

Abstract
Background
Pathological cardiac hypertrophy is a major cause of heart failure morbidity. The complex mechanism of intermolecular interactions underlying the pathogenesis of cardiac hypertrophy has led to a lack of development and application of therapeutic methods.
Methods and Results
Our study provides the first evidence that TRAF4, a member of the tumor necrosis factor receptor–associated factor (TRAF) family, acts as a promoter of cardiac hypertrophy. Here, Western blotting assays demonstrated that TRAF4 is upregulated in cardiac hypertrophy. Additionally, TRAF4 deletion inhibits the development of cardiac hypertrophy in a mouse model after transverse aortic constriction surgery, whereas its overexpression promotes phenylephrine stimulation‐induced cardiomyocyte hypertrophy in primary neonatal rat cardiomyocytes. Mechanistically, RNA‐seq analysis revealed that TRAF4 promoted the activation of the protein kinase B pathway during cardiac hypertrophy. Moreover, we found that inhibition of protein kinase B phosphorylation rescued the aggravated cardiomyocyte hypertrophic phenotypes caused by TRAF4 overexpression in phenylephrine‐treated neonatal rat cardiomyocytes, suggesting that TRAF4 may regulate cardiac hypertrophy in a protein kinase B–dependent manner.
Conclusions
Our results revealed the regulatory function of TRAF4 in cardiac hypertrophy, which may provide new insights into developing therapeutic and preventive targets for this disease.
Keywords: AKT pathway, cardiac hypertrophy, heart failure, phenylephrine, primary neonatal rat cardiomyocytes, TRAF4, transverse aortic constriction
Subject Categories: Hypertrophy, Remodeling, Myocardial Infarction
Nonstandard Abbreviations and Acronyms
- AKT
protein kinase B
- iAKT
inhibition of AKT phosphorylation
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- NRCMs
neonatal rat cardiomyocytes
- TAC
transverse aortic constriction
- WB
Western blotting
Research Perspective.
What Is New?
This study for the first time found the regulatory effect of TRAF4 (tumor necrosis factor receptor‐associated factor 4) knockout in mice and TRAF4 overexpression in cardiomyocytes on myocardial hypertrophy.
TRAF4 could regulate cardiac hypertrophy in a protein kinase B–dependent manner.
What Question Should Be Addressed Next?
According to the effect of TRAF4 on cardiac hypertrophy, it might be used as a new potential therapy target in the future clinical research for myocardial hypertrophy.
Cardiac hypertrophy is a compensatory response to various mechanical and neurohormonal stimuli, such as hypertension, chronic pressure overload, valvular heart diseases, myocardial infarction, and endocrine disorders. 1 , 2 , 3 However, sustained or excessive cardiac hypertrophy can progress to a decompensated state, which induces ventricular contractile dysfunction. This eventually leads to heart failure, one of the most common causes of mortality, and currently lacks efficient therapy. 4 During this intricate transition, cardiac hypertrophy is accompanied by fibrosis, arrhythmia, and valvular dysfunction owing to the inability to meet the biomechanical load. 5 Although the molecular mechanism involved in cardiac hypertrophy has been elucidated at multiple levels, effective pharmacological targets for treatment are still lacking. 6
Tumor necrosis factor receptor–associated factor (TRAF) family members, characterized by the TRAF homologous structural domain at the C‐terminus, RING structure at the N‐terminus, and zinc‐finger domains, are a group of linker molecules in the tumor necrosis factor superfamily and the Toll‐like receptor/interleukin‐1 receptor superfamily. 7 The regulatory function of the TRAF family is involved in a wide range of biological processes, including cell proliferation, differentiation, apoptosis, survival, and immune response. 8 TRAF4, an E3 ubiquitin ligase and member of the TRAF family, plays a crucial role in individual development. 9 Unlike other TRAF family members, TRAF4 migrates between the cytomembrane, cytoplasm, and nucleus through indirect action or formation of complexes to participate in the transduction of multiple signaling pathways, such as NF‐κB (nuclear factor kappa‐B), c‐Jun N‐terminal kinase, and AKT (protein kinase B). 10 , 11 , 12 However, the role of TRAF4 in cardiac hypertrophy has not yet been reported.
In this study, we identified that the expression of TRAF4 was increased in cardiac hypertrophy models both in vitro and in vivo. Genetic ablation of TRAF4 attenuated transverse aortic constriction (TAC)–induced cardiac hypertrophy in mice. In contrast, TRAF4 overexpression aggravates the hypertrophic phenotype induced by phenylephrine in neonatal rat cardiomyocytes (NRCMs). Furthermore, the AKT signaling pathway was confirmed to be strongly associated with the regulatory function of TRAF4 in cardiac hypertrophy process via systematic investigation. Importantly, inhibition of the AKT pathway reversed the exacerbated hypertrophic phenotype resulting from TRAF4 overexpression. Our results showed that TRAF4 activates the AKT signaling pathway to aggravate cardiac hypertrophy.
MATERIALS AND METHODS
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Traf4‐KO Mice Construction
The guide sequences of the target DNA region‐guide RNA1 (GGGTACTAAACGACCCACAAA) and RNA2 (CACCAGGCCAGTTTGCAAACGC) were predicted using the CRISPR online design tool (http://chopchop.cbu.uib.no/) and constructed into TRAF4‐sgRNA expression vectors with a skeleton vector of pUC57‐sgRNA (51132, Addgene). After mixing the purified products of in vitro transcription of the Cas9 expression vector pST1374‐Cas9 (Addgene 44758) and TRAF4‐sgRNA expression vector, the mixture was injected into a single cell of fertilized eggs from C57BL/6 mice using a FemtoJet 5247 microinjection system. Surrogate female mice were transplanted with the injected fertilized eggs and gave birth to F0 generation after ≈19 to 21 days of pregnancy. Genomic DNA was extracted from the toe tissue of 2‐week‐old mice and used for genotype identification using the following primers.
TRAF4‐check F1: 5′‐CAGCATGCCTGTTTCTTTCA‐3′,
TRAF4‐check R1: 5′‐ GCCCTCGTGACTCTGAAATC‐3′.
Mice TAC Surgery
TAC surgery was used to establish a pressure, overload‐induced cardiac hypertrophy model. Considering the protective effect of estrogen on myocardial hypertrophy, we selected male mice for the following study. Randomly selected male mice aged 9 to 11 weeks with body weights of 25.5 to 27 g were anesthetized. After the toe contractile reflex disappeared and the mice breathed smoothly and evenly, they were placed in the supine position on the automatic adjusting heating pad at 37 °C and fixed. After thoracotomy at the junction of the clavicle and thoracic vertebra, cutting open and using forceps to penetrate the incision and tear the muscles on both sides, separating the thymus glands on both sides and exposing the aortic arch, 7‐0 silk thread was passed through the aortic arch. The 26‐G cushion needle was placed parallel to the top of the aortic arch. After threading the cushion needle with silk thread and ligating the aortic arch, the cushion needle was pulled out to narrow the aortic arch. Mice in the sham group underwent identical operations in parallel without ligation. After the operation, the skin at the opening was sutured and the mice were placed in a 37 °C incubator to wake up.
All animal protocols were approved by the Animal Care and Use Committee of Zhongnan Hospital of Wuhan University.
Mice Echocardiography
Mice were anesthetized by inhalation of isoflurane (1.5%–2%) and then fixed in a supine position on a thermostatic plate. Echocardiography was performed using a small‐animal ultrasound imaging system (VEVO2100, FUJIFILM VISUALSONICS, Canada) with a 30‐MHz probe (MS400). Echograms of left ventricular volume and left ventricular wall thickness were obtained at the papillary muscle for 3 consecutive cardiac cycles, and parameters including left ventricular end‐systolic diameter, left ventricular end‐diastolic diameter, left ventricular ejection fraction, and fraction shortening were measured.
