of five and given ad libitum access to solid food and water and placed on a reverse light/dark cycle with lights ON from 10 PM to 10 AM.
Viral Vectors
AAV-DIO-ChR2-mCherry
AAV-DIO-ChR2-mCherry (DIO-ChR2, UNC Vector Core, serotype AAV5) is one of many Cre-based viral constructs that have been developed to transport retrogradely to a targeted brain region and genetically manipulate the activity of defined neuronal subpopulations to elucidate function and alter behavior [50]. VGAT-Cre/GAD67-GFP mice were injected bilaterally into the VTA with DIO-ChR2 (VGAT DIO-ChR2) to target VTA GABA neurons projecting to the NAc and allow the generation of optically evoked inhibitory postsynaptic currents (oIPSCs) specifically from VTA GABA neurons. For the procedure, mice were anesthetized with 2–4% isoflurane and injected bilaterally into the VTA (A/P, − 3.0; M/L, + 0.5; D/V, − 4.5 mm from bregma) under aseptic conditions with DIO-IRes-ChR2 (0.5 μL, UNC Vector Core, serotype AAV5) using a needle made from a fire-polished glass capillary. A microinjection pump (World Precision Instruments, Sarasota, FL) was used to deliver the virus into each hemisphere at a rate of 0.075 μL/min. The cannula was left in place following the injection for 7–10 min to ensure diffusion. Mice were sutured and housed in a single cage until full recovery (~ 7 days), and the virus was incubated for 3–4 weeks to reach peak expression in the NAc before experimentation.
AAV-flex-mRuby2-α6miRNA
Our hypothesis was that GABA terminals on VTA GABA neurons projecting to CINs in the NAc would express α6-nAChRs. We developed a Cre recombinase-microRNA (miRNA) construct that travels anterogradely to induce targeted knockdown of α6-nAChR mRNA (α6-miRNA). The miRNA plasmids used were adapted from pAAV-hSyn-FLEX-dsRed-shvgat (Addgene plasmid #67845, serotype AAV1) with the dsRed cassette replaced by mRuby2 to facilitate direct fluorescent visualization in tissue. The pPRIME system [51] was used to generate miRNA 22mers targeted against mouse Chrna6 designed online (http://katahdin.mssm.edu/siRNA/RNAi.cgi?type=miRNA), and a control scramble fragment that recognizes no known mammalian transcript was used as previously described [52]. The Chrna6 targeting sequence was GAG GTA GAA GAC GAC TGG AAAT, and the control scramble sequence was GAG CGC TTA GCT GTA GGA TTCT. Twenty two mers within the hairpin miRNA construct were cloned into the EcoRI/XhoI site of pAAV-hSyn-FLEX-mRuby2-shvgat to generate pAAV-hSyn-FLEX-mRuby2-scramble and pAAV-hSyn-FLEX-mRuby2-α6-miRNA. Cloning, subcloning, and AAV packaging was performed by the University of Minnesota Viral Vector and Cloning Core. Viral titers ranged from 2.31 × 1013 to 2.8 × 1014 gc/mL and were not diluted prior to injection. To dissect α6-nAChR-relevant VTA GABA neuron outputs to CINs, VGAT-Cre/GAD67-GFP mice were injected bilaterally into the VTA with AAV1-flex-mRuby2-α6miRNA (α6-miRNA) and control (0.5–1 μL/side). The virus was incubated for 3–4 weeks to reach peak expression before experimentation. Finally, to activate CINs directly, we used a ChAT-ChR2-eYFP mouse and stimulated the neurons optically with blue light.
Depletion of α6-nAChRs in VTA DA Neurons by α6-miRNA: qRT-PCR and RNAScope
Male and female heterozygous DA transporter (DAT)-Cre mice on a C57BL/6 J background were anesthetized with isoflurane, stereotaxically injected with 0.75 μL/side of AAV1-hSyn-FLEX-mRuby2-miRNA or AAV1-hSyn-FLEX-mRuby2-control targeting the VTA (coordinates: AP − 3.02, ML ± 0.52, DV − 4.75) and allowed to recover in their home cage. For quantitative reverse transcriptase polymerase (qRT-PCR) experiments, mice were euthanized 4 weeks after surgery, and micro-punches from the ventral midbrain were isolated and snap frozen on dry ice. RNA was isolated using QIAGEN RNeasy Plus kit (QIAGEN). 500–1000 ng of RNA per sample was reverse-transcribed using the Applied Biosystems high-capacity reverse transcriptase kit (Applied Biosystems). Samples were then processed for qPCR using Applied Biosystems Taqman probes for Chrna6, Chrna4, Chrnb3, and Gapdh transcripts. Expression levels relative to GAPDH were calculated using the delta dCt method, with values from control mice averaged for a relative expression value of 1. Each data point is from 1 mouse. For RNAscope, mice were euthanized 4–5 weeks after surgery, the brains were snap frozen, and the VTA sectioned and probed for tyrosine hydroxylase (TH), glutamate decarboxylase 67 (GAD67), and α6 (Chrna6) transcripts using multiplex RNAscope (Advanced Cell Diagnostics). DAPI staining was used to detect cell nuclei. Images were taken on a Keyence BZ-X700 epifluorescent microscope from one female DAT-Cre mice injected with the α6-miRNA virus and one female DAT-Cre mice injected with the control virus.
Chronic Intermittent Ethanol Exposure
Chronic intermittent EtOH (CIE) exposure was used to model EtOH-dependence and withdrawal. We have published many papers on this paradigm: Two recent papers describe the use of EtOH vapor chambers in mice to produce dependence [53, 54]. VGAT-Cre/GAD67-GFP mice were placed in EtOH vapor chambers combined with EtOH drinking to establish EtOH dependence and air vapor chambers combined with water as a control. We modified the vapor chamber system developed in the lab of Graeme Mason at Yale [55]. The six automated chamber system consisted of an air-pressurized, feedback-controlled EtOH flask with flow valves to each of three sealed chambers to regulate the flow of air (11 L/min) and concentration of EtOH to three of the six chambers placed in a ventilation hood. A breathalyzer (Drager Alcotest 6510) was used in a feedback loop to regulate the concentration of EtOH. The concentration of alcohol vapor was set in the feedback system with the breathalyzer at 3L/min flowmeter level to deliver 200 mg% blood alcohol levels (BALs), as previously reported [53]. In some mice, to confirm BALs at this target level, cheek blood samples were taken, and BALs were measured using an enzymatic kit (Sigma-Aldrich, St. Louis, MO). The target BAL level did not vary by more than 10% in the animals tested. In order to avoid overdosing during the first week of CIE vapor exposure, mice were exposed to 4, 6, and 8 h of vapor before exposing to 16 h vapor/day in order to induce tolerance. Mice were exposed to 5 days of vapors/week for at least 3 weeks in order to establish dependence, as demonstrated previously [53, 54]. Control animals were housed in three sealed chambers in the same ventilation hood but only received air.
Immunohistochemistry
Animals were anesthetized in 2–4% isoflurane then underwent transcardial perfusion with 4% paraformaldehyde (PFA). Once perfused, brains were carefully removed and placed in 4% PFA for at least 24 h to facilitate continued fixation. After incubation in PFA, brains were placed in a solution of 30% sucrose in phosphate-buffered saline (PBS) until the density of the brain matched that of the solution and the brains dropped to the bottom of the vial (~ 24–48 h). Brains were then flash frozen in dry ice and mounted on a cold microtome stage. Targeting the VTA and NAc, the brains were sliced at 40 μm, and slices were placed in cryoprotectant (30% ethylene glycol, 30% sucrose, 0.00002% sodium azide, in 0.1 M PBS) and kept at − 20 °C until mounting. To mount slides, sections were placed on microscope slides and dried for ~ 5 min. Once dried, a drop of VECTASHIELD (Vector Laboratories) was placed on the tissue, and a coverslip was placed on the slide. Slides were allowed to set overnight and then were kept at 4 °C until imaging. An Olympus FluoView FV1000 confocal microscope was used to image mounted slices. Brain slices were mounted on microscope slides and imaged under oil immersion at 60 × (Olympus, PlanApo 1.40 numerical aperture) or oil immersion at 40 × (Olympus, UPlanFLN 1.30 numerical aperture). To ensure consistent readings between samples, a constant photomultiplier tube voltage and gain were set between all acquired images.
Characterization of VTA GABA and NAc Neurons
GABA neurons in the VTA of brain slices were studied by visualizing GAD-67+ neurons in GAD-67 GFP mice, as reported previously [56, 57]. VTA neurons were further characterized using a GABA spike command waveform (spikes at 200 Hz for 500 ms), as GABA neurons will follow the spike command waveform with fidelity, while DA neurons will not [58]. Neurons that did not fluoresce in GAD-67 GFP mice and/or exhibit a non-cation-specific inward rectifying current (Ih) with low input resistance in C57BL/6 mice and did not follow the spike command waveform were assumed to be non-GABA putative DA neurons [57, 59–62]. VTA GABA neuron inhibition of CINs was the pathway focus for most of the electrophysiological recordings from this experiment performed on accumbal CINs. In ChAT-ChR2-eYFP mice, CINs could be visualized as YFP + mice. In non-ChAT mice, CINs in the NAc were characterized by their large size, shape, and the presence of an Ih current for whole-cell patch-clamp experiments. In the VGAT-Cre/GAD67-GFP mice, CINs were also visualized in the slice preparation as non-GFP neurons amongst predominantly GABA neurons in the striatum.
