

Abstract
Chromatin binding proteins are critical regulators of cell state in hematopoiesis1,2. Acute leukemias driven by rearrangements of the Mixed Lineage Leukemia gene (KMT2Ar) or Nucleophosmin (NPM1c) mutations require the chromatin adapter protein Menin, encoded by the MEN1 gene, to sustain aberrant leukemogenic gene expression programs3–5. In a phase 1 first-in-human clinical trial, the Menin-inhibitor revumenib (SNDX-5613), designed to disrupt the Menin-MLL1 interaction, induced clinical responses in leukemia patients with KMT2Ar or mutant NPM16. We identified somatic mutations in MEN1 at the revumenib/Menin interface in patients with acquired resistance to Menin inhibition. Consistent with the genetic data in patients, inhibitor/Menin interface mutations represent a conserved mechanism of therapeutic resistance in xenograft models and in an unbiased base-editor screen. These mutants attenuate drug-target binding by generating structural perturbations that impact small molecule binding but not the interaction with the natural ligand MLL1, and prevent inhibitor-induced eviction of Menin and MLL1 from chromatin. This study is the first to demonstrate that a chromatin-targeting therapeutic exerts sufficient selection pressure to drive evolution of escape mutants leading to sustained chromatin occupancy as a common mechanism of therapeutic resistance.
Menin is a chromatin adaptor protein that is critical for the formation and stability of highly conserved multiprotein complexes on chromatin, including Mixed Lineage Leukemia 1 (MLL1; KMT2A) and MLL2 (KMT2B) histone methyltransferase complexes and the JUND transcription factor complex4,7,8. Menin is critical for the development and maintenance of acute leukemias driven by rearrangements involving MLL1 (KMT2Ar) or mutations of the Nucleophosmin gene (NPM1c)3,5. A series of small molecule inhibitors that disrupt the Menin-MLL1 protein-protein interaction have been developed9–14 and demonstrate potent activity in pre-clinical models, including the ability to eradicate disease5,10,14. Based on this, several Menin-inhibitors recently entered phase 1 clinical trials (NCT04065399, NCT04067336, NCT04811560, NCT05388903, NCT04988555, NCT05153330). The Menin inhibitor revumenib has been reported to be safe and effective in patients with relapsed or refractory acute leukemia with an KMT2Ar or NPM1c mutation. In the ongoing phase 1/2 first-in-human study (AUGMENT-101) of revumenib, patients with KMT2Ar or NPM1c-mutant leukemia had an overall response rate of 53% with 30% of treated patients achieving a complete remission (CR) or complete remission with partial hematologic recovery (CRh)6. Here, we identified and characterized somatic mutations within the MEN1 gene that arise during Menin-inhibitor treatment in model systems and on the AUGMENT-101 trial (NCT04065399) which mediate therapeutic resistance.
MEN1 mutations in resistant leukemias
Despite significant single-agent activity of revumenib in a heavily pretreated KMT2Ar and NPM1c-mutant leukemia patient population, we identified patients who were treated on the AUGMENT-101 phase 1 study and subsequently developed acquired resistance following an initial response (Fig. 1a). Patients 1 and 4 with relapsed KMT2Ar AML achieved morphologic leukemia-free states (MLFS) after one cycle of treatment, followed by relapse despite continued exposure to study drug. Patient 2 with NPM1c-mutant AML had a reduction in circulating blast counts after 2 cycles of revumenib followed by disease progression, while Patient 3 with KMT2Ar AML achieved a complete remission with incomplete count recovery (CRi) and then progressed despite continued Menin-inhibitor treatment (Fig. 1a). Next-generation targeted sequencing of bone marrow specimens from these patients at diagnosis revealed a largely stable landscape of well characterized leukemia drivers but somatic mutations within the MEN1 gene were detected at time of relapse on revumenib (Extended Data Fig. 1a–c). In 3/4 patients the M327-residue was affected (M327V or M327I) and T349M, G331R and S160T were detected in each one patients at the end of treatment, respectively (Fig. 1a). Mutation allele frequencies (MAF) ranged from 5.9% to 28.2% (Extended Data Fig. 1a–c), owing to expected dilution effects from normal leukocytes. To quantitatively assess the frequency of these novel somatic mutations within the population of KMT2Ar and NPM1c-mutant patients that had been exposed to revumenib, we performed droplet digital PCR (ddPCR) for MEN1-M327V, -M327I, -G331R, -G331D and -T349M on DNA samples collected during the AUGMENT-101 trial at different study centers. We identified 31 patients, inclusive of responders and non-responders, who were treated with revumenib for more than 2 cycles of treatment (>56 days) and had DNA material available for analysis. Among these patients, 12 individuals (38.7%) carried one or more MEN1-mutations (Fig. 1b, Extended data Fig.1d). Of note, these mutations were not detected in the pre-treatment samples by ddPCR suggesting they were either not present or in very low abundance until Menin-inhibition established a selective fitness advantage (Fig. 1c). Longitudinal assessment of the detected mutations in single patients confirmed clonal outgrowth during drug treatment and suggested that selection for MEN1-mutant leukemia cells occurs after approximately 2 cycles of therapy (Fig. 1d, Extended data Fig.1e).
Figure 1: Menin inhibitor resistance is associated with emergence of MEN1 mutations.

a) Graphical depiction of the percentage (%) of leukemic blasts in the peripheral blood of patients during revumenib treatment on the AUGMENT-101 clinical trial. Clinical events are marked with arrows and labeled respectively. b) Schematic showing the fraction of patients in which MEN1-M327I, -M327V, -G331R, -G331D or -T349M was detected by droplet digital PCR (ddPCR). For this analysis we included patients that received at least two cycles of revumenib treatment (>56 days) and had 2 or more mutant droplets detected. c) Pie charts displaying the fraction of MEN1-mutant alleles measured by ddPCR at the time point of screening and relapse in 4 individual patients from the cohort shown in b). d) Longitudinal kinetics of MEN1-mutant selection in two patients from the cohort shown in b). Mutant allele frequencies at different timepoints during revumenib treatment were analyzed by ddPCR. e, f, g) Graphical display of the percentage (%) of human leukemia cells in the peripheral blood of individual NOG-mice during a long-term patient-derived xenograft (PDX) treatment trial with the Menin-inhibitor VTP-50469 (0.03% rodent diet). Blue bars in the background mark the time of oral Menin inhibitor exposure via drug-supplemented rodent diet. MEN1 mutations detected via targeted DNA-sequencing in individual animals of e) PDX 1 (MLL::AF6), f) PDX 2 (NPM1c) or g) PDX 3 (MLL::AF10) are labeled and marked with arrows.
In parallel, we conducted a pre-clinical study in which we treated 5 different patient-derived xenografts (PDX) derived from AML patients (4x KMT2Ar, 1x NPM1c) with the Menin-inhibitor VTP-50469, a close analog of revumenib that binds similarly to Menin14. We assessed long-term survival for up to 400 days after initiation of treatment. Out of 45 animals treated with the drug, 22 xenografted mice (from all 5 PDX models) relapsed during drug exposure or after treatment cessation or had human leukemia cells detected in the bone marrow at the end of treatment without overt manifestations of disease. Targeted DNA-sequencing and/or ddPCR was performed on 33 specimens (11 untreated and all 22 relapsed animals). MEN1 mutations were identified in 15 (68%) of those relapsed samples from 4 PDX models (Fig. 1e–g, Supplementary Table 2). Strikingly, MEN1-M327V, -M327I, -G331R, -G331D, -T349M and -S160C mutations were detected, demonstrating high recurrence and an exceptional predictive value of the PDX model system. In PDX1 (MLL::AF6) and PDX2 (NPM1c) we observed relapse in single animals, while disease was eradicated in a large proportion of the cohort (Fig. 1e, f). The MEN1 mutations identified in those animals were diverse and included M327I, M327V, G331D as well as T349M and arose after a relatively long period of drug exposure, suggesting de novo mutations were acquired during treatment. In contrast, all Menin-inhibitor treated recipients engrafted with sample PDX3 (MLL::AF10) relapsed after about 2 months of continuous drug treatment (Fig. 1g, Extended Data Fig. 2a–c). All mice possessed T349M mutations, possibly reflecting the selection of a pre-existent ultra-low frequency variant that could not be detected pre-therapy in bone marrow isolates (Fig. 1g, Extended data Fig. 2d, Supplementary Table 2). Interestingly, in PDX4 (MLL::AF9, AML) resistance developed in 4/9 animals without MEN1 mutations or other newly acquired genetic drivers detectable, indicating that adaptation and non-genetic processes may also lead to Menin-inhibitor resistance in individual cases. Similarly, disease persistence could be detected in 4/9 VTP-50469 treated animals in PDX5 (MLL::AF9, AML) while in only one of these animals a Menin mutation (T349M) could be detected. In summary, we identified recurrent somatic mutations within the MEN1-gene which affect the M327-, G331-, T349- and S160-residues of Menin in patients and PDX models that relapsed on Menin-inhibitor treatment. These mutations are distinct from known variants that disrupt Menin’s tumor suppressive function in MEN1-syndrome and none have been previously reported15.
Resistance mutations reduce drug binding
To gain an unbiased view on the spectrum of point mutations that may cause resistance to Menin-inhibitors we conducted a MEN1-focused CRISPR-Cas9 base-editor screen in MOLM13 (MLL::AF9) and MV4;11 (MLL::AF4) cells. A library of 518 single-guide RNAs (sgRNAs) targeting all MEN1-exons and flanking untranslated regions was designed and expressed along with a base-editor system to facilitate C→T base editing in proximity to the sgRNA binding sites16. Subsequently, cells were exposed to VTP-50469 or DMSO for 12 days and guide barcodes were counted by Next-Generation-Sequencing (NGS) to identify editing sites that confer resistance to drug treatment (Extended Data Fig. 3a). Using this approach base-editing affecting the majority of amino-acids within Menin could be achieved. Interestingly, the T349-, G331- and S160 residues were predicted as candidate drivers of Menin-inhibitor resistance in the screen (Fig. 2a, Extended data Fig. 3b, Supplementary Table 3), but the M327-residue was not amenable to a base edit due to the lack of a PAM sequence in this specific region.
Figure 2: Base-editor screening identifies recurrent MEN1 mutations mapping to the MLL1-binding pocket.
