

Abstract

There is an urgent need for new antibiotics given the rise of antibiotic resistance, and succinyl-diaminopimelate desuccinylase (DapE, E.C. 3.5.1.18) has emerged as a promising bacterial enzyme target. DapE from Haemophilus influenzae (HiDapE) has been studied and inhibitors identified, but it is essential to explore DapE from different species to assess selective versus broad-spectrum therapeutics. We have determined the structure of DapE from the ESKAPE pathogen Acinetobacter baumannii (AbDapE) and studied inhibition by known inhibitors of HiDapE. AbDapE is inhibited by captopril and sulfate comparable to HiDapE, but AbDapE was not significantly inhibited by a known indoline sulfonamide HiDapE inhibitor. Captopril and sulfate both stabilize HiDapE by increasing the thermal melting temperature (Tm) in thermal shift assays. By contrast, sulfate decreases the stability of the AbDapE enzyme, whereas captopril increases the stability. Further, we report two crystal structures of selenomethionine-substituted AbDapE in the closed conformation, one with AbDapE in complex with succinate derived from enzymatic hydrolysis of N6-methyl-l,l-SDAP substrate and acetate (PDB code 7T1Q, 2.25 Å resolution), and a crystal structure of AbDapE with bound succinate along with l-(S)-lactate, a product of degradation of citric acid from the crystallization buffer during X-ray irradiation (PDB code 8F8O, 2.10 Å resolution).
Introduction
The growing threat from antibiotic-resistant bacterial strains1 underscores the need to discover antibiotics with new mechanisms of action. The most widely encountered virulent species of antibiotic multidrug-resistant (MDR) microorganisms seen worldwide are known as the ESKAPE pathogens, which consist of Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.2 The ESKAPE pathogens are becoming increasingly resistant to many broad-spectrum antibiotics that are currently available due to a range of resistance mechanisms including mutations of the drug target, the use of efflux pumps, the ability to inactivate specific drugs, reduced cellular membrane permeability, and the growth of biofilms when the microbe becomes dormant in the host.2A. baumannii is an opportunistic bacterial pathogen3 primarily associated with hospital-acquired infections, but also plagues military personnel returning from conflict zones. Given that A. baumannii now exhibits a high incidence of MDR strains, there is an urgent need to develop novel and effective antibacterial agents against this pathogen.
The L-lysine biosynthetic pathway provides a wealth of opportunities toward new antibiotic targets, as it is required for bacterial growth and survival but is absent in humans.4 In particular, dapE-encoded N-succinyl-l,l-diaminopimelic acid desuccinylase (DapE, E.C. 3.5.1.18) is present in all Gram-negative and most Gram-positive bacteria and is one enzyme of the l-lysine biosynthetic pathway that is underexplored as a potential drug target.5 DapE is responsible for the synthesis of both lysine and meso-diaminopimelate (m-DAP),6 both of which are critical for peptidoglycan cell-wall synthesis. DapE catalyzes the hydrolysis of the substrate N-succinyl-l,l-diaminopimelic acid (l,l-SDAP), releasing succinate and providing l,l-diaminopimelic acid (L,L-DAP, Scheme 1).7
Scheme 1. Hydrolysis of l,l-SDAP and a substrate analogue by DapE.
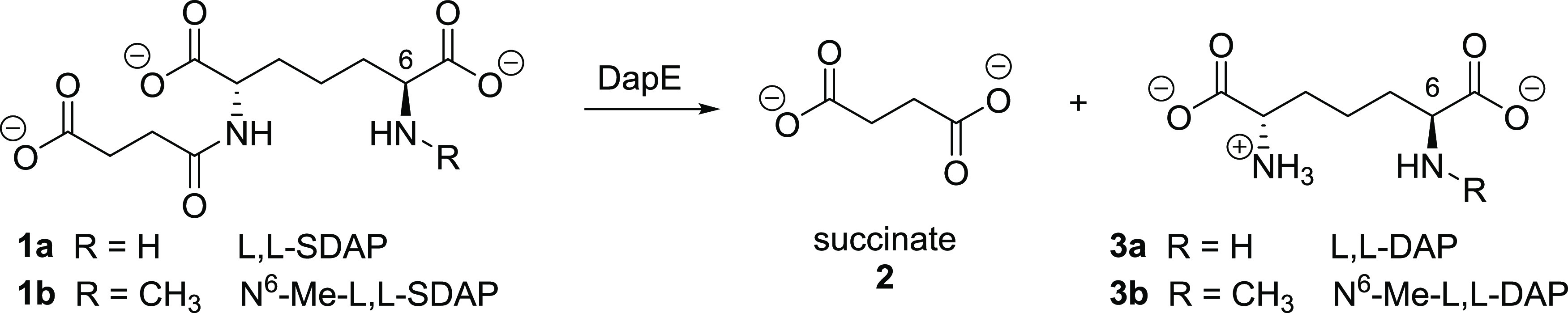
l,l-SDAP endogenous substrate (1a) and assay substrate N6-methyl-l,l-SDAP7 (1b) with formation of hydrolysis products succinate (2) and l,l-diaminopimelic acid derivatives 3a and 3b, respectively.