Patch-Clamp Electrophysiological Recordings
All brain slice preparations were performed in P28–120 day-old ChAT ChR2 eYFP or VGAT-Cre/GAD67-GFP mice in order to visualize ChAT + or GAD67 + neurons in the NAc by GFP and mCherry fluorescent imaging. The brains were extracted via anesthetization with 2–4% isoflurane. Upon extraction, the brain was glued onto a cutting stage. The brain was then sectioned in room temperature (ACSF; in mM: 124 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 12 glucose, 1.5 MgSO4, 2 CaCl2) and bubbled with 95% O2/5% CO2. Targeting the VTA or NAc, coronal slices (220 μM thick) were placed in an incubation chamber containing ACSF perfused with 95% O2/5% CO2 for at least 30 min. After 30 min, brain slices were then placed in a recording tissue chamber with ACSF continuously flowing at physiological temperatures (35 °C). Fluorescent cells were imaged on a Nikon Eclipse FNI microscope with a 40 × /0.80 n.a. objective lens. The filter cube for GFP detection was a Nikon C-FL ENDOW GFP 96343 cube (bandpass, 450–490 nm; barrier, 500–550 nm; dichroic, 495 nm). Excitation was performed with a Sutter Lambda TLED transmitted light source at 506 nm. Cells were then imaged using a differential interference contrast imaging in order to facilitate cell attached recordings. For low-frequency stimulation experiments, two hundred forty 0.4 ms blue light pulses with 1 Hz frequency were used (Thorlabs M470L3 – 470 nm, 650 mW mounted LED, 1000 mA). Intensity of light was determined by increasing light to evoke maximum IPSC amplitude and then decreasing light intensity until the IPSCs were 50% of maximum amplitude. Ethanol was only administered in animals older than day P28. All mice used in EtOH exposure groups were age-matched and were P28–120 days old with a median age at day P55.
Voltage-Clamp Recordings of Inhibitory Postsynaptic Currents
For voltage-clamp recordings in brain slices, electrodes were pulled from borosilicate glass capillary tubes (1.5 mm o.d.; A-M Systems, Sequim, WA) and filled with KCl pipette solutions (in mM: 128 KCl, 20 NaCl, 0.3 CaCl2, 1.2 MgCl2, 10 HEPES, 1 EGTA, 2 Mg-ATP, 0.25 Na-GTP, and 5 QX-314; pH 7.3) for recording mIPSCs. Pipettes having tip resistances of 2.5–5 MΩ, and series resistances typically ranging from 7 to 15 MΩ were used. Voltage-clamp recordings were filtered at 3 kHz with an Axon Instruments MultiClamp 700B amplifier and digitized at 10 kHz using Axon 1440A digitizers. Axon Instruments pClamp ver10, Mini Analysis (Synaptosoft: Decatur, GA), and Igor Pro (Wavemetrics: Oswego, OR) software packages were utilized for data collection and analysis. Mini IPSCs (mIPSCs) were recorded in the presence of 1.5 mM kynurenic acid to block GLU-mediated synaptic currents and isolate GABAAR-mediated synaptic currents and 100 μM lidocaine to block action potential-mediated IPSCs. Electrodes with a resistance of 3–5 MΩ between the pipette and external solutions were used to form tight seals (> 1 GΩ) on the cell surface, until suction was applied in order to convert to conventional whole-cell recording. A holding potential (VH) of − 60 mV was used and ionic currents measured in response to drug application.
Voltage-Clamp Recordings of CIN Firing Rate
While most striatal neurons are not autonomously active, CINs exhibit a regular spiking activity in absence of any synaptic inputs [63]. Cell-attached patch-clamp techniques were used to record CIN firing rate. Electrodes with a resistance of 3–5 MΩ between the pipette and external solutions were used to form tight seals (10 MΩ–1 GΩ) on the cell surface. Positive pressure was applied to the electrode when approaching the neuron. By applying suction to the electrode, a seal (10 MΩ–1 GΩ) was created between the cell membrane and the recording pipette. Spontaneous spike activity was then recorded in cell-attached mode with an Axon Instruments MultiClamp 700B amplifier, sampled at 10 kHz using NIDAW boards, and collected and analyzed using AxoGraph software. Neurons were recorded in voltage-clamp mode (voltage-clamp was set to 0 mV). Firing rate recordings in this study were performed in cell-attached mode in order to avoid dialyzing the contents of the cells and disrupting the cytoplasmic milieu (e.g., the Cl− ion gradient), which we have shown previously is perturbed with opioid dependence [64, 65]. A stable baseline recording of firing activity was obtained for 5–10 min before adding drugs. Neurons that did not achieve a stable baseline firing rate during this time were rejected from the study.
Carbon Fiber Electrodes and Fast Scan Cyclic Voltammetry
For voltammetry recordings, a 7.0 μm diameter carbon fiber was inserted into borosilicate capillary tubing (1.0 mm i.d.; A-M Systems, Sequim, WA) under negative pressure and subsequently pulled on a vertical pipette puller (Narishige, East Meadow, NY). The carbon fiber electrode (CFE) was then cut under microscopic control with 70–100 μm of bare fiber protruding from the end of the glass micropipette. The CFE was back-filled with 3 M KCl. For ex vivo voltammetry recordings, electrodes were positioned ~ 75 μm below the surface of the slice in the NAc core. Cholinergic interneuron DA release was evoked in the NAc core in ChAT-ChR2-eYFP mice every 2 min by a 4-ms pulse of blue light from the objective. The electrode potential was linearly scanned as a triangular waveform from − 0.4 to 1.2 V and back to − 0.4 V vs Ag/AgCl using a scan rate of 400 V/s. Cyclic voltammograms were recorded at the carbon fiber electrode every 100 ms (i.e., 10 Hz) by means of ChemClamp voltage-clamp amplifier (Dagan Corporation, Minneapolis, MN) or customized potentiostats. Voltammetry recordings were performed and analyzed using LabVIEW (National Instruments, Austin, TX)-based customized software (Demon Voltammetry, (Yorgason et al. 2011)). Dopamine levels were monitored for a stabilization period typically lasting 1 h. Once the stimulated DA response was stable for 4–5 successive collections and did not vary by more than 10%, baseline measurements were taken. Cholinergic interneuron-mediated DA releases was recorded during bath application of the increasing concentrations of EtOH (1, 10, 20, 40, 80 mM). Each concentration of EtOH was recorded for at least 15 min until the signal stabilized. To determine the mechanism of EtOH inhibition of DA, CIN-mediated DA release was also recorded in the presence of the α-conotoxin MII.
Drug Preparation and Administration
Ethanol (Decon Labs, King of Prussia, PA, USA) was added directly to ACSF to the desired concentrations (5 mM, 10 mM, 20 mM, 40 mM, 60 mM, 80 mM). All other drugs used in vitro were made fresh from powder in distilled water, then added to ACSF, and superfused onto brain slices: nicotine tartrate (Sigma‐Aldrich, St. Louis, MO, USA), TPMPA (Tocris Bioscience, Bristol, UK), and α-Ctx MII[H9A; L15A] (MII) were synthesized as previously described [66].
Statistical Analyses of Responses
Miniature IPSCs were recorded continuously and then organized into 5-min bins at baseline and after drug administration. The mean frequency and amplitude of mIPSCs was determined with MiniAnalysis (Synaptosoft) using a peak detector and temporal algorithm. Comparisons were made between 5 min of values before treatment and 5 min after drug treatment. Firing rates were determined from windowed and discriminated spike times and histogrammed into 10-s bins. Averaged firing rates of 5 min were determined on ratemeters by rectangular integration in Igor Pro (Wavemetrics, Oswego, OR). The last 5 min of integrated firing rate before treatment (i.e., drugs) was compared against 5 min of drug effects 5 min after administration unless otherwise indicated. All results were presented as raw mean values and percent control ± SEM. Results between groups were compared using a two-tailed unpaired t-test or ANOVA. Experiments relying on variance in time or current were analyzed using mixed models ANOVA with post hoc t-test at individual points. Dopamine release was analyzed in Demon Voltammetry software [67] and measured at peak oxidation currents. The software performs a running subtraction of capacitive currents to account for changes in extracellular DA levels and to reduce noise. Post-subtracted data was then compared across time against calibrated DA cyclic voltammograms, from which it calculates an r2 value to measure signal clarity. Statistics were performed in R studio. Single ANOVAs were used to compare concentration-specific differences between drug applications with Tukey HSD correction method for post hoc analysis. Statistical significance required ≥ 95% level of confidence (p ≤ 0.05). Analysis software included Microsoft Excel and Igor Pro (Wavemetrics, Oswego, OR). Significance levels are indicated on graphs with asterisks *,**, and *** and correspond to levels p < 0.05, 0.01, and 0.001, respectively. Figures were constructed with Igor Pro software (Wavemetrics, Lake Oswego, OR).
