

Abstract
Objectives:
To characterize the clinical manifestations and genetic basis of a familial cancer syndrome in patients with lipomas and BHD-like clinical manifestations including fibrofolliculomas and trichodiscomas and kidney cancer.
Methods:
Genomic analysis of blood and renal tumor DNA was performed. Inheritance pattern, phenotypic manifestations, and clinical and surgical management were documented. Cutaneous, subcutaneous and renal tumor pathologic features were characterized.
Results:
Affected individuals were found to be at risk for a highly penetrant and lethal form of bilateral, multifocal papillary renal cell carcinoma. Whole genome sequencing identified a germline pathogenic variant in PRDM10 (c.2029 T>C, p.Cys677Arg), which cosegregated with disease. PRDM10 loss of heterozygosity (LOH) was identified in kidney tumors. PRDM10 was predicted to abrogate expression of FLCN, a transcriptional target of PRDM10, which was confirmed by tumor expression of GPNMB, a TFE3/TFEB target and downstream biomarker of FLCN loss. In addition, a sporadic papillary RCC from the TCGA cohort was identified with a somatic PRDM10 mutation.
Conclusions:
We identified a germline PRDM10 pathogenic variant in association with a highly penetrant, aggressive form of familial papillary RCC, lipomas and fibrofolliculomas/trichodiscomas. PRDM10 LOH and elevated GPNMB expression in renal tumors indicate that PRDM10 alteration leads to reduced FLCN expression, driving TFE3-induced tumor formation. These findings suggest that individuals with BHD-like manifestations and subcutaneous lipomas, but without a germline pathogenic FLCN variant, should be screened for germline PRDM10 variants. Importantly, kidney tumors identified in patients with a pathogenic PRDM10 variant should be managed with surgical resection instead of active surveillance.
Introduction
Birt-Hogg-Dubé (BHD) syndrome is an autosomal dominant hereditary cancer disorder in which affected individuals are at risk for the development of cutaneous fibrofolliculomas and trichodiscomas, pulmonary cysts and renal tumors. BHD syndrome is caused by germline pathogenic variants in the tumor suppressor gene FLCN located on chromosome 17p11.2.1 2
Folliculin (FLCN), which functions as an amino acid sensor, is a RagC/D GTPase activating protein (GAP) that regulates the mechanistic target of rapamycin complex 1 (mTORC1) phosphorylation of the microphthalmia family transcription factors, TFE3 and TFEB. When FLCN is deficient, such as in BHD syndrome, TFE3/TFEB are no longer phosphorylated and translocate to the nucleus.3–8 TFE3/TFEB transcriptional activity in the nucleus increases RagC and RagD expression, which in turn hyperactivates mTORC1 through a feedback loop, resulting in a powerful growth signal via phosphorylation of S6K and 4EBP1.3, 9, 10 Increased nuclear localization of TFE3 and elevated expression of its downstream target, GPNMB, have been demonstrated in FLCN-deficient BHD renal tumors,8, 11 and TFEB and TFE3 have been shown to potentially be crucial drivers in BHD-associated renal tumorigenesis.3, 10 Most notably, we and others have seen papillary/clear and eosinophilic clear histologic features in some BHD renal tumors,11–15 histologies reminiscent of TFE3 and TFEB translocation fusion renal tumors.16–19
Although BHD syndrome was originally characterized by the inheritance of cutaneous fibrofolliculomas and trichodiscomas,20 we subsequently expanded the BHD clinical phenotype to include an association between the BHD dermatologic manifestations and familial renal carcinoma.21 In that initial study we identified 3 extended families in which 13 individuals presented with the BHD cutaneous manifestations, 7 of which developed renal tumors that co-segregated with BHD skin manifestations.21 Subsequent germline testing identified pathogenic FLCN variations in two of the three families; however, in Family 171, a missense FLCN variant of unknown significance (p.R239H) which did not co-segregate with the disease was found. The present study was conducted in order to characterize the clinical phenotype, management approach and genetic basis of the disease in this BHD-like family.
Here we report the identification a novel germline PRDM10 pathogenic variant, located in the same codon as was recently reported in a family with a BHD-like phenotype by van de Beek, et al.22 The PRDM10 gene encodes the PRDM10 transcription factor, a known regulator of many genes, including FLCN.23 We present the clinical manifestations and PRDM10 mutation analysis in Family 171 and demonstrate co-segregation of the PRDM10 variant with clinical phenotype in affected family members. We show homozygous loss of PRDM10 in tumors and discuss the potential relationship between PRDM10 loss and FLCN function. Here we describe the genetic basis and clinical and pathologic features a novel form of hereditary renal cell carcinoma characterized by cutaneous fibrofolliculomas and trichodiscomas, lipomas and a highly penetrant, rapidly growing, aggressive form of bilateral, multifocal papillary renal cell carcinoma.
Materials & Methods
Patients:
Patients in Family 171 were evaluated and managed at the Urologic Oncology Branch (UOB) of the National Cancer Institute (NCI), National Institutes of Health (NIH). This study was approved by the Institutional Review Board of the National Cancer Institute and patients provided informed consent on either Urologic Oncology Branch protocol NCI-89-C-0086 or NCI-97-C-0147. Patient clinical evaluation including imaging and dermatologic examination, and management, including surgical procedures were conducted at the Clinical Research Center, the Hospital at the National Institutes of Health, Bethesda, MD.
Whole Genome Sequencing:
Germline DNA from five family members with a history of renal cell carcinoma (II:4, III:2, III:5, III:7, and III:8) and tumor DNA from patients III:2 (a liver metastasis) and III:8 (two primary tumors from a left partial nephrectomy and a left flank metastasis) were subjected to whole genome sequencing (WGS). DNA was extracted from blood and tumor tissue using Promega Maxwell 16 Blood or Tissue DNA Purification Kits, or conventional phenol extraction. Human whole genome library preparation (350bp), hWGS (NovoSeq PE150, Q30≥85%, 30X coverage, 90G raw data per sample), and standard analysis were performed at Novogene Corporation Inc. (Sacramento, CA, USA). The Illumina NovaSeq 6000 sequencer (Illumina, Inc., San Diego, CA, USA) was used to carry out paired-end 150 bp sequencing.
PCR and Sanger DNA sequencing:
Additional DNA was extracted from blood and tumor tissue as above using the Maxwell RSC DNA FFPE Kit (Promega, WI, USA), or conventional phenol/chloroform extraction. PCR of PRDM10 exon 9 was performed using KOD DNA polymerase (EMD Millipore) according to the manufacturer’s specifications and utilizing the following PRDM10 primers: forward-AAACACGTGCGCAGCTTCCA; reverse-CCCAAAGAGTATCTCAGGTCCAGTTC. Bidirectional Sanger DNA sequencing of the PCR products was performed using the Big Dye Terminator v.1.1 Cycle Sequencing Kit (Applied Biosystems, CA, USA) according to the manufacturer’s specifications and run on an ABI 3130xl or 3730 Genetic Analyzer (Applied Biosystems, CA, USA). Sanger Sequencing was conducted at the CCR Genomics Core at the National Cancer Institute, NIH, Bethesda, MD 20892. Forward and reverse sequences were evaluated using Sequencher 5.0.1 (Genecodes, MI, USA) or Snap-Gene Viewer 6.1.2 (GSL Biotech, LLC).
Histology and Immunostaining:
Five-micron thick formalin-fixed paraffin-embedded sections were cut from all available tumor blocks. IHC staining and H&E staining were performed by either VitroVivo Biotech (Rockville, MD) or the Molecular Histopathology Laboratory at the Frederick National Laboratory for Cancer Research (Frederick, MD) using standardized methodologies. IHC staining was performed using either Human Osteoactivin/GPNMB anti goat (R&D Systems, Minneapolis, MN), TFE3 anti rabbit (Sigma Aldrich, Burlington, MA), Phospho-S6 Ribosomal Protein (Ser235/236) anti rabbit (Cell Signaling Technology, Danvers, MA), or Phospho-4-E-BP1 (Thr37/46) anti rabbit (Cell Signaling Technology, Danvers, MA). For selected tumors, an immunohistology panel was performed evaluating HMB45 (Agilent Dako, Carpinteria, CA), Melan-A (Abcam, Waltham, MA), Pan-cytokeratin (Novus Biologicals, Centennial, CO), Cytokeratin 7 (Abcam, Waltham, MA), Cathepsin K (Abcam, Waltham, MA), and Carbonic anhydrase IX (Cell Signaling Technology, Danvers, MA). Images were captured using an AxioScan.Z1 Slide Scanner (Zeiss, Oberkochen, DE).
