

Abstract
The understudied members of the druggable proteomes offer promising prospects for drug discovery efforts. While large-scale initiatives have generated valuable functional information on understudied members of the druggable gene families, translating this information into actionable knowledge for drug discovery requires specialized informatics tools and resources. Here, we review the unique informatics challenges and advances in annotating understudied members of the druggable proteome. We demonstrate the application of statistical evolutionary inference tools, knowledge graph mining approaches, and protein language models in illuminating understudied protein kinases, pseudokinases, and ion channels.
Keywords: protein evolution, orthology, network biology, machine learning, sequence embedding
Introduction
Protein kinases and ion channels are among the most prominent families of druggable proteins. They play fundamental roles in cellular functions and disease,(p1),(p2),(p3),(p4) and abnormal expression, mutation, or mis-regulation of these proteins is causally associated with various human disorders.(p5),(p6),(p7),(p8) However, despite the biomedical importance of these proteins, a significant portion of the human protein kinases and ion channels remains understudied and is referred to as ‘dark’ by the Illuminating Druggable Genome (IDG) consortium.(p9),(p10) The IDG Data and Resource Generation Centers (DRGCs) have generated valuable datasets, reagents, and chemical probes to illuminate understudied kinases, G protein-coupled receptors (GPCRs), and ion channels. Likewise, RNA interference (RNAi) screens of 260 conserved understudied genes have been performed in Drosophila, resulting in the generation of the ‘Unknome’ database.(p11), The International Mouse Phenotype consortium has also generated thousands of mutant lines, which can be leveraged to identify disease associations for understudied genes.(p12) While these and other ‘omic’ efforts have resulted in large volumes of data, effectively mining these data to annotate understudied proteins is a critical first step in identifying new drug targets. In particular, the wealth of sequence data available on protein kinases and ion channels from diverse organisms, combined with recent progress in structure prediction methods such as AlphaFold2,(p13) provides valuable information for predicting and prioritizing understudied targets for drug discovery efforts. Likewise, integrative mining of sequence data in the context of cellular pathways, protein–protein interactions, cell type-specific expression, and disease mutations can identify novel disease associations for understudied members of the protein kinase and ion channel superfamily.(p14)
This review highlights the informatics challenges and advances in illuminating understudied proteomes. We focus on three related but complementary approaches (Figure 1). We first describe the application of orthology-based inference tools and associated statistical methods for predicting understudied protein functions based on the evolutionary context (Figure 1a, left). Next, we describe the development and application of knowledge graph (KG) mining approaches for function prediction using the network context encoded in KGs (Figure 1b). Finally, we review recent advances in sequence embeddings generated from protein language models for annotating understudied protein kinases and ion channels (Figure 1a, right). Figure 1c provides an overview of how these tools can be combined to derive novel hypotheses for understudied proteins and aid drug discovery efforts. A list of tools, and links thereto, are also provided in Table 1 for easy access. The abbreviations used in this review are listed in Table 2 for ease of reference.
FIGURE 1.
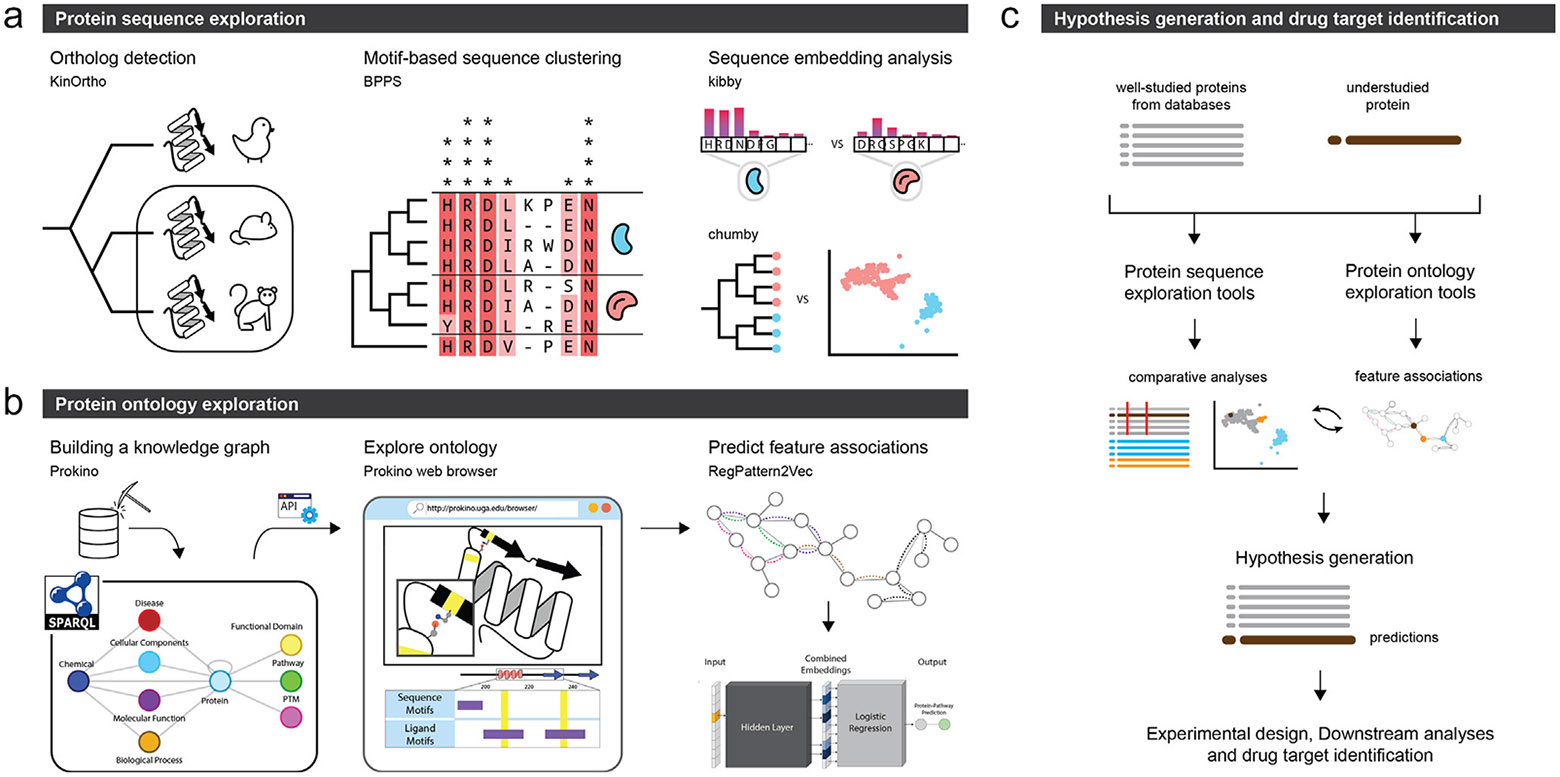
The panels represent the informatics approaches used for illuminating the druggable proteome. (a) Protein sequence exploration tools that enable analysis based on large collections of sequences, such as detection and collection of orthologs (KinOrtho), motif-based clustering of related sequences (BPPS), and embedding based-analyses (kibby and chumby). (b) Protein ontology exploration tools that rely on visualizing and querying prebuilt knowledge graphs to infer feature associations and predict functional and regulatory roles. (c) A schematic showing how the tools in panels (a) and (b) can be used iteratively to compare and contrast an understudied protein sequence with its well-studied counterparts to derive novel hypotheses and identify drug targets.