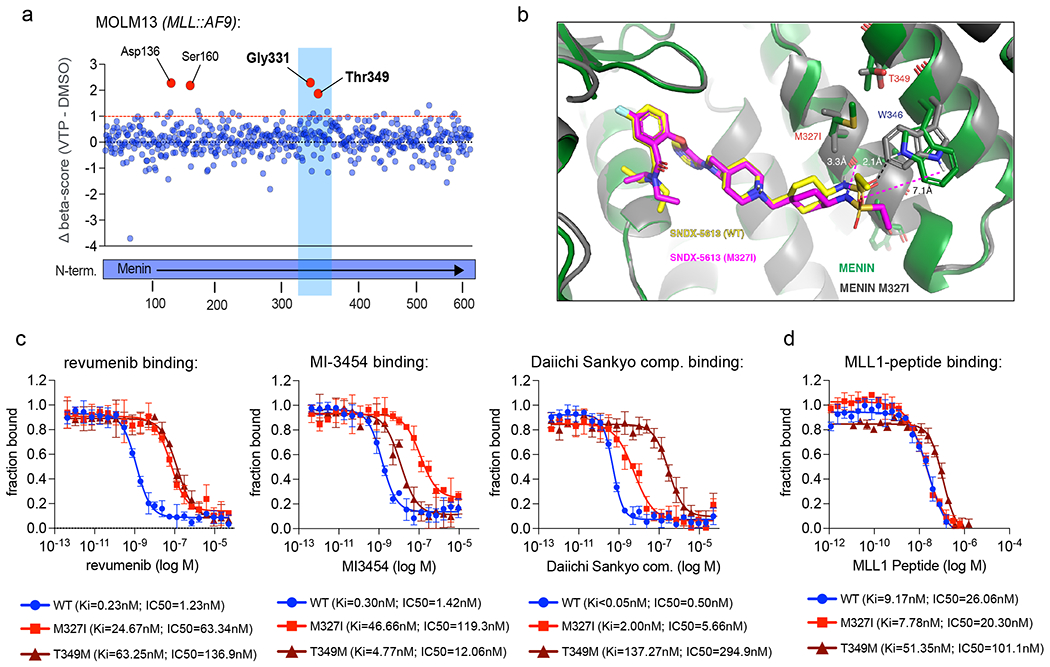
a) Dot-plot showing the results of a CRISPR-Cas9 base-editor screen in MOLM13 cells aiming to identify point mutations that cause resistance to Menin inhibitor treatment. Each dot represents a single guide RNA. Along the x-axis guide RNAs are sorted by their targeting location relative to the Menin-coding sequence. The y-axis shows differential CRISPR-beta-scores (DMSO-score subtracted from the VTP-50469-treatment score). Outstanding hits are marked in red and targeted amino acid residues are labeled. b) Structure alignment between co-crystal structure of revumenib bound to M327I-mutant Menin (PDB: 8E90) and revumenib bound to WT Menin (PDB: 7UJ4). revumenib is colored in yellow in WT Menin, and magenta in M327 mutant co-crystal structure. The magenta dashed lines indicate large distances, incapable of H-bond interactions, between revumenib in the M327I-mutant and the WT Menin protein and are in contrast to H-bond between W346 and revumenib in the WT Menin highlighted in black dashed line. c) Fluorescence polarization assay measuring dose-dependent displacement of an MLL1 peptide from WT, M327I- and T349M-mutant Menin under treatment with revumenib, MI-3454 or DS-25, a compound from the Daiichi-Sankyo Menin-inhibitor series. Data is presented as fraction bound of three independent replicates (N=3). Data point represent the mean +/− SD. d) Fluorescence polarization assay probing the binding affinity of a MLL1 peptide to WT, M327I- and T349M-mutant Menin. Data is presented as fraction bound of three independent replicates (N=3). Data point represent the mean +/− SD.
The X-ray co-crystal structure of revumenib bound to Menin revealed that residues M327, G331 and T349 are located in close proximity to the W346 residue (Fig. 2b, Extended data Fig. 3c,e, Supplementary Table 10). There is a strong hydrogen (H)-bond between the sulfonamide oxygen of revumenib and the indole N-H of W346 that contributes to inhibitor binding. This interaction was previously observed for the related inhibitor, VTP-5046914. In addition, we observe a hydrogen bond between the sulfonamide nitrogen of revumenib and the backbone ketone oxygen of M327. The key amino acids affected by MEN1-mutations (M327, G331, T349) are located around the W346 residue and do not overlap with the binding interface of MLL1 (Extended Data Fig. 3d,e), suggesting they likely perturb the distal revumenib sulfonamide interaction. Co-crystallography of revumenib with the Menin M327I mutant confirmed this effect. As seen in the overlay of the WT and mutant Menin revumenib structures (Fig. 2b), the branching methyl group of I327 projects toward the cyclohexyl ring of revumenib. The resulting steric clash displaces revumenib, leading to disruption of the H-bonds with W346, as well as with M327, reducing the inhibitor’s binding affinity, while leaving the molecule’s other interactions with Menin largely unchanged.
In parallel, we employed computational methods to assess dynamic structural changes in menin incorporating MEN1-T349M, -G331D, -G331R and -M327I/V for the SNDX5613/Menin interface, using the Folding@home distributed computing platform17. We launched all-atom molecular dynamics simulations of Menin-WT and individual mutant forms in both the presence and absence of revumenib (Extended Data Fig 4a–d) collecting an aggregate 5.469 milliseconds of simulation. Through a combination of Deep Learning methods18 and Markov State Models19, we predicted reductions in protein-ligand contacts between Menin and revumenib, perturbing the binding mode and decreasing affinity.
Consistent with the structural and computational results, competitive MLL1 fluorescence polarization (FP) binding assays20 showed that the M327I and T349M mutants increased the IC50-values of revumenib for MLL-displacement by 51- and 111-fold compared to WT Menin (Fig. 2c, Extended Figure 5a–c). The reduced affinity seen in the FP binding assay was consistent with a >30 fold increase in the dissociation off rate of revumenib from M327I vs WT Menin (Extended Figure 5b) and >50 fold change in mutant binding affinities (Kd) determined by isothermal calorimetry, ITC (Extended Figure 5c). Similarly, binding of MI-3454, a structural analog of KO-539, and DS-25, a potent compound from the Daiichi-Sankyo series21 were also severely affected by M327I, suggesting that Menin-inhibitor resistance at this site exhibit a broad, class effect (Fig. 2c). Interestingly, binding of MI-3454 and DS-25 were differentially affected by the M327I and T349M mutation, respectively (Fig. 2c). Moreover, this mutant effect is not specific to the Menin-MLL1 interaction, since the Menin-inhibitors’ability to block Menin-MLL2 binding was similarly affected by M327I and T349M mutations (Extended data Fig. 5d, e). Importantly, the ability of those mutants to bind the MLL1 peptide remained largely intact although a subtle decrease in binding affinity was observed for T349M mutant Menin (Fig. 2d).
Taken together, we independently reproduced the narrow spectrum of MEN1-hotspot mutations observed in patients and PDX by using an unbiased in vitro screening approach. Furthermore, we mapped these residues to an area of the binding pocket that is crucial for stabilizing binding of the Menin-inhibitor molecules but is dispensable for MLL1 binding.
Mutations confer resistance in vitro
We next assessed whether the mutations conferred resistance to Menin-inhibitor treatment in vitro. We expressed MEN1-M327I, -G331R and -T349M mutants as well as a -WT cDNA in MOLM13 (MLL::AF9), MV4;11 (MLL::AF4) and OCI-AML3 (NPM1c) cells using a lentiviral vector system. Dose-response assays performed by counting the number of viable cells after 10 days of drug treatment demonstrated a robust decrease in revumenib sensitivity in both KMT2Ar and NPM1c cells expressing these mutations (Fig. 3a, b, Extended Data Fig. 6a–c, f). Moreover, the induction of myeloid differentiation upon Menin-inhibitor treatment was impaired (Extended Data Fig. 6d, e). Of note, this resistance phenotype could similarly be observed when cells were treated with the structurally unrelated Menin-inhibitor MI-3454 (Extended Data Fig. 6f). The degree of drug resistance observed using this system may be dependent on the expression level of each construct (Extended Data Fig. 6g, h). To overcome the limitations of ectopic expression, we utilized CRISPR-Cas9 in conjunction with a homology directed repair template to edit M327I or T349M mutations into the endogenous MEN1 coding sequence of MV4;11 and OCI-AML3 cells (Fig. 3c). After nucleofection, M327I-edited or unedited bulk populations of cells were exposed to 50 nM of revumenib and cell counts were monitored over the course of a month (Fig. 3d). Unedited MV4;11 and OCI-AML3 cells stopped dividing shortly after exposure to the Menin-inhibitor, while CRISPR-Cas9-edited cells continued growing beyond 28 days which was accompanied by positive selection for the M327I mutation (Fig. 3e), providing in vitro support for the concept of inhibitor-driven clonal selection by this mutation. After screening of single cell clones, we established cell lines harboring M327I and T349M mutations (Extended Data Fig. 7a). MEN1M327I/M327I MV4;11 cells did not respond to revumenib at physiologically relevant doses with IC50 values shifting from the low nanomolar range to over 1 μM when the mutation was present (Fig. 3f, Extended Data Fig. 7b). MEN1M327I/WT cell lines retained some sensitivity to Menin-inhibition, however IC50 values were increased by 16-fold compared to MEN1-wild-type cells (Fig. 3f). Consistent with the findings from our binding assays, we observed reduced sensitivity to revumenib, MI-3454 and the Daiichi-Sankyo compounds in M327I- and T349M-mutant MV4;11 and OCI-AML3 cells (Fig. 3g, h Extended data Fig. 7 c–e). Of note, the observation that MI-3454 binding in the fluorescence polarization assay was less affected by T349M was reflected in the dose-response curves (Fig. 3h Extended data Fig. 7e). We further found that cells expressing MEN1-M327I were also resistant to the another Menin inhibitor M-89 and we confirmed that the more N-terminal located S160C mutation conferred a similar degree of revumenib resistance (Extended Data Fig. 7f, g). To answer the question whether the presence of recurrent MEN1 mutations may impact fitness of leukemia cells in the absence of a Menin-inhibitor we performed a cell competition assay (Extended Data Fig. 7h). As expected, revumenib exposure led to a rapid selection of M327I and T349M mutant cells while the chimerism between mutant and non-mutant cells decreased under DMSO treatment (Extended Data Fig. 7h). This effect was particularly pronounced for T349M mutant cells and only subtle when M327I was present, corresponding to the MLL1-binding assay (Fig. 2f). In summary, we demonstrate that endogenous or exogenous expression of recurrent MEN1 mutants in KMT2Ar or NPM1 mutant AML cell lines is sufficient to induce drug resistance to a panel of currently available Menin-inhibitors.
Figure 3: MEN1 mutations confer resistance to Menin-inhibitor treatment in vitro.

a, b) Dose-response curves of a) MOLM13 (MLL::AF9) and b) OCI-AML3 (NPM1c) cells to revumenib upon expression of MEN1-M327I, -G331R, -T349M or -WT. Cell counts were measured by flow cytometry and displayed relative to the DMSO control (mean +/− SEM, n=4, each 3 technical replicates). c) Schematic depicting the CRISPR-Cas9 gene editing strategy utilized to insert the M327I mutation into the endogenous MEN1-locus. d) Growth curves showing expansion of MV4;11 (MLL::AF4) and OCI-AML3 (NPM1c) bulk cell populations over time after nucleofection under treatment with 50nM revumenib (mean +/− SD, n=3, individual data points/no technical replicates). e) Curves depicting the fraction of the MEN1-M327I mutant allele detected by ddPCR in MV4;11 and OCI-AML3 bulk cell populations over time under treatment with 50nM revumenib (in vitro clonal selection assay) (mean, n=1, DNA pooled from 3 replicates). f, g) Dose-response curves showing the sensitivity of MV4;11 (MLL::AF4) cells harboring MEN1-M327I mutations to f) revumenib, g) MI-3454 and the Daiichi-Sankyo compound. h) Dose-response curves showing the sensitivity of MV4;11 (MLL::AF4) cells harboring MEN1-T349M mutations to revumenib, MI-3454 and the Daiichi-Sankyo compound. f-h) Cell counts were measured by flow cytometry and displayed relative to the DMSO control (mean +/− SEM, n=4, each 3 technical replicates). d, f-h) Statistical analysis was perfomed using an unpired t-test, two tailed, multiple comparisons. *** p<0.001; **p<0.01; *p<0.05.