Results
Members of Family 171 have been evaluated and managed at the National Cancer Institute since 1998 with multiple individuals presenting with fibrofolliculomas, lipomas and renal tumors.21 While the cutaneous phenotype of this family, including fibrofolliculomas and trichodiscomas, was consistent with that of Birt-Hogg-Dubé syndrome, a pathogenic variant of the FLCN gene which co-segregated with the disease was not identified (Supplementary Table 1).
Subsequent evaluation of this family was conducted in order to more precisely identify the clinical manifestations (Figure 1, Table 1). Five affected individuals were found to have biopsy confirmed fibrofolliculomas or trichodiscomas, the characteristic cutaneous manifestations described by Birt, Hogg and Dubé,20 with four of five affected family members presenting with multiple lesions (Table 1). One individual was found to have fibroepithelial polyps on the lip. Two affected members of the family presented with a single lung cyst and a third had a history of pneumothorax, demonstrating minimal pulmonary manifestations known to be associated with BHD. Five individuals across three generations of the family were found to have multiple subcutaneous lipomas, a phenotypic feature not traditionally associated with BHD (Table 1).
Figure 1: Family 171 pedigree.
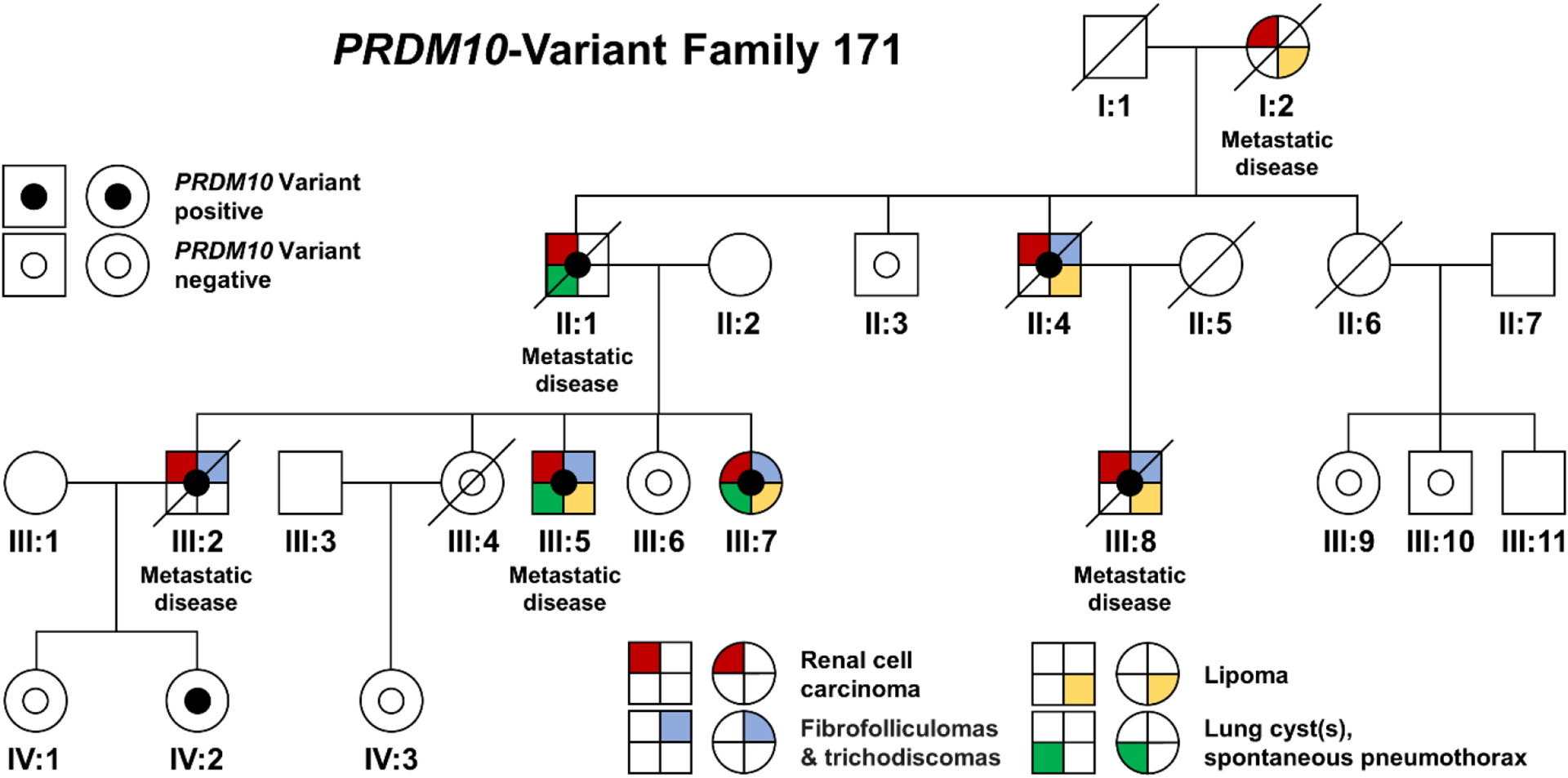
This family pedigree demonstrates the co-segregation of the p.Cys677Arg variant with clinical features. The presence of clinical features is shown by quadrants representing renal cell carcinoma (red), fibrofolliculoma or trichodiscoma (blue), lipoma (yellow), and presence of lung cysts or spontaneous pneumothorax (green). Two patients each presented with a single, unilateral lung cyst (III:5, III:7) and one patient (II:1) had a history of pneumothorax.
Table 1:
Genetic and Clinical Features of Family 171
| Patient ID | Germline PRDM10 Variant | Dermatologic and Pulmonary Manifestations | Pathology of Renal Tumors (age at surgery) | Additional Clinical Features |
|---|---|---|---|---|
| I:2 | Not tested | Lipoma | 16.0 cm left RCC with tubular cellular pattern (papillary) - No grade/stage given (68) | Metastatic disease, Died 69 yrs old |
| II:1 | p.Cys677Arg | Pneumothorax | 15.0 cm right RCC with rhabdoid morphology - Gr. 2, pT3a pN1 (69) 12.5 cm left clear cell RCC an associated tumor with vacuolated eosinophilic cytoplasm - Gr. 2, pT2b pN0 (69) |
Metastatic disease, Died 73 yrs old |
| II:3 | Negative | - | - | - |
| II:4 | p.Cys677Arg | 2 fibrofolliculomas*, 2 trichodiscomas*, lip fibro-epithelial polyp* & lipomas | 2.5 cm left papillary RCC with eosinophilic cytoplasm & clear cell features “TFE3-Like” - No grade, pT1a pNX (71) | Died 90 yrs old |
| III:2 | p.Cys677Arg | 3 fibrofolliculomas* | 4.0 cm right papillary RCC with eosinophilic cytoplasm & clear cell features - Gr. 3, pT1a pN0 (35) | Metastatic disease, Died 50 yrs old |
| III:4 | Negative | - | - | - |
| III:5 | p.Cys677Arg | 4 fibrofolliculomas* & multiple lipomas, 1 right lung cyst | 7.5 cm right RCC with papillary, eosinophilic, and focal clear cell features - Gr. 3, pT3a pN1 (69) | Metastatic disease |
| III:6 | Negative | - | - | - |
| III:7 | p.Cys677Arg | 1 fibrofolliculoma* & lipoma, 1 left lung cyst | 4.5 cm right papillary RCC with oncocytic and clear cell features (63) - Gr. 3, pT3b pN0 (63) Left 1.3 and 1.7 lesions on imaging |
- |
| III:8 | p.Cys677Arg | 2 fibrofolliculomas*, 1 trichodiscoma* & lipomas* | 2.3 cm left papillary RCC with clear cell features - Gr. 3, pT1a pNX (59) 1.7 cm left oncocytoid neoplasm - No grade , pT3 pNx (59) |
Metastatic disease, Died 61 yrs old |
| III:9 | Negative | - | - | - |
| III:10 | Negative | - | - | - |
| IV:1 | Negative | - | - | - |
| IV:2 | p.Cys677Arg | - | Left 1.5 cm cystic lesion on imaging | 30 yrs old |
| IV:3 | Negative | - | - | - |
Biopsy-proven
While hybrid oncocytic RCC (50%) and chromophobe RCC (34%) are the most common histologic tumor types known to be associated with BHD, we and others have shown that BHD patients are also at-risk for the development of papillary RCC with clear cell features and/or granular eosinophilic clear cell RCC11–13, 15, histologic subtypes commonly associated with TFE3 RCC and TFEB RCC respectively.19, 24 Individuals in Family 171 were found to have a high penetrance of the papillary/clear histologic subtypes (7 of 8 affected patients with tumors; 88%), which are much less commonly associated with BHD (Table 1). Immunohistochemical analysis performed on available tumor material showed that each tumor was positive for Cathepsin K and Pan-cytokeratin, several tumors were positive for Cytokeratin 7, and all were negative for HMB45 and Melan-A staining (Supplementary Table 2). Carbonic anhydrase IX was negative in most tumors, with the exception of a single clear cell RCC. The average age at diagnosis of kidney cancer in the family was 62 years old, ranging from 35 to 71 years old (Table 1).