TABLE 1.
List of informatics tools for illuminating understudied proteins
| Tool name | Description and location |
|---|---|
| KinOrtho | Full-length and domain-based orthology detection https://github.com/esbgkannan/KinOrtho |
| KinVieW | Visualization tool for correlating natural sequence variation with cancer variants and post-translational modifications in the protein kinase domain https://prokino.uga.edu/kinview/ |
| BPPS | Bayesian pattern-based hierarchical sequence partitioning https://www.igs.umaryland.edu/labs/neuwald/software/bpps/ |
| ProKinO | An integrated knowledge graph linking diverse forms of data on protein kinases https://prokino.uga.edu/ |
| RegPattern2Vec | A random walk-based graph embedding approach for link prediction tasks https://github.com/esbgkannan/RegPattern2Vec |
| chumby | Sequence embedding-based tree visualization https://github.com/esbgkannan/chumby |
| kibby | Alignment free embedding-based sequence conservation analysis https://github.com/esbgkannan/kibby |
| Pharos | Knowledge base for the Druggable Genome to illuminate understudied or poorly characterized portions of protein families https://pharos.nih.gov/ |
TABLE 2.
Abbreviations and their full forms provided for reference in the order they appear in the review
| Abbreviations | Referring to |
|---|---|
| IDG | Illuminating Druggable Genome |
| DRGCs | IDG Data and Resource Generation Centers |
| GPCRs | G protein-coupled receptors |
| RNAi | RNA interference |
| BPPS | Bayesian partitioning with pattern selection |
| ULK4 | Unc-51-like kinase 4 |
| PSKH2 | Serine/threonine-protein kinase H2 |
| TAOK1 | Thousand and one amino acid protein kinase 1 |
| PIK3C2A | Phosphatidylinositol 4-phosphate 3-kinase C2 domain-containing subunit alpha |
| GO | Gene ontology |
| NIAS | Novel Inferred Annotation Score |
| TCRD | Target Central Resource Database |
| TDL | Target development level |
| KGs | Knowledge graphs |
| RDF | Resource description format |
| ProKinO | Protein Kinase Ontology |
| SPARQL | SPARQL Protocol and RDF Query Language |
| PAK5 | p21-activated protein kinase 5 |
| Hsp90 | Heat shock protein 90 |
| ATP | Adenosine triphosphate |
| ESM2 | Evolutionary Scale Modeling 2 |
| VIBE | Variational Autoencoder Implemented Branch Support Estimation |
| UMAP | Uniform Manifold Approximation and Projection |
| GABRP | Gamma-aminobutyric acid receptor subunit pi |
| CHRNB1 | Acetylcholine receptor subunit beta |
| CHRNA10 | Neuronal acetylcholine receptor subunit alpha-10 |
| 5-HT3 | 5-Hydroxytryptamine type 3 |
| ZAC | Zinc-activated channel |
| GABA | Gamma-aminobutyric acid |
| nACh | Nicotinic acetylcholine |
Orthology-based inference tools to illuminate understudied proteomes
The understudied protein kinases and ion channels belong to a large superfamily of evolutionarily related proteins, several of which are functionally well illuminated. Thus quantitative comparisons of primary sequences and three-dimensional structures can provide critical evolutionary clues for predicting the functions of understudied members of the protein kinase and ion channel superfamily. Indeed, sequence and evolution-based approaches have proven crucial in the prioritization and functional characterization of understudied kinases(p15),(p16),(p17),(p18),(p19),(p20) and pseudokinases.(p16),(p18),(p19),(p21),(p22) However, the lack of dedicated tools to map orthologs across species, and the challenges in accurately aligning divergent sequences, present a significant bottleneck in the sequence-based annotation of understudied protein kinases and ion channels.