Results
Depletion of a6-nAChRs in Midbrain DA Neurons by a6-miRNA
Integral to this study was the verification of depletion of α6-nAChRs by the α6-miRNA viral construct for subsequent evaluation of EtOH effects in the VTA and NAc. We evaluated the expression of α6-nAChRs in midbrain DA neurons with qRT-PCR and fluorescent in situ hybridization using RNAscope following injection of α6-miRNA virus into the midbrain vs scrambled control miRNA virus. There was a significant decrease in Chrna6 transcript in male and female mice injected with the AAV containing α6-miRNA compared with scrambled control miRNA (Fig. 1A, B, C; F(1,26) = 12.64, p = 0.002; n = 15 mice, sexes combined, one replicate per animal). There were no significant gender effects F(1,26) = 0.96, p = 0.34. There was no effect of the α6-miRNA virus on α4 or β3 transcript in the same mice (Fig. 1C; all p > 0.05; n = 8–9 mice each, sexes combined, one replicate per animal).
Fig. 1.

Depletion of α6-nAChRs in midbrain DA neurons by α6-miRNA. A Illustration showing the experimental framework wherein AAV-flex-mRuby2-α6miRNA was injected into the VTA of DAT-Cre mice 3 weeks prior to experimentation. B Fluorescent in situ hybridization images of neurons (DAPI; blue) from the VTA showing knockdown of Chrna6 (red; α6-nAChRs) with DAergic neurons in the midbrain identified by TH (green) and distinguished from GAD67 + neurons (cyan). C α6-miRNA treatment depleted α6-nAChRs in TH + neurons, but not α4 or β3 nAChRs
EtOH Enhances GABAergic Transmission to VTA GABA Neurons via α6*-nAChRs
We have shown previously that α6*-nAChRs are expressed on GABA terminals on VTA GABA neurons and that GABA inhibition, likely from a subpopulation of VTA GABA neurons, mediates EtOH effects on VTA GABA neurons [40]. In this paper, we demonstrated that EtOH enhanced evoked IPSCs (eIPSCs) at dose levels 1–5 mM and reduced eIPSCs at dose levels 50–100 mM. Both the enhancement and reduction by EtOH were blocked by the α6*-nAChR antagonist α-conotoxin MII[H9A;L15A] (MII). Thus, EtOH enhancement of GABAergic transmission to VTA GABA neurons was mediated by α6*-nAChRs. Here, we sought to determine the effects of knockdown of α6*-nAChRs on GABA transmission to VTA GABA neurons in VGAT-Cre/GAD67-GFP mice with a Cre-dependent AAV-flex-mRuby2-α6miRNA (α6-miRNA) viral construct injected into the VTA of VGATCre/GAD67-GFP (Fig. 2A, B). Baseline eIPSC amplitude in the α6-miRNA knockdown experiments was 332.1 ± 32 pA (n = 6) and in the MII experiments was 421 ± 36 pA (n = 16). As we have demonstrated previously [40], low-dose EtOH (5 mM) significantly enhanced GABAergic inhibition to VTA GABA neurons (Fig. 2C, scrambled n = 6, α6-miRNA n = 5, MII n = 13). There was no effect of EtOH on paired-pulse responses (Fig. 2C), as previously reported [40]. Knockdown of α6*-nAChRs, or superfusion of MII, blocked EtOH enhancement of VTA GABA neuron eIPSCs at 5 mM (α6-miRNA: F(1,11) = 9.2, p < 0.002; Fig. 2D, EtOH n = 6, miRNA n = 5; MII: F(1,31) = 8.4, p < 0.003; Fig. 2E, control n = 16, MII n = 13, MII + EtOH n = 6). This experiment provides further confirmation that a subpopulation of VTA GABA neurons expressing α6*-nAChRs is involved in low-dose EtOH effects on GABAergic synaptic transmission to VTA GABA neurons and likely to VTA-NAc GABAergic projections.
Fig. 2.

EtOH enhances evoked VTA GABA neuron IPSCs with block by α-conotoxins or knockdown of α6*-nAChRs. A Illustration showing the experimental framework wherein AAV-flex-mRuby2-α6miRNA was injected into the VTA 3 weeks prior and VTA GABA neurons were recorded in whole-cell, voltage-clamp mode in VGAT-Cre/GAD67-GFP mice. B Immunohistochemical panel showing that some VTA GABA neurons express α6*-nAChRs. An antibody against tyrosine hydroxylase (TH) was used along with the GAD67-GFP in these mice. Imaged using oil immersion 40 × objective (Olympus, UPlanFLN 1.30 numerical aperture). C Low-dose EtOH (5 mM) enhanced eIPSC amplitudes in VTA GABA neurons in VGAT-Cre/GAD67-GFP mice injected with scrambled α6-miRNA into the VTA, but not mice injected with α6-miRNA. There was no effect on paired-pulse ratio (50 ms), as previously reported [40]. Knockdown (KD) of α6*-nAChRs (D) or superfusion of MII (E) significantly reduced EtOH enhancement of VTA GABA neuron eIPSCs. Values in parentheses are n values. Asterisks ** indicate significance level p < 0.01
α6*-nAChRs Mediate EtOH Inhibition of GABAergic Synaptic Transmission to CINs
We [48], and others [47], have shown that some VTA GABA neurons project to CINs. In our study, this was accomplished by injecting the α6-miRNA viral construct into the VTA of VGAT-Cre/GAD67-GFP mice and probing for mRuby puncta in the NAc and comparing against a control scrambled miRNA construct. We replicate this here (Fig. 3A, B). As mentioned above, we recently demonstrated that EtOH reduces VTA-NAc GABAergic oIPSCs to CINs via an atypical ρ−1 GABA receptor [48]. Interestingly, this was opposite to the excitatory effects of EtOH on GABAergic transmission to VTA GABA neurons, which are mediated by a typical GABAA receptor. We extended these studies to include the evaluation of EtOH effects on CIN mIPSCs, which facilitate the interpretation of presynaptic vs postsynaptic effects and the role of α6*-nAChRs on VTA-NAc GABA terminals to CINs. We hypothesized that since VTA-NAc GABAergic neurons express α6*-nAChRs and that they are likely expressed on GABA terminals to CINs, they might mediate EtOH effects on GABA release to CINs. We recorded CINs identified by fluorescence and an appreciable Ih current in whole-cell voltage-clamp mode (Fig. 3E) in 19 slices from 14 ChAT-ChR2-eYFP mice (Fig. 3C–E). Superfusion of EtOH (5–80 mM) significantly reduced mIPSC frequency (5 mM: F(1,21) = 4.2, p = 0.05; 10 mM: F(1,23) = 13.1, p = 0.001; 40 mM: F(1,17) = 5.22, p = 0.04; 80 mM: F(1,17) = 10.8, p = 0.005; baseline mean frequency = 1.29 ± 0.37 Hz; Fig. 3F, G), but not amplitude (p > 0.05; baseline mean amplitude = 50.1 ± 6.2 pA; n = 19; Fig. 3F, H). In separate experiments, we recorded CINs in 17 slices from 12 ChAT-ChR2-eYFP mice. Superfusion of MII did not significantly affect mIPSC frequency (p > 0.05; baseline mean frequency = 1.79 ± 0.30 Hz; n = 17; data not shown) or amplitude (p > 0.05; baseline mean amplitude = 27.9 ± 1.9 pA; data not shown). However, MII significantly blocked the inhibition of frequency produced by EtOH (F(1,16) = 7.24, p = 0.014 for EtOH vs MII + EtOH; Fig. 3F, I, Con + EtOH, n = 9, MII + EtOH, n = 8), but had no effect on amplitude (p > 0.05; Fig. 3F, J, Con + EtOH. n = 9, MII + EtOH, n = 8), suggesting a role for α6*-nAChRs in mediating EtOH effects on presynaptic release of GABA, likely VTA-NAc GABAergic inhibition of CINs.
Fig. 3.