Knockout of dapE is lethal to Helicobacter pylori and Mycobacterium smegmatis, revealing the essential role of this enzyme in bacterial survival.8,9 The lack of a similar pathway in humans suggests that inhibition of DapE should be selectively toxic to bacteria, making it a promising antibiotic target with a new mechanism of action.5 We previously reported an assay7 to assess inhibition of DapE employing the synthetic substrate N6-methyl-l,l-SDAP (1b), which when cleaved by DapE affords the primary amine product 3b (Scheme 1) that is quantified spectrophotometrically after treatment with ninhydrin.
The first X-ray crystal structure of an apo DapE from Neisseria meningitidis (NmDapE) was solved in 2005,10 and structures of mono- and dizinc forms from Haemophilus influenzae (HiDapE)11 and NmDapE12 were reported thereafter, including a structure of the DapE inhibitor captopril13 bound to the active site of NmDapE.12 We reported a DapE crystal structure revealing the previously unknown closed conformation of dimeric DapE with the products of enzymatic cleavage, succinate and diaminopimelic acid, bound in the HiDapE active site (PDB 5VO3).14 Interestingly, this substantial DapE conformational change was predicted prior to the report of the crystal structure by the elegant computational work of Mishra who employed principal component analysis of molecular dynamics trajectories of the DapE apo enzyme and the DapE-SDAP complex.15 Thereafter, we reported an atomic-resolution (1.3 Å) structure of the open conformation of NmDapE (PDB 5UEJ.)16 These DapE structures have enabled refinement of the understanding of the enzymatic mechanism of hydrolysis of DapE and further insight into the design of successful inhibitors toward new antibiotics.
Herein, we report the inhibition of AbDapE by captopril and sulfate using our ninhydrin-based assay and the effect of these compounds on the thermal melting temperature (Tm) of AbDapE proteins using thermal shift assays. We report a lack of inhibition of AbDapE by a synthetic micromolar inhibitor of HiDapE, indoline sulfonamide. Furthermore, we report two X-ray crystal structures of AbDapE in the closed conformation. Co-crystallization of AbDapE with the modified substrate N6-Me-l,l-SDAP, allowed us to capture the enzyme in the closed conformation in complex with succinate.
Materials and Methods
All chemicals were purchased from commercial sources and were of the highest quality available.
Preparation of Selenomethinoine and Native DapE Proteins
AbDapE was expressed in Escherichia coli and prepared according to a general protocol described previously.7,12,14 For protein crystallography and enzymatic characterization, bacteria were cultured in selenomethionine (SeMet) and M9-minimal media, respectively. Media were supplemented with 150 μg/mL ampicillin at 37 °C with shaking at 210 rpm until the OD600 reached a value of 1.5. The temperature was lowered to 18 °C, and isopropyl-d-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM. The culture was grown for 18 h and then centrifuged at 4,500 rpm for 10 min at 4 °C. The cell pellet derived from 3 L of culture was resuspended in 150 mL (1 g of cells/5 mL of buffer) of lysis buffer (10 mM Tris-HCl, pH 8.3), 500 mM NaCl, 5% glycerol, 20 mM imidazole, 5 mM tris (2-carboxyethyl) phosphine hydrochloride (TCEP) and stored at −30 °C. A frozen suspension of cells with expressed protein was thawed and sonicated at 50% amplitude in 5 s × 10 s cycles for 20 min at 4 °C. The lysate was cleared by centrifugation at 18,000g for 40 min at 4 °C, the supernatant was collected, and the protein was purified as previously described with some modifications. The supernatant was loaded onto a His-Trap FF (Ni-NTA) column using a GE Healthcare ÅKTA Pure system with a loading buffer (10 mM Tris-HCl (pH 8.3), 500 mM NaCl, 1 mM TCEP, and 5% glycerol). The column was washed with loading buffer followed by 10 mM Tris-HCl (pH 8.3), 500 mM NaCl, and 25 mM imidazole, and the protein was eluted with 10 mM Tris-HCl (pH 8.3), 500 mM NaCl and 500 mM imidazole. Eluate was loaded onto a Superdex 200 26/600 column and eluted with a loading buffer. Pure protein was collected and incubated with tobacco etch virus (TEV) protease overnight in buffer (10 mM Tris-HCl, pH 8.3, and 500 mM NaCl). The cleaved tag and TEV protease were separated from the protein by Ni-NTA-affinity chromatography using the loading buffer, and DapE protein was collected in two separate fractions: the flowthrough fraction and that with 25 mM imidazole in the loading buffer. Both fractions contained pure protein but were kept separate. Prior to crystallization, the protein was dialyzed into 150 mM NaCl, 10 mM Tris-HCl (pH 8.3), and 0.1 mM ZnCl2 for 3 h at room temperature. The protein was concentrated to 6–11 mg/mL, portions of which were incubated with 2 mM N6-methyl-l,l-SDAP for 30 min at room temperature and used for crystallization trials immediately or frozen in liquid nitrogen and kept at −80 °C for future use.