Chromatin and gene expression changes
We next investigated whether mutations that confer Menin inhibitor resistance attenuate the changes in chromatin and gene expression seen with inhibition of the Menin and MLL1 interaction in MEN1-WT leukemia cells. Chromatin Immunoprecipitation Sequencing (ChIP-Seq) was performed after treatment of the MV4;11 MEN1M327I/M327I and WT control cell lines with revumenib to assess genome-wide chromatin occupancy of Menin and MLL1 (Fig. 4a). As expected, treatment of MEN1-WT cells led to near-complete and global displacement of Menin from chromatin with exposure to as little as 100 nM of revumenib. In contrast, the M327I mutant cells retained Menin on chromatin and only showed a partial decrease in Menin chromatin occupancy even when exposed to 5 μM of revumenib (Fig. 4a, b). Consequently, inhibitor induced MLL-eviction from key target loci, including MEIS1, was largely abrogated in M327I mutant MV4;11 cells (Fig. 4c, Extended Data Fig. 8a). Consistent with these findings in the MLL::AF4-rearranged MV4;11 cells, displacement of Menin from chromatin was blunted both M327I in NPM1c-mutant OCI-AML3 cells (Extended data Fig. 8b–d). Furthermore, treatment of mice that were engrafted with the T349M-mutant PDX3 failed to displace Menin from chromatin after oral Menin-inhibitor treatment in vivo (Extended data Fig. 8e, f). The profound loss of MLL1 from promoters with the highest degree of Menin displacement (>80%) was blunted in T349M mutant cells (Extended data Fig. 8g, h). To delineate the consequences of the altered chromatin binding dynamics of Menin and MLL1 under revumenib treatment, we performed RNA-sequencing in MEN1-mutant MV4;11 cells and PDX3 samples. In line with our observations on chromatin, MEN1-M327I mutant MV4;11 cells showed a high degree of resistance to revumenib-mediated changes in gene expression (Fig. 4d). Specifically, the repression of canonical Menin-MLL1 target genes as well as the induction of gene programs that are associated with myeloid differentiation were largely abrogated in M327I-mutant cells and only a partial response could be observed under treatment with 5 μM of revumenib (Fig. 4h, Extended Data Fig. 9a, b). Of note, these effects on gene expression were consistent between the KMT2Ar MV4;11 and NPM1c-mutant OCI-AML3 cells (Extended Data Fig. 10a, b). Likewise, treatment of mice engrafted with T349M-mutant PDX3 failed to sufficiently repress Menin-MLL target genes or induce differentiation-associated gene signatures in human leukemia cells as compared to mice engrafted with isogenic MEN1-WT cells in response to Menin-inhibition (Fig. 4e). Quantitative real-time PCR analysis in leukemia cells from the NPM1c-mutant PDX2 (MEN1-T349M vs. WT) showed the same phenomenon (Extended Data Fig. 10c). Interestingly, the re-engagement of this canonical Menin-MLL1 driven gene expression program appears to distinguish MEN1-mutant cases from those that develop resistance in the absence of these mutations. We performed RNA-seq studies of matched relapse vs. pre-treatment samples from a patient and PDX4 that had developed Menin inhibitor resistance without MEN1 mutations and found significant transcriptional re-programming (Extended data Fig. 10d–f). In contrast to MEN1-mutant cases, these resistant leukemias had decreased expression of key MLL-target genes (including MEIS1 and HOX-genes) and increased expression of myeloid differentiation genes. This suggests the existence of alternative non-genetic mechanisms of resistance that establish a state that is tolerant of an attenuated Menin-MLL1 gene expression program. Overall, we demonstrate in both KMT2Ar and NPM1c model systems that MEN1 resistance mutations prevent Menin-inhibitor induced displacement of the Menin-MLL1 protein complex from chromatin at critical target genes, which consequently abrogate gene expression changes that are required to terminate leukemic self-renewal and induce myeloid differentiation.
Figure 4: Menin chromatin binding and aberrant gene expression is rescued by MEN1 mutations.

a) Torpedo-plots of total Menin signal intensity around transcription start sites (TSS) from ChIP-sequencing (ChIPseq) in MV4;11-MEN1-WT and -M327I-mutant cells treated with revumenib (0.1μM, 1μM, 5μM) or DMSO as control. b) Read-normalized Menin-TSS-signal at MLL1-target genes in MV4;11 cells under revumenib treatment (mean +/− SD, 3000 data points per condition). One-way ANOVA with correction for multiple comparisons was used for statistical analysis. c) ChIPseq tracks of Menin and MLL1 at the MEIS1-locus in MV4;11 cells under revumenib treatment (representative example of 3 replicates). d, e) Heatmaps of RNAseq data showing the expression dynamics of all genes that are differentially expressed (DEGs, as defined by DESeq2 with an adjusted p-value<0.05 and a fold change>2) under treatment with a Menin-inhibitor in d) MV4;11 cells or e) PDX3. Kmeans clustering (4) was applied to generate heatmaps (Morpheus Tool) based on z-scores of DEGs, representative genes are used for annotation.
Discussion
We report that somatic mutations in MEN1 that confer resistance to Menin inhibitor treatment are frequently acquired in KMT2Ar and NPM1c leukemia cells in patients and in preclinical PDX models of disease. These mutations lead to recurrent changes in amino acid residues M327, G331, T349 and S160 and induce drug resistance to structurally distinct classes of Menin-inhibitors, some of which, including revumenib, recently entered early-phase clinical trials. Remarkably, amino acids G331, T349 and S160 were also identified as putative targets for resistance-development in a base-editor screen of the MEN1 gene, strengthening the confidence in the predictive value of those experimental systems. These novel MEN1-mutations decrease the affinity of the Menin/inhibitor interaction, preventing drug-induced displacement of the Menin-MLL1-complex from chromatin and thereby abrogating critical gene expression changes. The affected amino acids are essential for small molecule binding, but not for MLL1 association with Menin which allows for continued oncogenic activity of the MLL1-Menin complex on chromatin. Most importantly, the discovery of acquired mutations in Menin validates the Menin/MLL1 interaction as a key oncogenic driver in patients with AML harboring KMT2A rearrangements or NPM1c mutations and, as such, represents a promising therapeutic target. However, the observation that these mutations confer resistance across multiple Menin inhibitors suggests that next-generation inhibitors will be required to circumvent this common mechanism of resistance. Structure-guided drug design to derive second-generation compounds that effectively block MLL1 binding while avoiding interactions with the residues affected by acquired MEN1 mutations, may be a feasible strategy to overcome acquired resistance to first generation Menin-inhibitors. Given these findings, prospective monitoring of MEN1 mutation status should be considered in patients receiving Menin-inhibitor therapy, particularly for patients with delayed or incomplete responses, to inform treatment decision-making. This is the first report of clinically occurring somatic mutations that mediate acquired resistance to small molecules targeting chromatin-binding protein complexes. These data support chromatin complexes and epigenetic mechanisms as critical therapeutic targets in cancer but also suggest that a common mechanism of resistance to therapies targeting these complexes may result from acquired mutagenesis of essential, non-driver epigenetic regulators.
Methods
Patient subjects and primary sample acquisition
The human subjects that are part of this report were enrolled at Memorial Sloan Kettering Cancer Center (MSKCC) or Dana-Farber Cancer Institute (DFCI) in the AUGMENT-101 trial, a Phase 1/2, open-label, dose-escalation and dose-expansion cohort study of SNDX-5613 in patients with relapsed/refractory leukemias, including those harboring an MLL/KMT2A gene rearrangement or Nucleophosmin (NPM1) mutation. Additional patient specimens were obtained from the centralized sample bank of Syndax pharmaceuticals. Patients provided oral and written consent to participate in the clinical trial and the sample banking and sequencing program at MSKCC or DFCI (IRB 19-448 and 06-107). Assessments of clinical response with bone marrow biopsies and peripheral blood assessment were performed monthly. Approval was obtained from the Institutional Review Board at each institution participating in these clinical trials. Additional consent was obtained from participants at Memorial Sloan Kettering Cancer Center with analyses performed on the institutional biobanking protocol approved by the Institutional Review Board. Patient biospecimens were anonymized by creating unique identifiers with no associated protected health information and keeping the key on a password-protected server. Data collection and research was performed in compliance with all relevant ethical regulations for human research participants.
Patient-derived xenografts (PDX)
For the assessment of long-term responses and the characterization of drug-resistance in the xenograft model system, we used 6 different PDX models of AML (5x KMT2Ar, 1x NPM1c-mutant), that were previously established and characterized in the “Center for Pediatric Cancer Therapeutics” (CPCT) at Dana-Farber Cancer Institute (DFCI). Studies were conducted in female NOD.Cg-PrkdcscidIl2rgtm1wjl/SzJ (NSG) mice (Jackson Laboratory) of 7-9 weeks. Cohorts of animals (under long-term treatent with Menin-inhibitor or control) consisted of 7-10 animals. Sample size of in vivo experiments was based on previous experimental data from preclinical trials using the Menin inhibitor. Animals in preclinical trial cohorts were randomly assigned to groups that were treated with the Menin-inhibitor. Patients were not randomized against a control cohort, since the first in human phase 1/2 trial (AUGMENT-101) is not designed and approved as randomized trial. Investigators on the clinical trial were not blinded as the design of AUGMENT-101 was not not designed and approved as a single- or double-blinded trial. Also during preclinical trials, investigators were not blinded. Detailed information on each graft is provided in Supplementary Table 2. In vivo experiments were performed at DFCI under the “Institutional Animal Care and Use Committee” (IACUC) protocol: #16-021. NOG-mice (Taconic, 10-14 weeks of age) were injected with 250.000 cryo-preserved leukemia cells of each graft without prior conditioning and engraftment was monitored in the peripheral blood by detecting the chimerism between human CD45 (PE; clone: HI30; Biolegend, 1:100 for staining) and mouse CD45 (APC-Cy7; clone: 30-F11; Biolegend, 1:100 for staining) using flow cytometry every 3-4 weeks (LSRFortessa™; BD Biosciences). When human cells were consistently detectable in all mice of a given graft, oral treatment with VTP-50469 was initiated (0.03% drug supplemented rodent diet; drug supplied by Syndax Pharmaceuticals; diet produced by ENVIGO). Oral treatment was given for 4 weeks or until no leukemia cells were detectable anymore in the peripheral blood of mice. After discontinuation, mice were closely monitored by peripheral blood chimerism, and treatment was re-initiated upon relapse. Animals that reached the study endpoint were euthanized using CO2 inhalation and subsequent cervical dislocation. Tibias, femurs, iliac crests, and lumbar vertebrates were cleaned using TX329 cotton wipes (Texwipe) and crushed using mortar and pestle to extract bone marrow cells. Spleen cells were extracted by straining the organ through a 40μM nylon cell filter (FALCON). The human leukemia cell burden in blood, spleen and bone marrow was measured by detecting the chimerism between human CD45 (PE; clone: HI30; Biolegend) and mouse CD45 (APC-Cy7; clone: 30-F11; Biolegend, 1:100 for staining) and the differentiation status of human cells was assessed using anti-CD13 (PerCp-Cy5.5; clone: WM-15; Biolegend, 1:100 for staining), anti-CD14 (PE-Cy7; clone: M5E2; Biolegend, 1:100 for staining) and anti-CD11b (FITC; clone: IRCF44; Biolegend, 1:100 for staining).