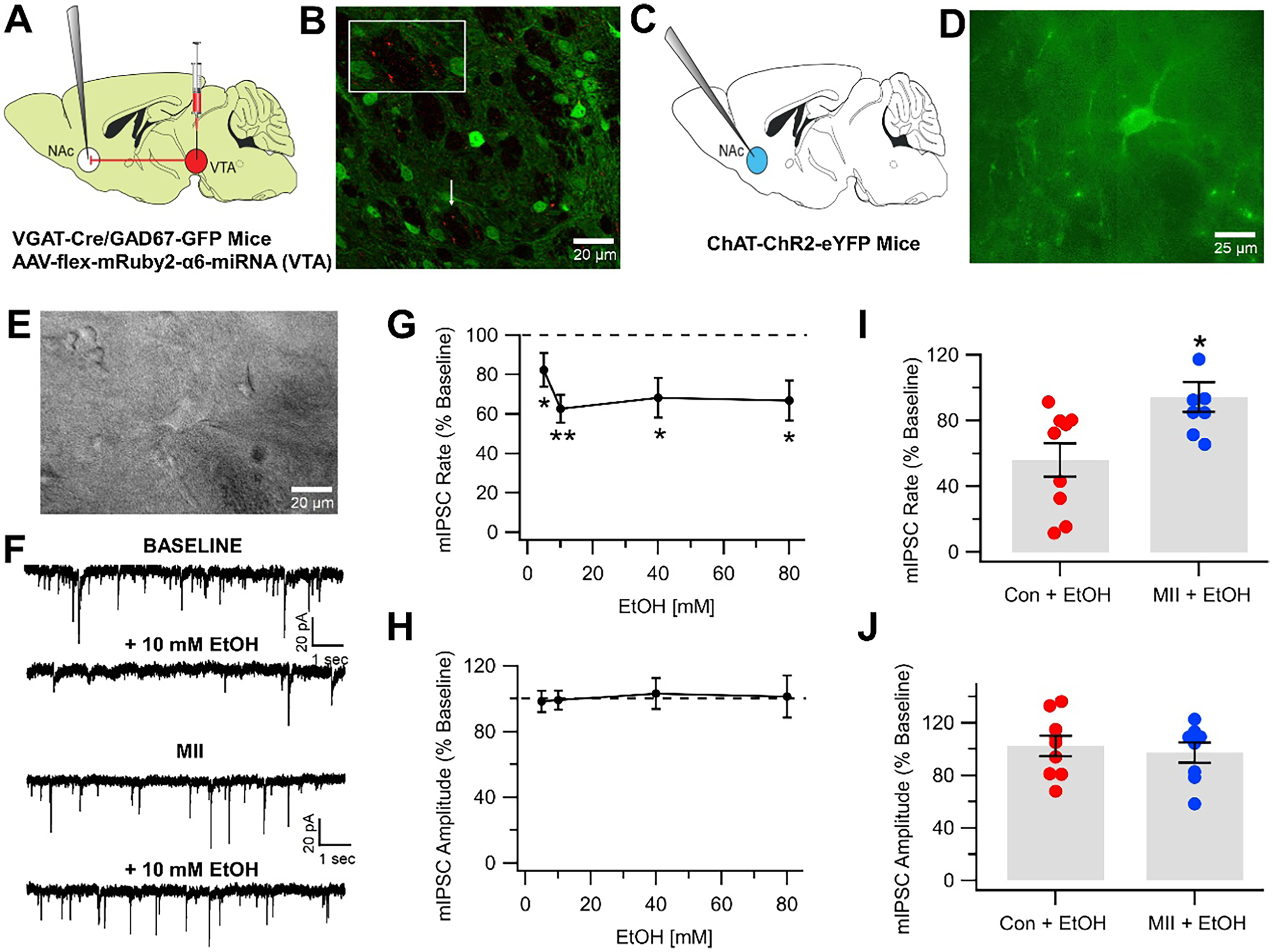
EtOH inhibits miniature IPSCs with block by α-conotoxins. A Illustration showing the experimental framework wherein AAV-flex-mRuby2-α6-miRNA was injected into the VTA 3 weeks prior and NAc CINs were recorded in whole-cell, voltage-clamp mode in VGAT-Cre/GAD67-GFP mice. B Immunohistochemical image showing that putative CINs in VGAT-Cre/GAD67-GFP mice are innervated by VTA-NAc GABAergic projections expressing α6*-nAChRs. Imaged using oil immersion 60 × objective (Olympus, PlanApo 1.40 numerical aperture). C Illustration showing the experimental framework wherein CINs were recorded in ChAT-ChR2-eYFP mice. D Image showing CINs in ChAT-ChR2-eYFP mice with fluorescence imaging (Olympus, UPlanFLN 1.30 numerical aperture). E Patch clamp recording of CINs in ChAT-ChR2-eYFP mice using infrared imaging. F Representative 10-s recordings of CIN mIPSCs. G Ethanol (5–80 mM) reduced CIN mIPSC frequency. H Ethanol (5–80 mM) did not affect CIN mIPSC amplitude. I, J Summary of effects of MII on EtOH inhibition of CIN mIPSCs. MII significantly reduced EtOH inhibition of CIN mIPSC frequency (I), but not amplitude (J). Asterisks * and ** indicate significance levels p < 0.05 and p < 0.01
Acute EtOH Enhances CIN Firing Rate via α6*-nAChRs
As CIN oIPSCs were inhibited by EtOH [48], but their firing rate enhanced by EtOH [48], and the EtOH inhibition of GABAergic mIPSCs was blocked by MII, we hypothesized that α6*-nAChRs would mediate low-dose EtOH effects on CIN firing rate, as the reduction of GABAergic inhibition to CINs might result in enhanced CIN firing rate. In VGAT-Cre/GAD67-GFP mice injected with α6-miRNA into the VTA, we confirmed the VTA-NAc GABAergic projection (Fig. 4A, B). As CINs could not be positively identified in these mice, we recorded GAD-negative CINs in the NAc using cell-attached, voltage-clamp mode. CINs were further identified by being inhibited by the muscarinic receptor agonist muscarine due to autoreceptor inhibition (Fig. 4C), which is a pharmacological way to distinguish them from other neurons in the NAc. As reported previously [48], EtOH (5–60 mM) significantly enhanced the firing rate of CINs (F(5,90) = 26.3, p < 0.001; baseline firing rate of CINs in the slice preparation was 0.47 ± 0.08 Hz; Fig. 4D). CIN firing rate was not affected by superfusion of MII (Fig. 4E, G; p > 0.05) but, interestingly, was significantly increased by injection of α6-miRNA into the VTA (2.05 ± 0.24 Hz, Fig. 4F, G; F(1,19) = 40.7, p < 0.001, control n = 11, MII n = 10, α6-miRNA n = 12). Ethanol enhancement of CIN firing rate was blocked by MII (Fig. 4E, H, control n = 11, MII n = 10, α6-miRNA n = 12) and injection of α6-miRNA into the VTA (Fig. 4F, H), suggesting that α6*-nAChRs on VTA-NAc GABA terminals from VTA GABA neurons are involved.
Fig. 4.

EtOH markedly enhanced CIN firing rate with block by α-conotoxins or knockdown of α6*-nAChRs. A Illustration showing the experimental framework wherein AAV-flex-mRuby2-α6-miRNA was injected into the VTA 3 weeks prior and NAc CINs were recorded in cell-attached, voltage-clamp mode in VGAT-Cre/GAD67-GFP mice. B Immunohistochemical panel showing that CINs in VGAT-Cre/GAD67-GFP mice are innervated by VTA-NAc GABAergic projections expressing α6*-nAChRs. Imaged using oil immersion 40 × objective (Olympus, UPlanFLN 1.30 numerical aperture). C CINs visualized in ChAT mice were characterized by autoreceptor inhibition by the M2 agonist muscarine. Insets are 5-s representative spike recordings before (a), during (b), and after (c) muscarine (wash). Times for (a, b, c) are indicated on the representative ratemeter below. D In putative CINs inhibited by muscarine in VGAT-Cre mice, firing rate is enhanced by increasing doses of EtOH. E Block of EtOH enhancement of CIN firing rate at high doses by MII. F Block of EtOH enhancement of CIN firing rate in mice injected with AAV-flex-mRuby2-α6-miRNA into the VTA. G Summary of effects of α6-miRNA or MII on CIN baseline firing rate. H Summary of EtOH effects in mice treated with MII or α6-miRNA. Values in parentheses are n values. Asterisks *, **, and *** indicate significance levels p < 0.05, p < 0.01, and p < 0.001, respectively
Adaptations in Acute EtOH Enhancement of Firing Rate During Withdrawal from Chronic Exposure to EtOH
We then sought to determine if acute EtOH activation of CIN firing rate might undergo adaptations in EtOH-dependent mice, similar to what we have demonstrated in VTA GABA neurons with chronic EtOH exposure [53, 54, 68]. Chronic intermittent exposure to EtOH for 3 weeks in vapor chambers resulted in a loss of EtOH activating effects on CINs (Fig. 5A). There was a significant difference between the effects of acute EtOH on CIN firing rate from 5 to 60 mM vs the effects of chronic EtOH (Fig. 5B; two-way ANOVA; drug, F(1,80) = 25.7, p < 0.001; concentration, F(5,80) = 28.95, p < 0.001; interaction, F(5,80) = 24.6, p < 0.001, acute EtOH, n = 6, chronic EtOH, n = 8).
Fig. 5.