Mice Dissection
The mice were euthanized and weighed 4 weeks after TAC or sham surgery. Then, the hearts of mice were excised and placed in a 10% KCl solution to stop in the diastolic phase. The heart was weighed and photographed before placing in 10% formalin or liquid nitrogen. Subsequently, the tibia was isolated, and its length was measured.
Histological Staining
After fixation in 10% formalin for 72 hours, left ventricles of mice were embedded in paraffin. The paraffin‐embedded mouse heart was cut into 5‐μm sections. After hematoxylin and eosin (hematoxylin, G1004, Servicebio; eosin, BA‐4024, Baso) and picrosirius red (PSR, 26357‐02, Hedebiotechnology) staining, the cardiomyocyte cross‐sectional areas and fibrotic collagen areas were calculated using a digital image analysis system (Image‐Pro Plus Version 6.0).
Western Blotting
The left ventricular tissues or cells were lysed with RIPA lysis solution (720 μL RIPA buffer, 20 μL phenylmethylsulfonyl fluoride, 100 μL complete protease inhibitor cocktail, 100 μL Phos‐stop, 50 μL NaF, and 10 μL Na3VO4 in a final volume of 1 mL), and centrifuged at high speed to obtain the supernatant containing total protein. After quantification using a BCA kit (Pierce), the equal quantities of protein were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to 0.45‐μm polyvinylidene difluoride membrane (IPVH00010, Millipore). The polyvinylidene difluoride membrane was incubated with indicated primary antibodies at 4 °C overnight after blocking with 5% skim milk for 1 hour at room temperature, followed by washing with Tris buffered saline with Tween 20 three times, 5 minutes each. After washing with Tris buffered saline with Tween 20 and incubation with secondary antibodies of the corresponding species (Jackson ImmunoResearch) for 1 hour at room temperature, protein signals were detected using a Bio‐Rad gel imaging system (ChemiDoc XRS+). Image Lab software (version 5.1) was used to analyze the Western blotting (WB) images. The antibodies used are listed in Table S1.
Quantitative Polymerase Chain Reaction
For the quantitative polymerase chain reaction (qPCR) assay, total RNA was extracted from the left ventricular tissues or cells using TRIzol reagent (15596–026, Invitrogen) and reverse‐transcribed into cDNA using the Transcriptor First Strand cDNA Synthesis Kit (04896866001, Roche). Subsequently, SYBR Green PCR Master Mix (04887352001, Roche) was used to detect the expression levels of the genes in the qPCR instrument (Roche), with GAPDH as the internal reference. Primers used for qPCR are listed in Table S2.
Identification of Adenovirus
The adenovirus used for TRAF4 overexpression in cardiomyocytes was purchased from Hanbio Technology Co., Ltd. (Shanghai, China). Using green fluorescent protein expression adenovirus as a control, cardiomyocytes were infected with adenovirus with a multiplicity of infection of 50 particles per cell for 12 hours, followed by identification.
Primary NRCM Isolation and Culture
After removing the blood vessels of isolated hearts, heart tissue from 1‐ to 2‐day‐old Sprague–Dawley neonatal rats were finely minced into 1 to 2 mm3 fragments and digested with 0.125% trypsin to obtain NRCMs. NRCMs were cultured in the DMEM/F12 (C11330500BT, Gibco) medium containing 10% fetal bovine serum (10099141C, GIBCO), 1% penicillin/streptomycin (15140‐122, GIBCO), and 5‐bromodeoxyuridine (0.1 mM, B5002, Sigma) to inhibit fibroblast proliferation. After 24 hours, the cells were infected with adenoviruses and cultured with serum‐free medium for 12 hours. Then, 50 μM phenylephrine (PHR1017, Sigma) or phosphate‐buffered saline, and the inhibitor of phosphorylated AKT (iAKT‐mk‐2206, 1 μmol/L, MedChemExpress) to inhibit phosphorylated AKT were added for 24 hours. The whole process of cell culture was under conditions of 37.0 °C and 5% CO2.
Immunofluorescence Staining of α‐Actinin
After NRCM were incubated with serum‐free medium for 12 hours and then stimulated with phenylephrine for 24 hours, NRCMs were fixed with 4% formaldehyde for 30 minutes, permeabilized with 0.2% Triton X‐100 for 5 minutes, and blocked with 8% goat serum at 37 °C. Next, NRCMs were incubated with primary antibodies against α‐Actinin (05‐384, Merck Millipore, 1:100 dilution) and secondary antibody (donkey anti‐mouse IgG [H + L] secondary antibody, A21202, Invitrogen, 1:200 dilution), respectively. Finally, the NRCMs were sealed with an antifade mounting medium containing DAPI. The images were analyzed using a digital image analysis system (Image‐Pro Plus Version 6.0) to calculate the cell surface area.
RNA Sequencing and Transcriptome Analysis
For RNA sequencing, total RNA extracted from the heart tissues of Traf4‐KO and wild‐type (WT) mice 4 weeks after TAC surgery was used to construct cDNA libraries. MGISEQ‐2000 RS was used for RNA sequencing of a single‐ended library with a reading length of 50 bp. HISAT2 software was used to map the sequence fragments to the mouse reference genome (MM10/GRCm38), and SAMtools then transformed the resulting files into Binary Alignment Map format that can store the comparison information. The fragments per kilobase of exon model per million mapped fragments value of each identified gene was calculated using StringTie with default parameters. DESeq2 was used to identify differentially expressed genes based on the following 2 criteria: (1) a fold change larger than 2, and (2) the corresponding adjusted P values <0.05.
Hierarchical Clustering Analysis
In the hierarchical clustering analysis, the unweighted pair group method with arithmetic mean algorithm was used to establish a hierarchical nested clustering tree by calculating the similarity between different samples, and the hclust function of the R package was used for visualization.
Gene Set Enrichment Analysis
In the Gene Set Enrichment Analysis, genes were sorted according to the level of differential expression, and gene sets based on the Gene Ontology database pathway were examined to determine whether they were concentrated at the top or bottom of the sorting list to investigate the overall expression changes. Java Gene Set Enrichment Analysis was used to perform Gene Set Enrichment Analysis with the Signal2Noise metric. Gene sets with P values <0.05 and false discovery rate <0.25 were considered statistically significant.
The Kyoto Encyclopedia of Genes and Genomes Enrichment Analysis
The Kyoto Encyclopedia of Genes and Genomes (KEGG) is a comprehensive database that integrates genomic, chemical, and phylogenetic functional information. KEGG pathway enrichment analysis was performed for all differentially expressed genes using Fisher exact test, and KEGG pathway annotations for all genes in the reference genome were downloaded from the KEGG database, with P<0.05 defined as significantly enriched pathways.
Statistical Analysis
All data statistics in this study are expressed as mean±SEM. When the data were normally distributed, a 2‐tailed t test was used for data comparison between 2 groups, 1‐way or 2‐way ANOVA was used for data comparison between multiple groups with 1 or 2 factors, and the Bonferroni test (assumed homogeneity of variance) or Tamhane's T2 (assumed heterogeneity of variance) was used for correction. Nonparametric tests were conducted by using Mann–Whitney test for 2 groups and Kruskal–Wallis test for multiple groups. Statistical analysis was performed by Statistical Package for the Social Sciences (SPSS) software (version 25.0).