Targeted DNA-sequencing of patient and PDX material
Diagnostic bone marrow aspirates were collected from patients before enrollment on the trial and at the time of relapse. Mononuclear cells were isolated via Ficoll gradient centrifugation and genomic DNA was extracted before CLIA-approved targeted sequencing was performed (MSKCC-IMPACT). Bone marrow from PDX (at relapse on or after VTP-50469 treatment or treatment naive as control) was extracted via crushing of the tibias, femurs, iliac crest, and lumbar vertebrates using mortar and pestle. Subsequently, murine cells were removed using the “Mouse Cell Depletion Kit” (Miltenyi Biotec) and genomic DNA was isolated from human cells using the DNeasy Blood and Tissue Kit™ (QIAGEN). Samples were subjected to targeted sequencing for oncogenic driver mutations using the MSK-IMPACT pipeline as previously described23.
Nomenclature of MEN1 mutations
All annotations of amino-acid changes inflicted by MEN1 mutations reported in this manuscript are based on the Menin-Isoform #1 (RefSeq: NM_000244.3).
Experimental procedure for the generation of the Menin:SNDX-5613 structures
Menin crystals were grown at 21°C using the sitting drop vapor diffusion method. Purified Menin at 11 mg/ml in 10 mM Tris pH 7.5, 50 mM NaCl, 1 mM TCEP was pre-incubated with inhibitor compounds dissolved in DMSO. The pre-incubation was done at 0.6 mM inhibitor concentration. For crystallization, 1.0 μL of the protein inhibitor complex was mixed 0.5 μL of seeds and 1.5 μL of a reservoir solution. SNDX-5613 was co-crystallized using as reservoir condition 0.1 M HEPES pH 7.9, 24% PEG 3350, 0.2 M Magnesium Nitrate and 20 % Ethylene Glycol. Crystals were flash cooled in cryo protection solution. For data acquisition, the crystal temperature was kept at 100 K. Diffraction data were collected at the Australian Synchrotron (beamline MX2) using a 16M pixel Dectris Eiger detector. Raw diffraction data were processed and scaled using XDS software (v. 18DEC2014). The structures were solved by molecular replacement with MOLREP (v. 11.7.03) in CCP4i (v. 7.1.018) using as search model the coordinates previously solved structures of menin (PDB ID 6PKC). The program REFMAC5 (v. 5.8.0267) was used for full structure refinement. The refined coordinates of the complex structures have been deposited in the RCSB Protein Data Bank.
Synthesis and cloning of MEN1 mutants into lentiviral constructs
The Menin-coding sequence was codon optimized and gene synthesis was performed via Twist Bioscience into a bacterial vector system (pTWIST-Amp). Small double-stranded DNA-blocks (gblocks, Integrated DNA Technologies) harboring the MEN1-M327I, M327V, G331R, T349M or D136N mutations were synthesized and cloned into pTWIST-Amp-MEN1 using a restriction enzyme mediated digest (BamHI+HindIII → M327I, M327V, G331R, HindIII+AfeI → T349M, SpeI+ClaI → D136N) and T4-ligase based cloning approach (all enzymes: New England BioLabs) in order to insert the mutations into the MEN1-coding sequence. The codons used were: M327I: ATG→ATC; M327V: ATG→GTG; G331R: GGT→CGT; T349M: ACC→ATG; D136N: GAC→AAC. Subsequently, MEN1 was amplified from the generated pTWIST template vectors using the Q5 High-Fidelity 2X Master Mix™ (New England BioLabs) and primers (forward: 5’-CCCAGGGGCTAGCATGGGTTTGAAAGCGGCGCAGA-3’; reverse: 5’-AGAGGTTGATTGTCGACTTAACGCGTTTATGCATAGTCCGGGACATCATACGGATAGCCGGCGTAGTCGGGCACGTCGTAGGGGTAAAGTCCCTTCCTTTGTCGTTTCAGAA-3’) under addition of a double-HA-tag to the C-terminus of Menin. The PCR product was gel-purified using the QIAquick Gel Extraction Kit™ (QIAGEN) and cloned into the pLEX-puro lentiviral vector system using T4-ligase after digest with NheI and MluI (New England BioLabs).
MEN1-Sequence codon optimized sequence used:
ATGGGTTTGAAAGCGGCGCAGAAAACTCTCTTTCCCCTCAGGAGCATTGATGATGTTGTCAGATTGTTCGCAGCGGAACTCGGTCGCGAAGAACCAGATCTCGTCCTACTAAGCCTCGTCCTTGGATTTGTTGAACACTTCTTGGCCGTGAATAGGGTGATTCCCACGAATGTGCCTGAATTGACGTTTCAACCGAGTCCTGCTCCAGATCCACCCGGTGGTTTGACTTATTTCCCTGTCGCTGATTTGTCCATTATTGCTGCACTGTACGCTAGGTTTACGGCTCAAATAAGAGGTGCAGTAGATCTCAGCCTGTACCCCCGCGAGGGCGGCGTGAGTTCCCGCGAACTAGTTAAGAAGGTATCTGACGTGATTTGGAATTCGCTGTCCCGTTCGTATTTTAAAGACAGAGCTCATATTCAATCATTGTTTTCTTTTATTACTGGCTGGTCACCGGTGGGTACTAAGCTGGATAGTTCGGGCGTCGCGTTCGCCGTCGTGGGAGCTTGTCAAGCTCTCGGACTACGCGACGTGCATTTGGCTCTCTCCGAAGACCACGCTTGGGTCGTCTTCGGTCCAAACGGAGAACAAACCGCCGAAGTGACATGGCATGGAAAAGGAAATGAAGATCGACGAGGACAAACTGTGAACGCTGGCGTTGCAGAACGATCCTGGCTTTATTTGAAGGGTTCGTATATGAGGTGCGATCGTAAAATGGAAGTTGCCTTTATGGTATGCGCAATAAATCCGAGCATCGATTTGCATACTGATTCTCTAGAACTATTGCAACTACAACAAAAGCTACTTTGGCTATTGTACGATCTCGGTCACCTAGAGAGATATCCGATGGCACTGGGAAATCTCGCTGACCTGGAAGAACTTGAACCTACTCCGGGACGTCCGGATCCTTTGACATTGTATCATAAAGGAATCGCTTCCGCTAAAACTTATTACCGTGACGAGCATATTTATCCTTATATGTATCTCGCCGGTTATCATTGCCGTAATAGGAACGTTCGTGAGGCGTTGCAAGCTTGGGCTGATACCGCTACAGTGATTCAAGATTATAATTATTGTCGTGAGGACGAAGAAATATATAAAGAATTCTTCGAGGTGGCTAACGACGTGATTCCTAATTTGCTTAAAGAAGCCGCATCCCTGCTTGAAGCTGGAGAAGAACGTCCAGGAGAACAGTCGCAAGGTACGCAATCTCAGGGAAGCGCTTTGCAAGATCCAGAATGTTTTGCTCATCTACTCAGATTTTATGATGGTATTTGTAAGTGGGAAGAAGGTTCCCCAACCCCCGTCCTTCACGTCGGATGGGCTACATTCCTGGTTCAAAGCCTTGGAAGGTTCGAAGGTCAAGTACGTCAGAAAGTTCGTATCGTCTCCAGGGAAGCTGAAGCTGCTGAAGCTGAAGAACCTTGGGGTGAAGAGGCTCGTGAGGGTCGTCGTCGCGGACCGCGCAGAGAATCGAAACCCGAAGAACCGCCTCCTCCTAAGAAGCCTGCCCTTGATAAAGGATTGGGTACTGGTCAAGGCGCCGTCAGTGGTCCGCCTAGGAAACCACCAGGCACTGTAGCGGGAACTGCACGTGGTCCCGAAGGCGGTTCCACAGCCCAAGTACCCGCTCCAACTGCTTCTCCACCGCCTGAAGGACCTGTACTAACCTTTCAATCTGAGAAAATGAAAGGAATGAAAGAACTCTTGGTCGCAACAAAGATTAATTCCAGTGCAATAAAATTGCAGCTGACCGCTCAATCACAGGTTCAAATGAAGAAGCAAAAGGTCAGCACACCATCAGATTATACCCTTAGTTTTCTGAAACGACAAAGGAAGGGACTTTAA
Purification of recombinant Menin
Wild-type human Menin was codon optimized for E. Coli expression, synthesized, and cloned in pET28+ derived vectors (Twist Biosciences) and the resulting vector was further subcloned with a synthesized gBlock (IDT) to produce a N-terminal StrepII-Avi-TEV Menin fusion. Mutant protein plasmids were generated by site-directed mutagenesis using the Q5® Site-Directed Mutagenesis Kit (NEB) following manufacturer instructions, all resulting plasmids were sequence verified. Recombinant proteins were expressed as N-terminal StrepII-Avi-TEV fusions in BL21-DE3 Rosetta cells following standard protocols. Briefly, each expression was performed at 4 L scale in LB media. 10 mL of overnight starter cultures was added to each liter of LB at 37°C, and protein expression induced by addition of 1 mM IPTG at OD600 of 0.6 following change of temperature to 18°C overnight. The overnight expression cultures were harvested by centrifugation at 4000 g for 20 min and resuspended in buffer containing 50 mM tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl) pH 8.0, 200 mM NaCl, 2 mM tris(2-carboxyethyl)phosphine (TCEP), 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 μM Bestatin, 2 μM E-64, 1 μM Pepstatin and 10 μM Leupeptin, and lysed by sonication. Following ultracentrifugation, the soluble fraction was passed over Strep-Tactin XT (IBA) affinity resin and eluted with wash buffer (50 mM Tris-HCl pH 8.0, 200 mM NaCl, 2 mM TCEP) supplemented with 50 mM biotin (MCE). The affinity-purified protein was subject to ion exchange chromatography (Poros 50HQ) followed by size exclusion chromatography (Superdex 200 10/300 GL) in 50 mM HEPES pH 7.4, 200 mM NaCl and 2 mM TCEP. The protein-containing fractions were concentrated using ultrafiltration (Millipore) and flash frozen in liquid nitrogen at 10 μM concentration.
Crystallography methods
Menin (WT)-SNDX-5613
Menin protein at 11 mg/ml in 10 mM Tris pH 7.5, 50 mM NaCl, 1 mM TCEP was used for setting up crystallization. Menin-5613 crystal was obtained using cross-seeding from other Menin co-crystals. For Menin-SNDX-5613 crystallization, 1.0 μL of the protein inhibitor complex was mixed 0.5 μL of seeds and 1.5 μL of a reservoir solution using the sitting drop vapor diffusion method. The reservoir solution contains 0.1 M HEPES pH 7.9, 24% PEG 3350, 0.2 M Magnesium Nitrate and 20 % Ethylene Glycol. Crystals were obtained within 10 days of incubation at 21°C.
Menin (M327I)-SNDX-50613
Menin(M327I) protein at 10.7 mg/ml in 10 mM Tris pH 7.5, 50 mM NaCl, 1 mM TCEP was used for setting up crystallization. Menin(M327I)-SNDX-50613 crystals were obtained using sitting drop vapor diffusion method with 1 ul of protein-inhibitor mix and 1 ul of reservoir buffer. The reservoir buffer contains 0.1 M MES pH 6.2, 16% PEG 3350, 0.2 M Potassium thiocyanate and 20% Ethylene Glycol. Crystals were obtained within 7 days of incubation at 21°C.
While most annotations of Menin mutations are based on isoform 2, which encodes 610 amino acid residues, some annotations are based on an alternate transcript with 615 amino acids. Biancaniello et al.24 have proposed that the 615-amino acid isoform be used as the standard reference for Menin, which was the reference used to report the crystallography data. References made to the 610-residue isoform in some instances will result in a 5-residue shift in register.
