

Abstract
Neurogenic bladder is caused by disruption of neuronal pathways regulating bladder relaxation and contraction. In severe cases, neurogenic bladder can lead to vesicoureteral reflux, hydroureter, and chronic kidney disease. These complications overlap with manifestations of congenital anomalies of the kidney and urinary tract (CAKUT).
To identify novel monogenic causes of neurogenic bladder, we applied exome sequencing (ES) to our cohort of families with CAKUT.
By ES, we have identified a homozygous missense variant (p.Gln184Arg) in CHRM5 (cholinergic receptor, muscarinic, 5) in a patient with neurogenic bladder and secondary complications of CAKUT. CHRM5 codes for a seven transmembrane-spanning G-protein-coupled muscarinic acetylcholine receptor. CHRM5 is shown to be expressed in murine and human bladder walls and is reported to cause bladder overactivity in Chrm5 knockout mice.
We investigated CHRM5 as a potential novel candidate gene for neurogenic bladder with secondary complications of CAKUT. CHRM5 is similar to the cholinergic bladder neuron receptor CHRNA3, which Mann et al. published as the first monogenic cause of neurogenic bladder. However, functional in vitro studies did not reveal evidence to strengthen the status as a candidate gene. Discovering additional families with CHRM5 variants could help to further assess the genes’ candidate status.
Keywords: CAKUT, neurogenic bladder, whole-exome sequencing, CHRM5
INTRODUCTION
Congenital anomalies of the kidney and urinary tract (CAKUT) represent the most frequent birth defect (~30%) and are the leading cause of chronic kidney disease in the first three decades of life [Ingelfinger et al., 2016], [Calderon-Margalit et al., 2018], [Queisser-Luft et al., 2002], [Connaughton et al., 2015]. The identification of over 40 monogenic causes of CAKUT in humans has led to the understanding that defects in nephrogenesis signaling pathways often result in urogenital malformations [van der Ven et al., 2018b], [van der Ven et al., 2018a], [Verbitsky et al., 2019]. Even though the genetic causes and molecular pathophysiology of these processes are not fully known, animal studies have shown that intrauterine obstruction to urine flow can secondarily result in CAKUT [van der Ven et al., 2018a]. Recently, we made the surprising discovery that CAKUT may be caused by recessive CHRNA3 variants. Unlike other CAKUT genes, CHRNA3 is not highly expressed in kidney, ureter, or bladder tissue, but rather in bladder neurons [Mann et al., 2019]. As a nicotinergic acetylcholine receptor CHRNA3, together with other receptors of the acetylcholine receptor group, acts in bladder innervation.
Muscarinic acetylcholine receptors (mAChR) as the M5 mAChR, are G-protein-coupled receptors with seven transmembrane-spanning domains. They are widely expressed in dopaminergic regions of the brain and the vascular system [Ishii and Kurachi, 2006]. However, Bschleipfer et al. showed expression of CHRM5 in all three cell layers of the human urothelium [Bschleipfer et al., 2007]. Likewise in mice, Zarghooni et al. showed Chrm5 expression in all layers of the murine urothelium [Zarghooni et al., 2007]. Interestingly, Deckmann et al. also showed that mice lacking Chrm5 demonstrate symptoms of bladder overactivity, making CHRM5 a strong candidate gene for human bladder voiding defects, if mutated [Deckmann et al., 2018].
Recently, we discovered a gene involved in the neuronal regulation of bladder contraction, CHRNA3, as the first neuronally expressed gene to cause CAKUT in humans [Weber et al., 2011]. Here, we describe the discovery of a CAKUT patient with a homozygous variant in a gene encoding yet another acetylcholine receptor, CHRM5, which is highly expressed in bladder neurons, and whose loss of function induces micturition defects in a knockout mouse model.
MATERIALS AND METHODS
Whole exome sequencing
Approval for human subjects’ research was obtained from the Institutional Review Boards at the Boston Children’s Hospital and the respective institutions where patient samples were acquired. Following written informed consent, samples from the patients and their family members were collected [Seltzsam et al., 2022] [Connaughton et al., 2019].
We applied exome sequencing (ES) and homozygosity mapping to 963 families with CAKUT. [Mann et al., 2019] [Seltzsam et al., 2022] [Connaughton et al., 2019]. DNA sequencing and variant analysis for genes known to cause isolated and syndromic CAKUT as well as for novel genes under a recessive hypothesis were performed as previously described and according to the ACMG guidelines. Homozygosity mapping was applied to the whole cohort of mixed consanguineous and non-consanguineous families to determine unknown and quantify known consanguinity and enable a focused analysis of runs of homozygosity, if significant. [van der Ven et al., 2018a] [Warejko et al., 2018] [Vivante et al., 2017] [MacArthur et al., 2014] [Connaughton et al., 2019] [Richards et al., 2008] (Supplementary Figure 1 and 2).
Filtering of variants was performed as previously described to retain only rare variants with deleterious in silico prediction and high evolutionary conservation, using available data from multiple online databases [Bamshad et al., 2011] [Lee et al., 2014] [Connaughton et al., 2020].
Lastly, to confirm phenotype-genotype segregation of the remaining variants we used Sanger sequencing in all affected and unaffected family members.
The same protocol was applied to an in-house control population of 1382 families with steroid-resistant nephrotic syndrome where no recessive variants meeting all filtering criteria were found.
Functional studies on CHRM5
Generation and Maintenance of Cell Lines
The materials for the generation of cell lines were purchased as previously described [Burger et al., 2021] [van der Westhuizen et al., 2015] [Haider et al., 2022]. Following previously described protocols, M5 mAChR DNA constructs were cloned into a pEF5/FTR/V5 vector and stably expressed in FlpIn CHO cells. Maintenance of cell lines in DMEM and Mycoplasma testing was performed as previously described. [Vuckovic et al., 2019] [Burger et al., 2021] [Keov et al., 2014] [Khajehali et al., 2018].
Equilibrium Radioligand Binding Experiments and IP Accumulation Assay
First, we tested equilibrium radioligand binding for the WT M5 mAChR or Q184R mutant cell line, following previously described protocols. Like in previous studies, saturation binding experiments were used to determine the affinity of [3H]-N-Methylscopolamine ([3H]-NMS) for M5 mAChR receptor constructs, as well as the competition between acetylcholine (ACh) and [3H]-NMS [Burger et al., 2021].
Second, we analyzed IP1 accumulation to test the variants’ downstream effects, as previously described [Burger et al., 2021].
Data Analysis
For data analysis with statistical analysis and nonlinear regression curve fitting we used GraphPad Prism (San Diego, CA) and equations as previously described for radioligand saturation binding experiments with [3H]-NMS and functional IP1 accumulation experiments using Ach [Burger et al., 2021] [Motulsky and Brown, 2006] [Draper-Joyce et al., 2018].