RESULTS
TRAF4 Expression Is Upregulated in Cardiac Hypertrophy Models
To investigate the role of TRAF4 in the development of cardiac hypertrophy, we determined the expression levels of TRAF4 in experimental cardiac hypertrophy models. First, we established a hypertrophic myocyte model through phenylephrine treatment of NRCMs, verified by enlarged cell size and upregulated RNA levels of cardiac hypertrophy markers, ANP (atrial natriuretic peptide) and brain natriuretic peptide, using immunofluorescence and qPCR, respectively (Figure 1A and 1B). In NRCMs, the protein level of TRAF4 was increased remarkably by phenylephrine but showed no significant difference at the RNA level (Figure 1B and 1C). Furthermore, a mouse model of TAC‐induced cardiac hypertrophy was developed. Four weeks after TAC surgery, TAC‐treated mice showed reduced left ventricular fraction shortening and left ventricular ejection fraction on echocardiographic assessments and increased RNA levels of ANP and brain natriuretic peptide in the myocardium tissue (Figure 1D and 1E). Consistently, compared with the sham group, the expression of TRAF4 was upregulated at the protein level, but not at the RNA level, in TAC‐treated mice (Figure 1E and 1F). In addition, immunohistochemistry was conducted on mice heart tissues between the sham and TAC group. The result showed that the protein level of TRAF4 was significantly upregulated in hypertrophic mice hearts induced by TAC surgery compared with the sham group, and TRAF4 mainly located in cardiomyocytes (Figure 1G). These results demonstrate that TRAF4 was upregulated in hypertrophic models, suggesting that TRAF4 might be involved in the development of cardiac hypertrophy.
Figure 1. TRAF4 expression is upregulated in cardiac hypertrophy models.
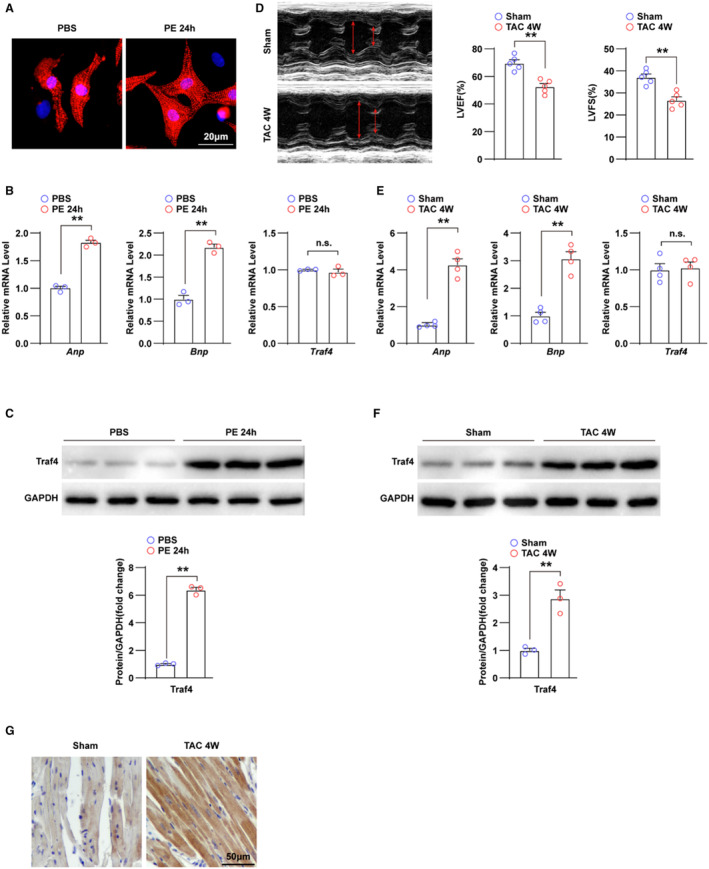
A, Representative immunofluorescence images of α‐actinin (red)‐ and DAPI (blue)‐stained NRCMs treated with phosphate buffered saline (PBS) or phenylephrine (PE) for 24 h, respectively. B, qPCR analysis of the mRNA levels of hypertrophic markers (atrial natriuretic peptide [Anp] and brain natriuretic peptide [Bnp]) and tumor necrosis factor receptor–associated factor 4 (Traf4) in NRCMs treated with PBS or PE for 24 h. C, WB analysis for protein levels of TRAF4 in NRCMs treated with PBS or PE for 24 h. D, Representative echocardiographic images and left ventricular fraction shortening (LVFS) and left ventricular ejection fraction (LVEF) statistics of mice at 4 weeks after sham or transverse aortic constriction (TAC) surgery (n=5 mice in each group). E, qPCR analysis of the RNA levels of hypertrophic markers and Traf4 in left ventricular tissues of mice at 4 weeks after sham or TAC surgery (n=4 mice in each group). F, WB analysis for protein levels of TRAF4 in the myocardium of mice at 4 weeks after sham or TAC surgery (n=3 mice in each group), and GAPDH served as a loading control. G, Immunohistochemistry images of TRAF4 in mice heart tissues of the sham and TAC group (n=4 mice in each group). Statistical analysis was carried out with 2‐tailed Student t test. **P<0.01 vs PBS or sham group. DAPI indicates 4,6‐diamidino‐2‐phenylindole; NRCMs, neonatal rat cardiomyocytes; n.s., not significant; qPCR, quantitative polymerase chain reaction; TAC, transverse aortic constriction; and WB, Western blotting.
TRAF4 Deficiency Ameliorates TAC‐Induced Cardiac Hypertrophy
To investigate the role of TRAF4 in the regulation of cardiac hypertrophy, we established Traf4 knock‐out (KO) mice, and WB was performed to confirm the generated mice (Figure 2A). During surgery, the heart rate and pressure data were monitored and recorded to ensure the effectiveness of TAC (Table S3). Four weeks after surgery, echocardiograms of TAC‐treated WT and KO mice and their control sham groups were assessed. Under basal conditions (sham), Traf4‐KO mice did not exhibit apparent abnormalities compared with the WT group (Figure 2B through 2E). However, 4 weeks after TAC surgery, Traf4‐KO mice showed improved myocardial contractile function, as evidenced by reduced left ventricular end‐diastolic dimension and left ventricular end‐systolic dimension, accompanied by elevated left ventricular fractional shortening and left ventricular ejection fraction (Figure 2B). Subsequently, parameters of cardiac remodeling, including heart weight, heart weight/body weight, and heart weight/tibia length ratios, decreased in Traf4‐KO mice compared with WT mice after 4 weeks of TAC surgery (Figure 2C through 2E). Moreover, TAC‐treated Traf4‐KO mice had a decreased size of both the gross heart and cardiomyocytes. The quantitative cell surface area based on hematoxylin and eosin staining of left ventricle sections from KO TAC mice was significantly lower than that of WT TAC mice (Figure 2F). Consistently, Traf4‐KO diminished the abnormally elevated RNA expression levels of cardiac fetal and prohypertrophic genes (Anp, Bnp, and Myh7) induced by TAC surgery (Figure 2G). In addition, relieved cardiac fibrosis was observed in TAC‐treated Traf4‐KO mice, as identified by picrosirius red staining and quantitative left ventricle collagen volume (Figure 2H). The RNA levels of collagen synthesis–related genes, including Collagen Iα1, Collagen IIIα1, and Ctgf, were also reduced in Traf4‐KO mice compared with WT controls after TAC surgery (Figure 2I). In a mouse model, Traf4‐KO exhibited protective effects against myocardial contractile dysfunction, cardiac remodeling, and fibrosis induced by TAC surgery.
Figure 2. TRAF4 deficiency ameliorates TAC‐induced cardiac hypertrophy.