Crystallization and Structure Determination
Crystallizations were set up with freshly purified AbDapE-SeMet protein at various concentrations of 6–11 mg/mL in 150 mM NaCl, 10 mM Tris-HCl (pH 8.3), and 2 mM N6-Me-l,l-SDAP using the sitting drop method in Corning 96-well plates with Classics II, PEGs II and Anions crystallization screens (QIAGEN) at 20 °C.
Diffraction quality crystals were screened, and data sets were collected at the 21-ID-G and 21-ID-D beamline of the Life Science Collaborative Access Team (LS-CAT) at the Advanced Photon Source, Argonne National Laboratory. Images were indexed, integrated, and scaled using the HKL3000 suite.17 Structures were determined by Molecular Replacement using Phaser18 from the CCP4 Suite.19 The crystal structure of the DapE from N. meningitidis MC58 (PDB code 5UEJ) was used as a search model. Initial solutions were refined in REFMAC,20 and manual model corrections were made in Coot.21 At this point, structures were carefully examined, and the two with the highest resolution and the best quality electron density maps near the substrate binding sites were selected for further refinement. Water molecules were generated using ARP/wARP,22 and ligand molecules were fit into electron density maps in Coot. Structures were further refined in REFMAC, and TLS corrections were applied during the final stages of refinement. Molprobity23,24 was used for monitoring the quality of the model during refinement and for the final validation of the structure. X-Ray data quality and structure refinement statistics are listed in Table S1. Coordinates of the final models and experimental structure factors for AbDapE-SeMet/succinate and AbDapE-SeMet/succinate + l-(S)-lactate complexes were deposited in the Protein Data Bank (PDB) as PDB entries 7T1Q and 8F8O, respectively.
Enzyme assay
A discontinuous kinetic assay was performed utilizing a Techne PCR Thermal Cycler System with a modified ninhydrin assay protocol.7 The volume of each component was adjusted to fit a total reaction volume of 100 μL. The final potential inhibitor concentration was 5 μM for captopril and 5 mM for Li2SO4. One indoline sulfonamide HiDapE inhibitor was assayed for inhibition of AbDapE at 100 μM. Inhibitors were dissolved in neat dimethylsulfoxide (DMSO, stock stored at −10 °C), and the preassay concentrations were adjusted to give a final concentration of 5% DMSO in the assay. The final AbDapE or HiDapE concentration was 8 nM. To a 50 mM HEPES (pH 7.5) buffered solution at 30 °C was added the selected inhibitor followed by AbDapE or HiDapE and incubated for 10 min. N6-Methyl-l,l-SDAP (2 mM) was added, and the mixture was allowed to react for 10 min followed by heating at 100 °C for 1 min and cooling to 0 °C. A 2% ninhydrin solution (100 μL) was added, and the mixture was vortexed or mixed well by rapid pipetting while cooled at 0 °C. The reaction was then heated at 80 °C for 15 min. The absorbance of an 80 μL aliquot was recorded at 570 nm on a BioTek Synergy 2 microplate reader.
Kinetic studies
A discontinuous kinetic assay was performed utilizing a Techne PCR Thermal Cycler System with a modified ninhydrin assay protocol.7 The volume of each component was adjusted to fit the total reaction volume of 100 μL, and the final enzyme concentration was 8 nM. Inhibition of AbDapE or HiDapE with various concentrations of inhibitor was studied in triplicate while changing the substrate concentration from 0.5 to 6.0 mM. The amount of enzymatic product N6-Me-l,l-DAP that was formed over 10 min was monitored by measuring the absorbance of the complex formed from the reaction of N6-Me-l,l-DAP with 2% ninhydrin in a similar fashion as stated above. The enzymatic activity was reported as the rate of formation of the product N6-Me-l,l-DAP in velocity (mM/sec). The kinetic constants (Ki) were found using the Michaelis–Menten equation in Microsoft Excel and GraphPad Prism using a nonlinear regression.
DapE Enzyme Inhibition: Ki Determination
The inhibition of AbDapE and HiDapE by captopril and sulfate was assessed following the protocol detailed by us previously7 with modifications as detailed above. The formation of N6-Me-l,l-DAP was detected by measuring the change in absorbance at 570 nm, which was then converted into velocity by finding the path length of the well-plate using a compound with a known molar absorption coefficient (ascorbic acid, ε = 2.8 mM–1 cm–1)25 at a known compound concentration, 80 mM. Once the path length was found, the absorbance was converted into velocity using nonlinear regression and the Ki values were determined using Microsoft Excel and GraphPad Prism.
Determination of kcat/KM with AbDapE
A discontinuous kinetic assay was performed utilizing a Techne PCR Thermal Cycler System with a modified ninhydrin assay protocol.7 The volume of each component was adjusted to fit the total reaction volume of 100 μL, and the final enzyme concentration was 8 nM. The activities of AbDapE and HiDapE were assessed by changing the substrate concentration from 0 mM to 6.5 mM. The amount of enzymatic product N6-Me-l,l-DAP that was formed over 10 min was monitored by measuring the absorbance of the complex formed from the reaction of N6-Me-l,l-DAP with 2% ninhydrin in a similar fashion as stated above. The enzymatic activity was reported as the rate of formation of the product, N6-Me-l,l-DAP, in velocity (mM/sec). The kinetic constants (kcat/KM) were found by using GraphPad Prism using a nonlinear regression using the Michaelis–Menten model.