Protein sequence data have been successfully employed to predict novel drug targets using methods such as prioritization based on gene essentiality,(p23) identification of common targets across multiple pathogens,(p24) or using a subtractive genomics approach.(p25) Given the widespread popularity of these methods, several tools have emerged to identify orthologs, albeit with slight variations in how they define computationally defined orthologs. Most methods rely on an initial all vs all pairwise protein similarity search to identify reciprocal best hits, and then on phylogenetic, matrix-based, or distance-based clustering methods to define orthologous gene sets.(p26),(p27),(p28),(p29),(p30),(p31),(p32),(p33),(p34),(p35),(p36),(p37) Resources such as ECOdrug(p38) further enhance evolutionary conservation-based drug discovery by curating drugs and their protein targets across different species. As a common theme across these comparative genomics approaches, they traditionally begin by identifying and comparing evolutionarily related sequences across diverse organisms using multiple sequence alignment methods. For example, the KinVieW visualization tools enables consistent and reproducible evolutionary comparisons using a consistent set of curated alignments spanning protein kinase families from diverse organisms.(p17) Likewise, statistical tools such as Bayesian partitioning with pattern selection (BPPS)(p39) group evolutionarily related sequences based on the patterns of conservation and variation in large multiple-sequence alignments. In so doing, they enable the functional annotation of understudied members by identifying patterns shared among understudied and illuminated members of a protein superfamily. Indeed, these statistical sequence classification methods and visualization tools have been successfully employed for predicting and experimentally testing the functions of several understudied kinases, such as Unc-51-like kinase 4 (ULK4) and the dark pseudokinase, serine/threonine-protein kinase H2 (PSKH2).(p15),(p18),(p19),(p22)
More recently, an orthology mapping tool, KinOrtho, was developed to alleviate the challenge of leveraging evolutionary data for protein kinase function prediction.(p20) KinOrtho employs a combination of query- and graph-based techniques, utilizing full-length and domain-based strategies to accurately map one-to-one kinase orthologs across an extensive set of 17 000 species. The effectiveness of KinOrtho is demonstrated through rigorous metrics, showcasing its superior performance compared to existing methods. KinOrtho enhances accuracy by identifying potential false positives and emphasizing sequences lacking proper kinase domains for further scrutiny, thus possessing great utility in dark kinase illumination. For example, KinOrtho detected a domain fusion event between the understudied TAOK1 and a functionally well-studied PIK3C2A kinase in a subset of nine nematode species. Subsequent coexpression analysis revealed a strong association between the two kinases extending well beyond nematodes to other organisms. This suggests a possible physical interaction between the two proteins and a functional association involving communication between the cell membrane and the cytosol.
Further, machine learning models have been leveraged to use the KinOrtho-defined orthologs for well-studied kinases, their sequence similarities, and manually curated Gene Ontology (GO) annotations as training features to derive functional predictions for the understudied kinases. This framework has predicted biological functions for various understudied kinases.(p20),(p40) Moreover, depending on the model’s ability to assign putative functions, a quantitative score, called the Novel Inferred Annotation Score (NIAS) score, is also assigned to each understudied kinase, which prioritizes them for functional studies. Thus, KinOrtho is a valuable tool for hypothesis generation using evolutionary information encoded in protein sequences and can be extended to any druggable protein family of interest. This framework is being adopted to perform an orthology-based analysis of the human ion channelome. A similar study of full-length ion channel sequences and their isolated pore domains could shed light on the conserved functional associations of understudied ion channel sequences across diverse species. Identifying associated binding partners or tandem domains could be of interest for directing drug design efforts to provide fine-tuned control over channel functions and their underlying disease mechanisms.
Data integration and knowledge graph mining approaches for function prediction and target identification
Data related to protein kinases and ion channels are stored in disparate data sources and formats, posing significant data integration and mining challenges. Thus, harmonized data resources linking related data on druggable gene families can be valuable for hypothesis generation and testing.(p10) For example, Target Central Resource Database (TCRD)(p41) and Pharos(p14) are centralized resources with several built-in tools for users to identify and prioritize new targets for functional illumination or target discovery. These resources offer user-friendly GraphQL application programming interface (API) for enrichment analysis and tools for data visualization. Importantly, these resources provide an annotation that classifies targets based on their available data, known as the target development level (TDL), allowing researchers to prioritize understudied proteins for drug discovery efforts. Likewise, DrugCentral(p42) aggregates data on new drug approvals and standardizes drug information, including preclinical research and clinical practice data such as chemical structures, molecular physicochemical descriptors, and patent status. Multiple additional resources have been developed that provide information on understudied targets’ structural(p43) and pathway contexts. Of note is the Reactome knowledgebase, a curated knowledgebase of biological pathways successfully employed for predicting the biological functions and therapeutic potential of understudied dark proteins.(p44) Likewise, the dark kinase knowledgebase(p40) and LinkedOmics database(p45) capture additional experimental, proteomic, and disease-related data on understudied dark proteins.
While aggregated databases and harmonized resources serve as a valuable starting point for target identification, identifying hidden patterns in harmonized data resources requires data minability. KGs are a powerful solution to data representation and integration challenges. They semantically link disparate data sources into a structured resource description format (RDF), enabling efficient data sharing, storage, and mining.(p46),(p47),(p48),(p49) One such example is the Protein Kinase Ontology (ProKinO), which integrates data related to sequence, structure, function, pathway, gene expression, and ligand binding sites on protein kinases(p50),(p51) in human- and machine-readable formats. Both graph mining techniques and machine learning on ProKinO data have enabled the illumination of understudied kinases using network context. For example, ProKinO mining using the SPARQL protocol and RDF query language (SPARQL) identified p21-activated protein kinase 5 (PAK5) as a frequently mutated dark kinase in human cancers, including a previously unrecognized role in acute myeloid leukemia.(p51) Furthermore, the impact of oncogenic mutations on PAK5 structure and function was predicted using the structural visualization tools built into the ProKinO browser.(p51) In another example, effective integration of informatics and experimental approaches resulted in the functional illumination of the understudied pseudokinase, PSKH2. Specifically, quantitative comparisons of PSKH2 orthologs from diverse organisms identified primate-specific ‘pseudogenization’ of PSKH2 based on variations in the active site. Subsequent analysis of AlphaFold2 models and cell-based assays established the role of the N and C-terminal segments flanking the pseudokinase domain in cellular localization and interaction with the Hsp90 molecular chaperone.(p22) Furthermore, mining of ProKinO KG using the link prediction algorithm, RegPattern2Vec, predicted multiple pathways for PSKH2 including a role for PSKH2 in cilium assembly.(p52)
More recently, the addition of drug (ligand) classes and their relationships to protein kinase structural features using semantic relationships (edges) with structural motifs has enabled queries and hypothesis generation regarding the mode of binding of kinase drugs within the ATP or allosteric pockets (Figure 2), making ProKinO a valuable tool for drug discovery efforts.