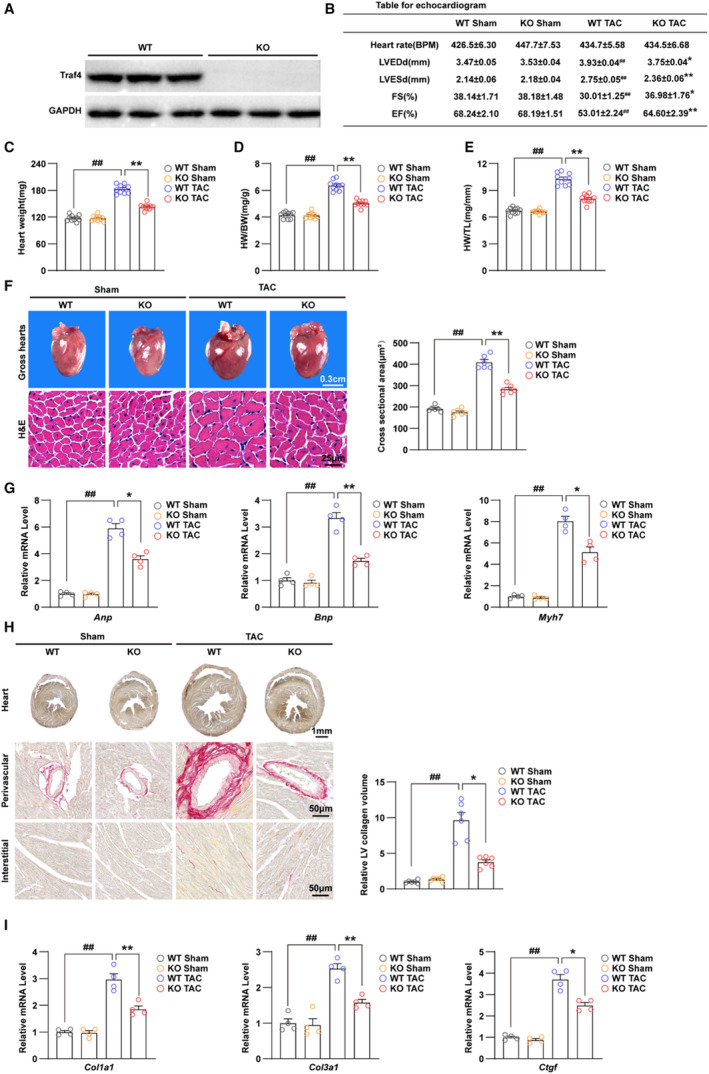
A, WB analysis for protein levels of TRAF4 (tumor necrosis factor receptor–associated factor 4) in myocardium tissues from wild‐type (WT) and Traf4‐knock‐out (KO) mice (n=3 mice/group), and GAPDH served as a loading control. B, Echocardiogram parameters (left ventricular end‐diastolic dimension [LVEDd], left ventricular end‐systolic dimension [LVESd], fraction shortening [FS] ejection fraction [EF]) of WT and Traf4‐KO mice 4 weeks after sham or transverse aortic constriction (TAC) surgery (n=10 mice/group). Heart weight (C), heart weight (HW)/body weight (BW) ratio (D), and HW/tibia length (TL) ratio (E) in WT and Traf4‐KO mice 4 weeks after sham or TAC surgery (n=10 mice/group). F, Representative images of gross hearts, hematoxylin and eosin (H&E)–stained sections from the left ventricle (LV) (left) and quantitative cross‐sectional area based on H&E staining (right) of WT and Traf4‐KO mice 4 weeks after sham or TAC surgery (n=6 mice/group). G, The mRNA levels of hypertrophic markers (atrial natriuretic peptide [Anp], brain natriuretic peptide [Bnp], and myosin heavy chain 7 [Myh7]) in myocardium of WT and Traf4‐KO mice 4 weeks after sham or TAC surgery (n=4 mice/group). H, Representative images of perivascular and interstitial PSR‐stained LV sections (left) and quantitative LV collagen volume base on PSR staining (right) of WT and Traf4‐KO mice at 4 weeks after sham or TAC surgery (n=6 mice/group). I, qPCR analysis of mRNA levels of collagen synthesis–related genes (collagen type I alpha 1(Col1a1), collagen type III alpha 1(Col3a1), and connective tissue growth factor (Ctgf)) in myocardium tissues from WT and Traf4‐KO mice at 4 weeks after sham or TAC surgery (n=4 mice/group). Statistical analysis was carried out by 2‐way ANOVA. ## P<0.01 vs WT sham; *P<0.05 or **P<0.01 vs WT TAC. PSR indicates picrosirius red; qPCR, quantitative polymerase chain reaction; and WB, Western blotting.
Overexpression of TRAF4 Exacerbates Phenylephrine‐Induced Cardiomyocyte Hypertrophy
Given that TRAF4 deficiency protected against cardiac hypertrophy in a mouse model, to further evaluate the effects of TRAF4 on cardiomyocytes during cardiac hypertrophy, we constructed Traf4‐overexpressed NRCMs using adenovirus vector (AdTraf4) infection and confirmed the elevated RNA and protein levels of TRAF4 through qPCR and WB (Figure 3A and 3B). Subsequently, a model of cardiomyocyte hypertrophy was established by phenylephrine stimulation. Results of immunofluorescence with anti‐α‐actinin showed that NRCMs infected with AdTraf4 had enlarged cell size under the condition of phenylephrine treatment compared with adenoviruses expressing the empty vector (AdVector) (Figure 3C). The AdTraf4‐ and AdVector‐infected phosphate‐buffered saline–treated control NRCMs showed no apparent differences (Figure 3C). These results were accompanied by upregulated RNA and protein levels of ANP, brain natriuretic peptide, and MYH7, induced by TRAF4 overexpression in phenylephrine‐treated NRCMs (Figure 3D and 3E). Overall, these in vitro data validated that TRAF4 promotes cardiomyocyte hypertrophy induced by phenylephrine treatment.
Figure 3. Overexpression of TRAF4 exacerbates phenylephrine‐induced cardiomyocyte hypertrophy.
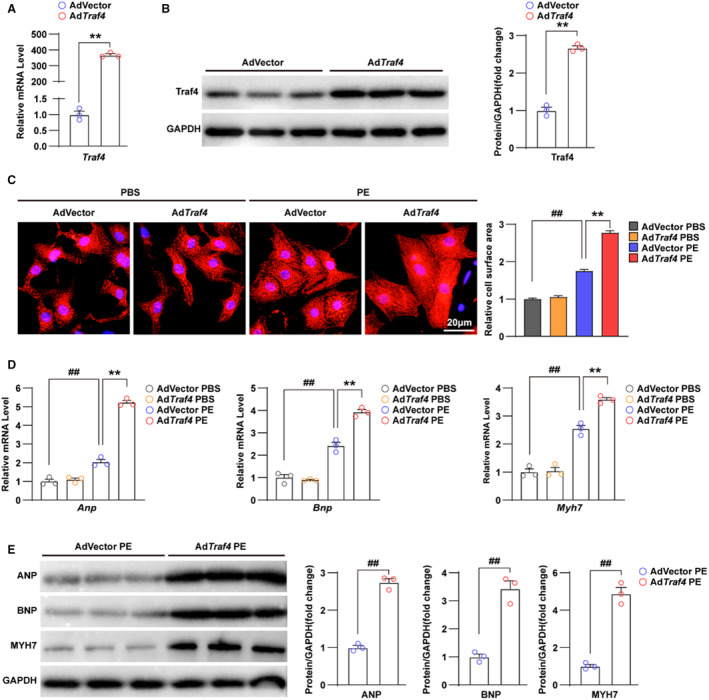
A, qPCR analysis of mRNA level of Traf4 (tumor necrosis factor receptor–associated factor 4) in adenovirus vector (AdVector) and AdTraf4 infected NRCMs. B, WB analysis for protein level of TRAF4 in AdVector and AdTraf4 infected NRCMs, and GAPDH served as a loading control. C, Representative immunofluorescence images (left) and quantitative relative cell surface area (right) of α‐actinin (red)‐ and DAPI (blue)–stained AdVector and AdTraf4 infected NRCMs treated with phosphate buffered saline (PBS) or phenylephrine (PE) for 24 h. D, qPCR analysis for mRNA levels and (E) WB analysis for protein levels of hypertrophic markers (Atrial natriuretic peptide (Anp), Brain natriuretic peptide (Bnp) and Myosin heavy chain 7(Myh7)) in AdVector and AdTraf4 infected NRCMs treated with PBS or PE for 24 h, respectively. Statistical analysis of (A, B, and E) was carried out by 2‐tailed Student t test, and (C and D) was carried out by nonparametric Kruskal–Wallis test and 2‐way ANOVA, respectively. ## P<0.01 vs AdVector PBS and **P<0.01 vs AdVector PE. DAPI indicates 4,6‐diamidino‐2‐phenylindole; NRCMs, neonatal rat cardiomyocytes; qPCR, quantitative polymerase chain reaction; TRAF4, tumor necrosis factor receptor–associated factor 4; and WB, Western blotting.
