

Graphical Abstract
A synthetic platform has been developed that provides access to platinum(IV) prodrugs of highly cytotoxic platinum−acridine anticancer agents and allows them to be incorporated into conjugation-ready prodrug−payloads (PPLs). The PPLs can be conveniently assembled in highly efficient microscale reactions utilizing strain-promoted azide−alkyne cycloaddition chemistry. Model reactions were performed to study the stability of the PPLs in buffers and media and to assess their compatibility with cysteine−maleimide Michael addition chemistry. Amide coupling was a successful strategy to generate a conjugate containing integrin-targeted cyclo[RGDfK] peptide. Reactions with ascorbate were performed to mimic the reductive activation of the PPLs and the latter conjugate, and a cyanine (Cy5) fluorophore-labeled PPL was used to probe the reduction of platinum(IV) in cancer cells by confocal microscopy. The PPL concept introduced here should be evaluated for treating solid tumors with PAs using cancer-targeting vehicles, such as antibody−drug conjugates.
INTRODUCTION
Anticancer drug development is witnessing a growing interest in DNA-targeted chemotherapies.1 Efficacious and better tolerated treatment modalities that can overcome the resistance observed with current oncology drugs, particularly in solid tumor malignancies, remain a high priority.2 This includes the safe and target-selective delivery of highly cytotoxic agents, such as DNA minor groove alkylators, topoisomerase I poisons, including their active metabolites, and anthracycline derivatives in the form of antibody−drug conjugates (ADCs).3−5 While highly potent in cell lines, DNA-targeted drugs are often too toxic for direct systemic administration. Their delivery as cytotoxic payloads that can be targeted at specific antigens expressed in solid tumors has recently resulted in several successful treatment options. These include the FDA-approved ADCs sacituzumab govitecan and trastuzumab deruxtecan, which contain the topoisomerase I poisons SN-38 and exatecan, respectively, as well as several ADCs in late-phase clinical trials.3,6
DNA-targeted platinum−acridine hybrid agents (PAs) represented by compound 1 (Figure 1) have demonstrated a unique mechanism of action and high potency in several models of solid tumors,7−10 which translates into promising efficacy in xenograft models in vivo.10,11 As in other cases of highly cytotoxic drugs, the suboptimal drug-like properties of PAs have prompted the evaluation of more efficient and safer forms of delivery of these agents to tumor tissue, including passively targeted nanoliposomes.11 Unlike the classical platinum-based drug cisplatin, which typically kills cancer cells at micromolar concentrations, PAs have demonstrated up to 3 orders of magnitude higher activity at single-digit nanomolar concentrations. This is significant since major advantages are observed for PAs in several solid tumors that do not respond well to current clinical treatments.8,12 Furthermore, cytotoxic drugs able to kill cancer cells in the nanomolar concentrations range hold promise as ADC deliverable payloads.3 To accelerate the preclinical evaluation of PAs, a versatile synthetic platform has been developed that allows for their optimization and efficient screening.13 The modular approach also provides access to derivatives functionalized with cleavable and noncleavable linkers, rendering PAs compatible with conventional click and conjugation chemistries.14,15
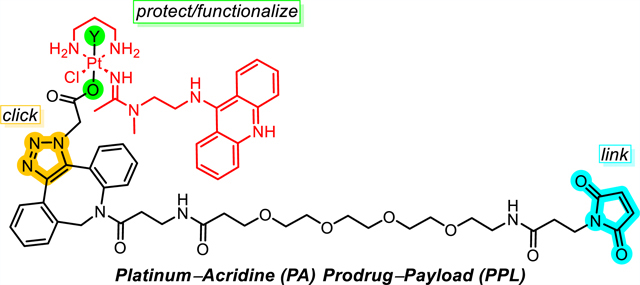
Figure 1.

Structure of platinum−acridine agent 1 and schematic of platinum(IV) prodrug−payloads developed in this study. Y and Y′ are axial ligands, with an extendable linker featuring a protein-reactive group (PRG) installed on Y′.
The nanomolar cytotoxicity and chemical versatility of PAs make a strong case for their delivery as bioconjugates directed at specific targets/antigens in solid tumors, for instance, in the form of ADCs or as conjugates incorporating alternative receptor-binding smaller peptides.16 Given the well-established promiscuous reactivity of Pt(II)-based drugs with components in blood,17 the delivery of PAs as chemically more inert Pt(IV) prodrugs18 can be expected to provide major advantages. The axial (trans) ligands present in Pt(IV) prodrugs do not only control the pharmacological properties of these agents but can also be used to introduce new functionalities allowing for modification and codelivery with other bioactive or diagnostic agents.19−22 Following this concept, in the present study, prodrug−payloads (PPLs) of compound 1 were generated containing a click chemistry-enabled axial ligand Y′ that can be conveniently extended with a strained alkyne linker bearing a terminal protein-reactive group (PRG) (Figure 1). The ease of preparation, chemical robustness, and predictable reductive activation chemistry observed for the new Pt(IV)-based payloads hold the promise of providing a new modality of delivering highly cytotoxic PAs more effectively.