Fluorescence polarization
The following peptides were synthesized by GenScript: N-terminal FITC-conjugated MLL14-43(C-A) peptide probe (SARWRFPARPGTTGGGGGGGRRGLGGAPRQRVPALLLPPGY) with a C-A modification that improves binding to Menin20, MLL14-43(C-A) (SARWRFPARPGTTGGGGGGGRRGLGGAPRQRVPALLLPPGY) and MLL14-43(WT) (SCRWRFPARPGTTGGGGGGGRRGLGGAPRQRVPALLLPPGY). N-terminally FITC-labelled and unmodified MLL2 peptides (MLL2( 15-48) SARGRFPGRPRGAGGGGGRGGRGNGAERVRVALR) were synthesized at Genscript. To derive the FITC-MLL14-43(C-A) peptide Kd, FITC-conjugated MLL14-43(C-A) peptide probe at final concentration of 1 nM was mixed with increasing concentration of purified StrepII-Avi-TEV-Menin (5 μM final top concentration, 2-fold, 23-point dilution and a buffer control) in an assay buffer (50 mM Tris pH 7.5, 200 mM NaCl, 0.1% Pluronic F-68 solution (Sigma)) in 384-well microplates at 15 μL. assay volume (Corning, 4514) and incubated for 30 min at room temperature (RT). For all assays change in fluorescence polarization was monitored using a PHERAstar FS microplate reader (BMG Labtech). The FITC-conjugated MLL14-43(C-A) peptide Kd was obtained from a fit in GraphPad Prism 9 using one site total model and averaged from three independent runs with three replicates (n=3, Extended data Figure 4). In competitive titration experiments Menin proteins (1 nM WT, 1 nM M327I or 3 nM for T349M) and 1 nM FITC-conjugated MLL14-43(C-A) peptide probe were incubated in an assay buffer for 30 min. SNDX-5613 was then dispensed to 384-well microplate (Corning, 4514) containing 15 μL of the assay mix using D300e Digital Dispenser (HP) and normalized to 1% DMSO followed by 90 min incubation. The fluorescence polarization was monitored by PHERAstar FS microplate reader (BMG Labtech). The MLL14-43(WT) and MLL14-43(C-A) peptide titrations were performed by addition of 7.5 μL of 2-fold 23-point serial dilution into 7.5 μL of assay mix with the final concentrations of 1 nM FITC-conjugated MLL4-43 peptide probe, 1 nM WT Menin or 1 nM Menin M327I or 3 nM Menin T349M in assay buffer. The fluorescence polarization was monitored by PHERAstar FS microplate reader (BMG Labtech). For analysis of the competitive titration experiments the last 10 cycles of the data were averaged to obtain technical replicates. Data from three independent replicates (n=3) was plotted and IC50 values estimated using variable slope equation in GraphPad Prism 9. The calculation of Ki values was performed using Ki calculator available at http://websites.umich.edu/~shaomengwanglab/software/calc_ki/index.html25. The assay parameters used for calculation of Ki were: 1 nM probe for all assays, 1 nM WT Menin, 1 nM Menin M327I, 3 nM Menin T349M, with Kd (mean ± S.D., N=3) of 0.76 ± 0.77 nM for WT Menin, 0.91 ± 0.99 nM for Menin M327I, 2.99 ± 1.62 nM for Menin T349M.
Fluorescence-based kinetic binding assays
Off-rates for test compounds were determined using jump dilution experiments in the HTRF assay measuring FITC-MLL4-43 binding to HIS-Menin. Briefly, test compounds (30 nM) were pre-incubated for 1 hr with 30 nM HIS-Menin prebound to 30 nM Lanthascreen anti-HIS-Tb antibodies in 10 uL of menin assay buffer (50 mM Tris, pH 7.4, 50 mM NaCl, 5 mM DTT, 0.01% TX100) containing 0.02% fatty acid free BSA. After pre-incubation, 1 uL of the assay mixture was transferred to 100 uL of menin assay buffer containing 30 nM FITC-MLL-4-43 (1000X Ki) in white, opaque 384-well plates. The HTRF signal was measured at various times using a 320 nm excitation and 520 and 620 nm emission wavelengths with a 50 us delay and a 200 us window with the gain set to 2100 using a BMGlabTech ClarioStar Plus plate reader. The data was normalized to % inhibition at each time point, where 0% is the HTRF signal in the presence of 0.3 nM HIS-menin without test compound, and 100% is the HTRF signal in the absence of HIS-menin. The resulting % inhibition values were fit to a one-site exponential dissociation model using XlFit to determine the dissociation rate.
Isothermal titration calorimetry
All calorimetric experiments were carried out in ITC buffer (25 mM Hepes (pH 7.5), 200 mM NaCl, 1.0 mM TCEP) at 25°C using an Affinity ITC from TA Instruments (New Castle, DE) equipped with autosampler. SNDX-5613 was dissolved in DMSO and diluted with the ITC buffer to final concentrations (50-200 μM, 1% DMSO). Protein sample was adjusted to contain 1% DMSO final concentration. The calorimetric cell, containing buffer or Menin (10μM WT Menin, 5μM M327I Menin, or 10μM T349M Menin) was titrated by injecting 2.5 μl of SNDX-5613 solution with the concentration of 100μM, 50μM or 200μM, respectively, 24 times with stirring speed at 75 rpm. The resulting isotherms were subtracted against buffer runs and fitted with a single-site model to yield thermodynamic parameters of ΔH and ΔS, stoichiometry, and KD using NanoAnalyze software (TA Instruments).
Cell culture and lentiviral transductions
Cell lines were obtained from the “American Type Culture Collection” (ATCC) or the “Deutsche Sammlung von Mikroorganismen und Zellkulturen” (DSMZ) and cultured in RPMI-1640™ (for MOLM13, MV4;11 and OCI-AML3) or DMEM™ (for HEK-293T) + 10% fetal bovine serum (FBS) + 1% penicillin/streptomycin (P/S) (Life Technologies/Thermo Fisher Scientific) at 37°C, 5% Co2 atmosphere and 95% humidity. Cell lines were tested and maintained mycoplasma negative throughout all experiments. Cell line authentication for MOLM13, MV4;11, and OCI-AML3 cells was performed via STR analysis by the MSKCC core facility. For the production of lentivirus HEK-293T cells were seeded in 10cm tissue culture treated dishes (Corning) 24 h before transfection to achieve 80% confluence. pLEX-puro plasmids (5 μg) containing a MEN1-WT, -M327I, -M327V, -G331R, -T349M, -D136N or the MEN1-CRISPR-Cas9-base-editor library were transfected along with lentiviral packaging plasmids (2 μg pMD2G + 5 μg psPAX2) using 30 μl XtremeGene9™ DNA transfection reagent (Sigma-Millipore) in 1.8ml Opti-MEM per 10 cm dish containing 4 ml DMEM + 10% FBS (Life Technologies/Thermo Fisher Scientific). Medium was exchanged with 10 ml fresh DMEM + 10% FBS and viral supernatants were harvested 24 h later by filtering through a 0.45 μm Nalgene syringe filter (Thermo Fisher Scientific). Viral supernatants were frozen at −80°C or directly used to infect target cells. For lentiviral transduction target cells were resuspended in crude viral supernatants and spin-infection was done in 50 ml Falcon-tubes for 2 h at 2000 rpm in a centrifuge heated to 37°C. Subsequently, viral supernatants were removed, and cells were resuspended in RPMI-1640™ + 10% FBS + 1% P/S. Puromycin was added to pLEX-puro transduced cell lines at a final concentration of 1μg/ml 48h after transduction to select for cells expressing the respective constructs.
CRISPR-Cas9 base-editor screening
An sgRNA-library of 518 single-guide RNAs (sgRNAs) targeting all MEN1-exons and flanking untranslated regions (Supplementary table 2) was designed, synthesized, and cloned into the pRDA_256 vector system at the Broad Institute of Harvard and MIT via the Genetic Perturbation Platform (GPP). Production of lentiviral particles and viral transduction was performed as described above. MOLM13 and MV4;11 cells were transduced with the base-editor library and selected with Puromycin 1μg/ml, Thermo Fischer Scientific) for 6 days. After selection, a baseline sample (5x 106 cells) was harvested for DNA-extraction and the remaining cells were split into 10 separate non-tissue culture treated T75 culture flasks. Each 5 of these flasks were treated with 50 nM of VTP-50469 or DMSO as control for 12 days and split every 3 days during this time. At the end of the experiment, cells were harvested and DNA was extracted using the DNeasy Blood and Tissue Kit™ (QIAGEN). Library construction, Next-Generation sequencing, and generation of an sgRNA count matrix were performed via the GPP at the Broad Institute. Beta-scores were calculated for each condition (baseline vs. endpoint) using the MaGECK-MLE pipeline. Differential beta scores for each guide-RNA were calculated by subtracting the control beta-score from the VTP-50469 values.
CRISPR-Cas9-based gene-editing of MEN1 by homology directed repair (HDR)
MV4;11 and OCI-AML3 cell lines were passaged 24 h prior to nucleofection. The guide RNA (150 pmol) (sequence: 5’-CATCTACCCCTACATGTACC-3’) was added to Alt-R S.p. HiFi Cas9 nuclease (Integrated DNA Technologies) (125 pmol) followed by an incubation at room temperature (RT) for 20 min to allow formation of ribonucleoprotein (RNP) complexes. 2 x 105 cells were sedimented at 200 g for 10 min at RT, washed with PBS and resuspended in the SG Cell Line Nucleofector™ Solution (Lonza). Cells, Alt-R™ Cas9 Electroporation Enhancer (Lonza) (120 pmol) and the HDR donor oligonucleotide containing the MEN1-M327I mutation as well as a silent mutation at the protospacer adjacent motif (PAM) site (120 pmol) (sequence:5’-TCCCGCACATTGCGGTTGCGACAGTGGTAGCCAGCGAGGTAGATGTAGGGGTAGATGTGTTCATCCCGATAGTAGGTCTTGGCT-3‘) were added to the RNP complex. For wildtype (WT) control, phosphate buffered saline (PBS) (Gibco™, Fisher Scientific) was added to the resuspended cells. Nucleofection was performed using a 16-well 20 μl Nucleocuvette™ stripe (Lonza) in a 4D-Nucleofector™ X Unit (Lonza) (program FF-100). Cells were incubated overnight in RPMI-1640 + 10% FCS + 1% P/S containing 1 μM Alt-R™ HDR enhancer V2 (Lonza) (or DMSO for WT control). The following day, the medium was replaced by RPMI-1640 + 10% FBS + 1% P/S. Single clones were selected in methylcellulose (MethoCult™ M3234, Stemcell™ Technologies) supplemented with 10% RPMI-1640, 10% FBS, 1% P/S and 25 nM SNDX-5613 (or DMSO for WT control), starting 3 days after nucleofection, and expanded in RPMI-1640 + 10% FCS + 1% P/S supplemented with 25 nM SNDX-5613 (or DMSO for WT control). For the in vitro clonal selection assay of bulk nucleofected cells over time, 5 x 103 cells (3 replicates per condition) were seeded in RPMI-1640 + 10% FBS + 1% P/S supplemented with 50 nM SNDX-5613 (or DMSO for WT control) 3 days after nucleofection. Cells were replated in fresh medium + drug every 3 days and viable cells were counted using the LSRFortessa™ (BD Biosciences) flow cytometer. Cumulative cell growth was calculated over 28 days. At each timepoint, cell pellets were frozen and genomic DNA was extracted subsequently using the DNeasy Blood and Tissue Kit™ (QIAGEN). Digital droplet-based PCR was performed from the genomic DNA to assess editing efficiency in the bulk cells over time. To analyze HDR efficiency in single cell clones, genomic DNA was PCR amplified using the Q5 High-Fidelity 2X Master Mix™ (New England BioLabs) and primers (forward: 5’-CCCTCAGCCCTGCCTTTTCTGC-3’; reverse: 5’-AGTCCTGGACGAGGGTGGTTGG-3’), and the resulting 641 bp fragment containing the Cas9 cut site was gel-purified using the QIAquick Gel Extraction Kit™ (QIAGEN). Sanger sequencing was used to analyze HDR efficiency (primer 5’-CTGGGATCTTCCTGTGGCCCCT-3’).