Sequence Comparisons
The nonrepetitive sequence database was searched for homologues of HiDapE using the blastp algorithm.26 The sequences for the fiveDapE proteins found were aligned using the Clustal Omega algorithm27 and ESPript (Figure S9).41
Thermal Shift Assay
Thermal shift studies were conducted on a Step One Real-Time PCR System and the associated QuantStudio software. Protein denaturation was measured by detecting the change in fluorescence of the SYPRO Orange dye. HiDapE was used at a final concentration of 200 nM, and SYPRO Orange was used at a final concentration of 10X. AbDapE-native enzyme was used at a final concentration of 16 μM and 10× SYPRO Orange dye. The AbDapE-SeMet was tested at a final concentration of 6 μM and SYPRO Orange was used at a final concentration of 10×. There was no change in Tm or inhibition by DMSO if the concentration of DMSO was kept lower than 5%. Dye concentrations higher than 12× denatured the enzyme. The experiment was carried out in 10-μL triplicates in 50 mM HEPES buffer at pH 7.5 in nanopure water, and the inhibitor concentrations were selected based on the half-maximal inhibitory concentration (IC50) values of the inhibitor. Sample solutions were dispensed into a 96-well optical reaction plate (Thermo Fisher Scientific), and the plate was sealed with an optical PCR plate sheet (Thermo Fisher Scientific). After equilibrating the system for 2 min at 25 °C, the temperature was continuously increased at a rate of 0.05 °C/s for 25 min and finally maintained at 99 °C for 2 min. The samples were scanned from 330 to 350 nm every 8 s (0.4 °C). Melting curves were obtained from the negative derivative and exported from the instrument to Microsoft Excel. The negative first derivatives of the melting curves were differentiated into the third derivative. Tm values were plotted against the log [concentration], and the Ki values were calculated using a derived Van’t Hoff equation according to Bhayani.28
Results and Discussion
Enzyme Characterization and Inhibitors of AbDapE
We determined that N6- methyl-l,l-SDAP can serve as a substrate for AbDapE, and it has a kcat/KM = 3.4 ± 0.9 × 105 M–1s–1 (Figure S1) compared to the kcat/KM = 4.4 ± 0.2 × 105 M–1s–1 (Figure S2) for the hydrolysis of N-methyl-l,l-SDAP by HiDapE, thus the N6-methyl modified substate is turned over at the same rate by both AbDapE native enzyme and HiDapE. We also assayed the SeMet derivative of AbDapE (AbDapE-SeMet) for hydrolysis of N6-methyl-l,l-SDAP and found that the kcat/KM of AbDapE-SeMet is 4.6 ± 0.98 × 105 M–1s–1 (Figure S3), which is not statistically different.
To compare inhibition of AbDapE to HiDapE, we ran kinetic assays using our modified ninhydrin assay7 using three known inhibitors of HiDapE: captopril29 lithium sulfate (Li2SO4),16 and the indoline sulfonamide 1-acetyl-5-chloro-N-isopentylindoline-6-sulfonamide.30 We found that both captopril and sulfate are competitive inhibitors of AbDapE, as previously shown for HiDapE.16,29 Captopril inhibited AbDapE with an IC50 of 1.2 μM versus an IC50 of 3.3 μM against HiDapE (Table 1). The saturation curves are reported in the Supporting Information as Figures S4A–D. Thus, we observed that captopril is a single-digit μM inhibitor of AbDapE, similar to HiDapE. Surprisingly, we found that the indoline sulfonamide, 1-acetyl-5-chloro-N-isopentylindoline-6-sulfonamide, did not inhibit AbDapE significantly at 100 μM, although this compound inhibits HiDapE with an IC50 = 54.0 μM.30 The lower potency of this inhibitor toward AbDapE relative to its inhibition of HiDapE is surprising and underscores the challenge of synthesizing a broad-spectrum DapE inhibitor. Sulfate inhibits AbDapE with an IC50 of 13,500 μM, compared to the reported16Ki of 23,900 μM forHiDapE.
Table 1. Inhibition of HiDapE and AbDapE.

Thermal Shift Assay (TSA) Results of HiDapE, AbDapE, and AbDapE-SeMet
A thermal shift assay (TSA) was conducted to observe the Tm of DapE from H. influenzae and A. baumannii. As the indoline sulfonamide did not inhibit AbDapE, we focused on the HiDapE inhibitors captopril29 and sulfate (Li2SO4).16 In the absence of inhibitor, the melting curves of HiDapE exhibit two melting temperatures, (Tm1) at 51.5 °C and (Tm2) at 78.2 °C, as does AbDapE, (Tm1) at 41.9 °C and (Tm2) at 63.2 °C, both of which are notably lower temperatures. As we had AbDapE-SeMet for the X-ray structure work, we also measured its Tm and compared it with the native enzyme. AbDapE SeMet exhibited only one Tm at 65.2 °C. The significantly higher Tm relative to Tm1 for HiDapE and for AbDapE demonstrates the stabilizing effect of the SeMet, consistent with observation in the literature.31
Using the thermal shift data, we determined the Ki values for the inhibitors using a derived Van’t Hoff Equation28 and plotting the Tm vs log [concentration]. The thermal shifts occurred at concentrations very near the Ki values of the inhibitors for HiDapE and AbDapE reported for kinetic assays using the ninhydrin assay. Captopril and sulfate bind to HiDapE with Ki values of 0.68 and 18,400 μM, respectively, and this thermal shift data are in agreement with previously published results determined via enzyme inhibition for HiDapE, the reported Ki = 1.82 μM, and the IC50 for inhibition of HiDapE by sulfate was 13,800 ± 2,800 μM.16,29 The Ki found by the thermal shift for captopril versus AbDapE was 0.79 μM, comparable to the Ki of 0.68 μM for captopril versus HiDapE (Table 2).