FIGURE 2.
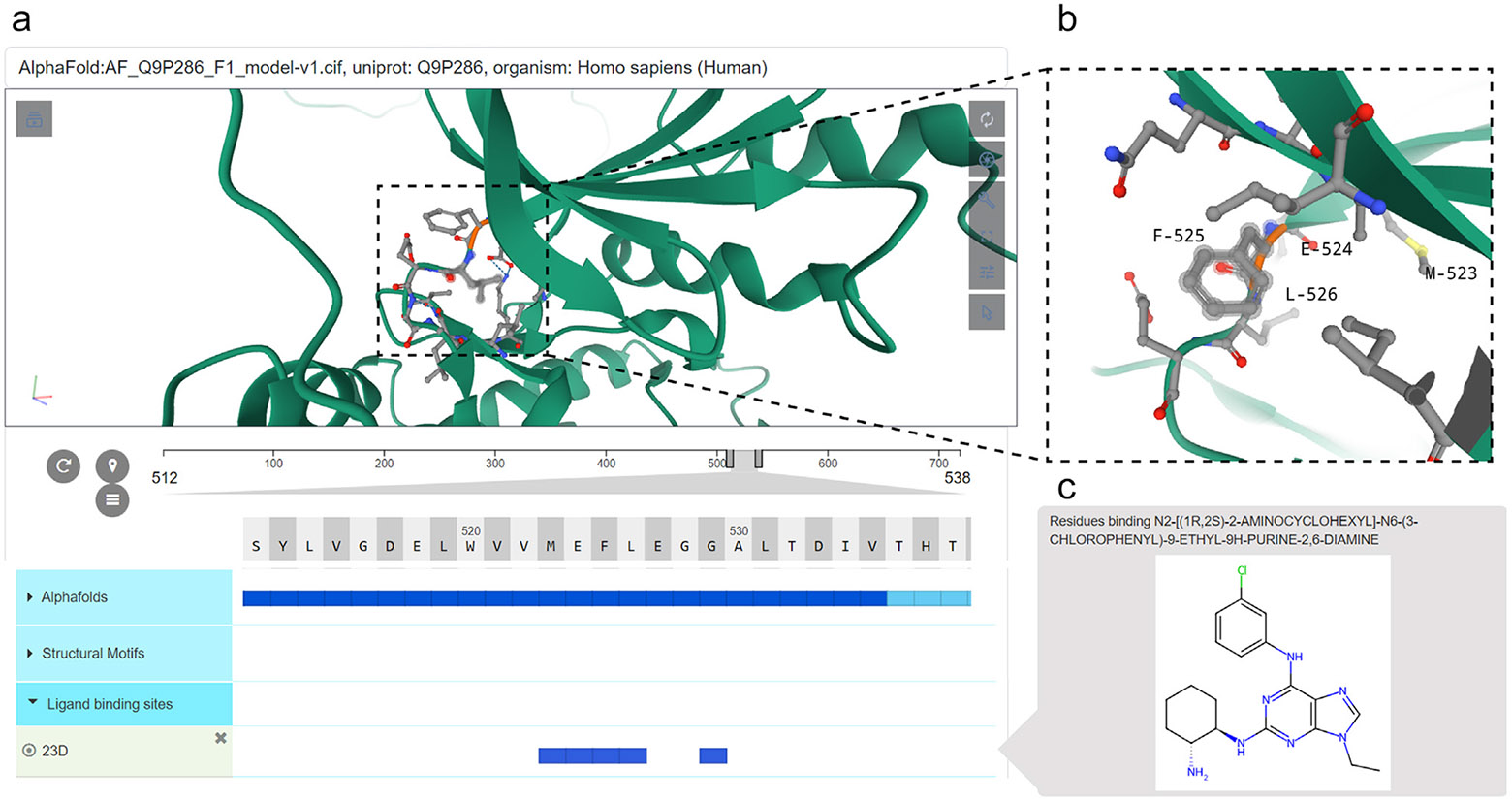
A demonstration of the ProtVista viewer within the ProKinO browser for mapping sequence annotations to 3D models using the p21 activated kinase 5 (PAK5) as an example. (a) Snapshot of the ProtVista viewer showing key sequence annotations within the ligand binding site of PAK5. (b) Zoomed-in view of the ATP/ligand binding residues. (c) Schematic of a small molecule targeting the active site of PAK5.
In addition to knowledge discovery through graph mining, KGs can be leveraged for machine learning tasks. In particular, graph representation learning through network embedding approaches(p53) offers the possibility of identifying hidden patterns or relationships (links) in large heterogeneous datasets. Indeed, various groups and IDG data management centers have successfully employed machine learning on KGs for predicting relationships between protein kinase inhibition and cancer;(p54),(p55) placing kinases in a pathway context;(p52) and predicting kinase substrates,(p56),(p57) drug and disease associations,(p58),(p59) and COVID-19 responses;(p49) among other applications.(p60),(p61),(p62) A significant challenge in the practical application of machine learning on KGs is the development of vector representations that effectively capture both local and latent contexts encoded in KGs. Various approaches for graph representation learning have been proposed,(p63) including the random walk-based method, RegPattern2Vec, which utilizes regular pattern-constrained random walks to capture multiple aspects of the node context within the KG.(p52),(p64) Learning functional representations from a kinase-centric KG establishes contextual information for kinases, interacting partners, modifications, pathways, localization, and chemical interactions. These known representations are then used to predict pathway associations for understudied kinases. Using RegPattern2Vec, Salcedo et al. predicted pathway associations for 30 dark kinases,(p52) and the predictions aligned well with pathway enrichment data obtained from experimentally generated protein proximity networks.(p40) A unique aspect of the RegPattern2Vec approach is that it enables the biological interpretation of the predictions by listing the collected random walks used in vector representations, thereby enabling the functional annotation of understudied targets.
