

Abstract
Background
Abdominal aortic aneurysm (AAA) is a vascular disease with a mortality rate of >80% if ruptured. Mitochondrial dysfunction has been previously implicated in AAA pathogenesis. In this study, we aimed to characterize the mitochondrial genetic landscape in AAA.
Methods and Results
Whole mitochondrial genome sequencing and bioinformatics analysis were performed in comorbidity matched 48 cases without AAA and 48 cases with AAA, objectively diagnosed, and selected from a cohort of 65‐year‐old men recruited for a screening program. We identified differential mutational landscapes in men with and without AAA, with errors in mitochondrial DNA replication or repair as potential sources. Heteroplasmic insertions and overall heteroplasmy of structural rearrangements were significantly elevated in AAA cases. Three heteroplasmic variants were associated with risk factors of AAA: leukocyte concentration, plasma glucose, and cholesterol levels, respectively. Interestingly, mutations were more prevalent in regulatory part of the mitochondria, the displacement loop region, in AAA as compared with controls (P value <0.05), especially in the conserved and critical mitochondrial extended termination‐associated sequence region. Moreover, we report a novel 24 bp mitochondrial DNA duplication present exclusively in cases with AAA (4%) and 75% of the unmatched AAA biopsies. Finally, the haplogroup cluster JTU was overrepresented in AAA and significantly associated with a positive family history of AAA (odds ratio, 2.9 [95% CI, 1.1–8.1]).
Conclusions
This is the first study investigating the mitochondrial genome in AAA, where important genetic alterations and haplogroups associated with AAA and clinical risk factors were identified. Our findings have the potential to fill in gaps in the missing genetic information on AAA.
Keywords: aortic aneurysm, abdominal, computational biology, DNA, mitochondrial, genome, mitochondrial, heteroplasmy, organelle biogenesis, risk factors
Subject Categories: Genetics, Aneurysm, Vascular Disease
Nonstandard Abbreviations and Acronyms
- D‐loop
displacement loop
- MT‐TAS2
mitochondrial extended termination‐associated sequence
- mtDel
mitochondrial DNA deletion
- mtDNA
mitochondrial DNA
- mtDNA‐CN
mitochondrial DNA copy number
- mtDup
mitochondrial DNA duplication
Clinical Perspective.
What Is New?
We report, for the first time, differential mitochondrial mutational landscapes in men with and without abdominal aortic aneurysm (AAA), potentially induced by defects in DNA replication/repair rather than oxidative stress.
AAA cases had more mutations in the mitochondrial displacement loop, especially in the conserved and critical mitochondrial extended termination‐associated sequence (MT‐TAS2) region, and low mitochondrial copy number may exacerbate the pathological consequences of mutations in this region.
We report a novel 24 bp mtDNA duplication present exclusively in AAA cases (4%) and a majority of the unmatched AAA biopsies.
What Are the Clinical Implications?
Our findings indicate the potential of mitochondrial genome variation to provide information on the missing heritability of AAA and implicates defects in mtDNA replication/repair as a potential future clinical target.
MT‐TAS2 mutation status has the potential to be a biomarker of AAA, and increasing the mitochondrial copy number could have a therapeutic potential, especially in individuals with MT‐TAS2 region mutation(s).
Overall, our study encourages future large‐scale and comprehensive studies on mitochondrial genomic variation in chronic and age‐related diseases that may introduce innovative clinical implications.
Abdominal aortic aneurysm (AAA) is a progressive vascular disease with an irreversible, localized dilation of the infrarenal aorta (aortic diameter ≥30 mm). 1 AAA predominantly occurs in men >65 years of age, with a prevalence between 1.5% and 1.8%. 2 There is no specific pharmacotherapy to prevent or limit the growth of AAA; however, secondary preventive measures are undertaken to control some modifiable risk factors of AAA. 1 , 2 Some known major risk factors of AAA are smoking, male sex, old age, and positive family history. 1 , 2 , 3 Various biological processes have been implicated in the formation and progression of AAA, such as chronic inflammation, extracellular matrix degradation, increased apoptosis, and oxidative stress. 3 , 4
Mitochondrion is an essential cellular organelle that has essential roles in cellular energy production, respiration, regulation of reactive oxygen species balance (oxidative stress), and apoptosis. 5 Mitochondria carry their own genome and, unlike the nuclear genome with 2 fixed copies, can carry multiple copies of their 16.5 kb, circular DNA per cell. 6 The mitochondrial genome consists of a total of 37 genes, out of which 13 code for the protein components involved in the oxidative phosphorylation process. The displacement loop (D‐loop) is a region in the mitochondrial genome that contains regulatory elements like promoter and replication origins. 6
Given its essential roles in different homeostatic processes, mitochondrial dysfunction has been implicated in various pathological conditions. 7 Accumulation of pathogenic mutations in the mitochondrial genome can lead to mitochondrial dysfunction and various disease phenotypes. 8 Given the polyploid nature of the mitochondrial DNA (mtDNA), mutations can exist as either heteroplasmy (containing a mixture of both wild‐type and mutant mtDNA) or homoplasmy (either wild‐type or mutant mtDNA). 6 Oftentimes, severe mutations are heteroplasmic, and clinical presentations occur when the heteroplasmic mutation fraction reaches a certain threshold, depending on, for example, the tissue and age. 8 As mtDNA mainly follows a uniparental (maternal) mode of transmission, individual ancestry or haplogroups (major branch points on the mitochondrial phylogenetic tree) can be traced back using mtDNA mutation profiles. Haplogroups have been independently associated with multiple mitochondrial characteristics and diseases. 9 , 10
There is an increasing body of evidence implicating mitochondrial dysfunction in AAA pathophysiology. 11 Previous studies have reported increased mitochondrial reactive oxygen species, apoptosis, dysregulation of mitochondrial biogenesis, mitophagy, and aberrant mitochondrial dynamics in AAA pathogenesis. 11 , 12 , 13 However, no study so far has investigated the role of mitochondrial dysfunction (which is associated with accumulation of mutation in its genome) in AAA, although AAA has been suggested to have a substantial heritability, as shown by multiple studies. 14 , 15 In spite of continuous efforts, a major chunk of the specific hereditary component associated with AAA is yet to be uncovered. We hypothesized that investigating mitochondrial genetics in AAA could therefore fill in gaps in its missing heritability. Therefore, in the present case–control study, we aimed to characterize the mitochondrial genome in AAA by investigating the association between AAA (including its clinical risk factors) and mtDNA mutations (mutational signatures/context, haplogroups) through a comprehensive next‐generation sequencing analysis.