Assessment of proliferation in vitro
To determine Menin-inhibitor responses of MOLM13, MV4;11 or OCI-AML3 cell lines expressing MEN1 mutants, 3 x 103 cells were plated per well in a non-tissue culture treated 96-well plate. The Menin-inhibitors were titrated on these cells in the following dose-range: DMSO, 3.9 nM, 7.813 nM, 15.625 nM, 21.25 nM, 62.5 nM, 125 nM, 250 nM, 500 nM, 1000 nM. Cells were treated in triplicates on each plate and the experiment was repeated 4 times. After 6 days, cells were split 1:7 and counts of viable cells in each well were determined after 10d of treatment by flow cytometry (LSRFortessa™ with High-throughput sampler (HTS), BD Biosciences). Relative cell counts were calculated as % of DMSO control. The average of each triplicate was calculated and the mean of all replicates was plotted. Error bars represent the standard error of means (SEM).
Chromatin Immunoprecipitation Sequencing (ChIPseq)
Cells were sequentially cross-linked using 2 mM DSG disuccinimidyl glutarate for 30 min and a final concentration of 1% formaldehyde for 10 min at room temperature (20–25 °C) and stopped with 125 mM glycine. Cells were lysed in lysis buffer (20 mM Tris-HCl at pH 7.5, 300 mM NaCl, 2 mM EDTA, 0.5% NP40, 1% Triton X-100, 1 mM PMSF, PIC) and incubated on ice for 30 min. The resuspended cells were then dounced in an ice-cold homogenizer. Nuclear pellets were collected and resuspended in shearing buffer (0.1% SDS, 0.5% N-lauroylsarcosine, 1% Triton X-100, 10 mM Tris-HCl at pH 8.1, 100 mM NaCl, 1 mM EDTA, 1 mM PMSF, PIC). Isolated chromatin was fragmented to an average size of 200–600 bp with a bioruptor (Diagenode). Precleared chromatin was immunoprecipitated overnight at 4 °C (anti-Menin: Bethyl Laboratories, #A300-105A, Lot#11, 5μg/IP; anti-MLL1: Bethyl Laboratories, #A300-086A, Lot#7, 5μg/IP) and immunocomplexes were collected with protein A Dynabeads. The immunocomplexes were washed eight times in wash buffer (50 mM HEPES-KOH at pH 7.6, 500 mM LiCl, 1 mM EDTA, 1% NP40, 0.7% sodium deoxycholate, 1 mM PMSF, PIC), followed by two 1 × TE washes, and eluted in elution buffer (50 mM Tris-HCl at pH 8.0, 10 mM EDTA, 1% SDS), crosslinks were reversed at 65 °C for 4 h or overnight, and DNA was purified using DNA Clean & Concentrator Kit according to the manufacturer’s instructions. ChIP- or input-DNA was used for Illumina library construction using ThruPlex DNA-seq kit (Takara) with 12 to 14 cycles of amplification and the use of single indexing barcodes. Paired-end sequencing (37bp) was performed on a NextSeq500 platform (Illumina). Raw Illumina NextSeq BCL files were converted to FASTQ using Illumina bcl2fastq. Reads were aligned to human GRCh38/hg38 genome using STAR 2.7.5. Aligned BAM files were sorted, duplicate reads marked and removed, and deduplicated BAMs indexed using Broad picard tools v2.9.4. Signal intensities around transcription start sites (TSS, +/− 3000bp) were quantified using BEDtools (v. 2.28.0) and data visualizations were produced using IGVtools (TDF signal pileups; v2.3.75) and ngs.plot (pileup heatmaps/torpedo plots). ChIPseq peaks were called using MACS2 (v. 2.1.4.) with appropriate input samples used as controls.
RNA Sequencing (RNAseq)
Total RNA was isolated from cell lines or mouse cell depleted PDX material using the RNeasy™ Mini Kit (QIAGEN). Quality control was performed using RNA-Tape™ (Agilent) and all samples used for sequencing passed QC with a RINe-score greater than 8. Poly(A) mRNA enrichment and library preparation was performed using the NEBNext Poly(A) mRNA Magnetic Isolation Module and NEBNext Ultra II RNA Library Prep kit (New England BioLabs) according to the manufacturer’s instructions. Sequencing was done on an Illumina NextSeq500 platform as 37bp paired end sequencing. Raw Illumina NextSeq BCL files were converted to FASTQ using Illumina bcl2fastq. Reads were aligned to human GRCh38/hg38 genome using STAR 2.7.5. Aligned BAM files were sorted, duplicate reads marked and removed, and deduplicated BAMs indexed using Broad picard tools v2.9.4. Raw per-gene counts were calculated with HTSeq-count (v.0.6.1pl). Differential RNA-seq expression was calculated using the BioConductor DESeq2 package (v1.24,0), using raw unnormalized per-gene counts from deduplicated BAMs. Z-scores for the generation of heatmaps were calculated from regularized gene-counts (from DeSeq2 package).
Statistical analysis
Statistics were performed using GraphPad Prism software (GraphPad Software, San Diego, CA). Statistical analysis of Menin inhibitor dose-response differences between MEN1-WT and MEN1-mutant cell lines was performed using unpaired Student’s t-test (two-tailed) (Fig. 3, Extended Data Fig. 5, 6). The assessment of statistical differences in Menin and MLL1 TSS-occupancy from ChIPseq data was performed using Mann-Whitney-U-test (two-tailed), since this data is not normally distributed (Fig. 4, Extended Data Fig. 7). Statistical comparison of disease burden and induction of differentiation in PDX across different conditions was performed using one-way ANOVA (Extended Data Fig. 2, 3). P < 0.05 was considered statistically significant. Legend for stars used to indicate significance in dose-response curves (Fig. 3, Extended Data Fig. 5, 6): * p<0.05, ** p<0.01, *** p<0.001.
Code availability
Scripts for structure preparation, docking, and simulation can be found on Github (https://github.com/choderalab/men1). Software packages used for ChIP and RNAseq analysis are commonly used by the community and are publicly available: bcl2fastq (v. 2.20.0.422), STAR (v. 2.7.5a), picard (v. 2.9.4), SAMtools (v. 1.95), IGVtools (v. 2.3.75), htseq-count (v. 0.6.1pl), DESeq2 (v. 1.24.0), bedtools (v. 2.28.0), MACS2 (v. 2.1.4), parallel (v. 20061222), snakemake (v. 5.20.0).
Extended Data
Extended Data Figure 1: Novel MEN1 mutations detected in patients upon relapse on revumenib.

a-c) Tables showing the results of the IMPACT targeted DNA-sequencing panel from patient 1-4 at the time point of screening prior to enrolling on the AUGMENT-101 trial and at relapse on revumenib treatment. MEN1 mutations are highlighted in red. d) Pie charts displaying the fraction of MEN1-mutant alleles measured by droplet digital PCR (ddPCR) at the time point of relapse (or last available sampling time point before relapse) in all individual patients from the cohort shown in Fig. 1b. Relative mutation frequencies (number of MEN1-mutant / WT droplets) are labeled in white. Mutations which were detected in > 2 droplets were considered. e) Longitudinal kinetics of MEN1 mutant selection in two selected patients from the cohort shown in Fig. 1b. Mutant allele frequencies at different time points during revumenib treatment were analyzed by ddPCR.
Extended Data Figure 2: Development of Menin-inhibitor resistance in a KMT2Ar PDX.

a) Box-plot (median, box: 25th to 75th percentile, whiskers: range) showing the percentage (%) of human leukemia cells in the bone marrow of NOG-mice transplanted with PDX3 at baseline (n=4), 4 weeks (w) (n=4), 8w (n=5) and at 10-12w (n=5; symptomatic leukemia relapse) on Menin-inhibitor treatment. Dots represent individual animals. One-way ANOVA with correction for multiple comparisons was used for statistical analysis. b) Box-plots (median, box: 25th to 75th percentile, whiskers: range) showing the mean fluorescence intensity (MFI) of the myeloid differentiation markers CD11b, CD13 and CD14 on the cell surface of human cells detected in the bone marrow of NOG-mice transplanted with PDX3 at baseline (n=4), 4w (n=5), 8w (n=4) and at 10-12w (n=4) on Menin-inhibitor treatment. Dots represent individual animals. One-way ANOVA with correction for multiple comparisons was used for statistical analysis. c) Bone marrow cytology pictures (cytospins) from each 2 representative animals at baseline, 4w, 8w and 12w on Menin-inhibitor treatment. d) Pie charts showing the fraction of MEN1-T349M (red) as compared to MEN1-WT (blue) measured by droplet digital PCR (ddPCR) at baseline, 8 weeks and 12 weeks (fulminant clinical relapse) in human cells isolated from PDX3 mice and purified using magnetic cell sorting.
Extended Data Figure 3: Base-editor screening as a tool to identify point mutants in MEN1.

a) Schematic depicting the workflow of the MEN1-base editor screen performed in MOLM13 (MLL::AF9) and MV4;11 (MLL::AF4) cells b) Dot-plot showing the results of a CRISPR-Cas9 base-editor screen in MV4;11 cells aiming to identify point mutations that cause resistance to Menin inhibitor treatment. Each dot represents a single guide RNA. Along the x-axis guide RNAs are sorted by their targeting location relative to the Menin-coding sequence. The y-axis shows differential CRISPR-beta-scores (DMSO-score subtracted from the VTP-50469-treatment score). Outstanding hits are marked in red and targeted amino acid residues are labeled. c) X-ray co-crystal structure of revumenib bound to WT-Menin (PDB: 7UJ4). The hydrogen bonds between sulfonamide oxygen of revumenib and indole nitrogen of W346 or sulfonamide nitrogen of revumenib and backbone carbonyl oxygen of M327 are indicated with black dashed lines. Non polar hydrogens are shown for revumenib and the W346. d) X-ray co-crystal structure of Menin in complex with MLL14-15 peptide (PDB: 4GQ6). View corresponds to Extended data Figure 3c. Recurrently mutated amino acids are labeled in red. The W346 residue that builds up a strong hydrogen bond with revumenib to stabilize binding of the molecule is marked in blue. e) Alignment of the Menin bound revumenib (PDB: 7UJ4) with Menin bound MLL14-15 peptide (PDB: 4GQ6). Recurrently mutated amino acids are labeled in red. The W346 residue that builds up a strong hydrogen bond with revumenib to stabilize binding of the molecule is marked in blue.
Extended Data Figure 4: Atomic modeling of Menin and its mutations using equilibrium simulations.