Language models for illuminating druggable proteomes
Recent advances in deep learning open the exciting possibility of predicting understudied protein functions using protein language models trained on millions of sequences cataloged in sequence databases.(p65) These models represent inferred structural and functional properties of proteins as vector embeddings, capturing hidden patterns and nuances that traditional alignment-based methods may otherwise miss. Sequences represented as vectors, or sequence embeddings, can be leveraged for various function annotation tasks, such as predicting post-translational modifications,(p66) protein–protein interactions,(p67) protein–drug interactions,(p68) evolutionary conservation,(p69),(p70) and structure.(p71),(p72) Lin et al., for example, recently employed ESMFold, a fully end-to-end single-sequence structure predictor that relies on the ESM2 transformer language model, to internalize encoded evolutionary information and perform single-sequence structure predictions with high speed and accuracy. This task constitutes an alternative to AlphaFold2 structure-prediction(p73) by eschewing the need for a traditional sequence alignment, thereby making structure prediction robust even against proteins with minimal sequence representation (orphan proteins).
Alongside structure prediction, sequence embeddings generated from language models have also been successfully employed for alignment-free conservation estimation(p69),(p74),(p75) in three druggable protein families. These data, along with other protein families, can be visualized using the sequence annotation viewer in Pharos.(p14) These embeddings have also been successfully employed for alignment-independent embedding-based protein classification,(p70) protein structure prediction,(p71),(p72),(p76) homology detection, structural alignment,(p77),(p78) and predicting kinase–substrate associations.(p79) These advancements have significant implications for illuminating understudied ion channels, which have been challenging to study using traditional alignment-based approaches because of the extensive diversification of ion channel sequences and 3D structures. Many ion channels share little homology in sequence space, with the difficulties in structural characterization being a critical barrier to understanding ion channel function.(p80) Both issues hinder structure prediction approaches like AlphaFold2, which depend on accurate sequence alignments and experimentally derived models for training.(p81) Therefore, using sequence embeddings to capitalize on inferred structure–function properties from a protein language model may help overcome the challenges associated with alignment-based approaches.
As an illustration of the successful application of such a strategy, recent efforts focused on employing a sequence embedding-based classification and visualization method (chumby)(p70) to delineate the distant evolutionary relationships within the human Cys-loop ligand-gated ion channel families. An embedding-based tree (Figure 3a) was successfully generated using the conserved transmembrane segments within these sequences. Consistent with previous functional classifications,(p82),(p83) the embedding based classification clearly separates the cationic receptors [5-hydroxytryptamine type 3 (5-HT3), nicotinic acetylcholine (nACh), zinc-activated channel (ZAC)] from anionic receptors [glycine, γ-aminobutyric acid type a (GABAA)] (Figure 3b), with 98 % statistical support estimated by the Variational Autoencoder Implemented Branch Support Estimation (VIBE) scores.(p70) This separation is also observed in the Uniform Manifold Approximation and Projection (UMAP) plot (Figure 3a). The glycine receptor family contains one dark ion channel sequence [gamma-aminobutyric acid receptor subunit pi (GABRP)]. In contrast, the nACh family has two dark ion channel sequences [acetylcholine receptor subunit beta (CHRNB1) and neuronal acetylcholine receptor subunit alpha-10 (CHRNA10)] grouped with other Cys-loop family members (stars in Figure 3a).
FIGURE 3.
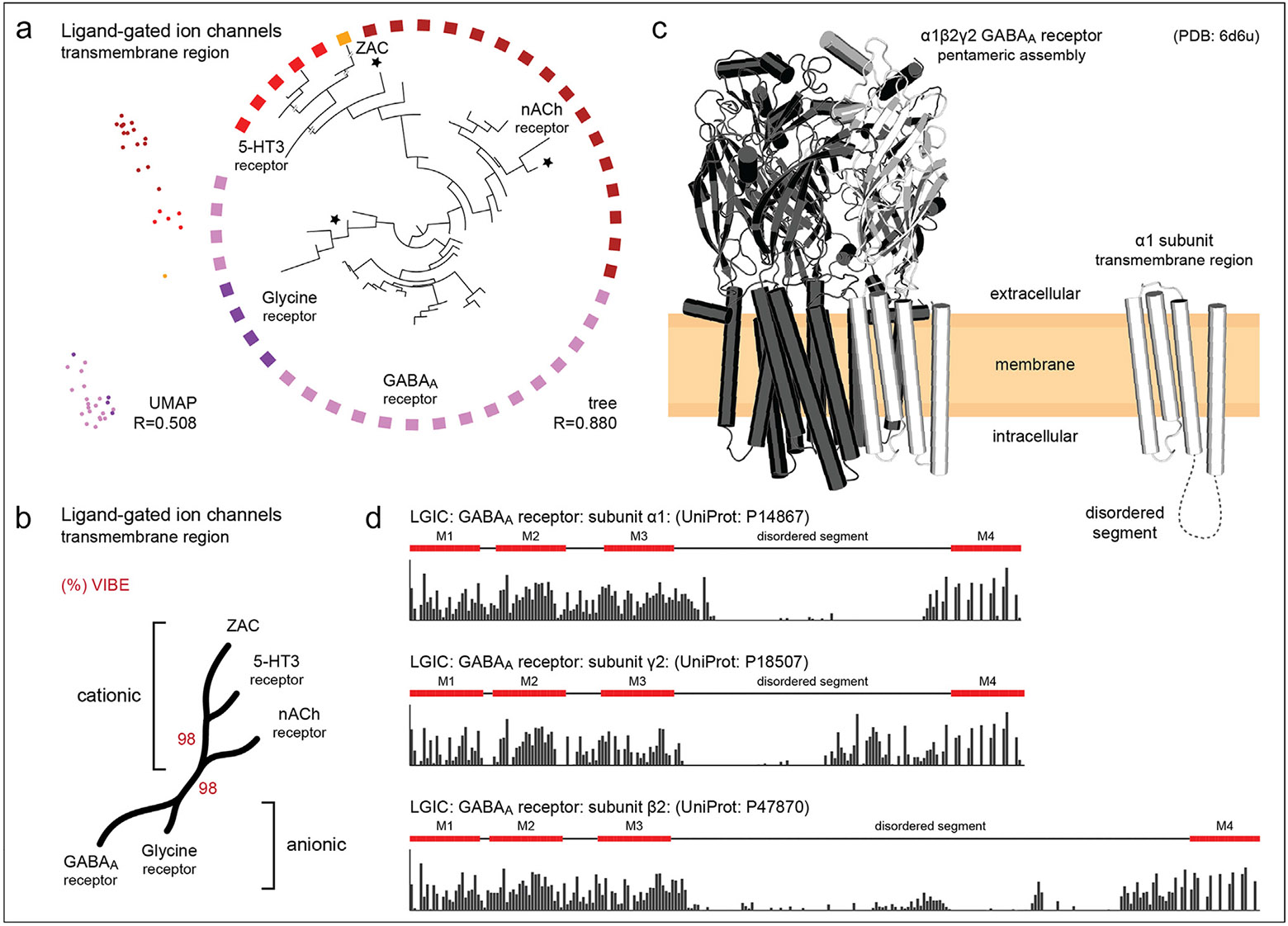