Tolerance to EtOH enhancement of CIN firing rate in EtOH-dependent mice. A This ratemeter shows a representative ratemeter recording of a CIN in a mouse during withdrawal from CIE exposure. B Chronic EtOH exposure produced tolerance to EtOH enhancement of CIN firing rate. Values in parentheses are n values. Asterisks *, **, and *** indicate significance levels p < 0.05, p < 0.01, and p < 0.001, respectively
α6*-nAChRs Mediate CIN-iLTD
Given the marked enhancement in baseline firing rate with α6-miRNA treatment (Fig. 4) and adaptations in EtOH effects on CIN firing rate in EtOH-dependent mice (Fig. 5), we hypothesized that α6*-nAChRs in VTA-NAc GABA projections to CINs would exhibit plasticity: Specifically, that long-term depression (LTD) of GABAergic input to CINs would switch to long-term potentiation (LTP) in EtOH-dependent mice. While other studies have investigated inhibitory LTD (iLTD) in medium spiny neurons (MSNs) [69, 70], no one has reported iLTD in accumbal CINs. We confirmed VTA GABA projections to accumbal CINs by eliciting oIPSCs from VTA GABA neurons using Cre-dependent DIO-ChR2-mCherry injected into the VTA of VGAT-Cre/GAD67-GFP mice, and we found robust iLTD of oIPSCs in accumbal CINs (CIN-iLTD) with the same low-frequency stimulation parameters (LFS; 1 Hz, 240 pulses, 4-ms pulses) that produce MSN-iLTD (Fig. 6A, B, control, n = 11). This indicates that CINs undergo GABAergic plasticity similar to MSNs and suggests that the activity-dependent lowering of inhibition from VTA GABA neurons constitutes a form of inhibitory GABAergic synaptic plasticity in the VTA-NAc mesolimbic pathway (i.e., VTA-NAc). To confirm that α6*-nAChRs play a key role in CIN-iLTD, we performed the same experiment in mice injected with α6-miRNA in VGAT-Cre/GAD67-GFP mice and found that CIN-iLTD was significantly reduced (Fig. 6C, F). Moreover, application of 100 nM MII significantly reduced CIN-iLTD, suggesting that α6*-nAChRs mediate CIN-iLTD (Fig. 6C, F; two-way ANOVA; drug, F(1,16) = 5.413, p = 0.0335; LTD, F(1,16) = 21.11, p < 0.001; interaction, F(1,16) = 7.278, p = 0.0158, control, n = 11, MII, n = 7, α6-miRNA n = 7). As mentioned previously, the VTA-NAc projection to CINs is mediated by atypical ρ−1 GABA receptors, which appear to mediate EtOH reduction of CIN IPSCs [48]. Thus, we evaluated their involvement in CIN-iLTD. Superfusion of the highly selective ρ−1 GABA antagonist TPMPA [71] blocked CIN-iLTD (F(1,17) = 20.9, p < 0.001; Fig. 6D, F, control, n = 11, TPMPA, n = 7). Finally, consistent with our hypothesis, chronic exposure to EtOH in the vapor chambers reduced CIN-iLTD with a trend towards LTP (Fig. 6E, F; two-way ANOVA; drug, F(1,15) = 3.66, p = 0.075; LTD, F(1,15) = 1.476, p = 0.2433; interaction: F(1,15) = 2.399, p = 0.1423, chronic air, n = 8, chronic EtOH, n = 8).
Fig. 6.

Role of α6*-nAChRs and atypical GABA receptors in CIN inhibitory plasticity. A Illustration showing the experimental framework, wherein Cre-dependent AAV-DIO-ChR2-mCherry was injected into the VTA in VGAT-Cre/GAD67-GFP mice and optically evoked IPSCs were recorded in NAc CINs in whole-cell, voltage-clamp mode. Activation of GABAergic inputs to NAc CINs from the VTA were obtained by blue light stimulation through the objective. B Inset shows representative oIPSCs evoked in NAc CINs before and after low frequency optical stimulation (LFS; 1 Hz, 240 pulses). LFS reduced GABAergic oIPSCs in NAc CINs, termed CIN-iLTD. Horizontal markers pre (green) and post (red) indicate times where comparisons between treatment conditions were performed. C Superfusion of MII or treatment with α6-miRNA reduced CIN-iLTD. D Superfusion of the GABA receptor ρ−1 antagonist TPMPA markedly reduced CIN-iLTD. E Chronic exposure to EtOH reduced CIN-iLTD and produced a mild LTP state. F Summary of drug and treatment effects on CIN-iLTD. Values in parentheses are n values. Asterisks ** and *** indicate significance levels p < 0.01 and p < 0.001, respectively, for comparisons to baseline. Hashtags #, ##, and ### represent significance levels p < 0.05, p < 0.01, and p < 0.001, respectively, for comparisons between control and treatment responses
α6*-nAChRs Mediate EtOH Inhibition of Evoked DA Release
We have demonstrated previously that acute EtOH inhibits evoked [41] and enhances spontaneous [42] DA release in the NAc, which is mediated by α6*-nAChRs, and that MII blocks both NIC and EtOH inhibition of evoked DA release in the NAc core via α6*-nAChRs [41]. Similar to electrically evoked DA release, superfusion of EtOH inhibited light-evoked CIN mediated DA release in ChAT-ChR2-eYFP mice (baseline DA release = 0.336 ± 0.0414 μM; Fig. 7B, D, n = 12), but only at high doses of EtOH (Fig. 7D) with block by MII (Fig. 7C, E), suggesting the involvement of α6*-nAChRs. Fitting a linear regression, we found that EtOH significantly inhibited CIN-mediated DA release in a concentration-dependent manner (F(1,65) = 21.62, p < 0.001, Fig. 7D), significantly inhibiting DA release at 60 and 80 mM to 69.7 ± 5.27% and 69.9 ± 7.54% of the baseline (t = − 4.191 and − 3.711, p < 0.001, Fig. 7D). Superfusion of MII significantly inhibited CIN-mediated DA release on its own to 71.2 ± 6.17% of the baseline, indicating a role of α6*-nAChRs in CIN-mediated DA release (MII DA release = 0.385 ± 0.106 μM, F(1,22) = 16.376, p < 0.001). Subsequent application of EtOH (60 mM) in the presence of MII had no significant effect on CIN-mediated DA release, indicating the involvement of α6*-nAChRs in EtOH-induced inhibition of CIN mediated DA release. Normalizing the effect of EtOH to the stabilized effect of MII, we observed no significant effect of EtOH in the presence of MII on CIN mediated DA release (F(1,16) = 3.4489, p = 0.0818; Fig. 7C, E). We also observed a significant difference in CIN-mediated DA release between EtOH with and without MII (F(1,15) = 6.9141, p = 0.01895, Fig. 7E; control, n = 12, EtOH, n = 12, MII, n = 11, MII + EtOH, n = 7).
Fig. 7.

Role of α6*-nAChRs in EtOH inhibition of evoked dopamine release in the NAc. A Illustration showing the experimental framework wherein CIN-mediated DA release was recorded following blue light stimulation in ChAT-ChR2-eYFP mice. B, C Current vs time plots show representative DA release evoked by light stimulation before and after superfusion of 60 mM EtOH under control (B) vs MII (C) conditions. Insets show superimposed cyclic voltammograms at the peak of DA release and color plots show cyclic voltammograms over time before and after Con + EtOH vs MII + EtOH. Ethanol inhibits CIN-mediated DA release with block by MII. D Concentration response for EtOH (1–80 mM) effects on CIN-mediated DA release. D Ethanol significantly inhibited DA release at 60–80 mM. E Summary of EtOH effects on CIN-mediated DA release. MII significantly reduces EtOH inhibition of CIN-mediated DA release. Asterisks * and *** indicate significance levels p < 0.05, and p < 0.001, respectively
Discussion
While it is well-known that both NIC and EtOH enhance DA neural activity and basal DA release in limbic structures, it has become increasingly evident that these effects are not mediated directly by DA neurons. The purpose of this study was to determine the effects of EtOH on the GABAergic projection from the VTA to NAc CINs, evaluate the role of α6*-nAChRs, and investigate plasticity in NAc CINs. Striatal CINs are powerful heterosynaptic regulators of DA terminal function. CIN activation results in DA release due to nAChR-mediated depolarization of DA terminals [42, 72, 73]. CINs receive both local circuit and distal GABAergic input, and our findings highlight the importance of VTA GABA neuron regulation of CIN activity in EtOH’s indirect actions at terminals in the NAc. We have shown previously that a subpopulation of VTA GABA neurons expresses α6*-nAChRs [40, 42] and that they modulate local GABA circuit in the VTA. Indeed, α6*-nAChR subunit mRNA levels are 16-fold higher than other nAChR subunits in the VTA [74]. We have reported previously [48], and replicated here with VTA-NAc knockdown of α6*-nAChRs with α6-miRNA, that VTA GABA neurons project to the NAc and synapse on CINs. Ethanol inhibition of GABAergic input to CINs from the VTA involves α6*-nAChRs, as either the potent α6-selective antagonist α-Ctx MII [66] or genetic knockdown of α6*-nAChRs in the GABAergic pathway to CINs with α6-miRNA blocks the inhibitory effects of EtOH on GABAergic inhibition of VTA GABA neurons. Thus, at least one population of VTA GABA neurons expresses α6*-nAChRs and presumably projects to the NAc and modulates CIN activity.