a) Trajectory length distributions for equilibrium simulations of Menin wild-type and mutants (rows). Simulations were all run simultaneously on Folding@home and the frequency distribution of their simulation lengths is indicated for each construct, both with and without revumenib (left and right columns, respectively. b) Equilibrium molecular dynamics simulations and Markov models reveal that helices contacting revumenib separate upon mutation. Distance distributions between the sulfonamide contacting helices were computed for WT Menin and each mutant. Error bands are computed by bootstrapping the markov state model using 10 random samples with replacement, generating standard errors for the Markov State Model populations. These errors were used to compute the histogram standard error ranges as shown above using 100 bins. Results are insensitive to changing the number of bootstrapped samples from 5 to 30. c) DiffNets analysis comparing WT to mutant Menin using backbone features showing helical separation (blue lines) around revumenib (magenta). Dashed lines indicate helical motion as a structural feature that significantly differs between WT and mutant Menin. Blue lines indicate that helices move further apart and separate upon mutation, while red dashed lines indicate that helices come closer together. d) Implied timescales after clustering all Folding@home simulations. Based on this plot, a lag time of 7 nanoseconds was chosen for MSM construction to ensure Markovanaity.
Extended Data Figure 5: MEN1 mutations impact binding affinity of revumenib to the MLL1/2 binding pocket.

a) Titration curves of WT-, M327I- and T349M-mutant Menin against a FITC-conjugated MLL1 4-43(C-A) peptide probe (N=3, each data point represents the mean of 3 technical triplicates +/− SD) for determination of equilibrium dissociation constant (Kd). b) Curves depicting the fraction of revumenib (left) or MLL1 (right) bound to Menin (WT or mutant) over time determining the molecule’s dissociation rates (off-rates) over time (N=8). Data point represent the mean +/− SD. c) Isothermal titration calorimetry assay measuring the binding of revumenib to WT-, M327I and T349M-mutant Menin confirming the mutation inflicted shift in affinity detected using the fluorescence polarization assay (Fig. 2e). d) Titration curves of WT-, M327I- and T349M-mutant Menin against a FITC-conjugated MLL2 (15-48) peptide probe at three peptide concentrations 0.5, 1 and 2 nM. Data is presented as fraction bound of three independent replicates (N=3). Data point represent the mean +/− SD. e) Fluorescence polarization assay measuring dose-dependent displacement of an MLL2 peptide from WT, M327I- and T349M-mutant Menin under treatment with revumenib or MI-3454. Data is presented as fraction bound of three independent replicates (N=3). Data point represent the mean +/− SD.
Extended Data Figure 6: Lentiviral expression of MEN1 mutants confers resistance in cell lines.

a) Western blot in MOLM13 cells showing expression of HA-tagged MEN l-WT and M327I-mutant construct. Representative Western Blot of 3 independent replicates. b) Dose-response curves of MOLM13 cells to revumenib upon expression of MEN1 mutants compared to -WT. Cell counts were measured by flow cytometry and displayed relative to the DMSO control (mean +/− SEM, n=4, each 3 technical replicates). c) b) Dose-response curves of MV4;11 and OCI-AML3 cells to revumenib upon expression of MEN1 mutants or -WT measured by Cell-titerGlo (mean +/− SD, n=3). d) Induction of differentiation marker expression by revumenib in OCI-AML3 cells expressing MEN1 mutants compared to WT measured by flow cytometry (MFI CD11b) (n=3, mean +/− SD). An unpaired, two-tailed t-test was used for statistical analysis. e) Quantification of the cytological assessment for blast morphology by a hematopathologist (n=5, median, box: 25th to 75th percentile, whiskers: range). One-way ANOVA with correction for multiple comparisons was used for statistical analysis. f) Dose-response curves of OCI-AML3 cells to revumenib (top panel) or MI-3454 (bottom panel) upon expression of MEN1 mutants compared to -WT. Cell counts were measured by flow cytometry and displayed relative to the DMSO (mean +/− SEM, n=4, each 3 technical replicates). g) Western blot in MV4;11 cells showing expression of HA-tagged MEN1-WT and -mutant constructs. Representative Western Blot of 2 independent replicates. h) Dose-response curves of MV4;11 cells to revumenib upon expression of MEN1 mutants compared to -WT. Cell counts were measured by flow cytometry and displayed relative to the DMSO (mean +/− SEM, n=4, each 3 technical replicates). b, f, h) Statistical analysis was perfomed using an unpired t-test, two tailed, multiple comparisons. *** p<0.001; **p<0.01; *p<0.05.
Extended Data Figure 7: MEN1-M327I endogenous gene-editing induces drug resistance to different Menin-inhibitors in leukemia cell lines.

a) Sanger-sequencing tracks showing gene-editing in MV4;11 and OCI-AML3 cells generating stable cell lines harboring the mutations indicated above the respective plots at the endogenous MEN1-locus. b) Dose-response curves of M327I homozygous or -WT MV4;11 cells to a high-dose range of revumenib. c) Dose-response curves of M327I heterozygous or -WT MV4;11 cells to MI-3454. d) Dose-response curves showing the sensitivity of OCI-AML3 (NPM1c) cells harboring the MEN1-M327I mutation to revumenib, MI-3454 and the Daiichi-Sankyo compound. e) Dose-response curves showing the sensitivity of OCI-AML3 (NPM1c) cells harboring the MEN1-T349M mutation to revumenib, MI-3454 and the Daiichi-Sankyo compound. f) Dose-response curves showing the sensitivity of MV4;11 cells harboring homozygous or heterozygous MEN1-M327I mutations to the covalent binder MI-89. g) Dose-response curves of S160C or -WT MV4;11 cells to revumenib. b-g) Cell counts were measured by flow cytometry and displayed relative to the DMSO control (mean +/− SEM, n=4, each 3 technical replicates). h) Fluorescence-based cell competition assay measuring relative cell fitness of MV4;11-MEN1-WT, -M327I or -T349M mutant cells in the presence or absence of revumenib (100nM) over the course of 21 days by flow cytometry (N=4, mean +/− SD). b-g) Statistical analysis was perfomed using an unpired t-test, two tailed, multiple comparisons. *** p<0.001; **p<0.01; *p<0.05.
Extended Data Figure 8: ChIPseq of Menin and MLL1 in MEN1-WT and -M327I-mutant cells.

a) ChIPseq tracks of Menin and MLL1 at the PBX3, MEF2C, JMJD1C-loci and the HOXA-cluster in MV4;11 cells under revumenib treatment (representative example of 3 replicates). b) Torpedo-plots of total Menin signal intensity around transcription start sites (TSS) from ChIP-sequencing (ChIPseq) in OCI-AML3-MEN1-WT and -M327I-mutant cells treated with revumenib (0.1μM, 1μM) or DMSO as control (N=2). Shown is one representative example. c) ChIPseq tracks of Menin at the MEIS1, PBX3, MEF2C, JMJD1C-loci and the HOXA-cluster in OCI-AML3 cells under revumenib treatment. d) Bar graphs showing Menin-ChIP-qPCR results at the MEIS1, MEF2C and HOXA10 transcription start sites after treatment with 100nM revumenib or DMSO as control (4 days treatment) (N=3, mean +/− SD). An unpaired, two-tailed t-test was used for statistical analysis. e) Torpedo-plots of Menin signal intensity around TSS from ChIPseq in MEN1-WT and -T349M-mutant PDX3 treated with VTP-50469 for 14 days. f) Menin-TSS-signal at MLL1-target genes in PDX3 (mean +/− SD, 3000 TSS data points per condition). Two-tailed Mann-Whitney-Tets was used for statistical analysis. g) Read-normalized MLL1-TSS-signal at sites that lose >80% of Menin in WT cells treated with VTP-50469 (mean +/− SD, 293 data points per condition). Two-tailed Mann-Whitney-Tets was used for statistical analysis. h) ChIPseq tracks of Menin and MLL1 at the MEIS1-locus and HOXA-cluster in PDX3.
Extended Data Figure 9: MEN1 mutations abrogate changes in gene expression signatures in MV4;11 cells upon revumenib treatment.

a) Geneset-enrichment analysis (GSEA) from revumenib (100nM, 1μM or 5μM) vs. DMSO treated MV4;11 cells harboring the MEN1-M327I mutation or -WT as control. Plotted are the False-discovery rate (FDR) q-values (y-axis) over the normalized enrichment scores (x-axis). Each dot represents a gene set. Relevant genesets covering MLL/HOX-related or myeloid differentiation associated terms were chosen for the analysis and selected terms are annotated. b) GSEA plots from revumenib (100nM, 1μM or 5μM) vs. DMSO treated MV4;11 cells harboring the MEN1-M327I mutation or -WT as control. GSEA was performed for MLL-fusion targets (Olsen et al., Mol. Cell, 2022) and the BROWN_MYELOID_CELL_CEVELOPMENT_UP geneset. Normalized enrichment scores and FDR q-values are indicated below each plot.
Extended Data Figure 10: MEN1 mutations blunt repression of key MLL-target genes upon revumenib treatment.

a) Bar graphs showing relative gene expression of MEIS1 and MEF2C in MEN1-M327I homozygous (left) and heterozygous (right) cells under treatment with a wide range of revumenib doses (mean +/− SD, n=3, each measured in triplicates) measured by quantitative real-time PCR using pre-validated Taqman® probes. b) a) Bar graphs showing relative gene expression of MEIS1 (left) and MEF2C (right) in MEN1-M327I and MEN1-T349M mutant cells under treatment with a wide range of revumenib doses (mean +/− SD, n=3, each measured in triplicates) measured by quantitative real-time PCR using pre-validated Taqman® probes. c) Bar graphs showing relative gene expression of MEIS1, PBX3 and HOXA7 in MEN1-T349M-mutant or WT PDX2 treated for 12 days with VTP-50469 (mean +/− SD, n=3, each measured in triplicates, replicates represent individual mice) measured by quantitative real-time PCR using pre-validated Taqman® probes. d) Graphical depiction of the percentage (%) of leukemic blasts in the peripheral blood and bone marrow of a patient that developed resistance without MEN1-mutations (or other somatic mutations detected by IMPACT-sequencing) during revumenib treatment on the AUGMENT-101 clinical trial. e) Cytology pictures (May-Grünwald/Giemsa staining) showing blast morphology of leukemia cells at screening and relapse under revumenib treatment of the same patient as shown in d). Representative cytology pictures from one individual patient sample. f) Volcano-plots showing gene expression changes in resistant leukemia cells from a patient (same as in d/e) and PDX-4 which developed non-genetic Menin-inhibitor resistance. Statistical determination of differentially expressed genes was performed using DESeq2.