Patient I:2
Patient I:2 underwent a left radical nephrectomy at 68 years of age in the 1960’s. Her tumor was >16 cm in size and demonstrated a “tubular” pattern, histologically consistent with the current diagnosis of papillary RCC. She died of metastatic renal cancer a year after her nephrectomy.
Patient II:1
Patient II:1 underwent right radical and left partial nephrectomies at 69 years of age. The right-sided tumor was a 15.0 cm RCC with rhabdoid morphology, concurrent with metastasis to an ipsilateral renal hilar lymph node; the left-sided surgery removed a 12.5 cm clear cell RCC with an associated tumor with vacuolated eosinophilic cytoplasm (a histologic subtype seen in TFEB RCC)19 (Figure 2).
Figure 2: Imaging, histopathology, and immunohistochemistry of Family 171 renal tumors.
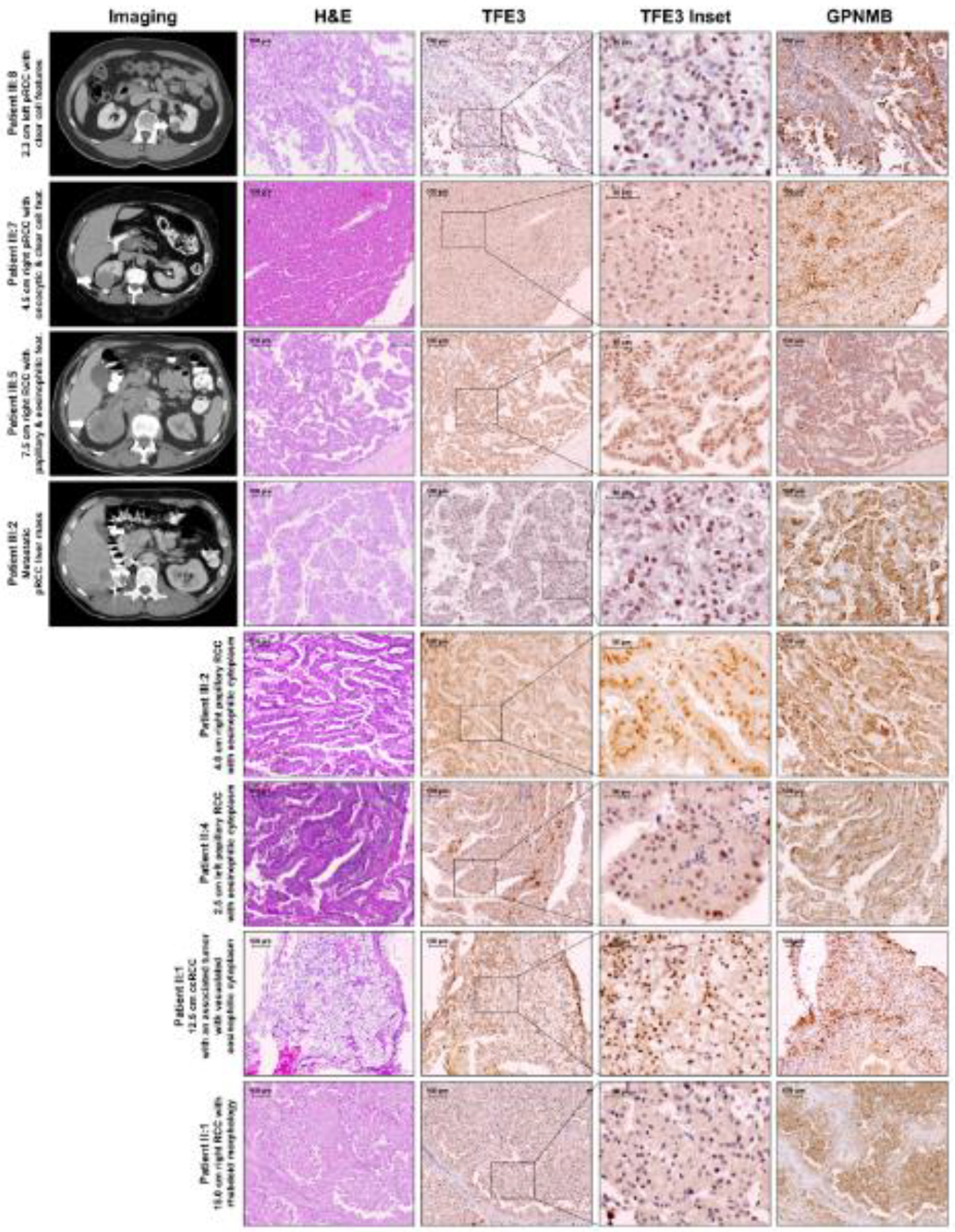
Axial imaging of patients from Family 171 showing both primary RCC (patients III:8, III:7, and III:5) and metastatic RCC (III:2) is matched to the histopathology of the excised tumors and immunohistochemistry for TFE3 and GPNMB. Histopathology, TFE3 and GPNMB staining are shown for 4 additional primary tumors for patients III:2, II:4, and II:1. These tumors demonstrate papillary histologies with clear cell and/or eosinophilic features, reminiscent of TFE3 and TFEB kidney cancer. All tumors demonstrated nuclear localization of TFE3, clearly visible by 4x magnification (TFE3 Inset) and positive GPNMB staining. pRCC – papillary renal cell carcinoma
Patient II:4
Patient II:4 underwent a left radical nephrectomy at an outside hospital at 71 years of age revealing a 2.5 cm papillary RCC with eosinophilic cytoplasm which was read at NIH as “looks like TFE3” RCC (Figure 2). Examination of the “normal” renal tissue revealed the presence of multiple small (“incipient”) lesions.
Patient III:2
Patient III:2 underwent a right radical nephrectomy at 35 years old for a 4.0 cm papillary RCC with eosinophilic cytoplasm which was noted to “resemble TFE3” RCC (Figure 2). Twelve years later multiple sites of metastasis were identified, including a 3.0 cm renal bed recurrence, a 2.7 cm hepatic mass and metastases in the diaphragm and right flank. The patient underwent surgical resection at NCI and the pathology was found to be papillary with clear cell features, similar to the primary (Figure 2, Supplementary Figure 1).
Patient III:5
Patient III:5 had a right radical nephrectomy at 69 years of age, revealing a 7.5 cm RCC with papillary, eosinophilic, and focal clear cell features. This tumor involved the renal sinus and the perinephric fat with vascular invasion and a 1.5 cm metastatic mass was identified within a hilar lymph node (Figure 2). The patient subsequently progressed to develop distant metastatic disease.
Patient III:7
Patient III:7 had a right partial nephrectomy at 63 years of age for removal of a 4.5 cm papillary RCC with oncocytic and clear cell features and focal perinephric fat invasion (Figure 2). In addition, she was found to have multiple lesions in the left kidney on abdominal imaging.