TRAF4 Promotes the Activation of the AKT Pathway During Cardiac Hypertrophy
To further investigate the mechanism of TRAF4 function in the development of TAC‐induced cardiac hypertrophy, the left ventricular tissues from Traf4‐KO and WT mice 4 weeks after TAC surgery were subjected to RNA‐sequence analysis to profile the gene expression differences. The original analysis data of RNA‐sequence was uploaded to the National Center of Biotechnology Information with an account number of PRJNA905727. Hierarchical cluster analysis showed that the WT and Traf4‐KO mice were clearly divided into 2 clusters according to their gene expression characteristics (Figure 4A). In order to further elucidate the regulatory role of TRAF4 on cardiac hypertrophy at the gene level, we performed Gene Set Enrichment Analysis on the RNA sequence data set. The results revealed that protein processing, heart function, and fibrosis process were all suppressed in Traf4‐KO samples (Figure 4B). Heart function, protein processing, and fibrosis‐related genes that were significantly downregulated in Traf4‐KO samples are shown in heat maps (Figure 4C). These results demonstrated the protective effect of TRAF4 deficiency on myocardial hypertrophy. Subsequently, to analyze the underlying signaling pathways that may be regulated by TRAF4, we performed KEGG signaling pathway enrichment analysis. The analysis revealed that molecules related to the AKT signaling pathway were the most altered, indicating that the AKT signaling pathway was the most significantly affected by TRAF4 deficiency during cardiac hypertrophy (Figure 4D).
Figure 4. RNA‐seq analysis reveals that TRAF4 influences AKT‐related pathways.

A, Clustering analysis result of the RNA‐seq data from wild‐type (WT) and tumor necrosis factor receptor–associated factor 4 (Traf4)‐knock‐out (KO) mice left ventricular tissues 4 weeks after transverse aortic constriction (TAC) surgery. B, Gene Set Enrichment Analysis (GSEA) pathway enrichment analysis of normalized enrichment score (NES) in pathways related to myocardial function, protein processing, and fibrosis. C, Heatmaps of downregulated myocardial function–related, protein processing–related, and fibrosis gene expression profiles based on the RNA‐seq data set. D, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis based on the RNA‐seq data set (PI3K‐AKT [phosphatidylinositol 3‐kinase‐protein kinase B], MAPK [mitogen activated kinase‐like protein], TNF [tumor necrosis factor], JAK–STAT [Janus kinase‐signal transducer and activator of transcription], AMPK [AMP‐activated protein kinase]). n=4 mice in each group. RNA‐seq indicates RNA sequencing.
To confirm the regulatory effect of TRAF4 on the AKT pathway in cardiac hypertrophy, we monitored the correlation between TRAF4 and AKT pathway proteins. WB results showed that the phosphorylation levels of AKT, mTOR, GSK3β, and p70S6K were attenuated in the myocardial tissues of Traf4‐KO mice compared with WT sham (Figure 5A). However, the phosphorylation of AKT, mTOR, GSK3β, and p70S6K was enhanced in AdTraf4 infected NRCMs compared with AdVector‐infected controls (Figure 5B). Altogether, our results demonstrate that TRAF4 promotes the activation of the AKT pathway in the development of cardiac hypertrophy.
Figure 5. TRAF4 overexpression promotes the activation of AKT signaling pathway.
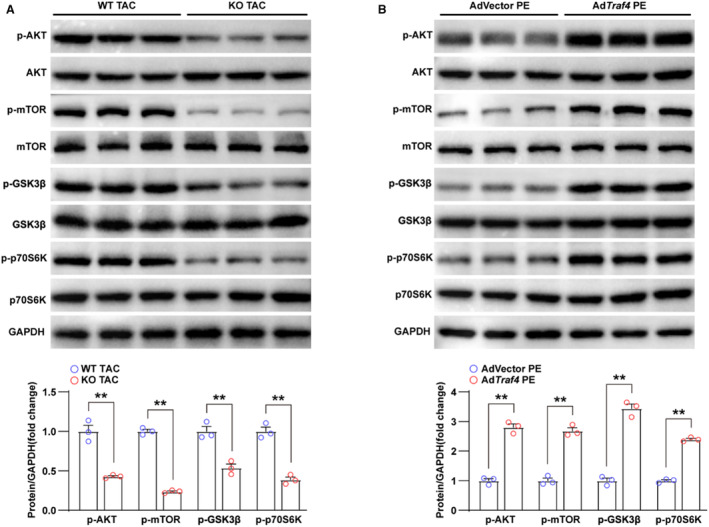
A, WB analysis of protein levels of total and p‐AKT (phosphorylated protein kinase B) pathway‐related proteins (p‐AKT, p‐GSK3β [phosphorylated glycogen synthase kinase 3 beta], p‐mTOR [phosphorylated mammalian target of rapamycin], p‐P70S6K [phosphorylated ribosomal protein S6 kinase], AKT, GSK3β, mTOR, and P70S6K) in left ventricular tissues from wild‐type (WT) and tumor necrosis factor receptor–associated factor 4 (Traf4) knock‐out (KO) mice at 4 weeks after transverse aortic constriction (TAC) surgery; GAPDH served as a loading control. B, Protein levels of total and phosphorylated AKT pathway‐related proteins in adenovirus vector (AdVector)‐ and AdTraf4‐infected NRCMs treated with phenylephrine (PE) for 24 h. Statistical analysis was carried out by 2‐tailed Student t test. **P<0.01 vs WT TAC or AdVector PE. NRCMs indicates neonatal rat cardiomyocytes; TRAF4, tumor necrosis factor receptor–associated factor 4; and WB, Western blotting.
Inhibition of AKT Eliminates the Promotive Function of TRAF4 in Cardiac Hypertrophy
It has been reported that TRAF4 is required for the recruitment of AKT to the cell membrane for its activation by mediating the Ub‐Lys63 ubiquitination of AKT. 9 Therefore, we investigated the functional correlation between TRAF4 and AKT in the regulation of cardiac hypertrophy. In the phenylephrine‐induced hypertrophic cardiomyocyte model, NRCMs infected with AdTraf4 and AdVector were treated with inhibitors of AKT phosphorylation (iAKT) or dimethyl sulfoxide as a control. WB results showed that the effectiveness of iAKT was evidenced by the suppressed phosphorylation level of AKT, whereas the basal and overexpressed protein levels of TRAF4 remained unaffected by iAKT (Figure 6A). To evaluate the hypertrophic phenotypes of each group, immunofluorescence and qPCR assays were performed to investigate the cell size and the expression of cardiac fetal and prohypertrophic genes. The results showed that iAKT reversed the relative cell surface area decrease and downregulated RNA levels of ANP, brain natriuretic peptide, and Myh7 promoted by TRAF4 overexpression (Figure 6B and 6C). The overexpression of TRAF4 promoted cardiomyocyte hypertrophy, which could be reversed by iAKT, indicating that TRAF4 might regulate cardiac hypertrophy in an AKT‐dependent manner.
Figure 6. AKT inhibitor abrogates the promoting effect of TRAF4 overexpression on cardiac hypertrophy.