RESULTS
Clinical phenotype of individual B2797_21 with CAKUT
By ES, we identified a homozygous missense variant in CHRM5 in patient B2797–21. He was born to non-consanguineous parents of Saudi-Arabian descent and presented at the age of nine months with acute kidney injury (Figure 1A). Renal ultrasound demonstrated severe bilateral hydronephrosis and hydroureter, which was not detected prenatally (Figure 1E–G). Voiding cystourethrogram (VCUG) revealed right-sided grade V vesicoureteral reflux (VUR), a small capacity of the urinary bladder with wall trabeculation, and associated diverticulum, and a dilated proximal urethra with normal appearance distally consistent with posterior urethral valve (PUV) (Figure 1 B–D). However, PUV could never be visualized during urologic interventions. The alleged PUV had been fulgurated twice within two years (at 9 months and 30 months of age). However, ultrasound imaging showed persisting severe bilateral hydronephrosis and hydroureter retrospectively, rendering the diagnosis of PUV insecure, retrospectively. VCUG re-demonstrated right-sided grade V VUR despite the operative resolution of the “PUV” features. The capacity of the urinary bladder improved. Kidney examination at the age of 2 years and 10 months showed bilateral obstruction of the kidneys in Tc99-Mg-3 renal scan with the left kidney contributing by 84% and the right kidney contributing by 16%. NM-GUS-Renogram (Diuretic) Scintigraphy (DTPA) demonstrated a dilated obstructed left kidney and a nonfunctioning right kidney (Figure 2). A FLU-micturition cystourethrogram (MCUG) revealed a grade V VUR with trabeculated bladder, likely related to neurogenic bladder. Renal ultrasound one year later showed increased severity of bilateral hydroureteronephrosis but stable appearance of the “neurogenic bladder”. For voiding management, clean intermittent catheterization (CIC) was recommended at the age of 2 years but was not applied until the age of 4. Medical control of CKD included the antihypertensive drugs captopril and amlodipine, supplementation of alfacalcidol, folic acid, iron, and sodium bicarbonate, oxybutynin as anticholinergic drug, and prophylactic cotrimoxazole. The patient did not have any clinical findings of familial dysautonomia, either in the initial clinical reports or after a specific request.
Figure 1. Pedigree and clinical information of individual B2797–21.
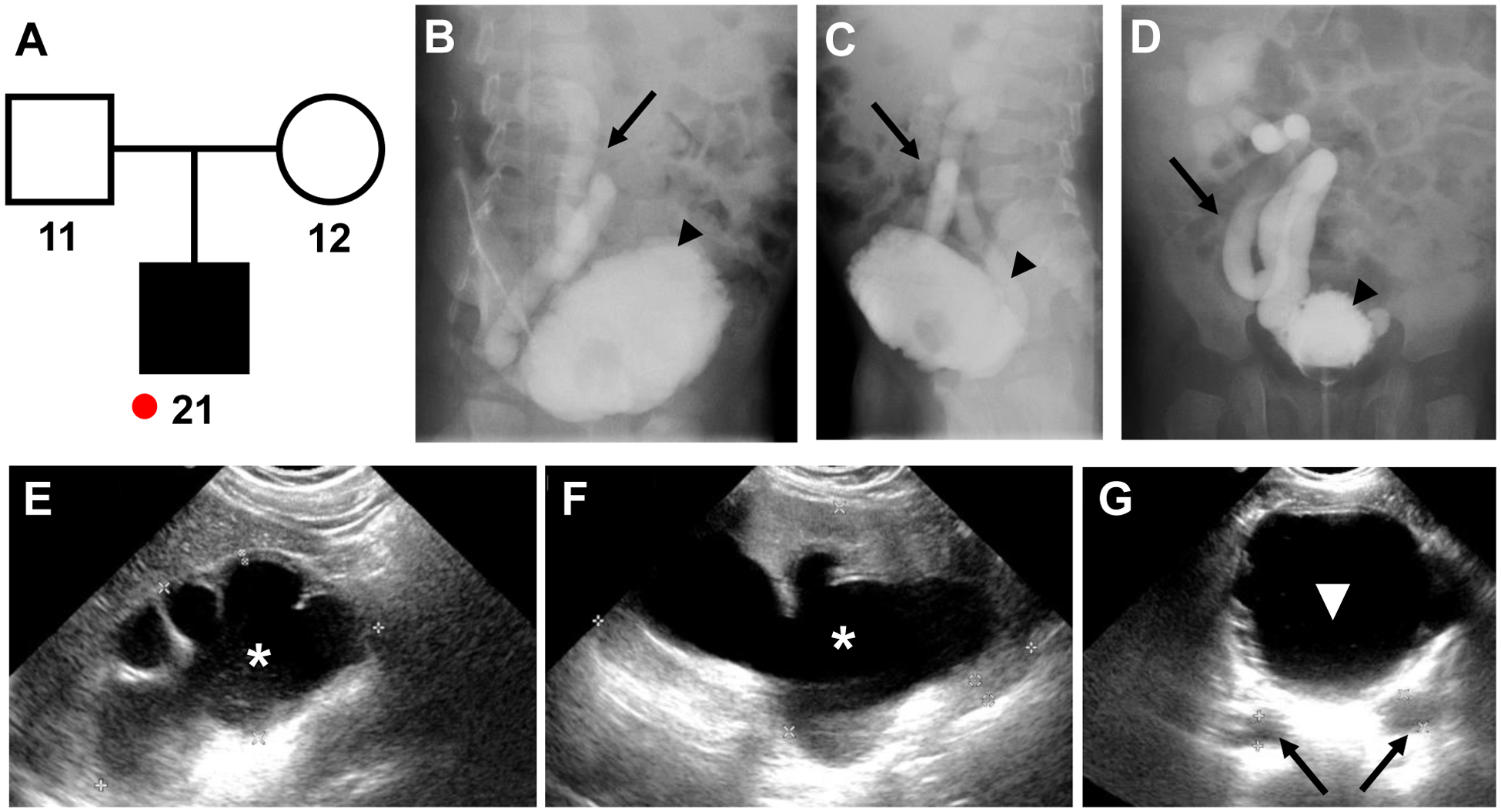
A) Pedigree of individual B2797–21: affected boy with neurogenic bladder, small trabeculated urinary bladder, bilateral severe hydronephrosis, grade V VUR right, chronic kidney disease (stage 4) and unaffected parents (−11 and 12); red dot, individual included in ES.
B-D) FLU-micturating cystourethrogram: lateral projection, right side (B), lateral projection, left side (C), and anterior-posterior projection (D) showing right-sided grade V vesicoureteric reflux (black arrow) with trabeculated urinary bladder (black arrowhead), likely caused by neurogenic bladder.
E-G) Ultrasound of right kidney (E), left kidney (F), and urinary bladder (white triangle) (G), showing severe hydronephrosis (white asterisk) and hydroureter (black arrows).
Figure 2. Tc99m-Mag3 Renal Scan at the age of 2 years and 10 months.