RESULTS AND DISCUSSION
Chemistry.
Payload-linker designs for efficient targeted delivery of highly cytotoxic drugs most importantly require that the linker moiety resist premature cleavage while the bioconjugate is in the systemic circulation. Conversely, the design must also allow for rapid release of the drug, triggered by a chemical stimulus or enzymatic cleavage, in its most cytotoxic form in the target tissue.5 The mechanism envisioned for the design pursued in this study (Figure 1) features chemically robust Pt(IV) precursors of compound 1 that can be modified with noncleavable linkers. Release of active payload in the form of compound 1 would be achieved by intracellular bioreductants in a process that promotes rapid and irreversible loss of axial ligands via the reductive trans-elimination of Y and Y′.
A critical step in designing the PPLs was to establish conditions under which axial ligands (Y, Y′) can be introduced into compound 1 in a stereoselective and stoichiometric manner using oxidative addition chemistry. A modular chemical approach was desired that requires minimal to no purification steps. To achieve this, reactions of compound 1 with suitable oxidizing agents had to be performed under stringently controlled conditions to avoid undesired reactions of the heteroaromatic acridine moiety. Initial reactions of compound 1 with chlorine gas and iodobenzene dichloride (PhICl2) under ambient conditions to introduce trans-chloro ligands following typical procedures used for classical Pt(II)-based drugs23 led to mixtures of polychlorinated products (data not shown). By contrast, the reaction of compound 1 with PhICl2 at −78 °C yields the desired trans-dichloro complex 2, which was characterized crystallographically as the dichloride salt (Figure 2A). While this result demonstrates that a Pt(IV) derivative of compound 1 can be generated by oxidative addition, axial dichloro ligands are usually considered too labile in Pt(IV) prodrug designs.18 Instead, the introduction of trans-hydroxo ligands was desirable, which also allows for further functionalization of the axial positions.23 This was achieved by reacting compound 1 with aqueous hydrogen peroxide followed by a nonaqueous workup of the reaction mixture and precipitation of the product from DMF, yielding compound 3. Alternatively, compounds with a mixed hydroxo−carboxylato coordination are accessible using this scheme if reactions with the above oxidant are performed in the presence of excess carboxylic acid. Compound 4, featuring trans hydroxo and butyrate ligands, was the major (∼70−80%) product in reactions conducted in this manner (Scheme 1). Unfortunately, major undesired side reactions leading to symmetrical trans-dibutyrate complexes and replacement of the equatorial chloride by butyrate render this method impractical. Instead, compound 3 had to be employed as the starting material for asymmetric axial functionalizations. With excess acetic anhydride, it can be cleanly converted to the stable diacetato derivative, 5, which was fully characterized in solution (see the SI) and by X-ray crystallography (Figure 2B) and can be considered a “classical” prodrug of compound 1. Most critically, a stoichiometric reaction with NHS-activated carboxylic acid allows for the introduction of a 2-azidoacetato (OAc2‑az) ligand, giving compound 6, in which the remaining hydroxo ligand can be subsequently converted into capping acetate or carbamate to generate derivatives 7 and 8, respectively. Compounds 6−8 were then used as “clickable” modules to access the desired PPLs by installing conjugatable linkers. Note that while the asymmetric axial ligation in 6−8 produces enantiomers, the chirality introduced into the PLLs is not expected to cause stereoselective pharmacological effects since reduction will generate 1, which is achiral.
Figure 2.

Crystal structures of compounds 2 (A) and 5 (B) with the lattice solvent molecules and counterions omitted, confirming the stereoselective introduction of axial ligands in 1. Nonhydrogen atoms are represented by 50% probability ellipsoids, and hydrogen atoms are represented by arbitrarily small spheres. Only heteroatoms of the asymmetric unit are labeled. For a summary of crystal parameters and a list of bond distances and angles, see the SI.
Scheme 1. Synthesis of Click Chemistry-Ready Pt(IV) Derivatives of 1a.