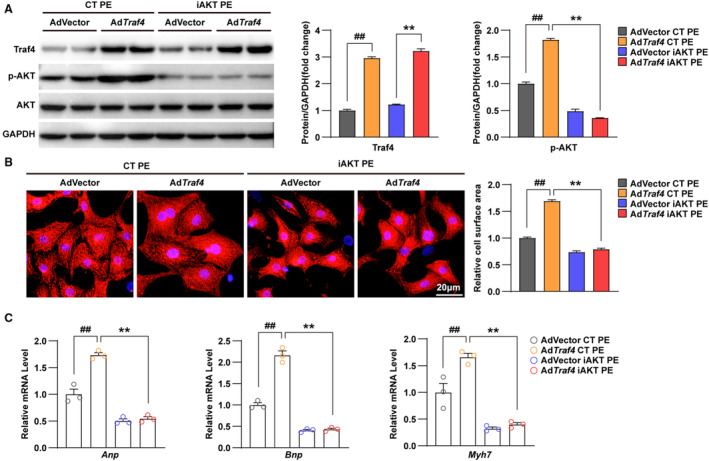
A, Protein levels of Traf4 (tumor necrosis factor receptor–associated factor 4), total AKT (protein kinase B), and phosphorylated AKT (p‐AKT) in DMSO‐treated control (CT) or inhibitors of AKT phosphorylation (iAKT)‐treated adenovirus vector (AdVector)‐ and AdTraf4‐infected NRCMs treated with phenylephrine (PE) for 24 h; GAPDH served as a loading control. B, Representative immunofluorescence images of α‐actinin (red)‐ and DAPI (blue)–stained CT or iAKT‐treated AdVector‐ and AdTraf4‐infected NRCMs treated with PE for 24 h. C, mRNA levels of hypertrophic markers (atrial natriuretic peptide (Anp), brain natriuretic peptide (Bnp), and myosin heavy chain 7(Myh7)) in DMSO‐ or iAKT‐treated AdVector‐ and AdTraf4‐infected NRCMs treated with PE for 24 h. Statistical analysis was carried out by 2‐ way ANOVA. ## P<0.01 vs AdVector CT PE; **P<0.01 vs AdTraf4 CT PE. DAPI indicates 4,6‐diamidino‐2‐phenylindole; DMSO, dimethyl sulfoxide; and NRCMs, neonatal rat cardiomyocytes.
DISCUSSION
Pathological cardiac hypertrophy has become one of the major causes of heart failure, which affects 1% to 2% of the adult population worldwide. 13 The heavy public health burden of heart failure demands effective therapeutic and preventative treatments emergently. 14 However, the pathogenesis of cardiac hypertrophy is extremely complex, comprising extensive alterations in various processes and functions, including collagen synthesis and fibrosis, cell volume enlargement, contractile function, protein synthesis, and inflammation, and involves a wide range of cell types, such as fibroblasts, vascular smooth cells, and leukocytes. 15 , 16 , 17 Suppression or reversion of cardiac hypertrophy is considered a promising approach for preventing the occurrence of heart failure. 5 Several signaling pathways and proteins have been identified as potential targets, including oxidative stress, serine/threonine phosphatases, nongated Ca2+ influx/Ca2+ signaling, downstream effectors of rapamycin or G‐protein‐coupled receptors, protein kinases, and chromatin remodeling agents. 18 This study identified TRAF4 as a stimulative regulator of pressure overload–induced cardiac hypertrophy. The effect of TRAF4 overexpression on cardiac hypertrophy depends on the AKT pathway. In our study, we found that the protein expression of TRAF4 upregulated significantly, but its mRNA levels were not changed in heart tissues of mice that undergo TAC surgery or cardiomyocytes stimulated by phenylephrine. This suggested that the altered expression of TRAF4 in hypertrophy is mediated by posttranslational modification rather than regulation in RNA level. The upregulation of TRAF4 may be due to increased protein stability or decreased degradation. Studies have shown that TRAF4 can be degraded by Smurf1 ubiquitination, 19 but whether the upregulation of TRAF4 in myocardial hypertrophy is caused by the downregulation of Smurf1‐induced ubiquitination still needs further study.
TRAF family members are multidomain proteins containing a C‐terminal TRAF domain, an N‐terminal RING domain, and several zinc‐finger domains in the central region, except TRAF1 and TRAF7. The RING domain is the core of the ubiquitin ligase catalytic structure, which enables TRAF family proteins E3 ubiquitin ligase activity. 20 The TRAF domain, the most prominent structure in the TRAF family, interacts with various membrane receptors via their intercellular regions. 21 These structural features make TRAF proteins act as adaptors and mediators in the signal transduction of tumor necrosis factor receptor, various cytokine receptors, and interleukin‐1 receptor/Toll‐like receptor. 21 TRAF4 is a unique member of the TRAF family with nuclear localization signals at the N‐terminus, which ensures that TRAF4 migrate to the nucleus. 22 This structure alteration enables TRAF4 to participate in broader processes, including neurogenesis, 23 cell polarity, 24 cell proliferation, 25 apoptosis, 26 oxidation, 27 osteogenic metabolism, 28 and repair, 29 while the majority of the TRAF family is mainly involved in immune‐related cell signaling. 30 TRAF4 has been reported to be involved in various types of cancer, ranging from solid tumors to hematological tumors 9 , 31 , 32 , 33 , 34 and inflammatory disease. 35 The present study has verified the promotive effect of TRAF4 in cardiac hypertrophy, which extends the understanding of the pathological function of TRAF4. Several studies have shown that in mice and pigs, adenovirus and adeno‐associated virus 9 have been widely used to knock down the expression of target genes in heart tissue, so as to achieve the effect of regulating heart failure, myocardial hypertrophy, and cardiac ischemia–reperfusion injury. 36 , 37 , 38 Therefore, this might provide some potential therapeutic opportunity in the treatment of heart failure by using adenovirus or adeno‐associated virus 9 to mediate TRAF4 knockdown.
During our investigation of the signaling pathways affected by TRAF4 in the development of cardiac hypertrophy, the AKT pathway was suppressed due to TRAF4 inhibition. Importantly, exacerbation of cardiomyocyte hypertrophy was blocked by an AKT inhibitor, indicating that AKT might be the downstream regulator of TRAF4 during cardiac hypertrophy. The AKT signaling pathway plays a crucial role in regulating numerous physiological functions and participates in the pathogenesis of various disorders such as cancer, 39 neurodegenerative disease, 40 angiogenesis, 41 and inflammatory response. 42 , 43 Sustained activation of AKT contributes to the decompensatory transition of cardiac hypertrophy, which leads to cardiac remodeling and, subsequently, heart failure. 44 , 45 , 46 The interaction between TRAF4 and AKT has been previously elucidated in different types of tumors. 9 , 47 , 48 , 49 , 50 This study provides evidence of TRAF4/AKT modulating cardiac hypertrophy for the first time.
This study has some limitations. In our study, TRAF4 knockout mice were used to prove that TRAF4 gene deletion can inhibit cardiac dysfunction, myocardial hypertrophy, and fibrosis induced by TAC surgery, indicating that TRAF4 can promote the process of myocardial hypertrophy. But because we used TRAF4 globally in knockout mice, we do not know through which types of cell that TRAF4 exerts its regulatory effects. To further investigate the regulation of TRAF4 on cardiomyocytes, we isolated primary cardiomyocytes and demonstrated that TRAF4 can promote the hypertrophy of cardiomyocytes induced by phenylephrine stimulation by using adenovirus‐mediated TRAF4 knockdown and overexpression. Our results demonstrate that TRAF4 can play its function by regulating cardiomyocyte hypertrophy both in vivo and in vitro, but whether TRAF4 has a regulatory effect on other cell types still needs to be further studied.