METHODS
The data generated during the present study are anonymized and submitted as Data S1 (list of complete mtDNA variants passing the quality control) and Data S2 (results from MitoSAlt analysis for deletions and duplications). Because of ethical considerations, individual clinical data cannot be disclosed. A detailed description of methods is provided in Data S3. The analysis code needed to reproduce the results in the article will be shared upon reasonable request to the corresponding author.
Study Population and Outcome Measurement
All practices were in accordance with the Declaration of Helsinki. The previous ethics committee in Lund approved the study plan (2010/239 and later amendments). The main cohort is a part of an AAA screening program where 65‐year‐old men from Malmö city and 15 neighboring municipalities in Sweden were invited for ultrasound screening. Participation of individuals diagnosed with AAA (aortic diameter of ≥30 mm) and individuals without AAA (aortic diameter <30 mm) was based on written informed consent. All participants were male and aged 65 years at the time of sampling. This hypothesis‐generating study follows a case–control design where 48 individuals with AAA were selected (including all individuals with positive family history of AAA). A number of cases were included based on a previous study investigating mitochondrial genetics in peripheral artery disease. Forty‐eight controls were then matched based on cardiovascular diseases and diabetes. Cardiovascular disease was defined as occurrences of stroke, myocardial infarction, heart failure, angina, atrial fibrillation, deep vein thrombosis, and pulmonary embolism. Of note, 2 additional individuals with diabetes were later identified in the study population. Blood sampling was done only at baseline screening. Eight postoperative, snap‐frozen AAA biopsies and 8 serum samples from the same individuals have also been collected. The larger cohort and the clinical and laboratory variables investigated and recorded in the study are explained in greater detail in a previous publication. 16 As a marker for oxidative stress, we included myeloperoxidase plasma protein levels that had been determined using a Proseek Multiplex CVD III96x96 (Olink Biosciences) panel in the baseline samples from the study population in a previous study from our group. 17
MtDNA Sequencing and Quantification
DNA was extracted from whole blood, biopsies, and serum samples (Data S3). For the specific amplification of the whole mitochondrial genome (16.5 kb), we employed long‐range polymerase chain reaction resulting in 2 overlapping amplicons, followed by sequencing library preparation using Nextera DNA flex kit and sequencing run on the iSeq100 system (Illumina, CA). Primer sequences are mentioned in Data S3. The workflow is based on the Human mtDNA Genome guide for the Illumina Sequencing Platform. The purified libraries' concentrations were determined using Qubit 4.0—1X ds DNA high sensitivity assay guidelines (Invitrogen), and library size (500–1000 bp) was verified on Experion (Bio‐Rad, Hercules, CA). The final library pool was diluted to 100 pmol/L (with 2% internal control, phiX) and was loaded into the iSeq100 system. Finally, dual‐index, paired‐end sequencing run was performed with a read length of 151 bp. Mitochondrial copy number (mtDNA‐CN) quantification protocol is mentioned in Data S3.
Variant Calling and mtDNA Structural Changes Detection
Following successful sequencing run, raw sequencing data (Fastq) were obtained, initial quality control was performed using FastQC, 18 and compiled report was generated using MultiQC v1.8. 19 Illumina sequencing adapters were removed by Trim Galore v0.6.5. 20 An open‐source bioinformatics pipeline, MToolBox‐v.1.0, 21 was adapted to analyze the mtDNA sequences using the revised Cambridge Reference Sequence (NCBI Reference Sequence: NC_012920.1) for alignment and variant calling. Using the pipeline, we additionally performed sequence realignment around known insertion/deletion sites, duplicate sequences marking, and base quality scores recalibration. Then, variant call format files, containing all variants identified in each sample were created. The further quality control and variant refinement criteria are further described in Data S3. To determine structural changes in the mtDNA (deletions: mtDel and duplications: mtDup), the MitoSAlt package was used. 22 Filtering criteria of Nie et al was applied. 23
Variant Annotations and mtDNA Haplogroup Assignment
The final merged variant call format file was annotated using R packages AnnotationDbi (v.1.54.1) 24 and GenomicFeatures (v.1.44.2). 25 Mutational signatures were assessed using SomaticSignatures. 26 The analytical framework, SigProfilerExtractor, was employed for de novo extraction of trinucleotide mutational signatures with known/unknown cause. 27 MtDNA haplogroups were determined and assigned using HaploGrep2. 28
Statistical Analysis
Statistical analyses were performed using R studio 29 and IBM SPSS statistics software version 27 (IBM, NY). The Wilcoxon‐Mann–Whitney test with exact distribution from the R package for permutation tests, coin 30 was used for analyzing differences in continuous variables, and Fisher's exact test was used for categorical variables. Quantitative variables are presented with mean±SD or median (25th and 75th percentiles). Categorical variables are stated as numbers and percentages. To infer the difference in mtDNA mutation frequency between groups with and without at different mitochondrial regions and contexts, Fisher's exact test was employed. Using Firth's small size correction, univariate logistic regression analysis was performed to calculate the odds ratios (OR), and multivariate logistic regression analysis was used to adjust the data for potential confounders/propensity score. Putative confounding covariates were selected based on their previously shown associations with both mtDNA variation and AAA outcome. For instance, smoking, cancer, high‐density lipoprotein, and blood cell composition (leukocyte/thrombocyte count) have been previously associated with both AAA and mtDNA changes. 31 , 32 , 33 We extracted the propensity score for 1 of our main outcomes, mitochondrial extended termination‐associated sequence (MT‐TAS2) mutation status, by accounting for smoking pack‐year, diabetes, cancer, plasma high‐density lipoprotein, leukocyte and thrombocyte count, plasma creatinine, plasma triglyceride, and family history of AAA. Of note, 2 additional individuals with diabetes were later identified in the study population and, hence, diabetes was adjusted for in the statistical analyses. To test the association between quantitative traits from the study cohort, with mtDNA variants, a linear regression method with permutation using the R package for linear models lmPerm, 34 controlling false discovery rate (Benjamini–Hochberg) was used. For analyzing significance of the difference/interaction between continuous variables with respect to 2 factor variables, 2‐way ANOVA, following linear regression with permutation from R package lmPerm was implemented. In the present study, an unadjusted P value of <0.05 was considered statistically significant. When multiple comparisons were made, the family‐wise error rate or false discovery rate was controlled using either the Bonferroni or Benjamini–Hochberg adjusted P value <0.05, respectively, chosen based on the availability in the applied R functions. The choice of correction method was noted for every analysis.