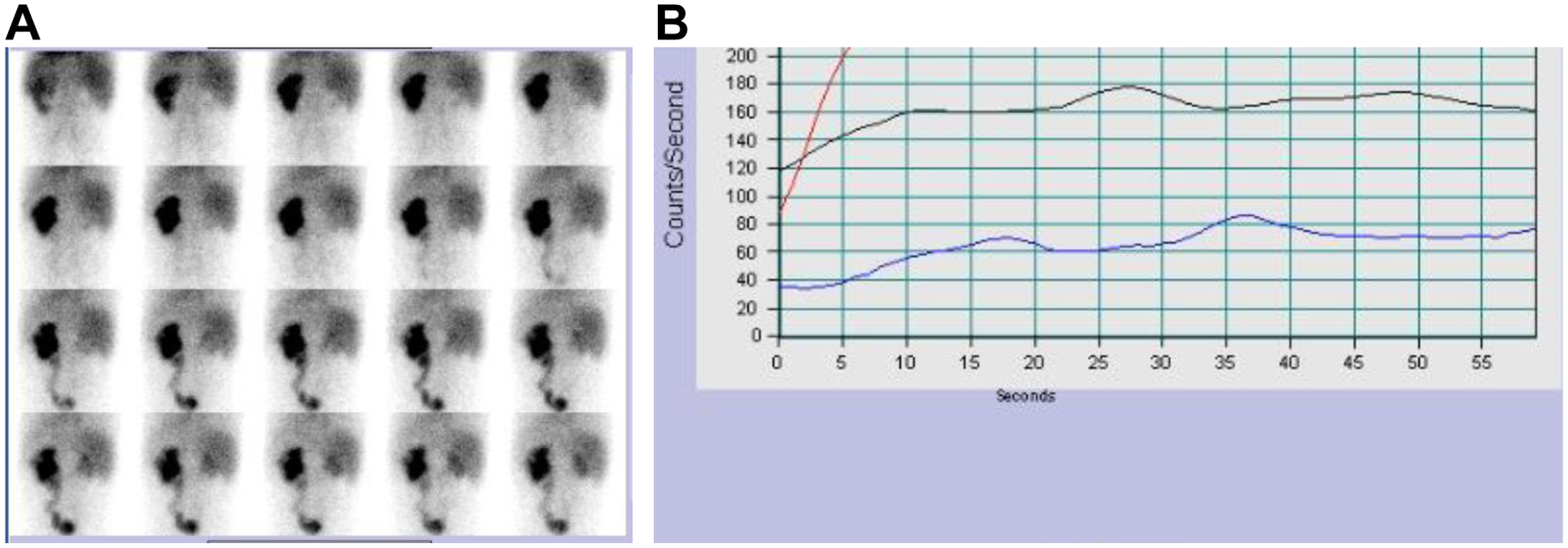
A) Tc99m-Mag3 Renal Scan and B) Flow study of the Tc99m-Mag3 Renal Scan both show the left kidney is contributing to renal function by 84%. The right kidney is contributing to renal function by 16%. Red line, left kidney; blue line, right kidney; black line, aorta.
Discovery of a biallelic variant in CHRM5
By ES in a worldwide cohort of 963 families with CAKUT, we identified a homozygous missense variant (NM_012125.3; c.551A>G, p.Gln184Arg) in exon 3 of the gene CHRM5 (Cholinergic Receptor Muscarinic 5) (Fig. 3 A). The variant is not reported in any of the consulted population databases and is predicted to be deleterious by two of three in-silico prediction tools (Table 1). CHRM5 encodes for the G-protein-coupled M5 mAChR which contains seven transmembrane domains. The variant p.Gln184Arg is located in the second extracellular loop between the fourth and fifth transmembrane-spanning domain (Fig. 3 B) [Vuckovic et al., 2019]. The variant was confirmed by Sanger sequencing to be homozygous in the index patient and heterozygous in both unaffected parents (Fig. 3 C). The evolutionary conservation of the amino acid residue includes species from Homo sapiens to Ciona intestinalis, as shown by the clustal alignment of the CHRM5 amino acid sequences (Fig. 3 D). Gene constraint metrics for CHRM5 show observed/expected (o/e) scores of 0.8 (0.72 – 0.89) for missense variants and o/e of 0.4 (0.22 – 0,74) for predicted loss of function variants indicating that CHRM5 is less tolerant for loss of function variants than for missense variants.
Figure 3. Exon and protein structure of human CHRM5 cDNA.
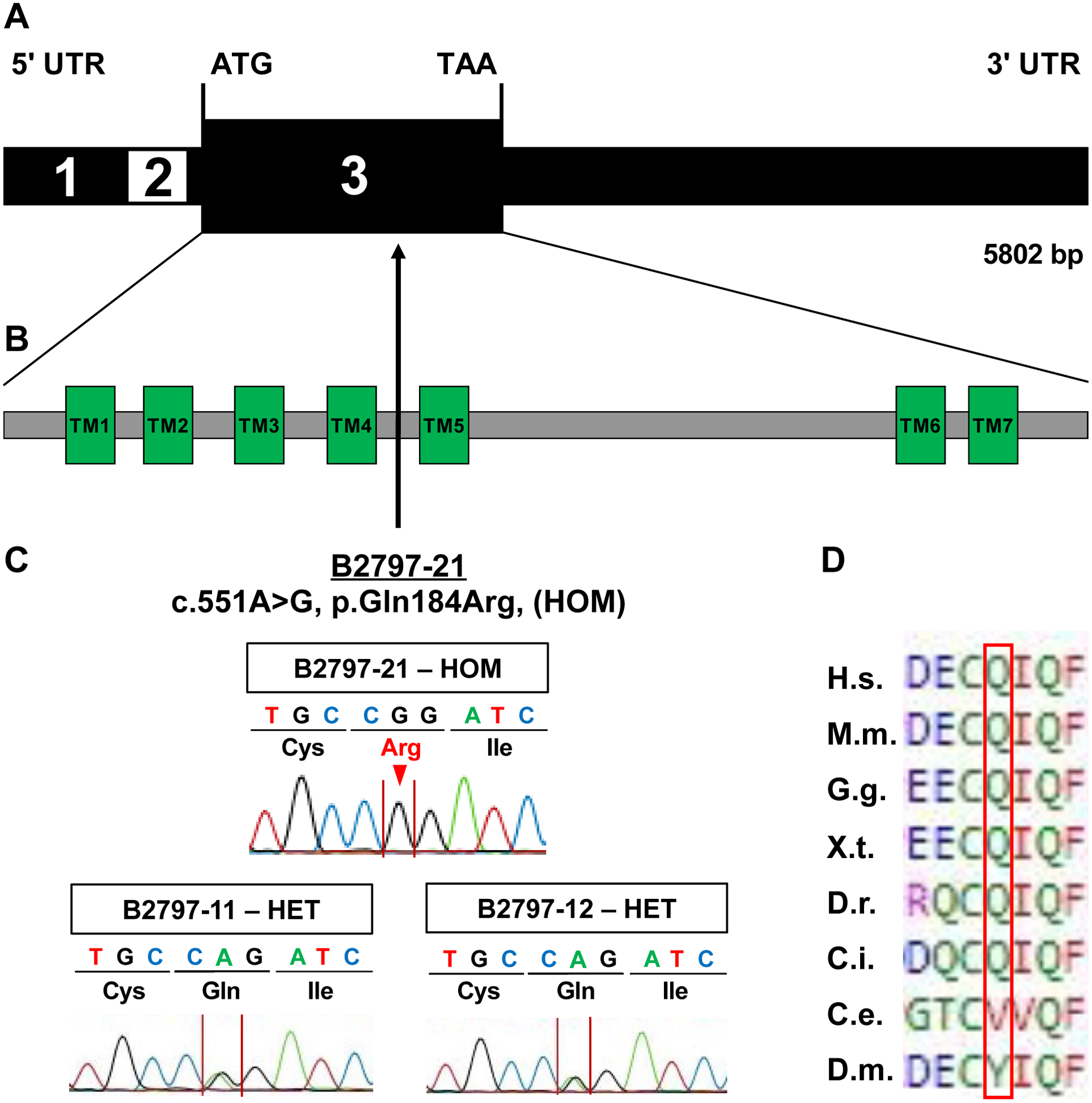
A) Exon structure of human CHRM5. Exon numbers are denoted in black and white.