aReagents and conditions: (a) PhICl2 (dropwise), MeOH, −78 °C to rt over 12 h; (b) 30% H2O2 (excess), H2O/DMF, 6 h, rt; (c) 30% H2O2/ butyric acid (excess), DMF, 24 h, rt; (d) Ac2O (excess), DMF, 12 h, rt; (e) 2-azidoacetic acid NHS ester, DMF, 1 h, rt; (f) Ac2O (excess), DMF, 24 h, rt; (g) pentyl isocyanate (excess), DMF, 24 h, rt.
One benefit of the chemistry devised for modifying the axial hydroxo ligands and the subsequent click chemistry-facilitated linker extensions (see below) is that these reactions occur without noticeable side products and usually in quantitative yields. Excess reagents and byproducts (e.g., NHS) can usually be removed by simple trituration. This allows for the assembly of microscale reactions that do not require laborious workup. Products with >95% purity by LC-MS analysis are conveniently obtained by simple reprecipitation, and extensive compound characterization can be limited to representative key intermediates.
From compounds 6 and 7 and the two dibenzoazacyclooctyne (DBCO) linker modules, DBCO-maleimide and DBCO-PEG4-maleimide, four PPLs (9−12) were generated using strain-promoted azide−alkyne cycloaddition (SPAAC) click chemistry24 (Scheme 2). The polyethylene glycol-based (PEG) linker was introduced to tune the hydrophilicity−hydrophobicity balance of the PPLs and to help prevent unfavorable stereoselective interactions between the racemic payloads and the chiral carrier in reactions of maleimide with cysteine residues.5 Taking advantage of a click chemistry-facilitated approach, stock solutions of the PPLs can be efficiently prepared by mixing the two components in DMF at ambient temperature. The reactions, which are quantitative and yield 9−12 in high purity, can be conveniently monitored by in-line LC-MS (see the SI for details) and supplemented with a limiting reagent, if necessary, to achieve stoichiometric conversion.
Scheme 2. Preparation of Prodrug-Payloads (PPLs)a.

aConditions: 1:1 stoichiometry, dry DMF, 24 h, dark, rt.
Prodrug−Payload Stability.
The stability of two representative PPLs (9 and 11) in the absence of reducing agents was assessed in various buffers and media, including those relevant to the preparation and MS characterization of the payloads and bioconjugates. Reactions were monitored by LC-MS for up to 72 h (see the SI for details). Major transformations observed for both derivatives are summarized in Scheme 3. The reactivity of both payloads is dominated by slow ligand substitution reactions of the Pt(IV) moieties. In PPL 9, the axial hydroxo ligand proved to be susceptible to slow substitution by chloride in PBS ([Cl−] = 140 mM; 100% after 72 h) and saline ([Cl−] = 154 mM; ∼10% after 72 h). The major transformation detected for PPL 11 in PBS, which is also observed in compound 5 featuring a trans-diacetato coordination, is the slow substitution of the equatorial chloro ligand, the leaving group in compound 1, with water, resulting in a hydroxo complex. Slow formation of the hydroxo species is also observed after 72 h of incubation in cell culture media (RPMI-1640, data not shown). Interestingly, this ligand exchange is not observed in saline, suggesting that phosphate (10 mM in PBS, 6 mM in the cell culture media) may catalyze this reaction by serving as an auxiliary leaving group. The normal level of inorganic phosphate in serum is ∼0.8−1.6 mM,25 potentially reducing the likelihood of this reactivity in circulation. After reduction to Pt(II) and release of the payload in the cell, an equatorial hydroxo ligand can be expected to convert to a labile aqua ligand due to the changed Lewis acidity of the metal center. Since the substitution of the chloro leaving group by water increases the DNA reactivity of Pt(II)-based drugs (including PAs)9 with DNA, loss of chloride in the PPL in circulation may be a beneficial activation step.
Scheme 3. Reactivity of PPLs in Media and Buffered Solutionsa.

aBased on product analysis by LC-MS after 72 h of continuous incubation in buffers at 37 °C. (i) PPL 9 in 1 × PBS (pH 7.4), saline (0.9% NaCl, pH 7.0), (ii) PPL 11 in 50 mM ammonium bicarbonate (ABC, pH 7.4), (iii) PPL 11 in 1 × PBS (pH 7.4).