Patient III:8
Patient III:8 presented at 59 years of age with a 2.3 cm left renal mass characterized by a recent rapid growth rate of up to 5.6 mm/yr (Supplementary Figure 2). The patient underwent a left partial nephrectomy for a papillary RCC with clear cell features and a 1.7 cm oncocytoid neoplasm (Figure 2, Supplementary Figure 1). Less than 10 months after his partial nephrectomy for the 2.3 cm renal mass, the patient was found to have multiple, recurrent ipsilateral retroperitoneal metastases (Figure 3), which were resected and were found to be papillary RCC with clear cell features (Supplementary Figure 1). He died of metastatic disease at age 61.
Figure 3: Tumor growth rate and PRDM10 sequence analysis in patient III:8.
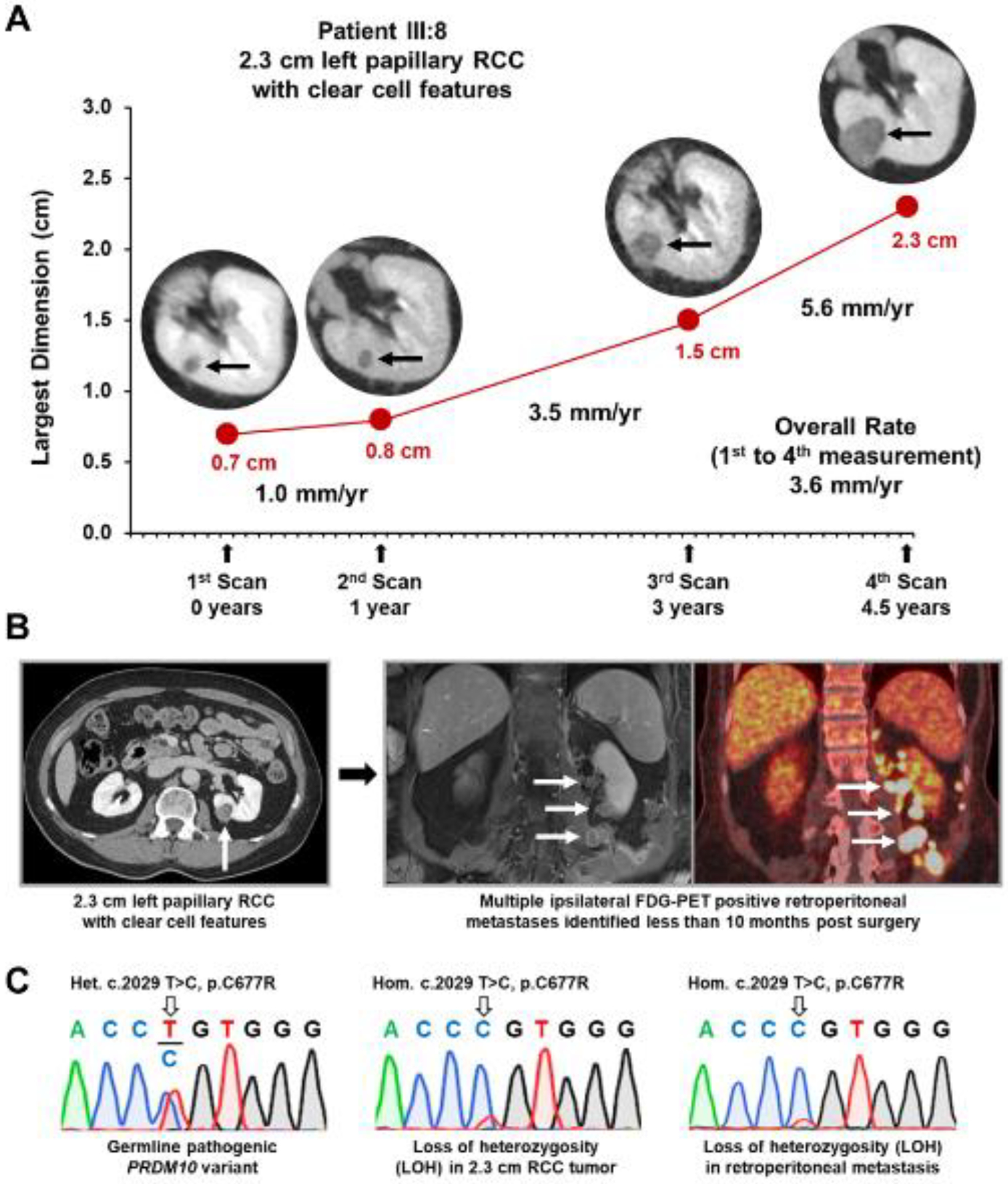
A) Patient III:8 had clinical imaging on 4 occasions before the excision of their 2.3 cm left pRCC with clear cell features. Measurements from these four events were used to calculate the overall growth rate for the tumor over the last four and a half years and demonstrated the increased growth rate that occurred in the last few years prior to surgery. B) Post-surgical surveillance imaging, including fluorodeoxyglucose (FDG)-positron emission tomography (PET), demonstrated the rapid development of multiple ipsilateral retroperitoneal metastases less than 10 months after the excision of the 2.3 cm left pRCC with clear cell features. C) Sanger sequencing demonstrates the germline nucleotide variant c.2029 T>C in PRDM10 that results in the p.Cys677Arg substitution in the PRDM10 protein in patient III:8. Tumor DNA analysis demonstrates somatic loss of heterozygosity (LOH) in both the 2.3 cm left pRCC with clear cell features and an example of the retroperitoneal metastases.
Whole genome sequencing, performed on germline DNA from 5 affected individuals, patients II:4, III:2, III:5, III:7 and III:8, led to identification of a germline PRDM10 missense variant p.Cys677Arg (c.2029 T>C) in all 5 patients (Supplementary Figure 2). No germline variants were identified in the PTEN gene that has been previously associated with familial lipomatosis (Supplementary Table 1).25 Whole genome sequencing confirmed that affected patients III:5 and III:7 did not harbor the FLCN p.R239H variant, as was previously seen by CLIA germline mutation analysis (Supplementary Table 1). Extended sequencing analysis and confirmation were carried out on germline DNA from 14 family members by Sanger sequencing (Supplementary Figure 2). Germline mutation carrier status was confirmed in 7 family members and cosegregation of the p.Cys677Arg variant with clinical manifestations was demonstrated in all 6 members over 30 years of age (Figure 1, Table 1). One family member, I:2, who had clinical phenotypic features passed away in the 1960’s, and no germline DNA was available for analysis.
The identification of a specific genetic alteration in a potential tumor suppressor gene led us to evaluate tumor tissues from this family for evidence of a “second hit” in PRDM10. Tumor DNAs from eight primary or metastatic RCCs derived from patients III:2 and III:8 were available for analysis. Loss of heterozygosity (LOH) was observed in both metastatic sites analyzed from patient III:2, and LOH was seen in 1 of 2 primary tumors and all 4 metastatic sites from patient III:8 (Figure 3, Supplementary Figure 2). Consistent with the recent in-vitro findings in HEK293 cells by van de Beek et al. and in a mouse embryo knockout model by Han et al., this study shows suppression of FLCN expression in in-vivo human PRDM10 renal tumors, as the consequence of combining the PRDM10 variant with loss of the wildtype PRDM10 allele (Figure 4A).22, 23 TaqMan PCR gene expression analysis was performed on two separate metastatic samples from patient III:2, and one primary 2.3 cm RCC and three distinct metastatic samples from patient III:8. This demonstrated a significant loss of FLCN expression in all tumor samples in comparison to the adjacent normal kidney tissue from patient III:8 and 4 unrelated normal kidney tissue samples (Figure 4B, Supplementary Figure 3). Furthermore, significant increases in expression of several known TFE3/B transcriptional target genes were demonstrated in the tumors, including RRAGC, GPNMB, NPC1, and SQSTM1. (Figure 4B, Supplementary Figure 3). Increased expression of GPNMB is a known downstream marker of FLCN loss due to activation of transcription factors TFE3/TFEB in BHD-associated RCCs and was observed in a cell line model that homozygously expressed a PRDM10 variant.8, 22, 26 Immunohistochemical staining of GPNMB was available for a broader range of cases, eight primary tumors and two metastases derived from six different patients, and all demonstrated strong positive tumor-specific staining of GPNMB with little GPNMB expression observed in adjacent normal tissues from two patients (Figure 2, Supplementary Figure 4). This observation correlated with predominately nuclear localization of TFE3 in these tumors, compared to the mostly cytoplasmic staining observed in the adjacent normal tissues (Figure 2, Supplementary Figure 4). Finally, selected tumors were evaluated for increased activity of the canonical mTOR pathway, due in part to the increased expression of RRAGC, by immunohistochemical staining for phospho-S6 and phospho-4E-BP1. All selected tumors demonstrated high levels of phospho-S6 staining that were increased in comparison to the adjacent normal tissue present in two cases (Supplementary Figure 5). The selected tumors showed varying degrees of positive phospho-4EBP1 staining that were still increased in comparison to the adjacent normal tissue present in two cases (Supplementary Figure 5).