B) Protein domain structure of the human CHRM5 protein with seven transmembrane regions (TM1-TM7).
C) Position of the homozygous missense variant p.Gln184Arg identified in individual B2797–21. Sanger sequencing showing the homozygous variant in the affected individual (B2797–21) compared to heterozygous variants in both parents (father B2797–11 and mother B2797–12).
D) Clustal alignment of amino acid sequences of CHRM5 to demonstrate evolutionary conservation from mammalia to insectae for each amino acid residue.
Glossary: UTR, untranslated region; ATG, start codon; TGA, stop codon; TM, transmembrane region; HOM, homozygous; HET heterozygous; H.s., Homo sapiens; M.m., Mus musculus; G.g., Gallus gallus; X.t., Xenopus tropicalis; D.r., Danio rerio; C.i., Ciona intestinalis; C.e., Caenorhabditis elegans; D.m., Drosophila melanogaster.
Table 1:
Homozygous variant in CHRM5 in individual (B2797_21) with neurogenic bladder.
| Family-Individual | B2797–21 | |
| Ethnic Origin | Saudi-Arab | |
| Sex | Male | |
| Gene | CHRM5 | |
| Nucleotide Change | c.551A>G | |
| Amino Acid Change | p.Gln184Arg | |
| Conservation | Conserved to C. intestinalis | |
| Segregation | Affected Son | Homozygous |
| Unaffected Mother | Heterozygous | |
| Unaffected Father | Heterozygous | |
| Population Frequency | GnomAD | Not reported |
| EVS | Not reported | |
| Biobase | Not reported | |
| Saudi Arab Database | Not reported | |
| Prediction Scores | Polyphen 2 | Deleterious (0.97) |
| Mutation Taster | Tolerated | |
| SIFT Score | Disease Causing | |
Polyphen 2: Polymorphism phenotyping v2 (http://genetics.bwh.harvard.edu/pph2/); SIFT: Sorting Intolerant from Tolerant algorithm (https://sift.bii.a-star.edu.sg/); EVS: Exome Variant Server (https://evs.gs.washington.edu/EVS/)
Subsequent focused evaluation of ES data for biallelic CHRM5 variants and a GeneMatcher submission did not yield further families with variants in CHRM5. However, the investigation for further families to establish causality is ongoing. Furthermore, in a control in-house cohort of 1382 families with nephrotic syndrome, we did not identify any biallelic variants in CHRM5.
Structural model of the variant and functional studies
The crystal structure of the M5 mAChR was recently determined [Vuckovic et al., 2019]. In the structure model, the residue Gln184 is located in the second extracellular loop of the receptor (Figure 4 A–C) and forms part of the conserved extracellular allosteric site [Burger et al., 2018]. The p.Gln184Arg variant replaces the polar amino acid glutamine with a positively charged amino acid arginine (Figure 4 B). Therefore, we examined if this variant could affect the binding or function of the endogenous orthosteric agonist, ACh, in a mutant CHO cell line. In radioligand binding experiments using the antagonist [3H]-NMS, we show that the Gln184Arg variant does not affect receptor expression or binding affinity of the radioligand [3H]-NMS in comparison to the wild-type (WT) M5 mAChR [Burger et al., 2018] (Figure 4 E, Table 2). We next determined the binding affinity of ACh for the Gln184Arg variant or WT M5 mAChR using a competition binding assay.
Figure 4. Structural model of the CHRM5 variant and functional studies.
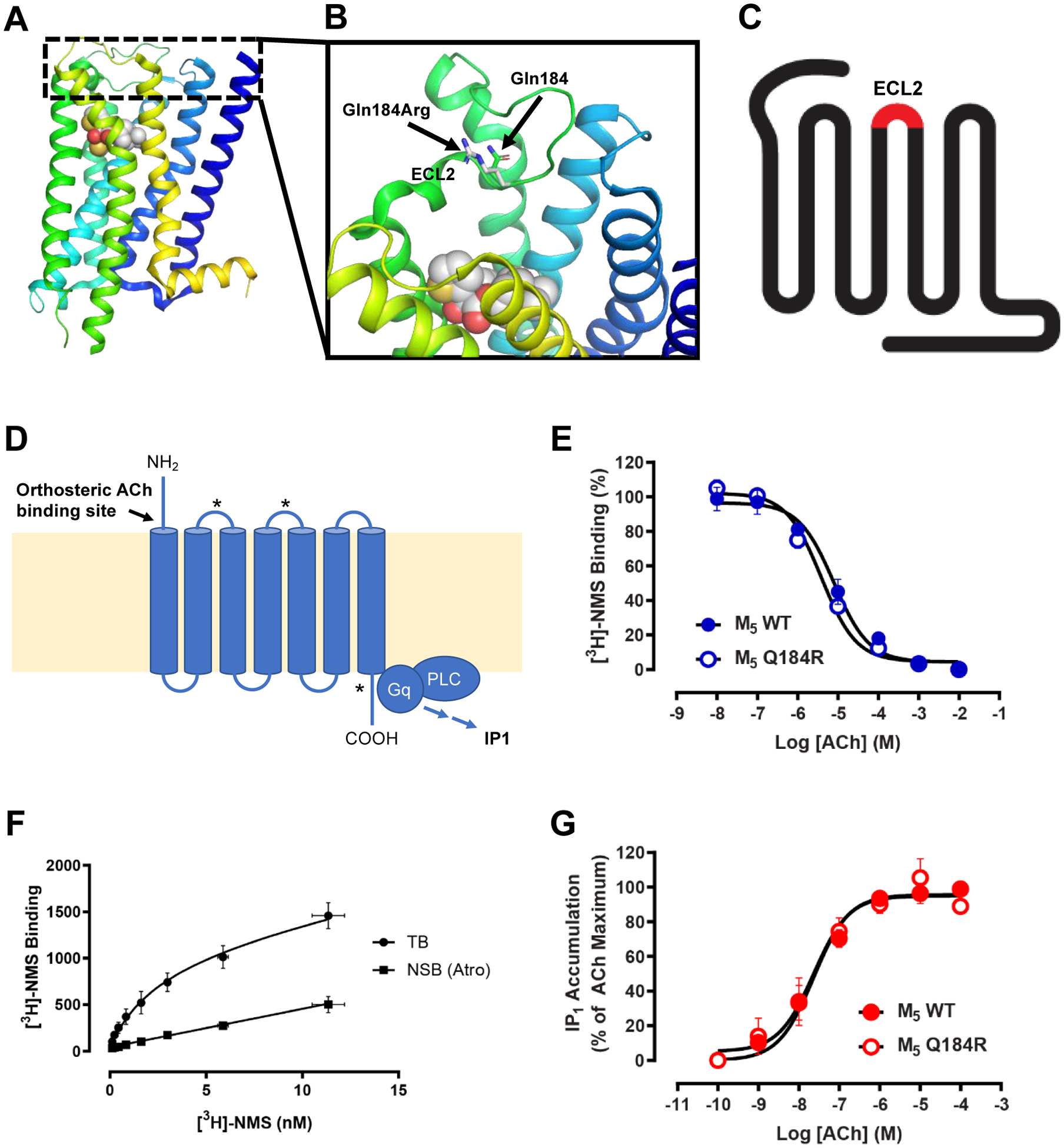
A-B) Crystal structure of CHRM5. B) The p.Gln184Arg variant is located in the second extracellular loop (ECL2) of the receptor and replaces the polar amino acid glutamine with a positively charged amino acid arginine.