RESULTS
Mitochondrial DNA Mutation Summary in Controls Without AAA and Cases With AAA
The baseline population characteristics are presented in Table S1. The average sequencing coverage of the mitochondrial genome in the 96 men was ~1168X, allowing reliable identification of mtDNA heteroplasmy ≥1% minor allele frequency. Following the quality control steps (Data S3), we identified 163 heteroplasmic variants at 137 unique positions, 867 homoplasmic variants at 332 unique positions, and 206 insertions/deletions at 19 unique sites. The population frequency (GeneBank Freq) of AAA‐associated mtDNA variants was slightly lower than the non‐AAA‐associated variants; however, the association was not statistically significant (Figure S1). A complete list of mutations after quality control can be found in Data S1.
Differential Mutational Landscape in AAA
We observed a differential mutation landscape across the mtDNA loci between men with and without AAA (Figure 1A and Figure S2). Cases with AAA had significantly more T>C homoplasmic transitions (P value=0.022, Figure 1E) and significantly less T>A heteroplasmic transversions (P value=0.019, Figure 1F). Upon comparison with men without AAA, cases with AAA carried higher homoplasmic mutation frequency in the gene‐coding component of mtDNA (P value=0.010, Figure 1C). On the other hand, heteroplasmic mutations were significantly more frequent in the regulatory D‐loop region of the mitochondrial genomes of AAA cases (P value=0.008, Figure 1D). The heteroplasmic mtDNA insertions were present at significantly higher frequency in AAA cases (P value <0.001, Figure 1B), and the insertion/deletions were exclusively heteroplasmic. We identified 2 known trinucleotide‐mutational signatures attributed to aging (clock‐like) and transcription‐coupled nucleotide excision repair (Table S2). Moreover, we observed 4 distinct associations between heteroplasmic mtDNA variants and quantitative traits known to be associated with AAA (false discovery rate <0.05; Table S3).
Figure 1. Differential mtDNA mutational landscape in AAA.
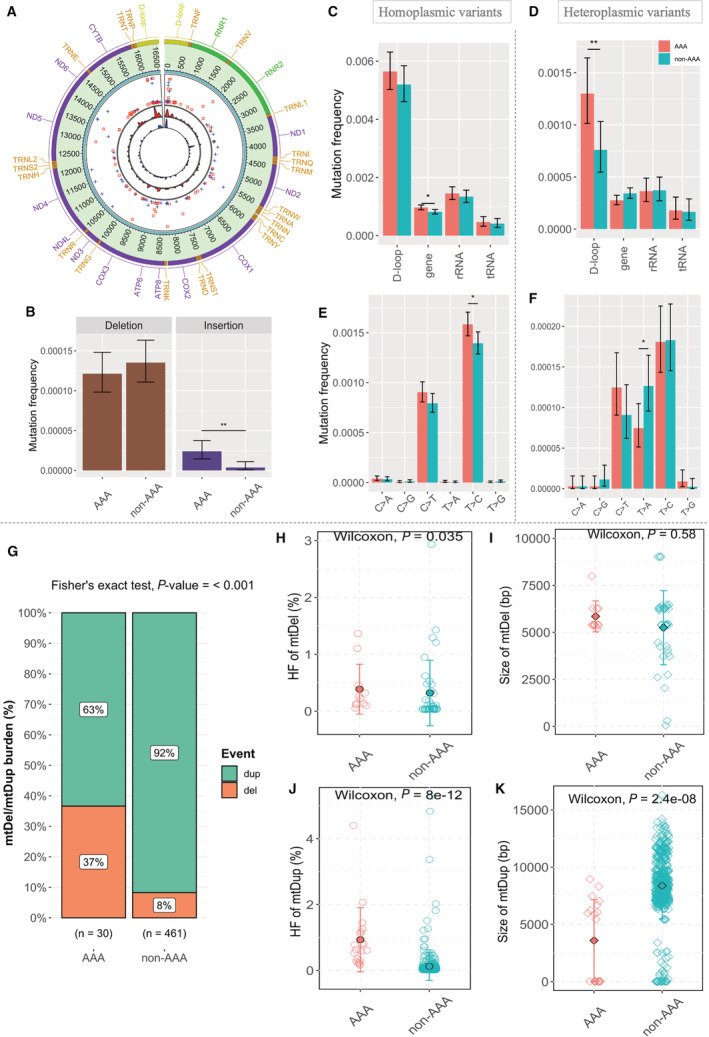
A, Overall distribution of heteroplasmic variants in the mitochondrial genomes of men with and without AAA. From the outermost track, (1) Genes on mitochondrial genome, colored by the type as yellow, D‐loop; purple, coding genes; green, rRNAs; orange, tRNAs. (2) Genomic position on the human mitochondrial genome. (3) The distribution of the heteroplasmic variants (the radial axis corresponds to the degree of heteroplasmic fraction going down from 100% to 0%). Red squares indicate cases with AAA, and blue pluses are controls. (4) The density of variants. To make the 2 tracks comparable, the density was adjusted to reflect the difference in the number of variants of the 2 groups: Red—AAA, Blue—non‐AAA. B through F, The bar charts compare mtDNA mutation frequency between cases with AAA and controls: (B) for INDELs, (C and D) in different mtDNA regions: D‐loop (1122 bp), protein‐coding (11 341 bp), tRNA (1504 bp), and rRNA (2513 bp), and (E and F) for single nucleotide substitution types. Mutation frequency was computed as the number of mutations (B–D) in each region divided by the product of the length of the region in mtDNA genome and the number of subjects (E and F) at each site divided by the product of the number of appearances of reference alleles in mtDNA genome and the number of subjects. Error bars indicate the 95% CIs of the mutation frequency assuming Poisson distribution. Statistical significance for the difference in mutation frequency between AAA cases and controls was marked with asterisks (**P value <0.01, *P value <0.05 by Fisher's exact test). G through K, MtDNA structural changes in cases with AAA and controls. G, Overall mtDel/mtDup burden shown as percentage in cases with AAA and controls. Total counts of mtDel/mtDup events for each group are mentioned at the x axis. H and J, HF of mtDel (top) and mtDup (bottom) in cases with AAA and controls. The black outlined dots denote mean HF in each group. I and K, Size in base pairs (bp) of mtDel (top) and mtDup (bottom) in cases with AAA and controls. The black outlined rhombi denote mean mtDel/mtDup size in each group. Red: cases with AAA; Green: controls. P=2‐sided P value determined by Wilcoxon‐Mann–Whitney test with exact distribution. AAA indicates abdominal aortic aneurysm; D‐loop, displacement loop; HF, heteroplasmic fraction; INDEL, insertion and deletion; mtDel, mitochondrial DNA deletion; mtDNA, mitochondrial DNA; mtDup, mitochondrial DNA duplication; RNA, ribosomal RNA; and tRNA, transfer RNA.