Table 2. Melt Temperatures from the Thermal Shift studies for HiDapE and AbDapE in the Absence and Presence of the Inhibitors Captopril and Lithium Sulfate.
| experiment | Tm1 HiDapEa (°C) | Ki (μM) | Tm2 HiDapEa (°C) | Tm1 AbDapEb (°C) | Tm2 AbDapEb (°C) | Ki (μM) | Tm AbDapE-SeMetc (°C) | Ki (μM) |
|---|---|---|---|---|---|---|---|---|
| enzyme only | 51.5 ± 0.3 | 78.2 ± 0.1 | 41.9 ± 0.3 | 63.2 ± 0.4 | 65.2 ± 0.1 | |||
| [Captopril] | ||||||||
| 0.005 μM | 51.5 ± 0.1 | 78.2 ± 0.1 | 41.9 ± 0.1 | 62.9 ± 0.1 | 65.3 ± 0.2 | |||
| 0.05 μM | 50.2 ± 0.6 | 78.6 ± 0.1 | 41.9 ± 0.2 | 63.3 ± 0.7 | 65.5 ± 0.1 | |||
| 0.1 μM | 50.0 ± 0.8 | 78.1 ± 0.1 | 41.9 ± 0.1 | 62.6 ± 0.3 | 65.3 ± 0.2 | |||
| 0.5 μM | 50.5 ± 0.1 | 78.3 ± 0.1 | 41.9 ± 0.2 | 63.1 ± 0.4 | 65.1 ± 0.1 | |||
| 0.8 μM | 50.3 ± 0.5 | 0.68 ± 0.65 | 77.9 ± 0.1 | 41.9 ± 0.1 | 62.3 ± 0.1 | 0.79 ± 0.61 | 66.1 ± 0.1 | |
| 1 μM | 51.2 ± 0.2 | 77.8 ± 0.1 | 41.7 ± 0.1 | 63.4 ± 1.0 | 65.7 ± 0.1 | |||
| 3 μM | 52.2 ± 0.1 | 78.0 ± 0.1 | 41.7 ± 0.2 | 65.1 ± 0.3 | 66.4 ± 0.3 | |||
| 5 μM | 53.2 ± 0.2 | 77.8 ± 0.1 | 41.4 ± 0.1 | 64.1 ± 0.2 | 67.2 ± 0.2 | 9.4 ± 0.3 | ||
| 10 μM | 52.9 ± 0.6 | 77.9 ± 0.1 | 41.1 ± 0.1 | 66.6 ± 0.2 | 68.5 ± 0.1 | |||
| 15 μM | 42.0 ± 0.1 | 65.5 ± 0.4 | ||||||
| [Li2SO4] | ||||||||
| 0.001 mM | 51.7 ± 0.3 | 75.6 ± 0.3 | 41.4 ± 0.1 | 63.1 ± 0.1 | 64.2 ± 0.7 | |||
| 0.005 mM | 51.4 ± 0.2 | 76.2 ± 0.5 | 41.8 ± 0.2 | 62.8 ± 0.2 | 64.1 ± 0.3 | |||
| 0.1 mM | 52.7 ± 0.4 | 76.1 ± 0.6 | 41.6 ± 0.1 | 62.2 ± 0.1 | 64.0 ± 0.2 | |||
| 0.5 mM | 51.2 ± 0.7 | 78.3 ± 0.1 | 41.7 ± 0.1 | 63.1 ± 0.1 | 64.9 ± 0.1 | |||
| 1 mM | 50.7 ± 0.2 | 77.8 ± 0.1 | 41.3 ± 0.1 | 63.0 ± 0.1 | 64.7 ± 0.5 | |||
| 5 mM | 51.2 ± 0.6 | 77.7 ± 0.1 | 41.6 ± 0.1 | 61.5 ± 0.2 | 65.2 ± 0.1 | |||
| 10 mM | 51.2 ± 0.6 | 18,400 ± 620 | 77.4 ± 0.1 | 41.5 ± 0.1 | 60.4 ± 0.1 | 65.0 ± 0.3 | ||
| 30 mM | 52.6 ± 0.5 | 74.6 ± 0.3 | 42.2 ± 0.2 | 59.1 ± 0.2 | ||||
| 50 mM | 53.3 ± 0.5 | 75.9 ± 0.1 | 42.1 ± 0.1 | 58.3 ± 0.1 | 63.1 ± 0.1 | |||
| 60 mM | 53.3 ± 0.3 | 73.8 ± 0.1 | 42.4 ± 0.2 | 57.5 ± 0.1 | ||||
| 70 mM | 54.4 ± 0.5 | 76.6 ± 0.1 | 42.4 ± 0.3 | 56.7 ± 0.2 | 61.8 ± 0.1 | |||
200 nM HiDapE + 10× dye.