C) Schema of seven transmembrane-spanning muscarinic acetylcholine (ACh) receptor. The red section marks the second extracellular loop (ECL2) where the variant p.Gln184Arg is located.
D) Schema of M5 mAChR activation. ACh activates the M5 mAChR causing activation of the Gq signaling pathway leading to production of the second messenger IP1. Asterisks mark allosteric binding sites.
E-G) Radioligand binding experiments using the antagonist [3H]-NMS at FlpIn CHO cells. E) The Gln184Arg variant does not affect receptor expression or binding affinity of the radioligand [3H]-NMS in comparison to the wild-type (WT) M5 mAChR as determined through saturation binding experiments. F) A modest 2-fold difference in binding affinity of ACh at the Gln184Arg variant is observed in comparison to WT M5 mAChR as determined through competition binding between a range of ACh concentrations and a KD concentration of [3H]-NMS. G) IP1 accumulation following activation of the Gln184Arg mutant in response to ACh is indistinguishable from the response observed at the WT M5 mAChR. For all experiments, data represent the mean ± S.E.M. of the three individual experiments performed in duplicate.
Table 2:
Binding parameters and IP1 accumulation for WT M5 mAChR, Q184R mutant.
| [3H]-NMS Saturation Binding | [3H]-NMS Competition Binding | IP1 accumulation ACh | ||
|---|---|---|---|---|
| Constructs | apKD | bBmax (fmol/mg) | cpKi (ACh) | dACh pEC50 |
| WT M5 mAChR | N.D. | N.D. | 5.16 ± 0.10 (3) | 7.63 ± 0.12 (3) |
| Q184R M5 mAChR | 8.58 ± 0.26 (3) | 1116 ± 150 (3) | 5.47 ± 0.08* (3) | 7.63 ± 0.20 (3) |
Data represent the mean ± S.E.M. of (n) independent experiments performed in duplicate. N.D. Not determined. N.R. No response.
significantly different from WT M5 mAChR, p < 0.05, un-paired T-test, Welch’s correction.
Negative logarithm of the radioligand equilibrium dissociation constant.
maximum density of binding sites.
Negative logarithm of the orthosteric agonist equilibrium dissociation constant as determined by a one-site competition binding model.
negative logarithm of the concentration of ACh required to give half-maximal response as determined by a three-parameter logistic equation.
The results from this experiment show a modest 2-fold difference in the binding affinity of ACh at the Gln184Arg variant compared to WT M5 mAChR (Figure 4 F, Table 2). Finally, we tested the Gln184Arg variant in a functional assay that measures the accumulation of the second messenger IP1 due to activation of the Gq signaling pathway resulting from activation of the M5 mAChR by ACh. In this assay, the IP1 accumulation of the Gln184Arg variant is indistinguishable from the WT M5 mAChR (Figure 4 G, Table 2). Collectively, these data indicate that the Gln184Arg variant does not impair M5 mAChR expression, nor the binding or signaling of ACh at the M5 mAChR in a recombinant cell line.
DISCUSSION
By ES, we identified CHRM5 as a novel potential candidate gene for neurogenic bladder in an individual with neurogenic bladder and secondary symptoms similar to CAKUT by identifying a homozygous missense variant (NM_012125.3; c.551A>G, p.Gln184Arg) in CHRM5. The genetic variant found in individual B2797–21 is absent in population frequency databases such as EVS, gnomAD, and the Saudi Population Database, is well conserved through species and is predicted to be deleterious by Polyphen2 and Mutation Taster. CHRM5 codes for the M5 mAChR, a seven transmembrane-spanning G-protein-coupled muscarinic acetylcholine receptor. The here-identified variant p.Gln184Arg is located in the second extracellular loop between the transmembrane-spanning alpha-helices 4 and 5. Overall, we propose CHRM5 as a potential new recessive monogenic cause for neurogenic bladder in humans. More families carrying variants in CHRM5 and expressing a phenotype of neurogenic bladder could confirm the observed genotype-phenotype relationship.
Protein expression and mouse model of CHRM5
The potential role of CHRM5 in bladder tone regulation can be supported by three findings: i) Chrm5 knockout mice show a bladder phenotype [Deckmann et al., 2018]. However, most of the reported knock-out mouse models focused on neurological symptoms and for example detected abnormal synaptic dopamine release [Bendor et al., 2010], [Yamada et al., 2001]. ii) CHRM5 is expressed in human and mouse bladder urothelium, even though other muscarinic receptors show stronger evidence for expression and functional relevance in bladder tone regulation [Bschleipfer et al., 2007], [Zarghooni et al., 2007]. iii) CHRM5 might act like the nicotinic acetylcholine receptor CHRNA3 (Cholinergic Receptor Nicotinic Alpha 3 Subunit) which is expressed in human bladder. We published biallelic variants in CHRNA3 as the first monogenic cause of neurogenic bladder with symptoms of familial dysautonomia in humans [Mann et al., 2019]. Nonetheless, our patient and the Chrm5 knockout mice did not show any signs of dysautonomia such as constant mydriasis. Further studies, like a point mutation animal knockout model, could clarify the role of the here-reported variant in the pathogenesis of neurogenic bladder.