MtDNA Structural Changes in Men With and Without AAA
We observed an overall increased burden of mtDel/mtDup in men without AAA as compared with cases with AAA (30 versus 461 events) (Figure 1G, P value <0.001 by Fisher's exact test). However, a majority of the mtDels and mtDups in the control group had very low‐level heteroplasmy, whereas the mtDel and mtDup heteroplasmy levels were significantly elevated in cases with AAA (P values=0.035 and 8.0E−12 respectively) (Figure 1H and 1J). AAA cases had smaller mtDup sizes (bp) compared with the control group (P value=2.4E−08; Figure 1K).We observed a similar mtDel/Dup: burden, heteroplasmy and size distribution in 8 AAA biopsies and matched serum samples, as the AAA cases (Figure S3). Furthermore, we identified a novel 24 bp duplication in the D‐loop region (16401–16 424) present in 2 out of the 48 AAA cases but present in 6 (75%) out of 8 of the unmatched AAA biopsies. A complete list of mtDNA structural changes detected can be found in Data S2.
MtDNA Mutations in the D‐Loop Region
Distribution and Context
As AAA cases had a significantly greater frequency of mtDNA variants (including single nucleotide variants and insertion/deletion) in the regulatory D‐loop region (Figure 1D), we further investigated the mutation patterns in the D‐loop. To get an insight into important region(s) in the D‐loop that may have a differential heteroplasmic burden than expected by chance, we performed separate and independent tests in the groups with and without AAA. In cases with AAA, out of the 17 subregions in the D‐loop (Figure 2A, bottom, purple and yellow horizontal bars), the number of heteroplasmic variants in 1 region (MT‐TAS2) was significantly higher than in the remainder of the D‐loop as analyzed by Fisher's exact test (with Bonferroni correction) per subregion (OR, 4.7; P value=0.0001, adjusted P value=0.0003). No such D‐loop region with increased heteroplasmic burden was observed in the group without AAA (OR, 1.1; P value=0.756, adjusted P value=1).
Figure 2. MtDNA mutations in D‐loop: distribution and context.
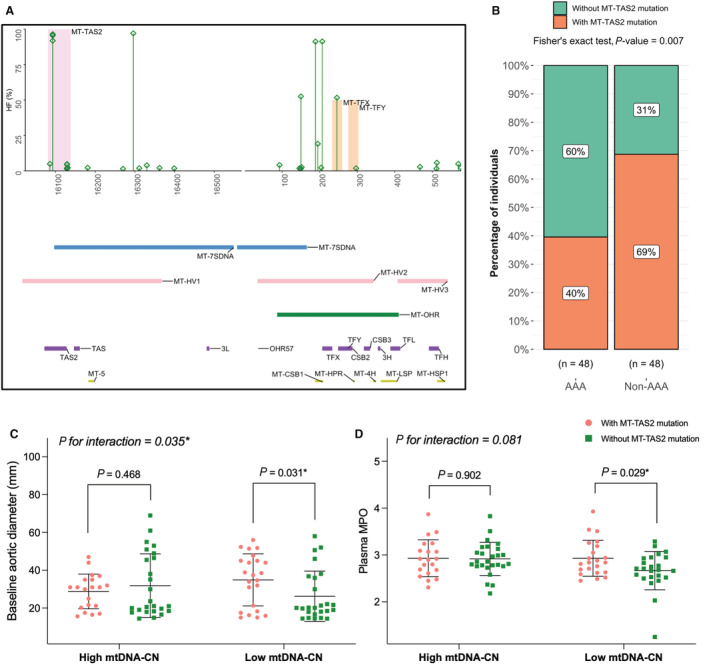
A, HFs observed in mtDNA sequences of cases with AAA in the D‐loop region. MT‐TAS2 is shadowed in light pink (OR, 4.7; adjusted P value=0.0003 by Fisher's exact test with Bonferroni correction). MT‐TFX and MT‐TFY are shadowed in light orange. The bottom panel shows horizontal bars, representing known subregions of the mitochondrial D‐loop region. B through D, Potential association(s) of MT‐TAS2 mutation status. (B) Distribution of individuals with and without MT‐TAS2 mutation(s) in AAA (left bar) and individuals without AAA (right bar), indicated as percentages. C and D, Mean comparisons of baseline aortic diameters (C) and MPO plasma levels (D) in individuals with and without MT‐TAS2 mutation(s), subgrouped based median mtDNA‐CN levels (low and high). The black horizontal line denote mean value in each group. Error bars denote SD. P=P value determined by linear model with permutation test and P for interaction (between MT‐TAS2 mutation status and mtDNA‐CN) is determined using 2‐way ANOVA with permutation. AAA indicates abdominal aortic aneurysm; D‐loop, displacement loop; HF, heteroplasmic fraction; MPO, myeloperoxidase; mtDNA, mitochondrial DNA; mtDNA‐CN, mitochondrial DNA copy number; MT‐CSB1/2/3, mitochondrial conserved sequence block 1/2/3; MT‐HPR, mitochondrial replication primer; MT‐HSP1, mitochondrial major H‐strand promoter; MT‐HV1/2/3, mitochondrial hypervariable segments 1/2/3; MT‐LSP, mitochondrial L‐strand promoter; MT‐OHR, mitochondrial H‐strand origins; MT‐OHR57, mitochondrial H‐strand origin; MT‐TAS2, mitochondrial extended termination‐associated sequence; MT‐TF(s), mitochondrial transcriptional factor 1 binding sites; MT‐TFX, mitochondrially encoded transcription factor binding site X; MT‐TFY, mitochondrially encoded transcription factor binding site Y; MT‐3H, mt3 H‐strand control element; MT‐3L, mitochondrial L‐strand control element; MT‐4H, mt4 H‐strand control element; and MT‐7SDNA, mitochondrial 7S DNA.