Figure 4: Proposed schematic of PRDM10 loss resulting in FLCN suppression and GPNMB expression.
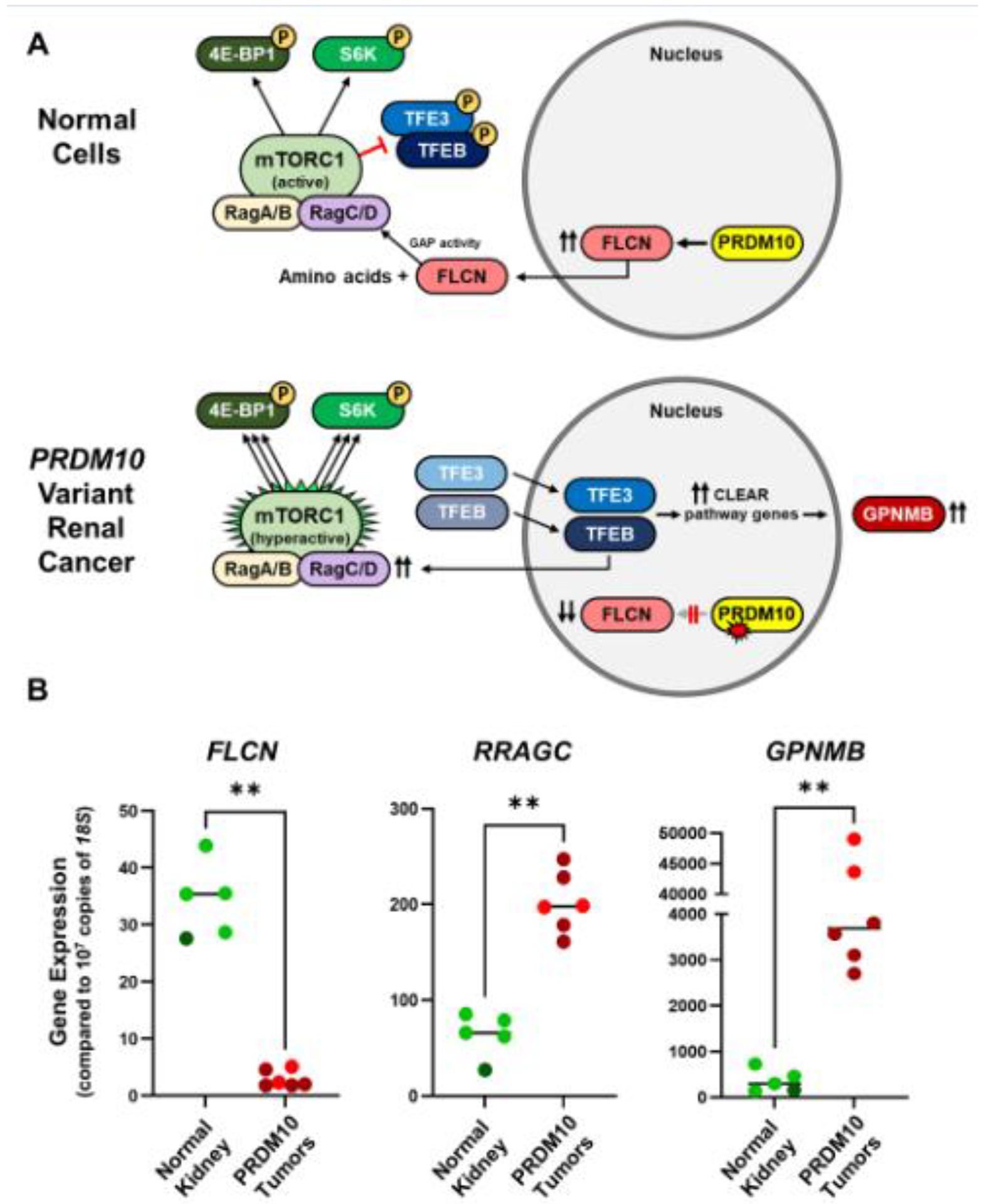
A) In normal cells, PRDM10 transcriptionally activates FLCN expression.23 The FLCN protein, in the presence of sufficient amino acids, is a GTPase activating protein (GAP) that targets RagC/D and regulates mTORC1 phosphorylation of TFE3 and TFEB resulting in their cytoplasmic sequestration. Alteration of PRDM10 in renal tumors has been shown to result in loss of FLCN expression, mimicking FLCN inactivation as seen in BHD syndrome.22 Loss of FLCN results in the inability of the mTORC1 complex to phosphorylate TFE3 and TFEB and their subsequent translocation to the nucleus to activate expression of the CLEAR (Coordinated Lysosomal Expression and Regulation) pathway genes. The known CLEAR genes, GPNMB, NPC1, SQSTM1, can be used as specific markers of CLEAR pathway activation. Overexpression of another CLEAR gene, RRAGC, results in increased levels of RagC protein, proposed to hyperactive the mTORC1 complex in BHD tumors resulting in a powerful growth signal via phosphorylation of S6K and 4EBP1.
B) Tumor cDNA was available for patients III:2 (red) and III: 8 (crimson) and this was compared to cDNA derived from the adjacent normal kidney tissue from patient III:8 (dark green ) and 4 unrelated normal kidney tissue samples (light green). Dot plots demonstrate statically significant decrease in FLCN expression and increase in RRAGC and GPNMB expression in the tumors. Mann Whitney test, ** < 0.01.
The identification of PRDM10 as a potential renal cell carcinoma predisposition gene led to a search of the RCC Cancer Genome Atlas (TCGA) project database for evidence of sporadic PRDM10 alterations. Investigation of the 283 papillary RCC tumors with DNA sequencing data available from the TCGA RCC study27 identified a sporadic type 2 papillary RCC with a somatic missense variant in the same amino acid as reported herein (p.Cys677), but resulting in a different amino acid substitution, p.Cys677Ser (c.2029 T>A) (Supplementary Figure 6). In addition, copy number analysis demonstrated a shallow deletion of this gene, consistent with LOH. This 4 cm, T3N0M0 tumor had invaded into the capsule and, post-surgery, the patient was reported as alive and free of disease at last evaluation (Supplementary Figure 6). The RNAseq data available for this cohort demonstrated that this tumor had a higher-than-average expression of GPNMB in comparison to either normal tissue or other type 2 papillary tumors (Supplementary Figure 6). The GPNMB expression level in the sporadic tumor with the PRDM10 p.Cys677Ser alteration was equivalent to RCC tumors characterized by TFE3 or TFEB fusions that are known to transcriptionally upregulate GPNMB. Notably, another papillary RCC in this cohort was found to have a somatic splice site variant in FLCN in combination with shallow gene deletion (LOH). This tumor, harboring a FLCN variant, also demonstrated a dramatic increase in GPNMB expression in a manner similar to the sporadic tumor with the PRDM10 variant (Supplementary Figure 6).
Discussion
In this report we describe a familial cancer syndrome, characterized by a germline pathogenic variant in PRDM10 and provide evidence of somatic loss of the remaining wild-type copy of PRDM10 in renal tumors, consistent with a tumor suppressor role for the PRDM10 gene. The pathogenic PRDM10 variant phenotype was associated with the canonical dermatologic features associated with BHD syndrome, fibrofolliculomas and trichodiscomas; however, we found little evidence of the pulmonary manifestations that are typically a highly penetrant feature of the BHD phenotype. The presence of subcutaneous lipomas represents a feature in PRDM10 families not found to be associated with BHD. We found no evidence of a germline pathogenic PTEN variant in Family 171, ruling out PTEN as responsible for the lipoma manifestations.