16 μM AbDapE + 10× dye.
6 μM AbDapE + 10× dye.
X-ray Crystal Structure of DapE from A. baumannii
We crystallized AbDapE-SeMet in the presence of the N6-methyl-l,l-SDAP substrate, which yielded two structures of AbDapE-SeMet in complex with a product of N6-methyl-l,l-SDAP hydrolysis, succinic acid (crystal 1, PDB code 7T1Q), and with succinic acid and lactic acid (crystal 2, PDB code 8F8O). Both crystals belong to space group P212121 with similar unit cell parameters (Table S1). Both structures were solved by using molecular replacement. The coordinates of NmDapE (PDB code 5UEJ) were used to solve the structure of crystal 1 (7T1Q), and this refined model of AbDapE-SeMet was used to solve the structure of crystal 2 (8F8O). Each structure is composed of two polypeptide chains in the asymmetric unit, which form a dimer (Figure 1A). Each AbDapE-SeMet chain consists of two domains: a catalytic domain comprising residues 1–177 and 299–378 and a dimerization domain comprising residues 178–298 (Figure 1A). Each AbDapE-SeMet protomer retains two canonical and fully occupied dinuclear-Zn(II) binding sites, in which zinc ions are coordinated at site one by residues His68, Asp101, and Glu164 and at site two by residues Asp101, Glu136, and His350 (Figure 1B). As expected, these residues are conserved across all Gram-negative DapE enzymes examined (Figure S9).
Figure 1.

The structure of A. baumannii DapE-SeMet. (A) Overall structure of the AbDapE-SeMet dimer from crystal 2 (PDB code 8F8O). Chain A is colored wheat, chain B yellow, and active site zinc ions are magenta. Atoms of the succinic acid (lime green carbons) and l-(S)-lactate (light gray carbons) are colored oxygen in red and nitrogen in blue. (B) Zoomed-in view of the zinc-binding site from chain A with atoms colored as in (A). Zinc-coordinating residues are shown as sticks. Atoms of succinic acid and l-(S)-lactate are also colored carbon in wheat, oxygen in red, and nitrogen in blue. (C) Superposition of the AbDapE-SeMet structure of crystal 2 (8F8O, chain A in wheat, chain B in yellow) and the structure of crystal 1 (7T1Q, chain A in teal, chain B in cyan) active sites. Succinic acid, l-(S)-lactate (8F8O, wheat carbons), and acetate (7T1Q, teal carbons) are shown, and zinc ions are shown as spheres in magenta (8F8O) and orange (7T1Q).
The two AbDapE-SeMet structures are nearly identical and overlay with a root-mean-square deviation (RMSD) of 0.23 Å across all atoms. Furthermore, one of the products of N6-methyl-l,l-SDAP hydrolysis, succinate, is observed in identical positions in both structures (Figure 1C). The second product, N6-methyl-l,l-DAP, is likely lost from the active site due to steric interactions from the additional N-methyl group and is instead exchanged for acetate in crystal 1 (7T1Q) derived from the crystallization buffer. In crystal 2, we observe l-(S)-lactate in the active site with the carboxylate in the same position as the acetate in crystal 1. We suspect that N6-methyl-l,l-DAP was exchanged for citrate derived from the crystallization buffer and subsequently underwent X-ray induced degradation during the data collection process. The synchrotron X-ray induced degradation of amino acids and ligands is well precedented.32 In our crystal, we hypothesize that degradation of citrate to lactate occurred via familiar loss of carbon dioxide and in addition the loss of acetate leading to lactate as shown in Scheme 2. A full mechanistic hypothesis is included in the Supporting Information as Scheme S1.
Scheme 2. Hypothesized X-ray Induced Loss of Acetate and CO2 Forming Lactate in the Active Site.

Interestingly, we observe only l-(S)-lactate bound to the active site, which would require an enantioselective hydrogen atom transfer to the 2-hydroxypropanoate radical. This agrees with the chirality of the active site and the preference for (S)-stereochemistry in the position occupied by lactate, which corresponds to the (S)-stereochemistry of the substrates SDAP and N6-methyl-l,l-SDAP.