In ammonium bicarbonate (ABC) buffer, the chloro ligand is readily replaced by ammonia. Given the low normal blood levels of ammonia (<30 μM26), this mechanistically undesirable reaction can be considered pharmacologically irrelevant, but may be observed in mass spectrometric analysis of the bioconjugates, which typically utilizes volatile ammonium-based buffers.27 The hydrolytic loss of the equatorial chloro ligand observed in 11 is consistent with reactivity patterns reported in other Pt(IV) prodrugs,28,29 but occurs at a considerably slower rate than in the Pt(II)-based parent PAs (t1/2(aquation) < 30 min9). Most importantly, reductive degradation to Pt(II) over time, which would result in the loss of axial ligands and formation of compound 1, is not observed for any of the payloads. Likewise, no ligand substitution reactions or hydrolytic carboxylate (“ester”) cleavage is observed that would lead to loss of the axial linker-modified ligand Y′. Together, these observations support the notion that the PPLs are sufficiently stable to prevent significant premature nonreductive dissociation by ligand substitution of the cytotoxic payload from the protein carrier. Chemical robustness is a prerequisite for short-term (72 h) functional assays performed in cultured cells and ultimately in applications requiring extended circulation in vivo.
Conjugation and Reductive Activation.
With the maleimide-modified payloads in hand, we next modeled the bioconjugation of the PPLs with cysteine residues in carrier proteins using the tripeptide glutathione (GSH). Incubation of PPLs 9 and 11 with GSH in PBS (pH 7.4) at room temperature at a 1:1 stoichiometry (Scheme 4) resulted in clean conversion to the expected Michael adduct in less than 30 min (for details of the LC-MS analysis of reaction mixtures, see the SI). Importantly, under these conditions, the GSH thiol group reacts selectively with maleimide, and no competing ligand displacement by cysteine sulfur is observed in the Pt(IV) moiety. This outcome was expected as we previously demonstrated that Michael addition outcompetes ligand substitution even in the more reactive maleimide-modified Pt(II)-based payloads.14 However, excess GSH results in the reduction of the Pt(IV) moiety and subsequent substitution of the chloro-leaving group by nucleophilic GSH, a reaction observed in platinum(II)−acridine agents and classical platinum drugs.30 These results confirm that while the Pt(IV) payloads can be expected to undergo reduction by excess thiol (e.g., intracellularly), the preparation of bioconjugates via selective modification of cysteine thiol groups can be achieved using Michael addition chemistry under stoichiometric conditions.
Scheme 4. Model Reactions in Selected PPLsa.

aReagents and Conditions: (a) 100 μM, GSH (1 equiv), pH 7.4 (PBS), rt. (b) 10 equiv. sodium ascorbate, pH 7.4 (pH-adjusted water, PBS), 37 °C. Reactions were monitored by LC-MS.
While the new PPLs are expected to remain intact when attached to a carrier protein in circulation, their design requires rapid reduction in the reducing environment of target cells to release compound 1. To mimic the reductive activation of PPLs 9 and 11, both payloads were treated with excess sodium ascorbate in pH-adjusted water (pH 7.4) and PBS (pH 7.4) at 37 °C. In reactions monitored by LC-MS (see the SI for details), reductive elimination of the axial ligands Y and Y′ leads to clean conversion of the PPLs to intact compound 1. The reduction rates depend on the nature of Y and the media. In water, complete reduction of PPL 11 occurred within the first 30 min of incubation but took 2 h for PPL 9. The relative rates are consistent with previous observations that platinum(IV) complexes with axial hydroxo ligands are often more resistant to reduction than complexes featuring carboxylate ligands.31 A similar rapid rate of reduction is observed for PPL 11 in PBS, whereas PPL 9 requires approximately 48 h for complete reduction to occur under the same conditions.
The platinum moieties in the PPLs derived from compound 1 are cationic entities featuring one chloro-leaving group and three amine/amidine donors ([PtN3X]+). By contrast, classical Pt(IV) prodrugs are electroneutral and contain two equatorial am(m)ine nonleaving groups and anionic leaving groups X ([PtN2X2]). These differences may contribute to the relative lability of axial hydroxo ligand in PPL 9 and the rapid reduction observed for PPL 11 with ascorbate, which appears to proceed faster than typically observed in Pt(IV) prodrugs with the same type of axial ligands under the same conditions.21 Additional studies in an extended library of derivatives are needed to firmly establish relationships between structure, reactivity, and electron transfer rates29,32 and tailor the stability of the PPLs to a specific delivery application.