Potential Functional Association(s) of MT‐TAS2 Region Mutation(s)
Based on the results, MT‐TAS2 was selected as a region of interest for further analysis. Mutations in the MT‐TAS2 region mainly occurred once in an individual and rarely twice. Comparing individuals with or without MT‐TAS2 mutations (MT‐TAS2 mutation status) showed that the proportion of individuals harboring mutation(s) in the MT‐TAS2 region was significantly higher in those with AAA as compared with those without AAA (Figure 2B, P value=0.007 by Fisher's exact test). The association between MT‐TAS2 mutation status and AAA remained significant after adjusting for potential confounders and propensity score (Tables S4 and S5). We found slight trends but no statistically significant association between individuals with MT‐TAS2 mutation(s), marker of mitochondrial function (mtDNA‐CN), baseline aortic diameter, and oxidative stress marker (myeloperoxidase) levels in the whole study population (Figure S4A through S4C). However, subgroup analysis based on median mtDNA‐CN cutoff revealed that the presence of mutation(s) in the MT‐TAS2 region is associated with significantly larger baseline aortic diameter (P=0.031, P for interaction (MT‐TAS2 mutation status and mtDNA‐CN) = 0.035) and higher plasma myeloperoxidase levels (P=0.029, P for interaction [MT‐TAS2 mutation status and mtDNA‐CN] = 0.081) in individuals with low mtDNA‐CN (Figure 2C and 2D and Table S6). Of note, we did not see significant associations between mtDNA copy number and AAA disease status (Figure S5).
Haplogroup Distribution in AAA and Family History of AAA
Haplogroups represent inherited mitochondrial genetic background of an individual. Based on the distribution pattern we observed in men with and without AAA and phylogenic relatedness, we combined JT and U haplogroups and performed comparisons against the background and common European HV haplogroup cluster (Figure S6). Men belonging to the JTU haplogroup cluster had higher odds for positive family history of AAA, as compared with men with haplogroup HV (Figure S6B, OR, 2.9 [95% CI, 1.1–8.1]). Diastolic blood pressure (P value=0.007) at baseline was increased in men belonging to the JTU haplogroup cluster compared with HV (Table S7). The association between diastolic blood pressure and JTU haplogroup cluster was independent of antihypertension medication use (OR, 1.2 [95% CI, 1.0–1.2]).
DISCUSSION
There is already an increasing body of evidence implicating mitochondrial dysfunction in AAA. 11 However, no study to date has examined mitochondrial dysfunction in terms of the mtDNA mutational profile in AAA. In the current study, we compared the mitochondrial genetic profiles in age and comorbidity matched men with and without AAA. We found that individuals with AAA have a distinct mtDNA mutation landscape as compared with individuals without AAA.
According to previous literature, mtDNA transitions (T:C>C:T) are often explained by errors/defects in replication, whereas oxidative damage is implicated in transversion mutations (G:C>T:A). 35 We observed greater frequency of homoplasmic transitions in AAA and a lesser frequency of heteroplasmic transversions in AAA (Figure 1E and 1F). In addition, we found 2 known trinucleotide mutational signatures that were associated with aging (clock‐like) and transcription‐coupled nucleotide excision repair in both individuals with and without AAA. This suggests that the main mechanism for accumulation of mutations in AAA may involve the replication or repair defects rather than reactive oxygen species mediated mtDNA damage, which is in line with recent studies that provide evidence against the free‐radical theory of aging and mtDNA damage. 36 We also found significant associations between heteroplasmic mtDNA variants and risk factors of AAA (Table S3). More large‐scale studies are needed to confirm whether mtDNA variants mediate the association(s) between AAA and its risk factors.
D‐loop is a regulatory region that carries essential promoters and origins for normal mtDNA replication. Moreover, heteroplasmic mutations are often associated with pathological state as homoplasmic mutations at essential sites are not tolerated during selection. 37 That could in part explain the higher heteroplasmic mutation frequency in the D‐loop region in men with AAA as compared with those without (Figure 1D). Interestingly, we found that heteroplasmic variants were significantly enriched in the MT‐TAS2 compared with the rest of the D‐loop region, in cases with (Figure 2A). Presence of mutation(s) in the conserved MT‐TAS2 D‐loop subregion was significantly associated with AAA (Figure 2B). MT‐TAS2 region corresponds with a replication fork barrier that promotes mitochondrial genomic stability, 38 and mutations in this region can possibly cause genomic instability in cases with AAA. We chose this subregion for further investigation into its potential association with baseline aortic diameter, mtDNA‐CN (surrogate marker of mitochondrial function), and plasma myeloperoxidase (oxidative stress marker). 39 , 40 Interestingly, we observed that the positive association between MT‐TAS2 mutation status, baseline aortic diameter, and the oxidative stress marker myeloperoxidase was present only in individuals with low mtDNA‐CN (Figure 2C and 2D and Table S6). This observation suggests that having a low mtDNA‐CN at baseline might exacerbate the impact of MT‐TAS2 conserved region mutation in AAA pathology. This observation is in line with a study by Filograna and colleagues, where they showed that improving the absolute mtDNA‐CN can decrease the pathological effect of mtDNA mutations in mice and that a decrease in mtDNA‐CN can worsen it. 41 Based on these findings, further studies investigating this region and its interaction with mtDNA‐CN in detail are needed.