Comparison of AbDapE-SeMet with Homologues from H. influenzae and N. meningitidis
Upon binding of l,l,-SDAP to DapE, the catalytic domain shifts to obstruct access to the active site in a significant transition from the DapE “open” conformation to the “closed” conformation.14 These changes bring two important residues, His195 and Tyr198, from the neighboring chain closer to the active site and help them directly interact with the substrate. We aligned the AbDapE-SeMet structure to that of HiDapE (5VO3, 58% identity) and compared the positions of reaction products in the active sites. The structures superimpose well with an RMSD of 0.87 Å (Figure 2). Close comparison of the AbDapE-SeMet dinuclear Zinc(II) active site to that of HiDapE shows that residues involved in substrate binding and catalysis are 100% conserved and almost all these residues are conserved across all Gram-negative DapE proteins (Figures 2 and S9,S10A). In our structures, specific residues from chain A, including Glu135, Arg179, Arg259, Gly325, and Thr326, and from chain B, including His195, Tyr198, and Asn246, form contacts with succinic acid, l-(S)-lactate and acetate (Figure S10B). Importantly, in agreement with what was previously observed for the HiDapE closed-state structure,14 the Zn(II) cluster, Arg179, and Gly325 from chain A, and His195 and Tyr198 of chain B form direct contacts with succinic acid. As expected, the distances between key residues His195 (3.0 Å) and Tyr198 (2.6 Å) and the oxygen atoms of succinic acid are also in agreement with the HiDapE structure. Lastly, the positions of zinc ions and succinate are almost identical in these structures, and the position of l-(S)-lactate in AbDapE-SeMet closely matches the position of the diaminopimelic moiety of the l-l-DAP product observed in the HiDapE active site. We conclude that these conserved interactions between the reaction products and AbDapE active site residues stabilize the protein in the closed conformation after substrate hydrolysis.
Figure 2.

Close-up view of the active sites of superimposed structures of HiDapE (PDB 5VO3, chain A pale green, symmetry-related chain of the dimer in pale violet) and AbDapE (PDB 8F8O chain A wheat, chain B yellow). Residues His195 and Tyr198 of chain B from AbDapE and corresponding residues from HiDapE and products of the reactions are shown as sticks (carbons match the colors of the chains, oxygen in red, nitrogen in blue), and zinc shown as spheres (magenta for 8F8O and pink for 5VO3). Bonds between His195.B and Tyr198.B of AbDapE and oxygen atoms of the succinate are shown by dashed lines with distances provided in angstroms.
DapE Conformational Changes Result from Hinge Region Flexibility
We previously compared DapE structures available in the PDB and described the flexibility of DapE from different bacteria species.16 At that time, there was only one closed ligand-bound structure known (PDB 5VO3) but, as noted above, the AbDapE structures represent two new ligand-bound forms. An overlay of the NmDapE (5UEJ) and HiDapE (3IC1) structures highlights the open conformation of the dimer in ligand-free structures, whereas AbDapE structures are more compact. The distance between C-terminal residues of α3 in open HiDapE and NmDapE structures averages 123 Å (Figure 3A) versus that in the closed AbDapE structures, which is 108 Å (Figure 3B). These data support the finding that binding of a ligand to the active site stabilizes a specific quaternary structure, the closed conformation. Furthermore, alignment of the catalytic and dimerization domains of open and closed structures shows no significant changes in their conformations (Figure 3C). In contrast, alignment of the hinge regions of open and closed structures demonstrates the flexibility of this region, suggesting that changes in quaternary structure are determined by movements in the hinge region rather than changes in the structures of other domains (Figure 3D). These overlays complement the analyses of DapE performed by Díaz-Sánchez.33
Figure 3.

Closed and open states of DapE result from movement in the hinge region. Overlaps of (A) open NmDapE and HiDapE and (B) closed AbDapE structures. The distances between c-terminal residues of α-helix 3 are shown with dashed lines. (C) Superimposed catalytic and dimerization domains of AbDapE (7T1Q), HiDapE, and NmDapE. (D) Superimposed hinge regions of AbDapE (7T1Q), HiDapE, and NmDapE. The α-helix 7 of the regions was aligned to emphasize movement of hinge residues. Coloring scheme is the same as in panels A and B.
Mishra34 very recently reported the binding energies of the products of DapE cleavage, with succinate bound very tightly (−25.87 and −22.26 kcal/mol in DapE monomer chains A and B, respectively), while DAP is less tightly bound (−19.14 kcal/mol and −17.51 kcal/mol in chains A and B, respectively). Moreover, he showed through molecular dynamics studies that DapE is very stable and remains closed with both products bound (starting with the products-bound structure PDB 5VO3), but removing both products allows DapE to open in a molecular dynamics run on the order of about 44 ns. The crystal structures we are reporting herein retain succinic acid in the active site, suggesting that this product is key for stabilizing the closed conformation of DapE.
Conclusions
We have enzymatically and structurally characterized DapE from A. baumannii, an important ESKAPE pathogen that exhibits significant antibiotic resistance. We assessed three known inhibitors of HiDapE: captopril, lithium sulfate, and 1-acetyl-5-chloro-N-isopentylindoline-6-sulfonamide for inhibition of AbDapE in our ninhydrin-based assay.14 Captopril inhibited AbDapE with an IC50 value of 1.2 μM, comparable to the IC50 of 3.3 μM for captopril versus HiDapE. Sulfate inhibited AbDapE with an IC50 of 13.5 mM, comparable to the IC50 of 23.9 mM for HiDapE, but the indoline sulfonamide did not measurably inhibit AbDapE, yet has an IC50 of 54.0 μM for HiDapE. This suggests that a broad-spectrum DapE inhibitor may be inaccessible. However, broad-spectrum antibiotics can disrupt the composition of a healthy microbiome, so greater antibiotic specificity may prove to be an advantage.