In summary, we identified TRAF4 as a promoter of cardiac hypertrophy. TRAF4 activates the AKT signaling pathway to enhance cardiomyocyte enlargement, fibrosis, and the expression of cardiac fetal and prohypertrophic genes. Reversion of TRAF4 overexpression exacerbated hypertrophic phenotypes induced by AKT phosphorylation inhibitor, suggesting that TRAF4 probably affects cardiac hypertrophy in an AKT‐dependent manner. These findings may provide new insights and potential methods for the pharmaceutical therapy for cardiac hypertrophy.
Sources of Funding
This work was supported by grants from the National Institutes of Health.
Disclosures
None.
Supporting information
Tables S1–S3
This manuscript was sent to Barry London, MD, PhD, Senior Guest Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.028185
For Sources of Funding and Disclosures, see page 13.
Contributor Information
Ke‐Qiong Deng, Email: dengkeqiong@whu.edu.cn.
Huo‐Ping Li, Email: lihuoping@hgyy.org.cn.
References
- 1. Rame JE, Dries DL. Heart failure and cardiac hypertrophy. Curr Treat Options Cardiovasc Med. 2007;9:289–301. doi: 10.1007/s11936-007-0024-3 [DOI] [PubMed] [Google Scholar]
- 2. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407. doi: 10.1038/s41569-018-0007-y [DOI] [PubMed] [Google Scholar]
- 3. Miao R, Lu Y, Xing X, Li Y, Huang Z, Zhong H, Huang Y, Chen AF, Tang X, Li H, et al. Regulator of G‐protein signaling 10 negatively regulates cardiac remodeling by blocking mitogen‐activated protein kinase‐extracellular signal‐regulated protein kinase 1/2 signaling. Hypertension. 2016;67:86–98. doi: 10.1161/HYPERTENSIONAHA.115.05957 [DOI] [PubMed] [Google Scholar]
- 4. Savarese G, Lund LH. Global public health burden of heart failure. Card Fail Rev. 2017;3:7–11. doi: 10.15420/cfr.2016:25:2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB [DOI] [PubMed] [Google Scholar]
- 6. Leong DP, McMurray JJV, Joseph PG, Yusuf S. From ACE inhibitors/ARBs to ARNIs in coronary artery disease and heart failure (part 2/5). J Am Coll Cardiol. 2019;74:683–698. doi: 10.1016/j.jacc.2019.04.068 [DOI] [PubMed] [Google Scholar]
- 7. Ruan X, Zhang R, Li R, Zhu H, Wang Z, Wang C, Cheng Z, Peng H. The research progress in physiological and pathological functions of TRAF4. Front Oncol. 2022;12:842072. doi: 10.3389/fonc.2022.842072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal. 2013;8:7. doi: 10.1186/1750-2187-8-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li W, Peng C, Lee MH, Lim D, Zhu F, Fu Y, Yang G, Sheng Y, Xiao L, Dong X, et al. TRAF4 is a critical molecule for Akt activation in lung cancer. Cancer Res. 2013;73:6938–6950. doi: 10.1158/0008-5472.CAN-13-0913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang A, Wang J, Ren H, Yang F, Sun L, Diao K, Zhao Z, Song M, Cui Z, Wang E, et al. TRAF4 participates in Wnt/beta‐catenin signaling in breast cancer by upregulating beta‐catenin and mediating its translocation to the nucleus. Mol Cell Biochem. 2014;395:211–219. doi: 10.1007/s11010-014-2127-y [DOI] [PubMed] [Google Scholar]
- 11. Abell AN, Johnson GL. MEKK4 is an effector of the embryonic TRAF4 for JNK activation. J Biol Chem. 2005;280:35793–35796. doi: 10.1074/jbc.C500260200 [DOI] [PubMed] [Google Scholar]
- 12. Zhang L, Zhou F, García de Vinuesa A, de Kruijf EM, Mesker WE, Hui L, Drabsch Y, Li Y, Bauer A, Rousseau A, et al. TRAF4 promotes TGF‐β receptor signaling and drives breast cancer metastasis. Mol Cell. 2013;51:559–572. doi: 10.1016/j.molcel.2013.07.014 [DOI] [PubMed] [Google Scholar]
- 13. Tanai E, Frantz S. Pathophysiology of heart failure. Compr Physiol. 2015;6:187–214. doi: 10.1002/cphy.c140055 [DOI] [PubMed] [Google Scholar]
- 14. Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, Finkelstein EA, Hong Y, Johnston SC, Khera A, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123:933–944. doi: 10.1161/CIR.0b013e31820a55f5 [DOI] [PubMed] [Google Scholar]
- 15. Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu J, Li W, Deng KQ, Tian S, Liu H, Shi H, Fang Q, Liu Z, Chen Z, Tian T, et al. The E3 ligase TRIM16 is a key suppressor of pathological cardiac hypertrophy. Circ Res. 2022;130:1586–1600. doi: 10.1161/CIRCRESAHA.121.318866 [DOI] [PubMed] [Google Scholar]
- 17. Zhang Y, Huang Z, Li H. Insights into innate immune signalling in controlling cardiac remodelling. Cardiovasc Res. 2017;113:1538–1550. doi: 10.1093/cvr/cvx130 [DOI] [PubMed] [Google Scholar]
- 18. McKinsey TA, Kass DA. Small‐molecule therapies for cardiac hypertrophy: moving beneath the cell surface. Nat Rev Drug Discov. 2007;6:617–635. doi: 10.1038/nrd2193 [DOI] [PubMed] [Google Scholar]
- 19. Li S, Lu K, Wang J, An L, Yang G, Chen H, Cui Y, Yin X, Xie P, Xing G, et al. Ubiquitin ligase Smurf1 targets TRAF family proteins for ubiquitination and degradation. Mol Cell Biochem. 2010;338:11–17. doi: 10.1007/s11010-009-0315-y [DOI] [PubMed] [Google Scholar]
- 20. Yang XD, Sun SC. Targeting signaling factors for degradation, an emerging mechanism for TRAF functions. Immunol Rev. 2015;266:56–71. doi: 10.1111/imr.12311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arkee T, Bishop GA. TRAF family molecules in T cells: multiple receptors and functions. J Leukoc Biol. 2020;107:907–915. doi: 10.1002/JLB.2MR1119-397R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Régnier CH, Tomasetto C, Moog‐Lutz C, Chenard MP, Wendling C, Basset P, Rio MC. Presence of a new conserved domain in CART1, a novel member of the tumor necrosis factor receptor‐associated protein family, which is expressed in breast carcinoma. J Biol Chem. 1995;270:25715–25721. doi: 10.1074/jbc.270.43.25715 [DOI] [PubMed] [Google Scholar]
- 23. Blaise S, Kneib M, Rousseau A, Gambino F, Chenard MP, Messadeq N, Muckenstrum M, Alpy F, Tomasetto C, Humeau Y, et al. In vivo evidence that TRAF4 is required for central nervous system myelin homeostasis. PLoS One. 2012;7:e30917. doi: 10.1371/journal.pone.0030917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kédinger V, Alpy F, Baguet A, Polette M, Stoll I, Chenard MP, Tomasetto C, Rio MC. Tumor necrosis factor receptor‐associated factor 4 is a dynamic tight junction‐related shuttle protein involved in epithelium homeostasis. PLoS One. 2008;3:e3518. doi: 10.1371/journal.pone.0003518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cai G, Zhu L, Chen X, Sun K, Liu C, Sen GC, Stark GR, Qin J, Li X. TRAF4 binds to the juxtamembrane region of EGFR directly and promotes kinase activation. Proc Natl Acad Sci USA. 2018;115:11531–11536. doi: 10.1073/pnas.1809599115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sax JK, El‐Deiry WS. Identification and characterization of the cytoplasmic protein TRAF4 as a p53‐regulated proapoptotic gene. J Biol Chem. 2003;278:36435–36444. doi: 10.1074/jbc.M303191200 [DOI] [PubMed] [Google Scholar]