Next, we demonstrated the utility of an azide-functionalized Pt(IV) derivative in generating a peptide conjugate using a combination of click and coupling chemistries. Here, the cyclic RGD pentapeptide cyclo[RGDfK] (RGD = arginine-glycine-aspartate, f = D-phenylalanine, K = lysine) was chosen as the targeting ligand (Scheme 5). cyclo[RGDfK] targets the heterodimeric integrin αvβ3,33 which is highly expressed in cancer cells and in the vasculature of many tumors and is being widely evaluated in cancer diagnosis and targeted drug delivery.34,35 Efficient preparation of model conjugate cyclo[RGDfK]-C6-DBCO-8 (14) is accomplished by a two-step process involving amide coupling to modify the lysine-NH2 residue in cyclo[RGDfK] with a DBCO linker (13) and subsequent SPAAC chemistry to attach Pt(IV) derivative 8 (see Scheme 1). Alternatively, 14 can also be assembled by first modifying click chemistry-enabled 8 with the linker module (DBCO-C6-NHS ester), followed by coupling to cyclo[RGDfK], but major degradation and reduction of the Pt(IV) moiety are observed under the basic conditions required to promote amide coupling.
Scheme 5. Preparation and Reductive Activation of an RGD Peptide Conjugatea.

aReagents and Conditions: (a) 1. DIPEA, DMF, 6 h, rt; 2. purification by SEC. (b) 8, DMF, 15 h, rt. (c) 10 equiv sodium ascorbate, PBS (pH 7.4), 37 °C. Reactions were monitored by LC-MS.
Similar to the trans-dicarboxylato-coordinated PPL 11, conjugate 14 with a mixed trans-carbamato/carboxylato coordination is readily reduced by excess ascorbate in PBS (Scheme 5). An LC-MS trace recorded for a sample withdrawn from the reaction mixture (Figure 3A) shows that approximately 40% reduction of model conjugate 14 to compound 1 had occurred after <10 min (based on integrated peak intensities in the UV−vis trace). This observation is corroborated by the concomitant appearance of peaks indicating the release of the axial cyclo[RGDfK]-DBCO-C6-carboxylato ligand. After approximately 40 min (Figure 3B), the LC-MS traces indicate complete, clean, and stereoselective reduction of 14, yielding compound 1 as the only detectable PA species in the mixture (see the caption of Figure 3 for details).
Figure 3.

Reduction of bioconjugate 14 by ascorbic acid monitored by LC-MS after ∼10 min (A) and ∼40 min (B) of incubation in PBS (pH 7.4) at 37 °C. The top panels in parts A and B show base peak chromatograms (top) and UV−vis chromatograms (bottom) monitored at an acridine-specific wavelength (413 nm). The bottom panels in A and B show positive-ion mode electrospray mass spectra of HPLC fractions. Characteristic HPLC peaks and the corresponding MS features are highlighted with boxes for intact 14 (blue; calc. m/z for [M + H]2+ = 873.37, [M+2H]3+ = 582.25), the reduction product 1 (red; calc. m/z for [M]+ = 597.19, [M + H]2+ = 299.09), and the released axial cyclo[RGDfK]-DBCO-carboxylato ligand (green, two SPAAC regioisomers; calc. m/z for [M]+ = 1020.47). See Scheme 5, reaction c, for color coding.
Activation in Cancer Cells.
The PPL design requires not only efficient delivery of PAs in their more inert Pt(IV) form to tumor tissue but also their rapid reductive release in target cells. The success of PAs as anticancer agents relies on the rapid formation of DNA adducts, which can only be achieved with the significantly more reactive Pt(II) form of the drug.9 Thus, the premise of the current design is that only compound 1 will be able to produce cytotoxicity at nanomolar concentrations. As a first step in establishing the viability of the design in vitro, we labeled a trans-dicarboxylato-based PPL surrogate with an axial fluorophore and monitored its reductive activation in NCI-H460 nonsmall-cell lung cancer cells by confocal fluorescence microscopy. The probe (15) was generated by reacting click chemistry-enabled compound 7 with the noncleavable DBCO linker-modified red-fluorescent cyanine dye Cy5 (Figure 4A). Together with the intrinsic blue fluorescence of the acridine chromophore, this reporter molecule provides insight into the cellular reductive activation of the probe in colocalization assays.
Figure 4.

Colocalization assay in NCI-H460 lung cancer cells. (A) Structure of the cyanine dye-modified probe, Cy5-DBCO-7 (15), generated from 7 and Cy5-DBCO, with the blue emitting acridine and red emitting Cy5 chromophores highlighted. (B) Confocal microscopy images captured of NCI-H460 cells (top panel) treated with 15 (20 μM, 12 h). Fluorescence associated with the acridine and Cy5 fluorophores was recorded in the blue and red channels, respectively. The arrow in the blue channel of a single cell (bottom panel) indicates blue fluorescence localized to the nucleolus, consistent with the accumulation of compound 1 in this region of the nucleus.