(a) An embedding tree of the human Cys-loop ligand-gated ion channels using the transmembrane region. Nodes with a black star indicate the dark ion channels in this subfamily. To the left of the tree, we plot a UMAP projection using the same dataset. (b) A condensed tree showing various families of Cys-loop ligand-gated ion channels. The red percentage values indicate VIBE scores. (c) Cryo-electron microscopy structures of an example membrane-bound ligand-gated ion channel, human α1β2γ2 GABAA receptor.(p86) The structural model on the left shows the heteropentameric assembly built from three unique subunits: α1, β2, and γ2. Each monomer consists of an N-terminal extracellular domain, which adopts a β-sandwich, followed by the C-terminal transmembrane domain. Within the pentamer, an α1 subunit is colored white. The structure on the right depicts the transmembrane domain of an α1 subunit. (d) Embedding-based sequence conservation for the transmembrane region of GABAA receptor subunits α1, β2, and γ2. Each consists of four transmembrane helices, designated M1–4, and a disordered segment on the M3–4 loop. Bars indicate the level of conservation at that sequence position, with taller bars indicating higher conservation.
Interestingly, CHRNA10 is placed closest and basal to the 5-HT3 and ZAC families. In the cellular context, the Cys-loop ligand-gated receptor superfamily members are known to form homo- or heteropentameric assemblies. For instance, the human α1β2γ2 GABAA receptor is a pentameric assembly of three unique subunits: α1, β2, and γ2 (Figure 3c). Additionally, we estimate sequence conservation across these three subunits using sequence embeddings(p69) which reveal highly conserved regions corresponding to the four conserved transmembrane helices. Low conservation scores indicate fast-evolving regions (Figure 3d), such as those observed between the M3 and M4 helices, which reflect variable functions among Cys-loop members such as receptor modulation, sorting, and trafficking. In contrast, the high conservation observed in the transmembrane domain reflects shared functions, such as multimeric assembly and gating mechanisms.(p84) Thus, such alignment-free identification of fast- and slow-evolving sites can be employed for uncovering functionally important regions of members of the dark channel subfamily (GABRP, CHRNB1, CHRNA10).
Concluding remarks
The IDG consortium has generated valuable datasets and resources to characterize understudied protein kinases, ion channels, and GPCRs. The next challenge is translating these data and resources into knowledge for drug discovery efforts. Here, we highlight the informatics challenges and advances in annotating understudied proteomes. Focusing on understudied protein kinases and ion channels, we demonstrate the application of evolutionary inference, KG mining, and protein language models in the functional annotation of understudied members of these protein families. In particular, sequence representations generated from protein language models can be leveraged for the functional annotation of understudied ion channels, which have been challenging to study using traditional alignment-based approaches. Likewise, fine-tuning of large language models with experimentally derived substrate-specific profiles of kinases generated from peptide-library studies(p85) holds excellent promise in mapping the phosphorylation networks of the entire kinome and placing understudied kinases in a pathway context. Finally, approaches integrating KGs and protein language models can be powerful new tools for translating genomic discoveries into therapeutic strategies.
Acknowledgments
Aarya Venkat, Mariah Salcedo, Rayna Carter, and members of the NK lab are acknowledged for comments and suggestions. Funding for NK from NIH (U01CA271376 and U01CA239106) is acknowledged.
Footnotes
Declarations of interest
No interests are declared.
CRediT authorship contribution statement
Rahil Taujale: Conceptualization, Data curation, Formal analysis, Methodology, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. Nathan Gravel: Data curation, Formal analysis, Methodology, Resources, Software, Visualization, Writing – original draft. Zhongliang Zhou: Formal analysis, Investigation, Methodology, Software, Writing – original draft. Wayland Yeung: Formal analysis, Methodology, Software, Validation, Visualization. Krystof Kochut: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft. Natarajan Kannan: Conceptualization, Funding acquisition, Investigation, Project administration, Software, Supervision, Validation, Writing – original draft, Writing – review & editing.
Data availability
Data will be made available on request.