We found that patients with familial PRDM10 renal cell carcinoma (PRDM10 RCC) are at risk for the development of an aggressive and highly penetrant form of bilateral, multifocal papillary renal cell carcinoma. All seven of the affected patients over 30 years of age developed kidney cancer; five of the seven have died of metastatic disease and a sixth is currently in hospice care with metastatic RCC. In contradistinction with BHD-associated kidney cancers, PRDM10 RCC is characterized by rapid growth (up to 5.6 mm/yr). PRDM10 RCC, like TFE3 kidney cancer, has a propensity to metastasize when the primary tumor is small (2.3 cm). The high penetrance of renal cell carcinoma in Family 171 enabled a more comprehensive characterization of the renal cancer phenotype associated with a germline PRDM10 alteration and a comparison with the presentation of RCC observed in BHD patients with a germline FLCN alteration. During their lifetime, up to one-third of BHD patients are predicted to develop RCC. These patients are at risk to develop multiple tumors in both kidneys, most often with hybrid oncocytic or chromophobe histologies, with an average age of onset of 48–50 years old.2, 13, 14, 28 The affected PRDM10 variant carriers developed fewer, presumably faster growing, highly penetrant tumors at a later age of onset (average age at diagnosis 62 years old) with 7 out of 8 affected family members developing kidney tumors. In contrast to the hybrid oncocytic or chromophobe histologies that develop most often in BHD patients, the tumors that developed in family members with the PRDM10 variant were most often papillary RCCs, with some displaying clear cell and eosinophilic features reminiscent of TFE3 and TFEB kidney cancer. Moreover, patient III:8 had a 2.3 cm tumor with a rapid growth rate that subsequently metastasized. Typically, management of BHD patients involves active surveillance of slowgrowing tumors until the largest tumor reaches the 3 cm threshold, at which time surgical intervention is recommended.29 However, given rapid growth and the high rate of metastasis in this family and the fact that a small renal tumor did metastasize, we recommend consideration of careful management and early surgical intervention for patients affected with PRDM10 RCC. We recommend surgical resection with wider margins when a renal tumor is identified in a patient with this pathogenic, missense variant p.Cys677Arg. Although an aggressive form of papillary RCC was found in this family, it is possible that patients with other PRDM10 variants could have a more indolent clinical course with a less aggressive pathologic phenotype.
The missense variant p.Cys677Arg, identified in Family 171, lies in the seventh of ten predicted zinc-finger repeat domains in PRDM10. The previously reported variant, p.Cys677Tyr, located in the same codon, was demonstrated by van de Beek, at al. to cause a loss of binding affinity to the FLCN promoter, resulting in reduced expression of FLCN.22 Notably, homozygous expression of the p.Cys677Tyr variant in the HEK293T cell line model resulted in the complete loss of detectable FLCN protein, but retained expression of some other PRDM10 targets. The repeated identification of amino acid substitutions at Cys677, albeit to different amino acids, in the family described herein (p.Cys677Arg) and the sporadic tumor from the TCGA cohort (p.Cys677Ser), highlight the importance of this highly conserved amino acid residue. This suggests that alteration of this specific amino acid may be only affecting a subset of PRDM10 transcriptional targets rather than all of them. In a study by Han et al., complete knockout of Prdm10 in mouse cells showed significantly decreased expression of a wide selection of Prdm10 target genes including Flcn; however, this was associated with growth inhibition.23 Thus, it is possible that a more selective inhibition of PRDM10 transcriptional activity that includes FLCN suppression may result in a pro-tumorigenic effect.
The availability of tumor DNA from this family enabled the identification of “second hit” loss of heterozygosity in almost all samples, thereby confirming the tumor suppressive nature of this alteration. RNA expression analysis demonstrated loss of FLCN expression and subsequent increased expression of TFE3/B transcriptional target genes, including RRAGC, GPNMB, NPC1, and SQSTM1, for the first time in human RCC tumors. This was further confirmed at the protein level by immunohistochemistry demonstrating the nuclear localization of TFE3 and subsequent upregulation of GPNMB in the renal tumors from patients with the PRDM10 variant. This matches the previous evidence of elevated expression of GPNMB as a well-established marker of FLCN loss in BHD-associated renal tumors.8, 26 Evidence of increased phospho-S6 and phospho-4E-BP1 IHC staining also correlated with increased activity of the canonical mTOR pathway in response to TFE3/B transcriptional target upregulation.
The similarities in clinical presentation between individuals affected with BHD and Family 171 with germline PRDM10 alteration could be explained by loss of FLCN function in both cases. Conversely, the unique features of the clinical presentation (lipomas, unusual RCC histology) might be due to the effect of the PRDM10 variant protein on other genes in addition to FLCN. Functional analysis of the previously reported variant, p.Cys677Tyr, showed that the alteration induced increased levels of PRDM10 protein, possibly due to a negative feedback loop, and that the expression of some PRDM10 gene targets was not negatively affected by the variant but upregulated by the presence of increased PRDM10 protein.22 Notably, several gene fusions involving PRDM10 have been reported in cases of undifferentiated pleomorphic sarcoma that demonstrate distinct transcriptional signatures and suggest an oncogenic effect of increased PRDM10 activity.30 Thus, a combination of specific FLCN loss and upregulation of other PRDM10 target genes may induce the more aggressive kidney cancers observed in Family 171. The different amino acid substitutions at Cys677 observed within the two reported families may also subtly alter the effects on PRDM10 target genes and influence the clinical presentation.
This study highlights a new gene, PRDM10, responsible for a BHD syndrome-like phenotypic presentation and provides evidence supporting PRDM10 as a tumor suppressor in the setting of RCC. The phenotypic similarities suggest that individuals presenting with BHD cutaneous and renal manifestations, in whom no FLCN sequence variant has been identified, should be screened for PRDM10 germline variants, especially if lipomas are also present. The observation of a sporadic TCGA case of papillary RCC with somatic PRDM10 alteration, in combination with reports demonstrating somatic FLCN alterations31, suggests that somatic FLCN loss may also represent a rare but potentially important driver event in sporadic RCC that requires further investigation.
Conclusions
This study identified a germline alteration in the PRDM10 gene, p.Cys677Arg, in a family with an aggressive form of papillary RCC, lipomas and fibrofolliculomas/trichodiscomas. Loss of heterozygosity of the PRDM10 variant in combination with elevated GPNMB expression in the renal tumors supports the paradigm whereby PRDM10 alteration leads to reduced FLCN expression and increased TFE3/TFEB transcription driving tumor formation. These findings suggest that individuals with BHD-like manifestations should be screened for PRDM10 germline variants when no FLCN sequence variants are identified and that renal tumors identified in patients with PRDM10 variants should be managed with care and consideration given to early surgical intervention.
Supplementary Material
Supplementary Figure 1: Additional H&E staining for PRDM10 variant tumors and metastasis
H&E, GPNMB, and TFE3 staining for additional primary kidney tumor and a metastasis for patient III:8 show positive GPNMB staining and nuclear localization of TFE3, clearly visible by 4x magnification (TFE3 Inset).
Supplementary Figure 2: Sequence analysis of PRDM10 in germline and tumor DNA
Sanger sequencing evaluated the presence of germline nucleotide variant c.2029 T>C in PRDM10 that results in the p.Cys677Arg substitution in the PRDM10 protein. Patients II:1, II:4, III:2, III:5, III:7, III:8 and IV:2 all inherit the germline heterozygous variant. Patient II:4 inherits the wild-type PRDM10 allele. Metastatic tumor DNA from patients III:2 and III:8 was sequenced and demonstrated somatic loss of heterozygosity (LOH), although no LOH was observed in the patient III:8’s second lesion, a 1.7 cm oncocytoid neoplasm. Other family members, II:3, III:4, III:6, III:9, III:10, IV:1, and IV:3, did not inherit the germline variant in PRDM10. The metastatic lesions from both III:2 and III:8 all demonstrate loss of heterozygosity (LOH) of the PDRM10 variant.
Supplementary Figure 3: RT-PCR expression analysis of FLCN and TFE3/B transcription target genes in PRDM10 variant tumors
Tumor cDNA was available for patients III:2 (red) and III: 8 (crimson) and this was compared to cDNA derived from the adjacent normal kidney tissue from patient III:8 (dark green ) and 4 unrelated normal kidney tissue samples (light green). Histograms demonstrated the relative expression in comparison adjacent normal kidney tissue from patient III:8 of FLCN and 4 downstream TFE3/B transcriptional targets, RRAGC, GPNMB, NPC1, and SQSTM1 (that encodes p62). Dot plots for NPC1 and SQSTM1 demonstrate statically significant increase in expression in the tumors. Mann Whitney test, ** < 0.01.