A structural model of the variant and functional studies
At the M1 – M4 mAChR subtypes the residue that corresponds to Gln184 is a Tyr/Phe residue that is critically important for the binding of allosteric modulators that bind to the common extracellular allosteric site [Burger et al., 2018], [Prilla et al., 2006], [Dror et al., 2013]. Recent studies have suggested that orthosteric ligands may occupy this allosteric site during their transition to binding in the orthosteric site [Dror et al., 2011], [Jakubík et al., 2017]. As such, we tested if the Gln184Arg variant could affect the binding or signaling of the endogenous orthosteric agonist ACh. However, our characterization of the Gln184Arg variant in a mutant CHO cell line revealed negligible differences in receptor expression, ACh binding, or ACh signaling via an IP1 accumulation assay in comparison to the WT M5 mAChR. It is important to note that these in vitro experiments do not replicate the complexities of M5 mAChR physiology. Intriguingly, it has been speculated that there are endogenous mAChR allosteric modulators [van der Westhuizen et al., 2015], [Moo et al., 2018]. Given the importance of the position of Gln184 at the other mAChR subtypes for the activity of allosteric modulators, there could be an unappreciated endogenous ligand that binds near Gln184 at the M5 mAChR.
In summary, we identified a recessive variant in CHRM5 as a potential novel cause of neurogenic bladder in humans. The variant is rare and has been predicted to be deleterious by different in-silico prediction tools. In vivo Chrm5 knockout mouse models show bladder overactivity [Deckmann et al., 2018]. However, our functional in vitro studies did not provide any additional evidence to support the candidate status of CHRM5. Still, it is important to note that the role of other endogenous mAChR allosteric modulators has been discussed in different papers [van der Westhuizen et al., 2015], [Moo et al., 2018]. Considering the importance of the variants’ position for the activity of allosteric modulators, an unappreciated endogenous ligand effect could be discussed. Additional families with variants in CHRM5 and further functional research are required to determine the involvement of CHRM5 in the pathogenesis of neurogenic bladder. For future clinical application, established genes for neurogenic bladder could help to early diagnose neurogenic bladder in patients with voiding dysfunction and lead to a targeted therapy avoiding redundant diagnostic and therapeutic procedures.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge Geoff Thompson for generating the Q184R M5 mAChR stable Flp-In-CHO cell line.
We thank Wanxia Wu for excellent technical assistance and Leslie Spaneas for recruitment of patients at Boston Children’s Hospital.
FUNDING
C.-H.W.W. was supported by funding from the American College of Medical Genetics and Genomics Foundation (ACMG/Takeda Next-Generation Biochemical Genetics Award) and National Institutes of Health (grant T32-GM007748)
D.M.C was funded by Health Research Board, Ireland (HPF-206-674), the International Pediatric Research Foundation Early Investigators’ Exchange Program, and the Amgen® Irish Nephrology Society Specialist Registrar Bursary. She is now funded by the Eugen Drewlo Chair for Kidney Research and Innovation at the Schulich School of Medicine & Dentistry at Western University, London, Ontario, Canada.
D.M.T. is funded by a National Health and Medical Research Council of Australia (NHMRC) Project Grant APP1138448 (D.M.T.) and an NHMRC Early Career Investigator Grant APP1196951 (D.M.T.).
F.H. is the William E. Harmon Professor of Pediatrics at Harvard Medical School. His research was supported by grants from the National Institutes of Health to F.H. (DK076683). The Yale Centers for Mendelian Genomics funded by the National Human Genome Research Institute (U54 HG006504) performed sequencing and data processing.
F.H. and S.Sh. are supported by grants from the Begg Family Foundation. This research was also supported by the Isabella Forrest Julian Research Fund for Pediatric Post Kidney Transplant Research.
Footnotes
ETHICS DECLARATION
This study was approved by the institutional review board (IRB) of the University of Michigan and of Boston Children’s Hospital as well as IRBs of institutions where families were recruited. Before inclusion, informed consent of each individual or their legal guardians, respectively, was obtained.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest with the research performed.
DATA AVAILABILITY STATEMENT
The data underlying this article are available in the article and its online supplementary material.
REFERENCES
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet 12: 745–755. [DOI] [PubMed] [Google Scholar]
- Bendor J, Lizardi-Ortiz JE, Westphalen RI, Brandstetter M, Hemmings HC, Sulzer D, Flajolet M, Greengard P. 2010. AGAP1/AP-3-dependent endocytic recycling of M5 muscarinic receptors promotes dopamine release. EMBO J 29: 2813–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bschleipfer T, Schukowski K, Weidner W, Grando SA, Schwantes U, Kummer W, Lips KS. 2007. Expression and distribution of cholinergic receptors in the human urothelium. Life Sci 80: 2303–2307. [DOI] [PubMed] [Google Scholar]
- Burger WAC, Gentry PR, Berizzi AE, Vuckovic Z, van der Westhuizen ET, Thompson G, Yeasmin M, Lindsley CW, Sexton PM, Langmead CJ, Tobin AB, Christopoulos A, Valant C, Thal DM. 2021. Identification of a Novel Allosteric Site at the M5 Muscarinic Acetylcholine Receptor. ACS Chem. Neurosci 12: 3112–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger WAC, Sexton PM, Christopoulos A, Thal DM. 2018. Toward an understanding of the structural basis of allostery in muscarinic acetylcholine receptors. J. Gen. Physiol 150: 1360–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon-Margalit R, Golan E, Twig G, Leiba A, Tzur D, Afek A, Skorecki K, Vivante A. 2018. History of Childhood Kidney Disease and Risk of Adult End-Stage Renal Disease. N. Engl. J. Med 378: 428–438. [DOI] [PubMed] [Google Scholar]
- Connaughton DM, Bukhari S, Conlon P, Cassidy E, O’Toole M, Mohamad M, Flanagan J, Butler T, O’Leary A, Wong L, O’Regan J, Moran S, O’Kelly P, Logan V, Griffin B, Griffin M, Lavin P, Little MA, Conlon P. 2015. The Irish Kidney Gene Project--Prevalence of Family History in Patients with Kidney Disease in Ireland. Nephron 130: 293–301. [DOI] [PubMed] [Google Scholar]