References
- 1.Alexander SPH et al. The Concise Guide to Pharmacology 2019/20: Ion channels. Br J Pharmacol. 2019;176:S142–S228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moran MM. TRP channels as potential drug targets. Annu Rev Pharmacol Toxicol. 2018;58:309–330. [DOI] [PubMed] [Google Scholar]
- 3.Oyrer J et al. Ion channels in genetic epilepsy: from genes and mechanisms to disease-targeted therapies. Pharmacol Rev. 2018;70:142–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8:982–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen P. The role of protein phosphorylation in human health and disease. The Sir Hans Krebs Medal Lecture. Eur J Biochem. 2001;268:5001–5010. [DOI] [PubMed] [Google Scholar]
- 6.Dworakowska B, Dolowy K. Ion channels-related diseases. Acta Biochim Pol. 2000;47:685–703. [PubMed] [Google Scholar]
- 7.Felix R. Channelopathies: ion channel defects linked to heritable clinical disorders. J Med Genet. 2000;37:729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Litan A, Langhans SA. Cancer as a channelopathy: ion channels and pumps in tumor development and progression. Front Cell Neurosci. 2015;9:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.IDG. Illuminating the Druggable Genome Consortium. National Institutes of Health; 2014. [Google Scholar]
- 10.Sheils T et al. How to illuminate the druggable genome using pharos. Curr Protoc Bioinformatics. 2020;69:e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rocha JJ et al. Functional unknomics: systematic screening of conserved genes of unknown function. PLoS Biol. 2023;21:e3002222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munoz-Fuentes V et al. The International Mouse Phenotyping Consortium (IMPC): a functional catalogue of the mammalian genome that informs conservation. Conserv Genet. 2018;19:995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varadi M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50:D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelleher KJ et al. Pharos 2023: an integrated resource for the understudied human proteome. Nucleic Acids Res. 2023;51:D1405–D1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Picado A et al. A chemical probe for dark kinase STK17B derives its potency and high selectivity through a unique P-loop conformation. J Med Chem. 2020;63:14626–14646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shrestha S et al. Cataloguing the dead: breathing new life into pseudokinase research. Febs J. 2020;287:4150–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McSkimming DI et al. KinView: a visual comparative sequence analysis tool for integrated kinome research. Mol Biosyst. 2016;12:3651–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Preuss F et al. Nucleotide binding, evolutionary insights, and interaction partners of the pseudokinase Unc-51-like kinase 4. Structure. 2020;28:1184–1196.e1186. [DOI] [PubMed] [Google Scholar]
- 19.Shrestha S, Bendzunas G, Kannan N. Protein kinase inhibitor selectivity “hinges” on evolution. Structure. 2022;30:1561–1563. [DOI] [PubMed] [Google Scholar]
- 20.Huang LC et al. KinOrtho: a method for mapping human kinase orthologs across the tree of life and illuminating understudied kinases. BMC Bioinformatics. 2021;22:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Boyle B et al. Computational tools and resources for pseudokinase research. Methods Enzymol. 2022;667:403–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Byrne DP et al. Evolutionary and cellular analysis of the 'dark' pseudokinase PSKH2. Biochem J. 2023;480:141–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doyle MA et al. Drug target prediction and prioritization: using orthology to predict essentiality in parasite genomes. BMC Genomics. 2010;11:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abadio AK et al. Comparative genomics allowed the identification of drug targets against human fungal pathogens. BMC Genomics. 2011;12:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosen MI et al. Application of a subtractive genomics approach for in silico identification and characterization of novel drug targets in Mycobacterium tuberculosis F11. Interdiscip Sci. 2014;6:48–56. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lechner M et al. Proteinortho: detection of (co-) orthologs in large-scale analysis. BMC Bioinformatics. 2011;12:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altenhoff AM et al. Inferring hierarchical orthologous groups from orthologous gene pairs. PLoS One. 2013;8:e53786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sonnhammer EL, Östlund G. InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015;43:D234–D239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaduk M, Sonnhammer E. Improved orthology inference with Hieranoid 2. Bioinformatics. 2017;33:1154–1159. [DOI] [PubMed] [Google Scholar]
- 31.Train CM et al. Orthologous Matrix (OMA) algorithm 2.0: more robust to asymmetric evolutionary rates and more scalable hierarchical orthologous group inference. Bioinformatics. 2017;33:i75–i82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cosentino S, Iwasaki W. SonicParanoid: fast, accurate and easy orthology inference. Bioinformatics. 2019;35:149–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huerta-Cepas J et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mi H et al. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47:D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nevers Y et al. OrthoInspector 3.0: open portal for comparative genomics. Nucleic Acids Res. 2019;47:D411–D418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Derelle R, Philippe H, Colbourne JK. Broccoli: combining phylogenetic and network analyses for orthology assignment. Mol Biol Evol. 2020;37:3389–3396. [DOI] [PubMed] [Google Scholar]
- 38.Verbruggen B et al. ECOdrug: a database connecting drugs and conservation of their targets across species. Nucleic Acids Res. 2018;46:D930–D936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neuwald AF. A Bayesian sampler for optimization of protein domain hierarchies. J Comput Biol. 2014;21:269–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berginski ME et al. The Dark Kinase Knowledgebase: an online compendium of knowledge and experimental results of understudied kinases. Nucleic Acids Res. 2021;49:D529–D535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheils TK et al. TCRD and Pharos 2021: mining the human proteome for disease biology. Nucleic Acids Res. 2020;49:D1334–D1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avram S et al. DrugCentral 2023 extends human clinical data and integrates veterinary drugs. Nucleic Acids Res. 2023;51:D1276–D1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.PDBe- KB: a community-driven resource for structural and functional annotations. Nucleic Acids Res 2020;48:D344–D353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brunson T et al. VIGET: a web portal for study of vaccine-induced host responses based on Reactome pathways and ImmPort data. Front Immunol. 2023;14:1141030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vasaikar SV et al. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46:D956–D963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang S et al. Querying knowledge graphs in natural language. J Big Data. 2021;8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicholson DN, Greene CS. Constructing knowledge graphs and their biomedical applications. Comput Struct Biotechnol J. 2020;18:1414–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rossanez A et al. KGen: a knowledge graph generator from biomedical scientific literature. BMC Med Inform Decis Mak. 2020;20:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reese JT et al. KG-COVID-19: a framework to produce customized knowledge Graphs for COVID-19 Response. Patterns (N Y). 2021;2 100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gosal G, Kochut KJ, Kannan N. ProKinO: an ontology for integrative analysis of protein kinases in cancer. PLoS One. 2011;6:e28782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saber Soleymani NG et al. Dark kinase annotation, mining and visualization using the Protein Kinase Ontology. PeerJ. 2023;11:e16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salcedo MV, Gravel N, Keshavarzhi A, Kochut K, Kannan N. Predicting protein and pathway associations for understudied dark kinases using pattern-constrained knowledge graph embedding. PeerJ. 2023;11:e15815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scarselli F et al. The graph neural network model. IEEE Trans Neural Netw. 2009;20:61–80. [DOI] [PubMed] [Google Scholar]
- 54.Ravanmehr V et al. Supervised learning with word embeddings derived from PubMed captures latent knowledge about protein kinases and cancer. NAR Genom Bioinform. 2021;3 lqab113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang LC et al. Quantitative Structure-Mutation-Activity Relationship Tests (QSMART) model for protein kinase inhibitor response prediction. BMC Bioinformatics. 2020;21:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirchoff KE, Gomez SM. EMBER: multi-label prediction of kinase-substrate phosphorylation events through deep learning. Bioinformatics. 2022;38:2119–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gavali S et al. A knowledge graph representation learning approach to predict novel kinase-substrate interactions. Mol Omics. 2022;18:853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang JJ et al. Knowledge graph analytics platform with LINCS and IDG for Parkinson’s disease target illumination. BMC Bioinformatics. 2022;23:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Evangelista JE et al. Toxicology knowledge graph for structural birth defects. Commun Med (Lond). 2023;3:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuleshov MV et al. KEA3: improved kinase enrichment analysis via data integration. Nucleic Acids Res. 2021;49:W304–W316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeon M et al. Prioritizing pain-associated targets with machine learning. Biochemistry. 2021;60:1430–1446. [DOI] [PubMed] [Google Scholar]
- 62.Zhavoronkov A, Vanhaelen Q, Oprea TI. Will artificial intelligence for drug discovery impact clinical pharmacology? Clin Pharmacol Ther. 2020;107:780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuxiao D, Chawla N, Swami A. metapath2vec: scalable representation learning for heterogeneous networks. 2017. [Google Scholar]
- 64.Abbas Keshavarzi NK, Krys Kochut. RegPattern2Vec: link prediction in knowledge graphs. In: 2021 IEEE International IOT, Electronics and Mechatronics Conference (IEMTRONICS). IEEE; 2021, Vol. 21:1–7. [Google Scholar]
- 65.Bepler T, Berger B. Learning the protein language: evolution, structure, and function. Cell Syst. 2021;12:654–669.e653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yan Y et al. MIND-S is a deep-learning prediction model for elucidating protein post-translational modifications in human diseases. Cell Rep Methods. 2023;3 100430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yao Y et al. An integration of deep learning with feature embedding for protein-protein interaction prediction. PeerJ. 2019;7:e7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh R et al. Contrastive learning in protein language space predicts interactions between drugs and protein targets. Proc Natl Acad Sci U S A. 2023;120 e2220778120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yeung W et al. Alignment-free estimation of sequence conservation for identifying functional sites using protein sequence embeddings. Brief Bioinform. 2023;24:bbac599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yeung W et al. Tree visualizations of protein sequence embedding space enable improved functional clustering of diverse protein superfamilies. Brief Bioinform. 2023;24:bbac619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chowdhury R et al. Single-sequence protein structure prediction using a language model and deep learning. Nat Biotechnol. 2022;40:1617–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin Z et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science. 2023;379:1123–1130. [DOI] [PubMed] [Google Scholar]
- 73.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Diaz DJ et al. Using machine learning to predict the effects and consequences of mutations in proteins. Curr Opin Struct Biol. 2023;78 102518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marquet C et al. Embeddings from protein language models predict conservation and variant effects. Hum Genet. 2022;141:1629–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weissenow K, Heinzinger M, Rost B. Protein language-model embeddings for fast, accurate, and alignment-free protein structure prediction. Structure. 2022;30:1169–1177.e1164. [DOI] [PubMed] [Google Scholar]
- 77.Hamamsy T et al. Protein remote homology detection and structural alignment using deep learning. Nat Biotechnol. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaminski K et al. pLM-BLAST: distant homology detection based on direct comparison of sequence representations from protein language models. Bioinformatics. 2023;39 btad579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou Z et al. Phosformer: an explainable transformer model for protein kinase-specific phosphorylation predictions. Bioinformatics. 2023;39 btad046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu Y, Wang K. Exploiting the diversity of ion channels: modulation of ion channels for therapeutic indications. Handb Exp Pharmacol. 2019;260:187–205. [DOI] [PubMed] [Google Scholar]
- 81.Binder JL et al. AlphaFold illuminates half of the dark human proteins. Curr Opin Struct Biol. 2022;74 102372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ortells MO, Lunt GG. Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends Neurosci. 1995;18:121–127. [DOI] [PubMed] [Google Scholar]
- 83.Nemecz à et al. Emerging molecular mechanisms of signal transduction in pentameric ligand-gated ion channels. Neuron. 2016;90:452–470. [DOI] [PubMed] [Google Scholar]
- 84.Thompson AJ, Lester HA, Lummis SC. The structural basis of function in Cys-loop receptors. Q Rev Biophys. 2010;43:449–499. [DOI] [PubMed] [Google Scholar]
- 85.Johnson JL et al. An atlas of substrate specificities for the human serine/threonine kinome. Nature. 2023;613:759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhu S et al. Structure of a human synaptic GABA (A) receptor. Nature. 2018;559:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.