Supplementary Figure 4: Evaluation of GPNMB immunohistochemistry in adjacent normal tissues
Formalin fixed surgically excised tumor tissues from both patients II:4 and III:2 contained adjacent normal tissue that are shown by H&E staining. TFE3 immunohistochemistry shows that the adjacent normal tissues had positive cytoplasmic TFE3 staining in a fraction of the normal cells in both cases, compared to the stronger, mostly nuclear TFE3 staining in the tumors, GPNMB immunohistochemistry demonstrates a clear distinction in between the positive GPNMB staining in the tumor tissue and the negative staining in adjacent normal tissues in both patients. In patient III:2 the differences in TFE3 and GPNMB staining can be clearly seen at the tumornormal boundary.
Supplementary Figure 5: Evaluation of pS6 and p4E-BP1 immunohistochemistry in PRDM10 variant tumors and adjacent normal tissues
Formalin fixed surgically excised tumor tissues from patients II:1, II:4, III:2 and III:8 and adjacent normal tissue for patients II:1 and III:2 was immunohistochemical staining for phospho-S6 ribosomal protein (pS6) and phospho-eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (p4E-BP1). A) pS6 immunohistochemistry demonstrated strong positive staining in all tumors with less signal present in the adjacent normal tissues. B) p4E-BP1 immunohistochemistry demonstrated a varying degree of positive staining across the tumors with little signal present in the adjacent normal tissues. T – tumor, N – normal.
Supplementary Figure 6: TCGA case of pRCC with somatic PRDM10 variant and GPNMB expression
A. Evaluation of the Cancer Genome Atlas (TCGA) papillary renal cell carcinoma cohort identified a single type 2 pRCC tumor that had a somatic variant of PRDM10 (c.2029 T>A, p.Cys677Ser) in the same amino acid as observed in Family 171. PRDM10 was also reported to have a shallow deletion in this tumor, suggestive of a second hit. This 4 × 4 × 3.5 cm tumor invaded into the capsule and was staged as T3N0M0. The tumor was described as high density and composed of trabeculae or elongated tubules or papillary, back-to-back, varying in shape and size, lined by moderately stratified cylindrical or cuboidal cells. The nuclei were irregular large, hyperchromatic with prominent nucleoli and the cytoplasm is moderate, eosinophilic, or clear. Similarly, a single unclassified papillary tumor had a somatic FLCN splice site variant (X513_Splice) in combination with a shallow deletion, suggestive of somatic FLCN loss.
B. Comparison of the available RNAseq data demonstrated that the GPNMB expression in both the PRDM10 and FLCN variant tumors was increased to a level above all normal tissues and most type 1 and type 2 pRCC tumors, but similar to the levels shown for TFE3 or TFEB altered tumors.
Acknowledgements
This research was supported in part by the National Institutes of Health. This project has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261201500003I. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. We would like to thank the CCR Sequencing Facility at Frederick National Laboratory for Cancer Research for performing the RNA-Seq library preparation and sequencing. Sanger Sequencing was conducted at the CCR Genomics Core at the National Cancer Institute, NIH, Bethesda, MD 20892.
Declaration of Competing Interest
Manuscript #:URL-D-22-02221
Title: “PRDM10 RCC: A Birt-Hogg-Dubé-like Syndrome Associated with Lipoma and a Highly Penetrant, Aggressive Form of Papillary Renal Cell Carcinoma”
Corresponding Author: Dr. W. Marston Linehan
Remaining Authors: Laura S. Schmidt, Ph.D.; Cathy D. Vocke, Ph.D.; Christopher J. Ricketts, Ph.D.; Zoe Blake, M.D.; Kristin K. Choo; Deborah Nielsen, R.N., B.S.N.; Rabindra Gautam, Ph.D.; Daniel R. Crooks, Ph.D.; Krista L. Reynolds; Janis L. Krolus; Meena Bashyal; Baktiar Karim, D.V.M., Ph.D.; Edward W. Cowen, M.D.; Ashkan A. Malayeri, M.D.; Maria J. Merino, M.D.; Ramaprasad Srinivasan, M.D., Ph.D.; Mark W. Ball, M.D.; Berton Zbar, M.D.
Submitted to: UROLOGY
Please copy this Conflict of Interest form and paste into your word processing software, type in each author and either indicate “no conflict” or specify any conflicts; it will be required that you submit the completed form (with all authors indicated) with the revised manuscript. Please list only conflicts of interest specific to this manuscript.
Examples of Conflict of Interest:
Source of Funding
Paid consultant to Sponsor
Study Investigator Funded by Sponsor
Employee of Sponsor
Board Membership with Sponsor
Stock Holder for Mentioned Product/Company
Patent Inventor for Mentioned Product
Any Financial Relationship to Competitors of Mentioned Product
Other (please specify)
This information will be kept confidential. The Editor will discuss the method of disclosure of any potential conflict of interest with the corresponding author on an individual basis.
Author Specify “No Conflict” or indicate specific conflict e.g. John Doe no conflict
e.g. Jane Doe paid consultant to company “x”
| 1) Laura S. Schmidt | No Conflict |
| 2) Cathy D. Vocke | No Conflict |
| 3) Christopher J. Ricketts | No Conflict |
| 4) Zoe Blake | No Conflict |
| 5) Kristin K. Choo | No Conflict |
| 6) Deborah Nielsen | No Conflict |
| 7) Rabindra Gautam | No Conflict |
| 8) Daniel R. Crooks | No Conflict |
| 9) Krista L. Reynolds | No Conflict |
| 10) Janis L. Krolus | No Conflict |
| 11) Meena Bashyal | No Conflict |
| 12) Baktiar Karim | No Conflict |
| 13) Edward W. Cowen | No Conflict |
| 14) Ashkan A. Malayeri | No Conflict |
| 15) Maria J. Merino | No Conflict |
| 16) Ramaprasad Srinivasan | No Conflict |
| 17) Mark W. Ball | No Conflict |
| 18) Berton Zbar | No Conflict |
| I accept the responsibility for the completion of this document and attest to its validity on behalf of the co-authors. | |
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
We have nothing to declare.