NCI-H460 lung cancer cells were treated with conjugate 15 for 12 h and then fixed prior to the capture of confocal images. Treatment at a high concentration of 20 μM, but only for a short period of time, was necessary to produce a sufficiently strong fluorescence signal in cells without causing apoptosis. The confocal microscopy images show a high level of colocalization of the two fluorescence signals to cytoplasmic regions in cells, which most likely represent lysosomal vesicles (Figure 4A). Initial sequestration into acidified lysosomes is a hallmark of PAs.36 A closer view of individual cells (Figure 4B) shows that the red Cy5 fluorescence is confined to discrete punctuate vesicular structures, while around the areas of colocalization, highly diffuse blue acridine-associated fluorescence is detected. This observation is suggestive of dissociation and spatial separation of the Cy5 dye from the acridine chromophore, as would be expected following reductive elimination of the axial ligands and the formation of compound 1. Importantly, cells show accumulation of the released PA (but not Cy5) in the nuclear region, in particular, the nucleolus (Figure 4B, bottom panel). The low fluorescence intensity in the nucleus relative to other cytoplasmic regions is caused by quenching of the blue emission of intercalated acridine by approximately 90% in PAs when bound to chromatin.34 The observed subcellular distribution is consistent with the previously established intracellular trafficking of PAs, which involves endolysosomal escape from vesicular structures in the perinuclear region and entry into the nucleus.36 Thus, the confocal microscopy data support a mechanism by which 15 undergoes intracellular reduction to release the active platinum(II) drug, 1, and can be considered a first step in validating the PPL design at the cellular level.
CONCLUSIONS
In this study, we have developed a versatile synthetic platform that allows for the design of PPLs of highly cytotoxic compound 1 and tested their reactivity and functionality as potential components of targeted anticancer bioconjugates. Besides their high potency at low-nanomolar doses and unique mechanism of action, PAs show additional features that make them an attractive alternative to conventional cytotoxic payloads. Hundreds of PAs have been synthesized by varying key structural components, such as (non)leaving groups, platinum−acridine linker region, and intercalating moiety.9 These parameters, in addition to the axial ligation (Y/Y′), can be used to optimize the stability−reducibility balance of the PPL for a specific carrier-payload combination or therapeutic application. The hydrophilicity of the dicationic PAs and their prodrugs, which results in high water solubility, is a desired feature of ADC payloads.3 The charge state also allows for rapid and selective entry into cancer cells via secondary-active transport by multidrug and toxin extrusion protein 1 (MATE1/SLC47A1), which is highly expressed in many tumors regardless of tissue of origin.12 This mechanism would open the possibility for rapid accumulation from the extracellular tumor environment in applications utilizing noninternalizing monoclonal antibodies and related carriers (“bystander killing”).37−39 Lastly, the PAs have demonstrated a spectrum of anticancer activity distinct from that of other DNA-targeted warheads currently used in ADC payloads, including topoisomerase I poisons and anthracyclines.7,12 Given these promising features and the renewed interest in DNA-targeted warheads in ADC payloads,3 the PPL concept introduced here should be evaluated for targeted applications in cancer treatment with PAs.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Christopher M. Tracy (WFU) for the acquisition of the HRMS spectra. Resources for this project were provided by NIH/NCI Cancer Center Support Grant P30CA012197 and by Wake Forest Innovations.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.3c00368.
Experimental section, NMR spectra, LC-MS and HRMS data, details of model reactions, and X-ray crystallography results (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.bioconjchem.3c00368
The authors declare no competing financial interest.
Contributor Information
Ikeer Y. Mancera-Ortiz, Department of Chemistry, Wake Forest University, Wake Downtown Campus, Winston-Salem, North Carolina 27101, United States
Jiangxue Chen, Department of Chemistry, Wake Forest University, Wake Downtown Campus, Winston-Salem, North Carolina 27101, United States.
Tyler A. Slade, Department of Chemistry, Wake Forest University, Wake Downtown Campus, Winston-Salem, North Carolina 27101, United States
Xiyuan Yao, Department of Chemistry, Wake Forest University, Wake Downtown Campus, Winston-Salem, North Carolina 27101, United States.
Shenjie Zhang, Department of Chemistry, Wake Forest University, Wake Downtown Campus, Winston-Salem, North Carolina 27101, United States.
Cynthia S. Day, Department of Chemistry, Wake Forest University, Winston-Salem, North Carolina 27109, United States
Ulrich Bierbach, Department of Chemistry, Wake Forest University, Wake Downtown Campus, Winston-Salem, North Carolina 27101, United States.