Supplementary Material
Acknowledgements:
We thank the patients and families for their participation in this clinical trial. We thank Jennifer A. Perry for her support in banking and providing PDX material that was used in this study. We thank Omar Abdel-Wahab, Bobby L. Bowman, and Linde A. Miles for their critical review of the manuscript. We are thankful to the citizen-scientists of Folding@home for donating their computing resources for simulations. We thank Shipra Bijpuria and Brian McKeever for assisting with structure determination/PDB deposition and Joel Cassel for menin dissociation measurements. Illustrations in Fig. 3c and Extended data Fig. 3a have been created with BioRender.com. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748. S.F.C. was supported by a Scholar Award from the American Society of Hematology, a Momentum Fellowship Award from The Mark Foundation for Cancer Research, a Young Investigator Award from the Edward P. Evans Foundation, and a Career Development Award from the NCI K08 CA241371-01A1. S.A.A was supported by NIH grants CA176745, CA206963, CA204639, CA066996. S.A.A. and R.M.S. were supported by a SPORE grant in Myeloid Malignancies, P50CA206963. R.L.L. was supported by a Cycle For Survival Innovation Grant, National Cancer Institute R35 CA197594, National Cancer Institute R01 CA173636, a grant from the Samuel Waxman Cancer Research Foundation, and SCOR grants from the Leukemia and Lymphoma Society. R.L.L and S.A.A. were supported by an Alex’s Lemonade Stand Foundation Crazy 8 grant. F.P. was supported by the German Research Foundation (DFG, PE 3217/1-1), a Momentum Fellowship award by the Mark Foundation for Cancer Research and a research grant from the “Else Kröner-Fresenius-Stiftung” (2021-EKEA.111). C.M. was funded by a Momentum Fellowship award by the Mark Foundation for Cancer Research. J.D.C acknowledges support from NIH grant P30 CA008748 and NIH grant R01 GM121505. J.D.C., A.V., D.S., and S.S. acknowledge funding from the Stiftung Charité, the BIH Einstein Foundation, MSKCC, NIH grant R01GM121505, and Bayer. E.S.F. was supported by NIH grants CA214608 and CA066966. J.A.C. is supported by Ruth L. Kirschstein Postdoctoral Individual National Research Service Award (NIH F32CA250240-02). W.X. was supported by Alex’s Lemonade Stand Foundation and the Runx1 Research Program, the Cycle for Survival’s Equinox Innovation Award in Rare Cancers, MSK Leukemia SPORE Career Enhancement Program. and a career development award from the NCI K08CA267058. H. R. was supported by a Fellow award from The Leukemia & Lymphoma Society. Sukrit Singh (S.S.) is a Damon Runyon Quantitative Biology Fellow supported by the Damon Runyon Cancer Research Foundation DRQ-14-22.
Competing interests:
S.F.C. is a consultant for and holds equity interest in Imago Biosciences. J.G.D. consults for Microsoft Research, Servier, Abata Therapeutics, Maze Therapeutics, BioNTech, and Pfizer; JGD consults for and has equity in Tango Therapeutics. J.G.D. receives support via the Functional Genomics Consortium (Merck, Abbvie, Janssen, Vir, and Bristol Meyers Squibb). R.L.L. is on the supervisory board of Qiagen and is a scientific advisor to Loxo, Imago, C4 Therapeutics and Isoplexis. He receives research support from and consulted for Celgene and Roche, research support from Prelude Therapeutics, and has consulted for Novartis and Gilead. He has received honoraria from Lilly and Amgen for invited lectures. E.M.S. receives research support to his institution from Agios, Amgen, Astellas, Bayer, Biotheryx, Bristol Myers Squibb, Eisai Foghorn, Servier, Syndax, Syros and consulting fees from Novartis, PinotBio, Janssen, Bristol Myers Squibb, Agios, Jazz, Menarini, Genentech, Genesis, Abbvie, Neoleukin, Gilead, Syndax, OnCusp, CTI Biopharma, Foghorn, Servier, Calithera, Daiichi, Aptose, Syros, Astellas, Ono Pharma, Blueprint, Kura. Epizyme and Cellectis. E.M.S. also holds equity interest in Auron Therapeutics.
S.A.A. has been a consultant and/or shareholder for Vitae/Allergan Pharmaceuticals, Neomorph, Inc., Imago Biosciences, Cyteir Therapeutics, C4 Therapeutics, and Accent Therapeutics. S.A.A. has received research support from Janssen, and Syndax and is an inventor on a patent related to Menin Inhibition WO/2017/132398A1. J.D.C. is a current member of the Scientific Advisory Boards of OpenEye Scientific Software, Interline Therapeutics, and Redesign Science. The Chodera laboratory receives or has received funding from the National Institute of Health, the National Science Foundation, the Parker Institute for Cancer Immunotherapy, Relay Therapeutics, Entasis Therapeutics, Silicon Therapeutics, EMD Serono (Merck KGaA), AstraZeneca, Vir Biotechnology, XtalPi, Interline Therapeutics, and the Molecular Sciences Software Institute, the Starr Cancer Consortium, the Open Force Field Consortium, Cycle for Survival, a Louis V. Gerstner Young Investigator Award, and the Sloan Kettering Institute. A complete funding history for the Chodera lab can be found at http://choderalab.org/funding. W.X. has received research support from Stemline Therapeutics. A.J.S.’s spouse is an employee of Bristol-Myers Squibb. The remaining authors declare no competing interests. E.S.F. is a founder, member of the scientific advisory board (SAB), and equity holder of Civetta Therapeutics, Jengu Therapeutics, Proximity Therapeutics, and Neomorph Inc (board member), SAB member and equity holder in Avilar Therapeutics and Photys Therapeutics, and a consultant to Astellas, Sanofi, Novartis, Deerfield and EcoR1 capital. The Fischer laboratory receives or has received research funding from Novartis, Deerfield, Ajax, Interline, and Astellas. R.M.S. has received advisory or consulting fees from Abbvie, Actinium, Agios, Arog, Astellas, Biolinerx, Celgene, Daiichi-Sankyo, Elevate, Gemoab, Janssen, Jazz, Macrogenics, Novartis, OncoNova, Syndax, Syntrix, Syros, Takeda, Trovagene, BergenBio, Foghorn Therapeutics, GSK, Aprea, Innate, Amgen, CTI Pharmaceuticals, BMS and Boston Pharmaceuticals.
Data availability
All raw and processed sequencing data are accessible via the NCBI Gene-Expression Omnibus (GEO) under the accession number: GSE196037 (ChIPseq: GSE196036; RNAseq: GSE196035). X-ray crystal structures are publicly available via the Protein Data Bank (PDB: 8E90, 7UJ4, 4GQ6). For entry 8E90, the web address is https://www.wwpdb.org/pdb?id=pdb_00008e90. For entry 7UJ4, the web address is https://www.wwpdb.org/pdb?id=pdb_00007uj4. For entry 4GQ6, a structure previously resolved by Shi et al.22, the web address is https://www.wwpdb.org/pdb?id=pdb_0004gq6. All storage intensive files (ie. MSM structures, transition matrices, weights, strided trajectories, etc.) can be found on OSF (https://osf.io/uge5j/). The complete dataset of trajectories (450 GB total storage needed) is available upon request; due to data size restrictions, external hard drives will be shipped to fulfill these data requests.
References
- 1.Rodrigues CP, Shvedunova M & Akhtar A Epigenetic Regulators as the Gatekeepers of Hematopoiesis. Trends Genet (2020). 10.1016/j.tig.2020.09.015 [DOI] [PubMed] [Google Scholar]
- 2.Uckelmann HJ & Armstrong SA Chromatin Complexes Maintain Self-Renewal of Myeloid Progenitors in AML: Opportunities for Therapeutic Intervention. Stem cell reports 15, 6–12 (2020). 10.1016/j.stemcr.2020.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yokoyama A et al. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 123, 207–218 (2005). 10.1016/j.cell.2005.09.025 [DOI] [PubMed] [Google Scholar]
- 4.Yokoyama A & Cleary ML Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer cell 14, 36–46 (2008). 10.1016/j.ccr.2008.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuhn MW et al. Targeting Chromatin Regulators Inhibits Leukemogenic Gene Expression in NPM1 Mutant Leukemia. Cancer discovery 6, 1166–1181 (2016). 10.1158/2159-8290.CD-16-0237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stein EM et al. in American Society of Hematology Annual Meeting & Exposition 2021 (Atlanta, GA, 2021). [Google Scholar]
- 7.Huang J et al. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 482, 542–546 (2012). 10.1038/nature10806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hughes CM et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Molecular cell 13, 587–597 (2004). 10.1016/s1097-2765(04)00081-4 [DOI] [PubMed] [Google Scholar]
- 9.Borkin D et al. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer cell 27, 589–602 (2015). 10.1016/j.ccell.2015.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klossowski S et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. The Journal of clinical investigation 130, 981–997 (2020). 10.1172/JCI129126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lei H et al. Recent Progress of Small Molecule Menin-MLL Interaction Inhibitors as Therapeutic Agents for Acute Leukemia. Journal of medicinal chemistry 64, 15519–15533 (2021). 10.1021/acs.jmedchem.1c00872 [DOI] [PubMed] [Google Scholar]
- 12.Perner F & Armstrong SA Targeting Chromatin Complexes in Myeloid Malignancies and Beyond: From Basic Mechanisms to Clinical Innovation. Cells 9 (2020). 10.3390/cells9122721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uckelmann HJ et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 367, 586–590 (2020). 10.1126/science.aax5863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krivtsov AV et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer cell 36, 660–673 e611 (2019). 10.1016/j.ccell.2019.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemos MC & Thakker RV Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 29, 22–32 (2008). 10.1002/humu.20605 [DOI] [PubMed] [Google Scholar]
- 16.Hanna RE et al. Massively parallel assessment of human variants with base editor screens. Cell 184, 1064–1080 e1020 (2021). 10.1016/j.cell.2021.01.012 [DOI] [PubMed] [Google Scholar]
- 17.Zimmerman MI et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nature Chemistry 13, 651–659 (2021). 10.1038/s41557-021-00707-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward MD et al. Deep learning the structural determinants of protein biochemical properties by comparing structural ensembles with DiffNets. Nature Communications 12, 3023 (2021). 10.1038/s41467-021-23246-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun X, Singh S, Blumer KJ & Bowman GR Simulation of spontaneous G protein activation reveals a new intermediate driving GDP unbinding. eLife 7 (2018). 10.7554/eLife.38465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grembecka J, Belcher AM, Hartley T & Cierpicki T Molecular basis of the mixed lineage leukemia-menin interaction: implications for targeting mixed lineage leukemias. The Journal of biological chemistry 285, 40690–40698 (2010). 10.1074/jbc.M110.172783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai H et al. Menin-MLL protein-protein interaction inhibitors: a patent review (2014-2021). Expert Opin Ther Pat 32, 507–522 (2022). 10.1080/13543776.2022.2045947 [DOI] [PubMed] [Google Scholar]
- 22.Shi A et al. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 120, 4461–4469 (2012). 10.1182/blood-2012-05-429274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ross DS et al. Immunohistochemical analysis of estrogen receptor in breast cancer with ESR1 mutations detected by hybrid capture-based next-generation sequencing. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 32, 81–87 (2019). 10.1038/s41379-018-0116-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biancaniello C et al. Investigating the Effects of Amino Acid Variations in Human Menin. Molecules 27 (2022). 10.3390/molecules27051747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikolovska-Coleska Z et al. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem 332, 261–273 (2004). 10.1016/j.ab.2004.05.055 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed sequencing data are accessible via the NCBI Gene-Expression Omnibus (GEO) under the accession number: GSE196037 (ChIPseq: GSE196036; RNAseq: GSE196035). X-ray crystal structures are publicly available via the Protein Data Bank (PDB: 8E90, 7UJ4, 4GQ6). For entry 8E90, the web address is https://www.wwpdb.org/pdb?id=pdb_00008e90. For entry 7UJ4, the web address is https://www.wwpdb.org/pdb?id=pdb_00007uj4. For entry 4GQ6, a structure previously resolved by Shi et al.22, the web address is https://www.wwpdb.org/pdb?id=pdb_0004gq6. All storage intensive files (ie. MSM structures, transition matrices, weights, strided trajectories, etc.) can be found on OSF (https://osf.io/uge5j/). The complete dataset of trajectories (450 GB total storage needed) is available upon request; due to data size restrictions, external hard drives will be shipped to fulfill these data requests.