References
- 1.Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2:157–164. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dube syndrome. Nat Rev Urol. 2015;12:558–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Napolitano G, Di Malta C, Esposito A, et al. A substrate-specific mTORC1 pathway underlies Birt-Hogg-Dube syndrome. Nature. 2020;585:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jansen RM, Peruzzo R, Fromm SA, Yokom AL, Zoncu R, Hurley JH. Structural basis for FLCN RagC GAP activation in MiT-TFE substrate-selective mTORC1 regulation. Sci Adv. 2022;8:eadd2926 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Napolitano G, Di Malta C, Ballabio A. Non-canonical mTORC1 signaling at the lysosome. Trends Cell Biol. 2022;32(11):920–931. [DOI] [PubMed] [Google Scholar]
- 6.Shen K, Rogala KB, Chou HT, Huang RK, Yu Z, Sabatini DM. Cryo-EM Structure of the Human FLCN-FNIP2-Rag-Ragulator Complex. Cell. 2019;179:1319–1329 e1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lawrence RE, Fromm SA, Fu Y, et al. Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science. 2019;366:971–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong SB, Oh H, Valera VA, Baba M, Schmidt LS, Linehan WM. Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLoS One. 2010;5:e15793 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Malta C, Siciliano D, Calcagni A, et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017;356:1188–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li K, Wada S, Gosis BS, Thorsheim C, Loose P, Arany Z. Folliculin promotes substrate-selective mTORC1 activity by activating RagC to recruit TFE3. PLoS Biol. 2022;20:e3001594 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiMalta C, Granieri L, Vilardo C, et al. TFEB AND TFE3 DRIVE KIDNEY CYSTOGENESIS AND TUMORIGENESIS. EMBO Mol Med. 2023;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pavlovich CP, Walther MM, Eyler RA, et al. Renal tumors in the Birt-Hogg-Dub‚ syndrome. Am J Surg Pathol. 2002;26:1542–1552. [DOI] [PubMed] [Google Scholar]
- 13.Pavlovich CP, Grubb RL 3rd, Hurley K, et al. Evaluation and management of renal tumors in the Birt-Hogg-Dube syndrome. J Urol. 2005;173:1482–1486. [DOI] [PubMed] [Google Scholar]
- 14.Benusiglio PR, Giraud S, Deveaux S, et al. Renal cell tumour characteristics in patients with the Birt-Hogg-Dube cancer susceptibility syndrome: a retrospective, multicentre study. Orphanet J Rare Dis. 2014;9:163 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bakhati B, Perez Del Nogal G, Salinas I, Bajaj K. Birt-Hogg-Dube Syndrome: Two Patients With Different Initial Presentations. Cureus. 2022;14:e30578 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sidhar SK, Clark J, Gill S, et al. The t(X;1)(p11.2;q21.2) translocation in papillary renal cell carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene. Hum Mol Genet. 1996;5:1333–1338. [DOI] [PubMed] [Google Scholar]
- 17.Argani P, Olgac S, Tickoo SK, et al. Xp11 translocation renal cell carcinoma in adults: expanded clinical, pathologic, and genetic spectrum. Am J Surg Pathol. 2007;31:11491160. [DOI] [PubMed] [Google Scholar]
- 18.Zhong M, De Angelo P, Osborne L, et al. Translocation renal cell carcinomas in adults: a single-institution experience. Am J Surg Pathol. 2012;36:654–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis IJ, Hsi BL, Arroyo JD, et al. Cloning of an Alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc Natl Acad Sci U S A. 2003;100:6051–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Birt AR, Hogg GR, Dube WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674–1677. [PubMed] [Google Scholar]
- 21.Toro JR, Glenn G, Duray P, et al. Birt-Hogg-Dube syndrome: a novel marker of kidney neoplasia. Arch Dermatol. 1999;135:1195–1202. [DOI] [PubMed] [Google Scholar]
- 22.van de Beek I, Glykofridis IE, Oosterwijk JC, et al. PRDM10 directs FLCN expression in a novel disorder overlapping with Birt-Hogg-Dube syndrome and familial lipomatosis. Hum Mol Genet. 2022;ddac288 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han BY, Seah MKY, Brooks IR, et al. Global translation during early development depends on the essential transcription factor PRDM10. Nat Commun. 2020;11:3603 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Argani P, Lae M, Hutchinson B, et al. Renal carcinomas with the t(6;11)(p21;q12): clinicopathologic features and demonstration of the specific alpha-TFEB gene fusion by immunohistochemistry, RT-PCR, and DNA PCR. Am J Surg Pathol. 2005;29:230–240. [DOI] [PubMed] [Google Scholar]
- 25.Hanssen AM, Fryns JP. Cowden syndrome. J Med Genet. 1995;32:117–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furuya M, Hong SB, Tanaka R, et al. Distinctive expression patterns of glycoprotein nonmetastatic B and folliculin in renal tumors in patients with Birt-Hogg-Dube syndrome. Cancer Sci. 2015;106:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ricketts CJ, De Cubas AA, Fan H, et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018;23:313–326 e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zbar B, Alvord WG, Glenn G, et al. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dube syndrome. Cancer Epidemiol Biomarkers Prev. 2002;11:393–400. [PubMed] [Google Scholar]
- 29.Stamatakis L, Metwalli AR, Middelton LA, Marston Linehan W. Diagnosis and management of BHD-associated kidney cancer. Fam Cancer. 2013;12:397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofvander J, Puls F, Pillay N, et al. Undifferentiated pleomorphic sarcomas with PRDM10 fusions have a distinct gene expression profile. J Pathol. 2019;249:425–434. [DOI] [PubMed] [Google Scholar]
- 31.Khoo SK, Kahnoski K, Sugimura J, et al. Inactivation of BHD in sporadic renal tumors. Cancer Res. 2003;63:4583–4587. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Additional H&E staining for PRDM10 variant tumors and metastasis
H&E, GPNMB, and TFE3 staining for additional primary kidney tumor and a metastasis for patient III:8 show positive GPNMB staining and nuclear localization of TFE3, clearly visible by 4x magnification (TFE3 Inset).
Supplementary Figure 2: Sequence analysis of PRDM10 in germline and tumor DNA
Sanger sequencing evaluated the presence of germline nucleotide variant c.2029 T>C in PRDM10 that results in the p.Cys677Arg substitution in the PRDM10 protein. Patients II:1, II:4, III:2, III:5, III:7, III:8 and IV:2 all inherit the germline heterozygous variant. Patient II:4 inherits the wild-type PRDM10 allele. Metastatic tumor DNA from patients III:2 and III:8 was sequenced and demonstrated somatic loss of heterozygosity (LOH), although no LOH was observed in the patient III:8’s second lesion, a 1.7 cm oncocytoid neoplasm. Other family members, II:3, III:4, III:6, III:9, III:10, IV:1, and IV:3, did not inherit the germline variant in PRDM10. The metastatic lesions from both III:2 and III:8 all demonstrate loss of heterozygosity (LOH) of the PDRM10 variant.
Supplementary Figure 3: RT-PCR expression analysis of FLCN and TFE3/B transcription target genes in PRDM10 variant tumors
Tumor cDNA was available for patients III:2 (red) and III: 8 (crimson) and this was compared to cDNA derived from the adjacent normal kidney tissue from patient III:8 (dark green ) and 4 unrelated normal kidney tissue samples (light green). Histograms demonstrated the relative expression in comparison adjacent normal kidney tissue from patient III:8 of FLCN and 4 downstream TFE3/B transcriptional targets, RRAGC, GPNMB, NPC1, and SQSTM1 (that encodes p62). Dot plots for NPC1 and SQSTM1 demonstrate statically significant increase in expression in the tumors. Mann Whitney test, ** < 0.01.
Supplementary Figure 4: Evaluation of GPNMB immunohistochemistry in adjacent normal tissues
Formalin fixed surgically excised tumor tissues from both patients II:4 and III:2 contained adjacent normal tissue that are shown by H&E staining. TFE3 immunohistochemistry shows that the adjacent normal tissues had positive cytoplasmic TFE3 staining in a fraction of the normal cells in both cases, compared to the stronger, mostly nuclear TFE3 staining in the tumors, GPNMB immunohistochemistry demonstrates a clear distinction in between the positive GPNMB staining in the tumor tissue and the negative staining in adjacent normal tissues in both patients. In patient III:2 the differences in TFE3 and GPNMB staining can be clearly seen at the tumornormal boundary.
Supplementary Figure 5: Evaluation of pS6 and p4E-BP1 immunohistochemistry in PRDM10 variant tumors and adjacent normal tissues
Formalin fixed surgically excised tumor tissues from patients II:1, II:4, III:2 and III:8 and adjacent normal tissue for patients II:1 and III:2 was immunohistochemical staining for phospho-S6 ribosomal protein (pS6) and phospho-eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (p4E-BP1). A) pS6 immunohistochemistry demonstrated strong positive staining in all tumors with less signal present in the adjacent normal tissues. B) p4E-BP1 immunohistochemistry demonstrated a varying degree of positive staining across the tumors with little signal present in the adjacent normal tissues. T – tumor, N – normal.
Supplementary Figure 6: TCGA case of pRCC with somatic PRDM10 variant and GPNMB expression
A. Evaluation of the Cancer Genome Atlas (TCGA) papillary renal cell carcinoma cohort identified a single type 2 pRCC tumor that had a somatic variant of PRDM10 (c.2029 T>A, p.Cys677Ser) in the same amino acid as observed in Family 171. PRDM10 was also reported to have a shallow deletion in this tumor, suggestive of a second hit. This 4 × 4 × 3.5 cm tumor invaded into the capsule and was staged as T3N0M0. The tumor was described as high density and composed of trabeculae or elongated tubules or papillary, back-to-back, varying in shape and size, lined by moderately stratified cylindrical or cuboidal cells. The nuclei were irregular large, hyperchromatic with prominent nucleoli and the cytoplasm is moderate, eosinophilic, or clear. Similarly, a single unclassified papillary tumor had a somatic FLCN splice site variant (X513_Splice) in combination with a shallow deletion, suggestive of somatic FLCN loss.
B. Comparison of the available RNAseq data demonstrated that the GPNMB expression in both the PRDM10 and FLCN variant tumors was increased to a level above all normal tissues and most type 1 and type 2 pRCC tumors, but similar to the levels shown for TFE3 or TFEB altered tumors.