REFERENCES
- (1).Reuvers TGA; Kanaar R; Nonnekens J. DNA Damage-Inducing Anticancer Therapies: From Global to Precision Damage. Cancers 2020, 12, 2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zhong L; Li Y; Xiong L; Wang W; Wu M; Yuan T; Yang W; Tian C; Miao Z; Wang T; Yang S. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Conilh L; Sadilkova L; Viricel W; Dumontet C. Payload diversification: a key step in the development of antibody-drug conjugates. J. Hematol. Oncol. 2023, 16, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Yaghoubi S; Karimi MH; Lotfinia M; Gharibi T; Mahi-Birjand M; Kavi E; Hosseini F; Sineh Sepehr K; Khatami M; Bagheri N; Abdollahpour-Alitappeh M. Potential drugs used in the antibody-drug conjugate (ADC) architecture for cancer therapy. J. Cell. Physiol. 2020, 235, 31–64. [DOI] [PubMed] [Google Scholar]
- (5).Joubert N; Beck A; Dumontet C; Denevault-Sabourin C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).do Pazo C; Nawaz K; Webster RM The oncology market for antibody-drug conjugates. Nat. Rev. Drug Discovery 2021, 20, 583–584. [DOI] [PubMed] [Google Scholar]
- (7).Wu H; Bierbach U. Chemosensitivity-Gene Expression Correlations and Functional Enrichment Analysis Provide Insight into the Mechanism of Action of a Platinum-Acridine Anticancer Agent. ChemMedChem 2022, 17, No. e202200331. [DOI] [PubMed] [Google Scholar]
- (8).Zhang S; Yao X; Watkins NH; Rose PK; Caruso SR; Day CS; Bierbach U. Discovery of a Chiral DNA-Targeted Platinum-Acridine Agent with Potent Enantioselective Anticancer Activity. Angew. Chem., Int. Ed. 2020, 59, 21965–21970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Suryadi J; Bierbach U. DNA Metalating−Intercalating Hybrid Agents for the Treatment of Chemoresistant Cancers. Chem. − Eur. J. 2012, 18, 12926–12934. [DOI] [PubMed] [Google Scholar]
- (10).Ma Z; Choudhury JR; Wright MW; Day CS; Saluta G; Kucera GL; Bierbach U. A Non-Cross-Linking Platinum−Acridine Agent with Potent Activity in Non-Small-Cell Lung Cancer. J. Med. Chem. 2008, 51, 7574–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ding S; Hackett CL; Liu F; Hackett RG; Bierbach U. Evaluation of a Platinum-Acridine Anticancer Agent and Its Liposomal Formulation in an in vivo Model of Lung Adenocarcinoma. ChemMedChem 2021, 16, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yao X; Watkins NH; Brown-Harding H; Bierbach U. A membrane transporter determines the spectrum of activity of a potent platinum-acridine hybrid anticancer agent. Sci. Rep. 2020, 10, 15201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ding S; Qiao X; Kucera GL; Bierbach U. Using a build-and-click approach for producing structural and functional diversity in DNA-targeted hybrid anticancer agents. J. Med. Chem. 2012, 55, 10198–10203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yao X; Tracy CM; Bierbach U. Cysteine-Directed Bioconjugation of a Platinum(II)-Acridine Anticancer Agent. Inorg. Chem. 2019, 58, 43–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ding S; Bierbach U. Linker design for the modular assembly of multifunctional and targeted platinum(II)-containing anticancer agents. Dalton Trans. 2016, 45, 13104–13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Deonarain MP; Yahioglu G; Stamati I; Pomowski A; Clarke J; Edwards BM; Diez-Posada S; Stewart AC Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours? Antibodies 2018, 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Timerbaev AR; Hartinger CG; Aleksenko SS; Keppler BK Interactions of antitumor metallodrugs with serum proteins: advances in characterization using modern analytical methodology. Chem. Rev. 2006, 106, 2224–2248. [DOI] [PubMed] [Google Scholar]
- (18).Hall MD; Mellor HR; Callaghan R; Hambley TW Basis for design and development of platinum(IV) anticancer complexes. J. Med. Chem. 2007, 50, 3403–3411. [DOI] [PubMed] [Google Scholar]