Even though the mtDel/mtDup count was comparatively low in cases with AAA, the heteroplasmy level was significantly elevated as compared with controls (Figure 1G through J). It has been previously postulated that the low/moderately low‐level inherited heteroplasmic mutations can potentially intensify the effects of somatic mutations, especially in aging‐related disorders. 42 Therefore, even though the observed heteroplasmy levels of mtDel and mtDup were low in AAA (~1%–5%), they could still be associated with AAA pathophysiology. The novel 24 bp mtDup (16 401–16 424) that we observed in the majority of the AAA tissues and matched serum samples is present in the D‐loop region and falls under the MT‐7SDNA locus. Accumulation of the 7SDNA segment has been previously observed in Parkinson's disease 43 ; we speculate that duplications in the region could have similar pathological consequences in AAA.
The haplogroups distribution in our study fits with that reported in a representative Swedish population study. 44 We found that men belonging to the JTU haplogroup cluster had greater odds of having a positive family history of AAA as compared with men with an HV (Figure S6). Diastolic blood pressure was significantly elevated in individuals with JTU haplogroups as compared with HV (Table S7). Interestingly, a previous study showed a similar association between haplogroup cluster JTU and age‐related macular degeneration. 45 A previous Danish study reported that participants with a positive family history of AAA through female relatives had more than a 2.5 times increase in prevalence of AAA compared with individuals who had male relatives with AAA. 46 This is an interesting observation that might explain the association between maternally inherited mtDNA haplogroups and AAA family history found in the present study.
The main strength of this study is that we have investigated the whole mitochondrial genome in participants with objectively diagnosed AAA and individuals without AAA using next‐generation sequencing method. We have confirmed the Swedish haplogroup distribution and identified certain known mutations in the European population, which proves the robustness of the sequencing method. The sequencing quality control criteria employed are stringent and facilitated filtering of potential sequencing errors. The long‐range polymerase chain reaction amplification method used for mtDNA‐specific library preparation has been shown to limit the detection of nuclear‐embedded mitochondrial sequences, a potential error source while sequencing mitochondrial genome. 47 However, along with its strengths, the study has some limitations as well. For instance, owing to the relatively low sample size of the study, we did not have enough statistical power for making some conclusions, especially with respect to the association between individual mutations and disease status. However, we used permutation tests in our study that are more appropriate for small sample size analysis. Moreover, as this study is based on an exclusively male cohort, we could not take potential sex‐specific profiles of mitochondrial mutations/haplogroups into account. 48 However, AAA is more common in men. 1 , 2 , 32 Future studies with mixed sexes are needed to account for sex‐associated mitochondrial genomic profiles. Owing to the case–control and observational nature of the present study, potential casual relationship(s) and mechanistic insights could not be determined. Finally, owing to the case–control study design, even though matched for certain confounders, there still could be some residual confounding elements. Future studies with larger sample size and more rigorously matched groups or propensity matching are hence needed for validation of the current findings. Nevertheless, this is a hypothesis‐generating study, where we have, for the first time, characterized the whole mitochondrial genome in AAA.
CONCLUSIONS
In conclusion, to the best of our knowledge, this is the first study where the whole mitochondrial genome in AAA has been investigated. Our findings reveal a different mtDNA mutational landscape in men with AAA compared with those without AAA. The mutational signatures suggest errors in mitochondrial replication or repair process, instead of oxidative stress, as the main source of mutations in AAA. Additionally, we report a novel 24 bp mtDNA duplication in the D‐loop region present in 75% of the AAA biopsies and matched serum samples examined. We found that the haplogroup cluster JTU is overrepresented in AAA and is significantly associated with a positive family history of AAA. Finally, we observed that the regulatory D‐loop region of the mitochondrial genome had significantly higher heteroplasmic frequency in AAA, especially in the conserved MT‐TAS2 region and showed that low mtDNA‐CN may exacerbate the pathological consequences of mutations in the MT‐TAS2 region in AAA.
The present study suggests an important role of the mitochondrial D‐loop region in AAA that should be further investigated. Overall, our findings demonstrate that the oftentimes neglected mitochondrial genome has the potential to guide new strategies in diagnosis and risk stratification of AAA. Future studies (including functional) on larger populations and mixed sexes are required for a more comprehensive and confident analysis of the role of mitochondrial genetics in AAA and its interplay with clinical/environmental risk factors.
Sources of Funding
The Swedish Heart‐Lung Foundation to Dr Sundquist. Research Funds at Skåne University Hospital, Region Skåne (430751), the Hulda Ahlmroth Foundation, and from the Swedish Government under the LUA/ALF agreement to Dr Gottsäter.
Disclosures
The authors declare that they have no competing interests.
Supporting information
Data S1
Data S2
Data S3
Tables S1–S7
Figures S1–S6
Acknowledgments
Support by National Bioinformatics Infrastructure Sweden is gratefully acknowledged. The authors would like to thank Anna Hedelius for providing excellent technical support.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.029248
For Sources of Funding and Disclosures, see page 9.