- 27. Wu RF, Xu YC, Ma Z, Nwariaku FE, Sarosi GA Jr, Terada LS. Subcellular targeting of oxidants during endothelial cell migration. J Cell Biol. 2005;171:893–904. doi: 10.1083/jcb.200507004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cen S, Li J, Cai Z, Pan Y, Sun Z, Li Z, Ye G, Zheng G, Li M, Liu W, et al. TRAF4 acts as a fate checkpoint to regulate the adipogenic differentiation of MSCs by activating PKM2. EBioMedicine. 2020;54:102722. doi: 10.1016/j.ebiom.2020.102722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deng CC, Zhu DH, Chen YJ, Huang TY, Peng Y, Liu SY, Lu P, Xue YH, Xu YP, Yang B, et al. TRAF4 promotes fibroblast proliferation in keloids by destabilizing p53 via interacting with the deubiquitinase USP10. J Invest Dermatol. 2019;139:1925–1935. doi: 10.1016/j.jid.2019.03.1136 [DOI] [PubMed] [Google Scholar]
- 30. Park HH. Structural feature of TRAFs, their related human diseases and therapeutic intervention. Arch Pharm Res. 2021;44:475–486. doi: 10.1007/s12272-021-01330-w [DOI] [PubMed] [Google Scholar]
- 31. Yang F, Wang J, Ren HY, Jin J, Wang AL, Sun LL, Diao KX, Wang EH, Mi XY. Proliferative role of TRAF4 in breast cancer by upregulating PRMT5 nuclear expression. Tumour Biol. 2015;36:5901–5911. doi: 10.1007/s13277-015-3262-0 [DOI] [PubMed] [Google Scholar]
- 32. Yang L, Guo Y, Liu X, Wang T, Tong X, Lei K, Wang J, Huang D, Xu Q. The tumor suppressive miR‐302c‐3p inhibits migration and invasion of hepatocellular carcinoma cells by targeting TRAF4. J Cancer. 2018;9:2693–2701. doi: 10.7150/jca.25569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu Y, Duan N, Duan S. MiR‐29a inhibits glioma tumorigenesis through a negative feedback loop of TRAF4/Akt signaling. Biomed Res Int. 2018;2018:2461363. doi: 10.1155/2018/2461363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sharma S, Pavlasova GM, Seda V, Cerna KA, Vojackova E, Filip D, Ondrisova L, Sandova V, Kostalova L, Zeni PF, et al. miR‐29 modulates CD40 signaling in chronic lymphocytic leukemia by targeting TRAF4: an axis affected by BCR inhibitors. Blood. 2021;137:2481–2494. doi: 10.1182/blood.2020005627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shen J, Qiao Y, Ran Z, Wang T. Different activation of TRAF4 and TRAF6 in inflammatory bowel disease. Mediators Inflamm. 2013;2013:647936. doi: 10.1155/2013/647936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li L, Cui YJ, Liu Y, Li HX, Su YD, Li SN, Wang LL, Zhao YW, Wang SX, Yan F, et al. ATP6AP2 knockdown in cardiomyocyte deteriorates heart function via compromising autophagic flux and NLRP3 inflammasome activation. Cell Death Discov. 2022;8:161. doi: 10.1038/s41420-022-00967-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liao H, Gao W, Ma J, Xue H, Wang Y, Huang D, Yan F, Ye Y. GPR39 promotes cardiac hypertrophy by regulating the AMPK‐mTOR pathway and protein synthesis. Cell Biol Int. 2021;45:1211–1219. doi: 10.1002/cbin.11566 [DOI] [PubMed] [Google Scholar]
- 38. Liu S, Li K, Wagner Florencio L, Tang L, Heallen TR, Leach JP, Wang Y, Grisanti F, Willerson JT, Perin EC, et al. Gene therapy knockdown of hippo signaling induces cardiomyocyte renewal in pigs after myocardial infarction. Sci Transl Med. 2021;13:eabd6892. doi: 10.1126/scitranslmed.abd6892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang Z, Richmond A, Yan C. Immunomodulatory properties of PI3K/AKT/mTOR and MAPK/MEK/ERK inhibition augment response to immune checkpoint blockade in melanoma and triple‐negative breast cancer. Int J Mol Sci. 2022;23:7353. doi: 10.3390/ijms23137353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sayed FA, Kodama L, Fan L, Carling GK, Udeochu JC, Le D, Li Q, Zhou L, Wong MY, Horowitz R, et al. AD‐linked R47H‐TREM2 mutation induces disease‐enhancing microglial states via AKT hyperactivation. Sci Transl Med. 2021;13:eabe3947. doi: 10.1126/scitranslmed.abe3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kobialka P, Sabata H, Vilalta O, Gouveia L, Angulo‐Urarte A, Muixí L, Zanoncello J, Muñoz‐Aznar O, Olaciregui NG, Fanlo L, et al. The onset of PI3K‐related vascular malformations occurs during angiogenesis and is prevented by the AKT inhibitor miransertib. EMBO Mol Med. 2022;14:e15619. doi: 10.15252/emmm.202115619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol. 2015;27:286–296. doi: 10.1016/j.smim.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guan H, Cheng WL, Guo J, Chao ML, Zhang Y, Gong J, Zhu XY, She ZG, Huang Z, Li H. Vinexin beta ablation inhibits atherosclerosis in apolipoprotein E‐deficient mice by inactivating the Akt‐nuclear factor kappaB inflammatory Axis. J Am Heart Assoc. 2017;16(2):e004585. doi: 10.1161/JAHA.116.004585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, et al. The sirtuin SIRT6 blocks IGF‐Akt signaling and development of cardiac hypertrophy by targeting c‐Jun. Nat Med. 2012;18:1643–1650. doi: 10.1038/nm.2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deng KQ, Wang A, Ji YX, Zhang XJ, Fang J, Zhang Y, Zhang P, Jiang X, Gao L, Zhu XY, et al. Suppressor of IKKɛ is an essential negative regulator of pathological cardiac hypertrophy. Nat Commun. 2016;7:11432. doi: 10.1038/ncomms11432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yan L, Wei X, Tang QZ, Feng J, Zhang Y, Liu C, Bian ZY, Zhang LF, Chen M, Bai X, et al. Cardiac‐specific mindin overexpression attenuates cardiac hypertrophy via blocking AKT/GSK3beta and TGF‐beta1‐Smad signalling. Cardiovasc Res. 2011;92:85–94. doi: 10.1093/cvr/cvr159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang J, Li X, Yang W, Jiang X, Li N. TRAF4 promotes tumorigenesis of breast cancer through activation of Akt. Oncol Rep. 2014;32:1312–1318. doi: 10.3892/or.2014.3304 [DOI] [PubMed] [Google Scholar]
- 48. Chen H, Qian Z, Zhang S, Tang J, Fang L, Jiang F, Ge D, Chang J, Cao J, Yang L, et al. Silencing COX‐2 blocks PDK1/TRAF4‐induced AKT activation to inhibit fibrogenesis during skeletal muscle atrophy. Redox Biol. 2021;38:101774. doi: 10.1016/j.redox.2020.101774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim D, Nam GY, Seo E, Jun HS. Inhibition of ChREBP ubiquitination via the ROS/Akt‐dependent downregulation of Smurf2 contributes to lysophosphatidic acid‐induced fibrosis in renal mesangial cells. J Biomed Sci. 2022;29:31. doi: 10.1186/s12929-022-00814-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu K, Wu X, Zang X, Huang Z, Lin Z, Tan W, Wu X, Hu W, Li B, Zhang L. TRAF4 regulates migration, invasion, and epithelial‐mesenchymal transition via PI3K/AKT signaling in hepatocellular carcinoma. Oncol Res. 2017;25:1329–1340. doi: 10.3727/096504017X14876227286564 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.