- (19).Pichler V; Mayr J; Heffeter P; Domotor O; Enyedy EA; Hermann G; Groza D; Kollensperger G; Galanksi M; Berger W; Keppler BK; Kowol CR Maleimide-functionalised platinum(iv) complexes as a synthetic platform for targeted drug delivery. Chem. Commun. 2013, 49, 2249–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Deng Z; Zhu G. Beyond mere DNA damage: Recent progress in platinum(IV) anticancer complexes containing multi-functional axial ligands. Curr. Opin. Chem. Biol. 2023, 74, No. 102303. [DOI] [PubMed] [Google Scholar]
- (21).Babu T; Sarkar A; Karmakar S; Schmidt C; Gibson D. Multiaction Pt(IV) Carbamate Complexes Can Codeliver Pt(II) Drugs and Amine Containing Bioactive Molecules. Inorg. Chem. 2020, 59, 5182–5193. [DOI] [PubMed] [Google Scholar]
- (22).Wang X; Wang X; Jin S; Muhammad N; Guo Z. Stimuli- Responsive Therapeutic Metallodrugs. Chem. Rev. 2019, 119, 1138–1192. [DOI] [PubMed] [Google Scholar]
- (23).Wilson JJ; Lippard SJ Synthetic Methods for the Preparation of Platinum Anticancer Complexes. Chem. Rev. 2014, 114, 4470–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kim E; Koo H. Biomedical applications of copper-free click chemistry: in vitro, in vivo, and ex vivo. Chem. Sci. 2019, 10, 7835–7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Malberti F. Hyperphosphataemia: treatment options. Drugs 2013, 73, 673–688. [DOI] [PubMed] [Google Scholar]
- (26).Ali R; Nagalli S. Hyperammonemia. In StatPearls; StatPearls Publishing, 2023. Updated Apr 7, 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557504. [PubMed] [Google Scholar]
- (27).Xu T; Zhang F; Chen D; Sun L; Tomazela D; Fayadat-Dilman L. Interrogating heterogeneity of cysteine-engineered anti- body-drug conjugates and antibody-oligonucleotide conjugates by capillary zone electrophoresis-mass spectrometry. MAbs 2023, 15, No. 2229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kastner A; Poetsch I; Mayr J; Burda JV; Roller A; Heffeter P; Keppler BK; Kowol CR A Dogma in Doubt: Hydrolysis of Equatorial Ligands of Pt(IV) Complexes under Physiological Conditions. Angew. Chem., Int. Ed. 2019, 58, 7464–7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Xu Z; Wang Z; Deng Z; Zhu G. Recent advances in the synthesis, stability, and activation of platinum(IV) anticancer prodrugs. Coord. Chem. Rev. 2021, 442, No. 213991. [Google Scholar]
- (30).Ma Z; Rao L; Bierbach U. Replacement of a Thiourea-S with an Amidine-NH Donor Group in a Platinum−Acridine Antitumor Compound Reduces the Metal’s Reactivity with Cysteine Sulfur. J. Med. Chem. 2009, 52, 3424–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Choi S; Filotto C; Bisanzo M; Delaney S; Lagasee D; Whitworth JL; Jusko A; Li CR; Wood NA; Willingham J; Schwenker A; Spaulding K. Reduction and anticancer activity of platinum(IV) complexes. Inorg. Chem. 1998, 37, 2500–2504. [Google Scholar]
- (32).Hall MD; Hambley TW Platinum(IV) antitumour compounds: their bioinorganic chemistry. Coord. Chem. Rev. 2002, 232, 49–67. [Google Scholar]
- (33).Pierschbacher MD; Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [DOI] [PubMed] [Google Scholar]
- (34).Chen H; Niu G; Wu H; Chen X. Clinical Application of Radiolabeled RGD Peptides for PET Imaging of Integrin alpha(v)-beta(3). Theranostics 2016, 6, 78–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Danhier F; Le Breton A; Preat V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol. Pharmaceutics 2012, 9, 2961–2973. [DOI] [PubMed] [Google Scholar]
- (36).Ding S; Qiao X; Suryadi J; Marrs GS; Kucera GL; Bierbach U. Using fluorescent post-labeling to probe the subcellular localization of DNA-targeted platinum anticancer agents. Angew. Chem., Int. Ed. 2013, 52, 3350–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rossin R; Versteegen RM; Wu J; Khasanov A; Wessels HJ; Steenbergen EJ; Ten Hoeve W; Janssen HM; van Onzen A; Hudson PJ; Robillard MS Chemically triggered drug release from an antibody-drug conjugate leads to potent antitumour activity in mice. Nat. Commun. 2018, 9, 1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ashman N; Bargh JD; Spring DR Non-internalising antibody-drug conjugates. Chem. Soc. Rev. 2022, 51, 9182–9202. [DOI] [PubMed] [Google Scholar]
- (39).Staudacher AH; Brown MP Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.