References
- 1. Wanhainen A, Verzini F, Van Herzeele I, Allaire E, Bown M, Cohnert T, Dick F, van Herwaarden J, Karkos C, Koelemay M, et al. Editor's choice ‐ European Society for Vascular Surgery (ESVS) 2019 clinical practice guidelines on the management of abdominal aorto‐iliac artery aneurysms. Eur J Vasc Endovasc Surg. 2019;57:8–93. doi: 10.1016/j.ejvs.2018.09.020 [DOI] [PubMed] [Google Scholar]
- 2. Chaikof EL, Dalman RL, Eskandari MK, Jackson BM, Lee WA, Mansour MA, Mastracci TM, Mell M, Murad MH, Nguyen LL, et al. The Society for Vascular Surgery practice guidelines on the care of patients with an abdominal aortic aneurysm. J Vasc Surg. 2018;67:2–77. doi: 10.1016/j.jvs.2017.10.044 [DOI] [PubMed] [Google Scholar]
- 3. Kuivaniemi H, Ryer EJ, Elmore JR, Tromp G. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev Cardiovasc Ther. 2015;13:975–987. doi: 10.1586/14779072.2015.1074861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Golledge J. Abdominal aortic aneurysm: update on pathogenesis and medical treatments. Nat Rev Cardiol. 2019;16:225–242. doi: 10.1038/s41569-018-0114-9 [DOI] [PubMed] [Google Scholar]
- 5. Osellame LD, Blacker TS, Duchen MR. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab. 2012;26:711–723. doi: 10.1016/j.beem.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gustafsson CM, Falkenberg M, Larsson N‐G. Maintenance and expression of mammalian mitochondrial DNA. Ann Rev Biochem. 2016;85:133–160. doi: 10.1146/annurev-biochem-060815-014402 [DOI] [PubMed] [Google Scholar]
- 7. Diaz‐Vegas A, Sanchez‐Aguilera P, Krycer JR, Morales PE, Monsalves‐Alvarez M, Cifuentes M, Rothermel BA, Lavandero S. Is mitochondrial dysfunction a common root of noncommunicable chronic diseases? Endocr Rev. 2020;41:bnaa005. doi: 10.1210/endrev/bnaa005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park CB, Larsson N‐G. Mitochondrial DNA mutations in disease and aging. J Cell Biol. 2011;193:809–818. doi: 10.1083/jcb.201010024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marom S, Friger M, Mishmar D. MtDNA meta‐analysis reveals both phenotype specificity and allele heterogeneity: a model for differential association. Sci Rep. 2017;7:43449. doi: 10.1038/srep43449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kenney MC, Chwa M, Atilano SR, Falatoonzadeh P, Ramirez C, Malik D, Tarek M, del Carpio JC, Nesburn AB, Boyer DS, et al. Molecular and bioenergetic differences between cells with African versus European inherited mitochondrial DNA haplogroups: implications for population susceptibility to diseases. Biochim Biophys Acta. 2014;1842:208–219. doi: 10.1016/j.bbadis.2013.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Summerhill VI, Sukhorukov VN, Eid AH, Nedosugova LV, Sobenin IA, Orekhov AN. Pathophysiological aspects of the development of abdominal aortic aneurysm with a special focus on mitochondrial dysfunction and genetic associations. Biomol Concepts. 2021;12:55–67. doi: 10.1515/bmc-2021-0007 [DOI] [PubMed] [Google Scholar]
- 12. Cooper HA, Cicalese S, Preston KJ, Kawai T, Okuno K, Choi ET, Kasahara S, Uchida HA, Otaka N, Scalia R, et al. Targeting mitochondrial fission as a potential therapeutic for abdominal aortic aneurysm. Cardiovasc Res. 2021;117:971–982. doi: 10.1093/cvr/cvaa133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Navas‐Madroñal M, Rodriguez C, Kassan M, Fité J, Escudero JR, Cañes L, Martínez‐González J, Camacho M, Galán M. Enhanced endoplasmic reticulum and mitochondrial stress in abdominal aortic aneurysm. Clin Sci (Lond). 2019;133:1421–1438. doi: 10.1042/cs20190399 [DOI] [PubMed] [Google Scholar]
- 14. Larsson E, Granath F, Swedenborg J, Hultgren R. A population‐based case‐control study of the familial risk of abdominal aortic aneurysm. J Vasc Surg. 2009;49:47–51. doi: 10.1016/j.jvs.2008.08.012 [DOI] [PubMed] [Google Scholar]
- 15. Wahlgren CM, Larsson E, Magnusson PK, Hultgren R, Swedenborg J. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J Vasc Surg. 2010;51:3–7; discussion 7. doi: 10.1016/j.jvs.2009.08.036 [DOI] [PubMed] [Google Scholar]
- 16. Vats S, Sundquist K, Wang X, Zarrouk M, Ågren‐Witteschus S, Sundquist J, Gottsäter A, Memon AA. Associations of global DNA methylation and homocysteine levels with abdominal aortic aneurysm: a cohort study from a population‐based screening program in Sweden. Int J Cardiol. 2020;321:137–142. doi: 10.1016/j.ijcard.2020.06.022 [DOI] [PubMed] [Google Scholar]
- 17. Memon AA, Zarrouk M, Ågren‐Witteschus S, Sundquist J, Gottsäter A, Sundquist K. Identification of novel diagnostic and prognostic biomarkers for abdominal aortic aneurysm. Eur J Prev Cardiol. 2019;27:132–142. doi: 10.1177/2047487319873062 [DOI] [PubMed] [Google Scholar]
- 18. Andrews S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Cambridge, UK: Babraham Bioinformatics; 2010.. [Google Scholar]
- 19. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krueger F. Trim Galore: A Wrapper Tool Around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files. Cambridge, UK: Babraham Bioinformatics; 2015. [Google Scholar]
- 21. Calabrese C, Simone D, Diroma MA, Santorsola M, Guttà C, Gasparre G, Picardi E, Pesole G, Attimonelli M. MToolBox: a highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high‐throughput sequencing. Bioinformatics. 2014;30:3115–3117. doi: 10.1093/bioinformatics/btu483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Basu S, Xie X, Uhler JP, Hedberg‐Oldfors C, Milenkovic D, Baris OR, Kimoloi S, Matic S, Stewart JB, Larsson N‐G, et al. Accurate mapping of mitochondrial DNA deletions and duplications using deep sequencing. PLoS Genet. 2020;16:e1009242. doi: 10.1371/journal.pgen.1009242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nie Y, Murley A, Golder Z, Rowe JB, Allinson K, Chinnery PF. Heteroplasmic mitochondrial DNA mutations in frontotemporal lobar degeneration. Acta Neuropathol. 2022;143:687–695. doi: 10.1007/s00401-022-02423-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. P Hervé, C Marc, F Seth, L Nianhua. AnnotationDbi: manipulation of SQLite‐based annotations in Bioconductor. R package version 1.56.2. 2021.