- Connaughton DM, Dai R, Owen DJ, Marquez J, Mann N, Graham-Paquin AL, Nakayama M, Coyaud E, Laurent EMN, St-Germain JR, Blok LS, Vino A, Klämbt V, Deutsch K, Wu C-HW, Kolvenbach CM, Kause F, Ottlewski I, Schneider R, Kitzler TM, Majmundar AJ, Buerger F, Onuchic-Whitford AC, Youying M, Kolb A, Salmanullah D, Chen E, van der Ven AT, Rao J, Ityel H, Seltzsam S, Rieke JM, Chen J, Vivante A, Hwang D-Y, Kohl S, Dworschak GC, Hermle T, Alders M, Bartolomaeus T, Bauer SB, Baum MA, Brilstra EH, Challman TD, Zyskind J, Costin CE, Dipple KM, Duijkers FA, Ferguson M, Fitzpatrick DR, Fick R, Glass IA, Hulick PJ, Kline AD, Krey I, Kumar S, Lu W, Marco EJ, Wentzensen IM, Mefford HC, Platzer K, Povolotskaya IS, Savatt JM, Shcherbakova NV, Senguttuvan P, Squire AE, Stein DR, Thiffault I, Voinova VY, Somers MJG, Ferguson MA, Traum AZ, Daouk GH, Daga A, Rodig NM, Terhal PA, van Binsbergen E, Eid LA, Tasic V, Rasouly HM, Lim TY, Ahram DF, Gharavi AG, Reutter HM, Rehm HL, MacArthur DG, Lek M, Laricchia KM, Lifton RP, Xu H, Mane SM, Sanna-Cherchi S, Sharrocks AD, Raught B, Fisher SE, Bouchard M, Khokha MK, Shril S, et al. 2020. Mutations of the Transcriptional Corepressor ZMYM2 Cause Syndromic Urinary Tract Malformations. Am. J. Hum. Genet 107: 727–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connaughton DM, Kennedy C, Shril S, Mann N, Murray SL, Williams PA, Conlon E, Nakayama M, van der Ven AT, Ityel H, Kause F, Kolvenbach CM, Dai R, Vivante A, Braun DA, Schneider R, Kitzler TM, Moloney B, Moran CP, Smyth JS, Kennedy A, Benson K, Stapleton C, Denton M, Magee C, O’Seaghdha CM, Plant WD, Griffin MD, Awan A, Sweeney C, Mane SM, Lifton RP, Griffin B, Leavey S, Casserly L, de Freitas DG, Holian J, Dorman A, Doyle B, Lavin PJ, Little MA, Conlon PJ, Hildebrandt F. 2019. Monogenic causes of chronic kidney disease in adults. Kidney Int 95: 914–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckmann K, Rafiq A, Erdmann C, Illig C, Durschnabel M, Wess J, Weidner W, Bschleipfer T, Kummer W. 2018. Muscarinic receptors 2 and 5 regulate bitter response of urethral brush cells via negative feedback. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol 32: 2903–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper-Joyce CJ, Michino M, Verma RK, Klein Herenbrink C, Shonberg J, Kopinathan A, Scammells PJ, Capuano B, Thal DM, Javitch JA, Christopoulos A, Shi L, Lane JR. 2018. The structural determinants of the bitopic binding mode of a negative allosteric modulator of the dopamine D 2 receptor. Biochem. Pharmacol 148: 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, Baell JB, Sexton PM, Christopoulos A, Shaw DE. 2013. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503: 295–299. [DOI] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE. 2011. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. U. S. A 108: 13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider A, Deng X, Mastromihalis O, Pfister SK, Jeppesen TE, Xiao Z, Pham V, Sun S, Rong J, Zhao C, Chen J, Li Y, Connors TR, Davenport AT, Daunais JB, Hosseini V, Ran W, Christopoulos A, Wang L, Valant C, Liang SH. 2022. Structure–activity relationship of pyrazol-4-yl-pyridine derivatives and identification of a radiofluorinated probe for imaging the muscarinic acetylcholine receptor M4. Acta Pharm. Sin. B: S2211383522003203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelfinger JR, Kalantar-Zadeh K, Schaefer F, World Kidney Day Steering Committee. 2016. World Kidney Day 2016: Averting the legacy of kidney disease-focus on childhood. Pediatr. Nephrol. Berl. Ger 31: 343–348. [DOI] [PubMed] [Google Scholar]
- Ishii M, Kurachi Y. 2006. Muscarinic acetylcholine receptors. Curr. Pharm. Des 12: 3573–3581. [DOI] [PubMed] [Google Scholar]
- Jakubík J, Randáková A, Zimčík P, El-Fakahany EE, Doležal V. 2017. Binding of N-methylscopolamine to the extracellular domain of muscarinic acetylcholine receptors. Sci. Rep 7: 40381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keov P, López L, Devine SM, Valant C, Lane JR, Scammells PJ, Sexton PM, Christopoulos A. 2014. Molecular Mechanisms of Bitopic Ligand Engagement with the M1 Muscarinic Acetylcholine Receptor. J. Biol. Chem 289: 23817–23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajehali E, Valant C, Jörg M, Tobin AB, Conn PJ, Lindsley CW, Sexton PM, Scammells PJ, Christopoulos A. 2018. Probing the binding site of novel selective positive allosteric modulators at the M1 muscarinic acetylcholine receptor. Biochem. Pharmacol 154: 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M, Fox M, Fogel BL, Martinez-Agosto JA, Wong DA, Chang VY, Shieh PB, Palmer CGS, Dipple KM, Grody WW, Vilain E, Nelson SF. 2014. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA 312: 1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, Barrett JC, Biesecker LG, Conrad DF, Cooper GM, Cox NJ, Daly MJ, Gerstein MB, Goldstein DB, Hirschhorn JN, Leal SM, Pennacchio LA, Stamatoyannopoulos JA, Sunyaev SR, Valle D, Voight BF, Winckler W, Gunter C. 2014. Guidelines for investigating causality of sequence variants in human disease. Nature 508: 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann N, Kause F, Henze EK, Gharpure A, Shril S, Connaughton DM, Nakayama M, Klämbt V, Majmundar AJ, Wu C-HW, Kolvenbach CM, Dai R, Chen J, van der Ven AT, Ityel H, Tooley MJ, Kari JA, Bownass L, El Desoky S, De Franco E, Shalaby M, Tasic V, Bauer SB, Lee RS, Beckel JM, Yu W, Mane SM, Lifton RP, Reutter H, Ellard S, Hibbs RE, Kawate T, Hildebrandt F. 2019. CAKUT and Autonomic Dysfunction Caused by Acetylcholine Receptor Mutations. Am. J. Hum. Genet 105: 1286–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moo EV, Sexton PM, Christopoulos A, Valant C. 2018. Utility of an “Allosteric Site-Impaired” M2 Muscarinic Acetylcholine Receptor as a Novel Construct for Validating Mechanisms of Action of Synthetic and Putative Endogenous Allosteric Modulators. Mol. Pharmacol 94: 1298–1309. [DOI] [PubMed] [Google Scholar]
- Motulsky HJ, Brown RE. 2006. Detecting outliers when fitting data with nonlinear regression - a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 7: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prilla S, Schrobang J, Ellis J, Höltje H-D, Mohr K. 2006. Allosteric interactions with muscarinic acetylcholine receptors: complex role of the conserved tryptophan M2422Trp in a critical cluster of amino acids for baseline affinity, subtype selectivity, and cooperativity. Mol. Pharmacol 70: 181–193. [DOI] [PubMed] [Google Scholar]