Ethanol-induced reductions of GABA currents in CINs are associated with increases in CIN firing that we have shown recently [48] and confirmed here, with particular relevance to locus of effect. While in the previous study [48], we evaluated CIN oIPSCs, in this study, we evaluated mIPSCs, which enables the interpretation of pre- vs postsynaptic mechanism for EtOH inhibition of evoked IPSCs, as well as any involvement of α6*-nAChRs. Superfusion of EtOH inhibited mIPSC frequency, but not amplitude, suggesting a presynaptic role. This makes some sense since α6*-nAChRs would be expressed on GABA terminals to CINs. However, we have hypothesized in the past that EtOH was acting on atypical GABA receptors on CINs. However, there was no effect on amplitude of mIPSCs, which would be expected. As VTA-NAc GABA neurons express α6*-nAChRs, it is reasonable to speculate that EtOH is acting presynaptically on VTA-NAc GABAergic terminals to decrease GABA release, which would result in an enhancement of CIN firing rate via activation of atypical ρ−1 GABA receptors on CINs. Bicuculline is the benchmark antagonist for GABAARs, but not all ionotropic GABA(A) receptors are sensitive to bicuculline [75]. Bicuculline (10 μM) did not eliminate oIPSCs in NAc CINs [48]. However, TPMPA did block EtOH’s inhibition of oIPSCs. This supports our hypothesis but suggests a need for further investigation, as EtOH enhances typical GABAAR-mediated inhibition but reduces atypical GABAAR-mediated inhibition.
We then evaluated the role of α6*-nAChRs in mediating EtOH effects on CIN activity in the NAc, with the hypothesis that α6*-nAChRs mediate low-dose and high-dose EtOH effects on CIN firing rate. Both MII and genetic knockdown of α6*-nAChRs in the GABAergic pathway to CINs with α6-miRNA blocked EtOH’s enhancement of CIN firing rate which supports this hypothesis. From previous reports, blocking α6 subunit containing α6*-nAChRs attenuates both the high-dose inhibitory effects of EtOH [41, 76] and low-dose (5–10 mM) enhancement of spontaneous DA transients by EtOH [42], suggesting overlap in the mechanisms involved in these opposing effects and the GABAergic effects observed. The dogma is that α6*-nAChRs are only expressed on DA terminals. However, as mentioned above, at least one population of VTA GABA neurons projects to the NAc and expresses α6*-nAChRs [40]. At first glance, it would seem counterintuitive that EtOH would enhance CIN activity but inhibit DA release, at least at high doses. Mainly, would not the opposite effects accrue as cholinergic interneurons that contribute to DA release become more excitable? However, increases in ACh levels, which would be expected if CIN activity was enhanced, are often associated with decreases in evoked DA release. This effect is sometimes attributed to desensitization of nAChRs on DA neurons. Indeed, we [41], and others [77], have shown that acetylcholinesterase inhibitors and even NIC itself decrease evoked DA release presumably through nAChR desensitization [77]. However, the opposite could also be operational, as we have reported increases in spontaneous DA release at lower, reward-relevant, concentrations of EtOH [42], which is also mediated by α6*-nAChRs. Recently, CINs have been implicated in EtOH-mediated effects on DA levels in the NAc, as measured by microdialysis, with cholinergic antagonists preventing EtOH-induced DA increases [78]. Ethanol has also been shown to inhibit CINs in the dorsal striatum [79]. In contrast, we have demonstrated previously [48] and confirmed here that EtOH consistently and robustly enhances NAc CIN activity, suggesting that regional effects may exist that could be due to local GABA subunit composition on CINs amongst other factors. We have shown previously that VTA GABA neurons undergo a switch in GABAAR-mediated inhibition with opioid [65, 80, 81] and alcohol [53] dependence. As VTA GABA neurons project to CINs, express α6*-nAChRs, and there was a marked increase in baseline CIN firing rate with knockdown of α6*-nAChRs by α6-miRNA injection into the VTA of VGAT-Cre mice, we hypothesized that GABAergic transmission from VTA GABA neurons to CINs was neuroadaptive. We compared chronic vs acute EtOH effects on CIN firing rate, as well as on plasticity in VTA GABA neuron synaptic transmission to CINs. In mice treated with CIE in vapor chambers for 3 weeks, acute EtOH no longer enhanced CIN firing rate, suggesting changes in synaptic plasticity in EtOH-dependent mice. These differences could be due to changes in receptor expression after CIE, which would require further experimentation to determine. We then specifically evaluated GABAergic synaptic plasticity to CINs. Low-frequency stimulation of GABAergic input from the VTA to the NAc produced LTD of oIPSCs in NAc CINs, a novel finding which we termed CIN-iLTD. We hypothesized that LTD of GABAergic input to CINs would switch to LTP in EtOH-dependent mice. While EtOH-dependent mice did not exhibit CIN-iLTP as hypothesized, iLTD was blocked. The pharmacological antagonist MII and genetic knockdown of α6*-nAChRs with α6-miRNA blocked CIN-iLTD, demonstrating involvement of α6*-nAChRs in CIN-iLTD and α6*-nAChR mesolimbic plasticity associated with alcohol dependence. TPMPA abolished CIN-iLTD, suggesting the involvement of ρ−1 GABA receptors on CINs. Similarly, EtOH did not affect CIN firing rate in mice in withdrawal from chronic EtOH, suggesting tolerance to EtOH. Since TPMPA also blocked CIN-iLTD, further investigation is needed to establish the role of atypical GABA receptors in mediating the effects of chronic EtOH and associated CIN plasticity.
We sought to determine what these cellular and synaptic effects of EtOH on CINs might have on CIN-mediated DA release and the involvement of α6*-nAChRs. We have demonstrated previously that α6*-nAChRs mediate both NIC and high-dose EtOH inhibition of electrically evoked DA release in the NAc [41], that CINs mediate spontaneous DA release [72], and that α6*-nAChRs mediate low-dose EtOH enhancement of CIN-mediated spontaneous release of DA [42]. Why does EtOH have differential effects on evoked vs spontaneous DA release, but yet both involve α6*-nAChRs? In order to address this, we evaluated the effects of EtOH on CIN-mediated DA release evoked by direct activation of CINs. Similar to what we reported previously with electrically evoked DA release [41], EtOH inhibited optically evoked CIN-mediated DA release at high doses. We were expecting enhancement by EtOH at lower doses, since we have shown that spontaneous DA release was mediated by CINs and enhanced by EtOH but at low doses (i.e., 5–10 mM). Regardless, the high-dose EtOH effect, similar to what we found with electrically evoked DA release, was blocked by MII, suggesting a role for α6*-nAChRs. Considering our mIPSC studies above addressing pre- vs postsynaptic effects, future studies will need to address how presynaptic mechanisms involving α6*-nAChRs might reconcile low- and high-dose EtOH effects. However, since α6*-nAChRs are also hypothesized to be expressed on DA terminals, it may very well needed to be explained by dissecting these inputs further. Figure 8 illustrates an interpretation of our findings in the context of the mesolimbic DA system and α6*-nAChRs. Clearly, in vivo, EtOH will have effects on multiple substrates in the mesolimbic DA system. However, α6*-nAChRs appear to be integral to its effects in both the VTA and NAc. We are now conducting simulation studies to determine if α6*-nAChRs are a molecular target for EtOH but dependent on the subunit hodological signature (i.e., might there be different subunits expressed on DA vs GABA terminals).
Fig. 8.