- 25. Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gehring JS, Fischer B, Lawrence M, Huber W. SomaticSignatures: inferring mutational signatures from single‐nucleotide variants. Bioinformatics. 2015;31:3673–3675. doi: 10.1093/bioinformatics/btv408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Islam SMA, Díaz‐Gay M, Wu Y, Barnes M, Vangara R, Bergstrom EN, He Y, Vella M, Wang J, Teague JW, et al. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. Cell Genomics. 2022;2:100179. doi: 10.1101/2020.12.13.422570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kloss‐Brandstätter A, Pacher D, Schönherr S, Weissensteiner H, Binna R, Specht G, Kronenberg F. HaploGrep: a fast and reliable algorithm for automatic classification of mitochondrial DNA haplogroups. Hum Mutat. 2011;32:25–32. doi: 10.1002/humu.21382 [DOI] [PubMed] [Google Scholar]
- 29. Team R . RStudio: Integrated Development Environment for R. Boston, MA: RStudio, PBC; 2022. [Google Scholar]
- 30. Hothorn T, Hornik K, van de Wiel MA, Zeileis A. Implementing a class of permutation tests: the coin package. J Stat Softw. 2008;28:1–23. doi: 10.18637/jss.v028.i08 27774042 [DOI] [Google Scholar]
- 31. Hägg S, Jylhävä J, Wang Y, Czene K, Grassmann F. Deciphering the genetic and epidemiological landscape of mitochondrial DNA abundance. Hum Genet. 2021;140:849–861. doi: 10.1007/s00439-020-02249-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Altobelli E, Rapacchietta L, Profeta VF, Fagnano R. Risk factors for abdominal aortic aneurysm in population‐based studies: a systematic review and meta‐analysis. Int J Environ Res Public Health. 2018;15:2805. doi: 10.3390/ijerph15122805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cho H‐J, Yoo J‐H, Kim M‐H, Ko K‐J, Jun K‐W, Han K‐D, Hwang J‐K. Risk of various cancers in adults with abdominal aortic aneurysms. J Vasc Surg. 2023;77:80–88.e82. doi: 10.1016/j.jvs.2022.03.896 [DOI] [PubMed] [Google Scholar]
- 34. Torchiano BWAM. lmPerm: permutation tests for linear models; 2.1.0. 2016.
- 35. Ju YS, Alexandrov LB, Gerstung M, Martincorena I, Nik‐Zainal S, Ramakrishna M, Davies HR, Papaemmanuil E, Gundem G, Shlien A, et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife. 2014;3:e02935. doi: 10.7554/eLife.02935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bratic A, Larsson N‐G. The role of mitochondria in aging. J Clin Investig. 2013;123:951–957. doi: 10.1172/JCI64125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Craven L, Alston CL, Taylor RW, Turnbull DM. Recent advances in mitochondrial disease. Annu Rev Genomics Hum Genet. 2017;18:257–275. doi: 10.1146/annurev-genom-091416-035426 [DOI] [PubMed] [Google Scholar]
- 38. Shi Y, Posse V, Zhu X, Hyvärinen AK, Jacobs HT, Falkenberg M, Gustafsson CM. Mitochondrial transcription termination factor 1 directs polar replication fork pausing. Nucleic Acids Res. 2016;44:5732–5742. doi: 10.1093/nar/gkw302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kong AS, Lai KS, Hee CW, Loh JY, Lim SHE, Sathiya M. Oxidative stress parameters as biomarkers of cardiovascular disease towards the development and progression. Antioxidants (Basel, Switzerland). 2022;11:1175. doi: 10.3390/antiox11061175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Longchamps RJ, Yang SY, Castellani CA, Shi W, Lane J, Grove ML, Bartz TM, Sarnowski C, Liu C, Burrows K, et al. Genome‐wide analysis of mitochondrial DNA copy number reveals loci implicated in nucleotide metabolism, platelet activation, and megakaryocyte proliferation. Hum Genet. 2022;141:127–146. doi: 10.1007/s00439-021-02394-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Filograna R, Koolmeister C, Upadhyay M, Pajak A, Clemente P, Wibom R, Simard ML, Wredenberg A, Freyer C, Stewart JB, et al. Modulation of mtDNA copy number ameliorates the pathological consequences of a heteroplasmic mtDNA mutation in the mouse. Sci Adv. 2019;5(4):eaav9824. doi: 10.1126/sciadv.aav9824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Keogh M, Chinnery PF. Hereditary mtDNA heteroplasmy: a baseline for aging? Cell Metabolism. 2013;18:463–464. doi: 10.1016/j.cmet.2013.09.015 [DOI] [PubMed] [Google Scholar]
- 43. Podlesniy P, Puigròs M, Serra N, Fernández‐Santiago R, Ezquerra M, Tolosa E, Trullas R. Accumulation of mitochondrial 7S DNA in idiopathic and LRRK2 associated Parkinson's disease. EBioMedicine. 2019;48:554–567. doi: 10.1016/j.ebiom.2019.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lembring M, van Oven M, Montelius M, Allen M. Mitochondrial DNA analysis of Swedish population samples. Int J Legal Med. 2013;127:1097–1099. doi: 10.1007/s00414-013-0908-6 [DOI] [PubMed] [Google Scholar]
- 45. Kenney MC, Hertzog D, Chak G, Atilano SR, Khatibi N, Soe K, Nobe A, Yang E, Chwa M, Zhu F, et al. Mitochondrial DNA haplogroups confer differences in risk for age‐related macular degeneration: a case control study. BMC Med Genet. 2013;14:4. doi: 10.1186/1471-2350-14-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Joergensen TMM, Houlind K, Green A, Lindholt JS. Abdominal aortic diameter is increased in males with a family history of abdominal aortic aneurysms: results from the Danish VIVA‐trial. Eur J Vasc Endovasc Surg. 2014;48:669–675. doi: 10.1016/j.ejvs.2014.09.005 [DOI] [PubMed] [Google Scholar]
- 47. White EJ, Ross T, Lopez E, Nikiforov A, Gault C, Batorsky R, Darcy C, Campagna DR, Fleming MD, Thompson JF. Chasing a moving target: detection of mitochondrial heteroplasmy for clinical diagnostics. bioRxiv. 2017;222109. Preprint. doi: 10.1101/222109 [DOI] [Google Scholar]
- 48. Zhang FF, Cardarelli R, Carroll J, Fulda KG, Kaur M, Gonzalez K, Vishwanatha JK, Santella RM, Morabia A. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics. 2011;6:623–629. doi: 10.4161/epi.6.5.15335 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Data S2
Data S3
Tables S1–S7
Figures S1–S6