- Queisser-Luft A, Stolz G, Wiesel A, Schlaefer K, Spranger J. 2002. Malformations in newborn: results based on 30,940 infants and fetuses from the Mainz congenital birth defect monitoring system (1990–1998). Arch. Gynecol. Obstet 266: 163–167. [DOI] [PubMed] [Google Scholar]
- Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE, Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. 2008. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. Off. J. Am. Coll. Med. Genet 10: 294–300. [DOI] [PubMed] [Google Scholar]
- Seltzsam S, Wang C, Zheng B, Mann N, Connaughton DM, Wu C-HW, Schneider S, Schierbaum L, Kause F, Kolvenbach CM, Nakayama M, Dai R, Ottlewski I, Schneider R, Deutsch K, Buerger F, Klämbt V, Mao Y, Onuchic-Whitford AC, Nicolas-Frank C, Yousef K, Pantel D, Lai EW, Salmanullah D, Majmundar AJ, Bauer SB, Rodig NM, Somers MJG, Traum AZ, Stein DR, Daga A, Baum MA, Daouk GH, Tasic V, Awad HS, Eid LA, El Desoky S, Shalaby M, Kari JA, Fathy HM, Soliman NA, Mane SM, Shril S, Ferguson MA, Hildebrandt F. 2022. Reverse phenotyping facilitates disease allele calling in exome sequencing of patients with CAKUT. Genet. Med. Off. J. Am. Coll. Med. Genet 24: 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ven AT, Connaughton DM, Ityel H, Mann N, Nakayama M, Chen J, Vivante A, Hwang D-Y, Schulz J, Braun DA, Schmidt JM, Schapiro D, Schneider R, Warejko JK, Daga A, Majmundar AJ, Tan W, Jobst-Schwan T, Hermle T, Widmeier E, Ashraf S, Amar A, Hoogstraaten CA, Hugo H, Kitzler TM, Kause F, Kolvenbach CM, Dai R, Spaneas L, Amann K, Stein DR, Baum MA, Somers MJG, Rodig NM, Ferguson MA, Traum AZ, Daouk GH, Bogdanović R, Stajić N, Soliman NA, Kari JA, El Desoky S, Fathy HM, Milosevic D, Al-Saffar M, Awad HS, Eid LA, Selvin A, Senguttuvan P, Sanna-Cherchi S, Rehm HL, MacArthur DG, Lek M, Laricchia KM, Wilson MW, Mane SM, Lifton RP, Lee RS, Bauer SB, Lu W, Reutter HM, Tasic V, Shril S, Hildebrandt F. 2018a. Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract. J. Am. Soc. Nephrol. JASN 29: 2348–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ven AT, Vivante A, Hildebrandt F. 2018b. Novel Insights into the Pathogenesis of Monogenic Congenital Anomalies of the Kidney and Urinary Tract. J. Am. Soc. Nephrol 29: 36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbitsky M, Westland R, Perez A, Kiryluk K, Liu Q, Krithivasan P, Mitrotti A, Fasel DA, Batourina E, Sampson MG, Bodria M, Werth M, Kao C, Martino J, Capone VP, Vivante A, Shril S, Kil BH, Marasà M, Zhang JY, Na Y-J, Lim TY, Ahram D, Weng PL, Heinzen EL, Carrea A, Piaggio G, Gesualdo L, Manca V, Masnata G, Gigante M, Cusi D, Izzi C, Scolari F, van Wijk JAE, Saraga M, Santoro D, Conti G, Zamboli P, White H, Drozdz D, Zachwieja K, Miklaszewska M, Tkaczyk M, Tomczyk D, Krakowska A, Sikora P, Jarmoliński T, Borszewska-Kornacka MK, Pawluch R, Szczepanska M, Adamczyk P, Mizerska-Wasiak M, Krzemien G, Szmigielska A, Zaniew M, Dobson MG, Darlow JM, Puri P, Barton DE, Furth SL, Warady BA, Gucev Z, Lozanovski VJ, Tasic V, Pisani I, Allegri L, Rodas LM, Campistol JM, Jeanpierre C, Alam S, Casale P, Wong CS, Lin F, Miranda DM, Oliveira EA, Simões-E-Silva AC, Barasch JM, Levy B, Wu N, Hildebrandt F, Ghiggeri GM, Latos-Bielenska A, Materna-Kiryluk A, Zhang F, Hakonarson H, Papaioannou VE, Mendelsohn CL, Gharavi AG, Sanna-Cherchi S. 2019. The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat. Genet 51: 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivante A, Hwang D-Y, Kohl S, Chen J, Shril S, Schulz J, van der Ven A, Daouk G, Soliman NA, Kumar AS, Senguttuvan P, Kehinde EO, Tasic V, Hildebrandt F. 2017. Exome Sequencing Discerns Syndromes in Patients from Consanguineous Families with Congenital Anomalies of the Kidneys and Urinary Tract. J. Am. Soc. Nephrol. JASN 28: 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuckovic Z, Gentry PR, Berizzi AE, Hirata K, Varghese S, Thompson G, van der Westhuizen ET, Burger WAC, Rahmani R, Valant C, Langmead CJ, Lindsley CW, Baell JB, Tobin AB, Sexton PM, Christopoulos A, Thal DM. 2019. Crystal structure of the M5 muscarinic acetylcholine receptor. Proc. Natl. Acad. Sci. U. S. A 116: 26001–26007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warejko JK, Tan W, Daga A, Schapiro D, Lawson JA, Shril S, Lovric S, Ashraf S, Rao J, Hermle T, Jobst-Schwan T, Widmeier E, Majmundar AJ, Schneider R, Gee HY, Schmidt JM, Vivante A, van der Ven AT, Ityel H, Chen J, Sadowski CE, Kohl S, Pabst WL, Nakayama M, Somers MJG, Rodig NM, Daouk G, Baum M, Stein DR, Ferguson MA, Traum AZ, Soliman NA, Kari JA, El Desoky S, Fathy H, Zenker M, Bakkaloglu SA, Müller D, Noyan A, Ozaltin F, Cadnapaphornchai MA, Hashmi S, Hopcian J, Kopp JB, Benador N, Bockenhauer D, Bogdanovic R, Stajić N, Chernin G, Ettenger R, Fehrenbach H, Kemper M, Munarriz RL, Podracka L, Büscher R, Serdaroglu E, Tasic V, Mane S, Lifton RP, Braun DA, Hildebrandt F. 2018. Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. CJASN 13: 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber S, Thiele H, Mir S, Toliat MR, Sozeri B, Reutter H, Draaken M, Ludwig M, Altmüller J, Frommolt P, Stuart HM, Ranjzad P, Hanley NA, Jennings R, Newman WG, Wilcox DT, Thiel U, Schlingmann KP, Beetz R, Hoyer PF, Konrad M, Schaefer F, Nürnberg P, Woolf AS. 2011. Muscarinic Acetylcholine Receptor M3 Mutation Causes Urinary Bladder Disease and a Prune-Belly-like Syndrome. Am. J. Hum. Genet 89: 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Westhuizen ET, Valant C, Sexton PM, Christopoulos A. 2015. Endogenous allosteric modulators of G protein-coupled receptors. J. Pharmacol. Exp. Ther 353: 246–260. [DOI] [PubMed] [Google Scholar]
- Yamada M, Lamping KG, Duttaroy A, Zhang W, Cui Y, Bymaster FP, McKinzie DL, Felder CC, Deng CX, Faraci FM, Wess J. 2001. Cholinergic dilation of cerebral blood vessels is abolished in M(5) muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. U. S. A 98: 14096–14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarghooni S, Wunsch J, Bodenbenner M, Brüggmann D, Grando SA, Schwantes U, Wess J, Kummer W, Lips KS. 2007. Expression of muscarinic and nicotinic acetylcholine receptors in the mouse urothelium. Life Sci 80: 2308–2313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and its online supplementary material.