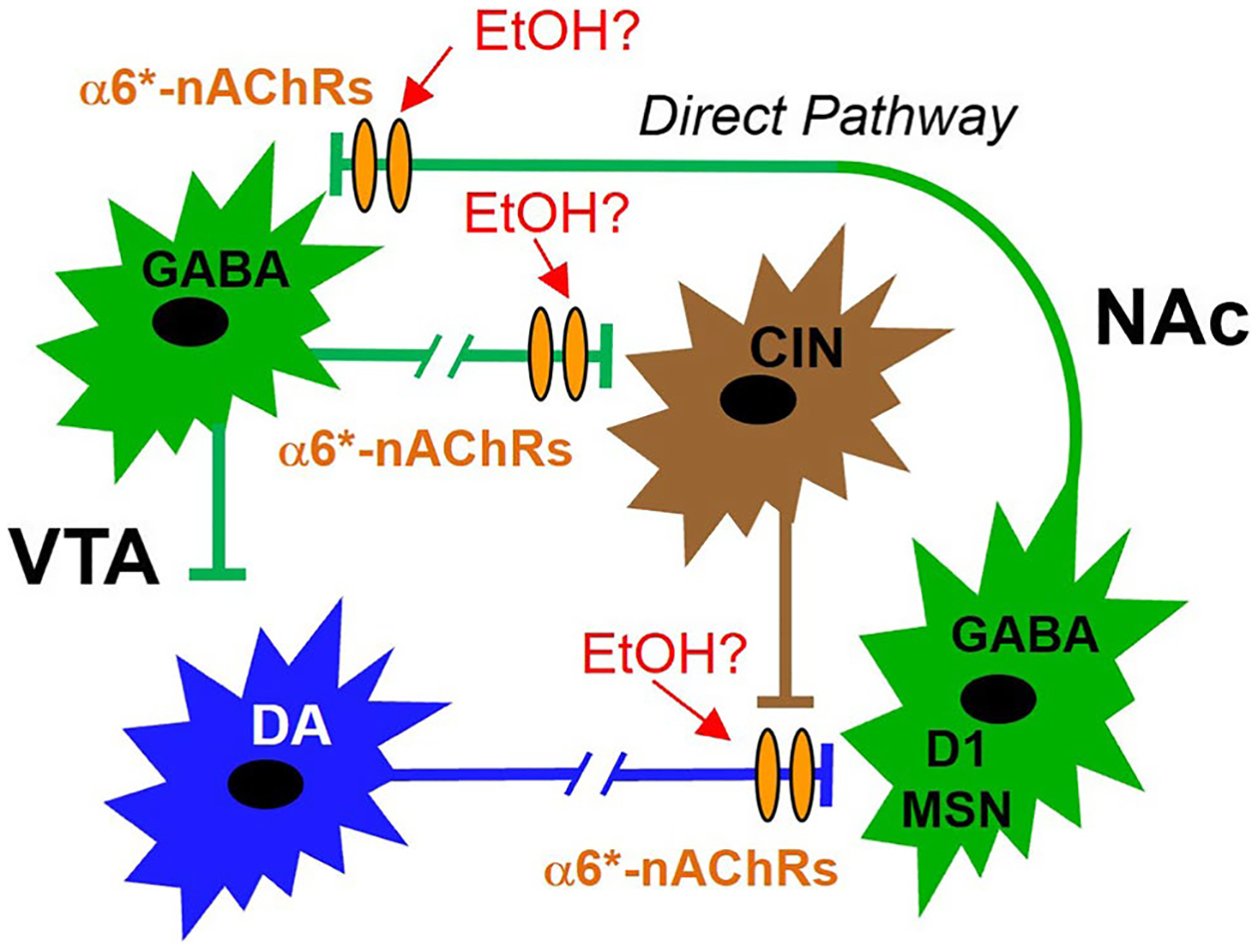
Theoretical framework for the involvement of α6*-nAChRs in EtOH effects on DA release in the mesolimbic pathway. Dopamine neurons in the VTA project to the NAc (blue). Dopamine release in the NAc is modulated by CINs (brown). Dogma maintains that α6*-nAChRs are located on DAergic terminals in the NAc, and we have shown previously that they are also expressed on GABA terminals to some VTA GABA neurons (green). Based on our findings demonstrating enhancement of CIN firing rate by EtOH via α6*-nAChRs, we hypothesize that the enhancement of DA release by EtOH, and ultimately EtOH reward, is a result increased NAc CIN activity subsequent to reduction of VTA-NAc GABAergic transmission
These findings have potential clinical implications. Alcohol use disorder and TUD are serious psychiatric disorders with high medical, psychiatric, and social consequences. To date, only 3 medications have been approved by the US Food and Drug Administration (FDA) to treat AUD: disulfiram, naltrexone, and acamprosate. A number of alternative medications are now being evaluated for treatment of AUD in human studies including varenicline (Chantix), a drug historically used to treat TUD. However, these medications have a range of side effects and mixed efficacy [82]. Elucidating a clear molecular target of EtOH and finding how EtOH changes the reward pathway of the brain on a molecular level is sine qua non to not only directly treat the disorder, but perhaps halt or even reverse dangerous changes in the brain associated with AUD. Understanding the role of α6*-nAChRs in AUD will also lead to a better understanding of NIC dependence and the development of safer and more effective treatments for AUD, particularly when comorbidity includes TUD.
Funding
This work was funded by NIH grants AA020919 and DA035958 to Scott C. Steffensen.
Footnotes
Ethics Approval Animals were treated in strict accordance with the Brigham Young University (BYU) Animal Research Committee (IACUC) guidelines, which incorporate and exceed current NIH guidelines. The BYU IACUC reviewed and approved the procedures detailed herein.
Consent for Publication All authors have given their consent to publish.
Competing Interests The authors declare no competing interests.
Some of the data presented here has been presented previously at scientific conferences and has appeared as a master’s thesis for coauthor Elizabeth Q. Anderson in pre-print form in BYU Scholars Archive.
Data Availability
The datasets generated and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
References
- 1.Bobo JK, Husten C (2000) Sociocultural influences on smoking and drinking. Alcohol Res Health 24(4):225–232 [PMC free article] [PubMed] [Google Scholar]
- 2.Matson LM, Grahame NJ (2015) Emotional reactivity to incentive downshift as a correlated response to selection of high and low alcohol preferring mice and an influencing factor on ethanol intake. Alcohol 49(7):657–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.SAMHSA (2016) National Survey on Drug Use and Health (NSDUH)
- 4.Sacks JJ et al. (2015) 2010 National and state costs of excessive alcohol consumption. Am J Prev Med 49(5):e73–e79 [DOI] [PubMed] [Google Scholar]
- 5.Report SG, National center for chronic disease prevention and health promotion (US) Office on smoking and health (2014) The Health Consequences of Smoking—50 Years of Progress
- 6.Xu X et al. (2015) Annual healthcare spending attributable to cigarette smoking: an update. Am J Prev Med 48(3):326–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koob GF (2003) Alcoholism: allostasis and beyond. Alcohol Clin Exp Res 27(2):232–243 [DOI] [PubMed] [Google Scholar]
- 8.Overstreet DH, Knapp DJ, Breese GR (2002) Accentuated decrease in social interaction in rats subjected to repeated ethanol withdrawals. Alcohol Clin Exp Res 26(8):1259–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blackburn JR et al. (1986) Increased dopamine metabolism in the nucleus accumbens and striatum following consumption of a nutritive meal but not a palatable non-nutritive saccharin solution. Pharmacol Biochem Behav 25(5):1095–1100 [DOI] [PubMed] [Google Scholar]
- 10.Wise RA, Bozarth MA (1987) A psychostimulant theory of addiction. Psychol Rev 94(4):469–492 [PubMed] [Google Scholar]
- 11.Koob GF (1992) Dopamine, addiction and reward. Semin Neurosci 4:139–148 [Google Scholar]
- 12.Wise RA (1996) Addictive drugs and brain stimulation reward. Annu Rev Neurosci 19:319–340 [DOI] [PubMed] [Google Scholar]
- 13.Wise RA (2004) Dopamine, learning and motivation. Nat Rev Neurosci 5(6):483–494 [DOI] [PubMed] [Google Scholar]
- 14.Wise RA (2008) Dopamine and reward: the anhedonia hypothesis 30 years on. Neurotox Res 14(2–3):169–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hyman SE, Malenka RC (2001) Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci 2(10):695–703 [DOI] [PubMed] [Google Scholar]
- 16.Hyman SE, Malenka RC, Nestler EJ (2006) Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29:565–598 [DOI] [PubMed] [Google Scholar]
- 17.Kauer JA, Malenka RC (2007) Synaptic plasticity and addiction. Nat Rev Neurosci 8(11):844–858 [DOI] [PubMed] [Google Scholar]
- 18.Nugent FS, Kauer JA (2008) LTP of GABAergic synapses in the ventral tegmental area and beyond. J Physiol 586(6):1487–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wooltorton JR et al. (2003) Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci 23(8):3176–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klink R et al. (2001) Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 21(5):1452–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang KC, Jin GZ, Wu J (2009) Mysterious alpha6-containing nAChRs: function, pharmacology, and pathophysiology. Acta Pharmacol Sin 30(6):740–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mansvelder HD, Keath JR, McGehee DS (2002) Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron 33(6):905–919 [DOI] [PubMed] [Google Scholar]
- 23.Larsson A et al. (2004) Is an alpha-conotoxin MII-sensitive mechanism involved in the neurochemical, stimulatory, and rewarding effects of ethanol? Alcohol 34(2–3):239–250 [DOI] [PubMed] [Google Scholar]
- 24.Jerlhag E et al. (2006) Role of the subunit composition of central nicotinic acetylcholine receptors for the stimulatory and dopamine-enhancing effects of ethanol. Alcohol Alcohol 41(5):486–493 [DOI] [PubMed] [Google Scholar]
- 25.Lof E et al. (2007) Nicotinic acetylcholine receptors in the ventral tegmental area mediate the dopamine activating and reinforcing properties of ethanol cues. Psychopharmacology 195(3):333–343 [DOI] [PubMed] [Google Scholar]
- 26.Powers MS et al. (2013) Nicotinic acetylcholine receptors containing alpha6 subunits contribute to alcohol reward-related behaviours. Genes Brain Behav 12(5):543–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y et al. (2014) Enhanced synthesis and release of dopamine in transgenic mice with gain-of-function alpha6* nAChRs. J Neurochem 129(2):315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Britt JP, Bonci A (2013) Alcohol and tobacco: how smoking may promote excessive drinking. Neuron 79(3):406–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doyon WM et al. (2013) Nicotine decreases ethanol-induced dopamine signaling and increases self-administration via stress hormones. Neuron 79(3):530–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doyon WM et al. (2013) Potential substrates for nicotine and alcohol interactions: a focus on the mesocorticolimbic dopamine system. Biochem Pharmacol 86(8):1181–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le AD et al. (2003) Nicotine increases alcohol self-administration and reinstates alcohol seeking in rats. Psychopharmacology 168(1–2):216–221 [DOI] [PubMed] [Google Scholar]
- 32.Ostroumov A et al. (2015) Cigarettes and alcohol: the influence of nicotine on operant alcohol self-administration and the mesolimbic dopamine system. Biochem Pharmacol 97(4):550–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith BR et al. (1999) Exposure to nicotine enhances acquisition of ethanol drinking by laboratory rats in a limited access paradigm. Psychopharmacology 142(4):408–412 [DOI] [PubMed] [Google Scholar]
- 34.Reus VI et al. (2007) Varenicline: new treatment with efficacy in smoking cessation. Drugs Today (Barc) 43(2):65–75 [DOI] [PubMed] [Google Scholar]
- 35.Rollema H et al. (2007) Pharmacological profile of the alpha4beta2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology 52(3):985–994 [DOI] [PubMed] [Google Scholar]
- 36.Mitchell JM et al. (2012) Varenicline decreases alcohol consumption in heavy-drinking smokers. Psychopharmacology 223(3):299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blomqvist O et al. (1996) Voluntary ethanol intake in the rat: effects of nicotinic acetylcholine receptor blockade or subchronic nicotine treatment. Eur J Pharmacol 314(3):257–267 [DOI] [PubMed] [Google Scholar]
- 38.Steensland P et al. (2007) Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, selectively decreases ethanol consumption and seeking. Proc Natl Acad Sci U S A 104(30):12518–12523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dopico AM, Lovinger DM (2009) Acute alcohol action and desensitization of ligand-gated ion channels. Pharmacol Rev 61(1):98–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steffensen SC et al. (2017) alpha6 subunit-containing nicotinic receptors mediate low-dose ethanol effects on ventral tegmental area neurons and ethanol reward. Addict Biol 23(5):1079–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schilaty ND et al. (2014) Acute ethanol inhibits dopamine release in the nucleus accumbens via alpha6 nicotinic acetylcholine receptors. J Pharmacol Exp Ther 349(3):559–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao F et al. (2019) Alpha6-containing nicotinic acetylcholine receptor is a highly sensitive target of alcohol. Neuropharmacology 149:45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen DJ et al. (2018) Pharmacological and functional comparisons of alpha6/alpha3beta2beta3-nAChRs and alpha4beta2-nAChRs heterologously expressed in the human epithelial SH-EP1 cell line. Acta Pharmacol Sin 39(10):1571–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zoli M et al. (2002) Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci 22(20):8785–8789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan KR et al. (2012) GABA neurons of the VTA drive conditioned place aversion. Neuron 73(6):1173–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Zessen R et al. (2012) Activation of VTA GABA neurons disrupts reward consumption. Neuron 73(6):1184–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown MT et al. (2012) Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature 492(7429):452–456 [DOI] [PubMed] [Google Scholar]
- 48.Yorgason JT et al. (2022) Modulation of dopamine release by ethanol is mediated by atypical GABAA receptors on cholinergic interneurons in the nucleus accumbens. Addict Biol 27(1):e13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamamaki N et al. (2003) Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol 467(1):60–79 [DOI] [PubMed] [Google Scholar]
- 50.Gompf HS et al. (2015) Targeted genetic manipulations of neuronal subtypes using promoter-specific combinatorial AAVs in wild-type animals. Front Behav Neurosci 9:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stegmeier F et al. (2005) A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A 102(37):13212–13217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lesscher HM et al. (2008) Amygdala protein kinase C epsilon regulates corticotropin-releasing factor and anxiety-like behavior. Genes Brain Behav 7(3):323–333 [DOI] [PubMed] [Google Scholar]
- 53.Nelson AC et al. (2018) Ventral tegmental area GABA neurons are resistant to GABA(A) receptor-mediated inhibition during ethanol withdrawal. Front Neurosci 12:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams SB et al. (2018) Glutamate Transmission to ventral tegmental area GABA neurons is altered by acute and chronic ethanol. Alcohol Clin Exp Res 42(11):2186–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J et al. (2012) An ethanol vapor chamber system for small animals. J Neurosci Methods 208(1):79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allison DW et al. (2011) Mefloquine effects on ventral tegmental area dopamine and GABA neuron inhibition: a physiologic role for connexin-36 gap junctions. Synapse 65(8):804–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steffensen SC et al. (2011) The role of connexin-36 gap junctions in alcohol intoxication and consumption. Synapse 65(8):695–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steffensen SC et al. (2008) Cocaine disinhibits dopamine neurons in the ventral tegmental area via use-dependent blockade of GABA neuron voltage-sensitive sodium channels. Eur J Neurosci 28:2028–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Allison DW et al. (2006) Connexin-36 gap junctions mediate electrical coupling between ventral tegmental area GABA neurons. Synapse 60(1):20–31 [DOI] [PubMed] [Google Scholar]
- 60.Allison DW et al. (2011) Mefloquine effects on ventral tegmental area dopamine and GABA neuron inhibition: a physiologic role for connexin-36 GAP junctions. Synapse 65(8):804–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson SW, North RA (1992) Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol 450:455–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Margolis EB et al. (2006) The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol 577(Pt 3):907–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bennett BD, Callaway JC, Wilson CJ (2000) Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci 20(22):8493–8503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ting-A-Kee R, van der Kooy D (2012) The neurobiology of opiate motivation. Cold Spring Harb Perspect Med 2(10):a012096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vargas-Perez H et al. (2014) BDNF signaling in the VTA links the drug dependent state to drug withdrawal aversions. J Neurosci 34(23):7899–7909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McIntosh JM et al. (2004) Analogs of alpha-conotoxin MII are selective for alpha6-containing nicotinic acetylcholine receptors. Mol Pharmacol 65(4):944–952 [DOI] [PubMed] [Google Scholar]
- 67.Yorgason JT, Espana RA, Jones SR (2011) Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J Neurosci Methods 202(2):158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gallegos RA et al. (1999) Adaptive responses of gamma-aminobutyric acid neurons in the ventral tegmental area to chronic ethanol. J Pharmacol Exp Ther 291(3):1045–1053 [PubMed] [Google Scholar]
- 69.Wang Z et al. (2006) Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 50(3):443–452 [DOI] [PubMed] [Google Scholar]
- 70.Augustin SM, Chancey JH, Lovinger DM (2018) Dual dopaminergic regulation of corticostriatal plasticity by cholinergic interneurons and indirect pathway medium spiny neurons. Cell Rep 24(11):2883–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pan Y et al. (2005) Pharmacology of GABAC receptors: responses to agonists and antagonists distinguish A- and B-subtypes of homomeric rho receptors expressed in Xenopus oocytes. Neurosci Lett 376(1):60–65 [DOI] [PubMed] [Google Scholar]
- 72.Yorgason JT, Zeppenfeld DM, Williams JT (2017) Cholinergic interneurons underlie spontaneous dopamine release in nucleus accumbens. J Neurosci 37(8):2086–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brimblecombe KR et al. (2018) Targeted activation of cholinergic interneurons accounts for the modulation of dopamine by striatal nicotinic receptors. eNeuro 5(5). 10.1523/ENEURO.0397-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang K et al. (2009) Distinctive nicotinic acetylcholine receptor functional phenotypes of rat ventral tegmental area dopaminergic neurons. J Physiol 587(Pt 2):345–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johnston GA (2013) Advantages of an antagonist: bicuculline and other GABA antagonists. Br J Pharmacol 169(2):328–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yorgason JT et al. (2015) Greater ethanol inhibition of presynaptic dopamine release in C57BL/6J than DBA/2J mice: role of nicotinic acetylcholine receptors. Neuroscience 284:854–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang T et al. (2009) Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine. J Neurosci 29(13):4035–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loften A et al. (2020) An acetylcholine-dopamine interaction in the nucleus accumbens and its involvement in ethanol’s dopamine-releasing effect. Addict Biol 26(3):e12959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blomeley CP et al. (2011) Ethanol affects striatal interneurons directly and projection neurons through a reduction in cholinergic tone. Neuropsychopharmacology 36(5):1033–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ting AKR et al. (2013) Ventral tegmental area GABA neurons and opiate motivation. Psychopharmacology 227(4):697–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vargas-Perez H et al. (2009) Ventral tegmental area BDNF induces an opiate-dependent-like reward state in naive rats. Science 324(5935):1732–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Litten RZ et al. (2016) Potential medications for the treatment of alcohol use disorder: an evaluation of clinical efficacy and safety. Subst Abus 37(2):286–298 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.