Both AbDapE and a SeMet derivative were characterized with a TSA using captopril and sulfate as ligands and compared to those of the more thoroughly studied HiDapE. AbDapE exhibited biphasic transitions at 41.9 °C and at Tm2 at 63.2 °C that were somewhat lower than those transitions for HiDapE (Tm1 = 51.5 °C and Tm2 = 78.2 °C). In contrast, AbDapE-SeMet exhibited a single phase transition, with Tm at 65.2 °C; the higher temperature is consistent with the known stabilization of proteins by substitution of methionine by SeMet. The negative shift in Tm2 may indicate that captopril and sulfate ions destabilize HiDapE by binding more tightly to the unfolded molten globular state of the enzyme,35−37 thus stabilizing the more globular state. In general, it is risky to make conclusions about Tm2, since having passed Tm1, the enzyme is no longer in its native state and is in some partly globular state.
Díaz-Sánchez et al. reported thermal shift (also referred to as thermofluorescence, TF) studies for DapE from E. faecium (EfDapE) and E. coli (EcDapE), and they observed an apparent melt temperature (app. Tm) of 43.0 ± 0.5 °C for EfDapE and 42.0 ± 0.6 °C for EcDapE.35 Further, they discovered two new inhibitors of DapE, disulfiram and orphenadrine, and reported a lowering of the Tm values of DapE in the presence of those inhibitors, whereas we observed a stabilization of Tm1 for HiDapE and AbDapE with both captopril and sulfate. While it is most common for a ligand binding to a protein to stabilize the structure and therein raise the Tm to higher temperatures, the lowering of Tm through destabilization has been observed as well.38−40
Differences in potencies of inhibitors as well as differences in thermal shift may be due to the somewhat lower homology between AbDapE and HiDapE (58.2%). By comparison with another important infectious bacteria species, Mycobacterium tuberculosis DapE (MtDapE) has only 24.4% homology with HiDapE, and hence, its structure–activity relationship should be quite distinct.
As previously noted, the native HiDapE and AbDapE enzymes exhibit two melt temperatures. We hypothesize that Tm1 may indicate the denaturation of the dimer, and Tm2 involves the denaturation of the dissociated monomer. These two temperatures corresponding to denaturation are consistent with our previously reported circular dichroism studies,7 which revealed that the α-helices of the enzyme begin to denature between 50 and 60 °C, corresponding to Tm1, and undergo further denaturation after 2–3 min of heating at 80 °C (≤10% of α-helices activity remaining), corresponding to Tm2. It has been reported31 that substitution of Met with SeMet stabilizes proteins; it is curious that SeMet has two Tm values, whereas AbDapE-SeMet exhibits only one Tm value.
Attempts to cocrystallize AbDapE with the modified substrate N6–methyl-l,l-SDAP substrate resulted in two AbDapE structures in the complex with reaction products, succinic acid and l-(S)-lactate (8F8O) and acetate (7T1Q). These new DapE structures are the only closed DapE conformers reported in the literature except for one previously reported structure of HiDapE in complex with succinic and diaminopimelic (DAP) acids (5VO3).14 Comparison of these structures shows that the position of succinic acid is retained in both H. influenzae and A. baumannii DapE enzymes. Furthermore, interactions between succinic acid, zinc ions, and key residues of DapE are conserved in our structures, and these interactions help to stabilize the closed conformation after catalysis. These interactions reveal conformational changes in the DapE hinge region that are noteworthy when comparing the closed and open conformers. Lastly, despite expulsion of the N6-methyl-l,l-DAP product from the AbDapE active site, carboxylates of l-(S)-lactate and acetate in AbDapE structures are in the same position as the carboxylate of DAP in HiDapE. These data support conservation of the DapE mechanism in another pathogenic bacterium, specifically an ESKAPE pathogen, further reason for continued efforts in developing novel therapeutics that target DapE.
Acknowledgments
The authors thank Ievgeniia Dubrovska for freezing crystals that led to the A. baumannii DapE structures. The authors also thank Sergii Pshenychnyi at Northwestern University for protein purification.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c08231.
X-ray collection and refinement statistics; plots of inhibition by captopril, sulfate (Li2SO4), 1-acetyl-5-chloro-N-isopentylindoline-6-sulfonamide, and TSA plots (PDF)
Accession Codes
Author Contributions
# E.H.K. and G.M. contributed equally to this work.
This research was supported in part by the National Institutes for Allergies and Infectious Diseases of the National Institutes of Health department of the Health and Human Services under contracts HHSN272201700060C and 75N93022C00035. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (grant 085P1000817).
The authors declare no competing financial interest.
Supplementary Material
References
- Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline, Including Tuberculosis; World Health Organization, 2017.
- Mulani M. S.; Kamble E. E.; Kumkar S. N.; Tawre M. S.; Pardesi K. R. Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: a review. Front. Microbiol. 2019, 10, 539 10.3389/fmicb.2019.00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg A. Y.; Seifert H.; Paterson D. L. Acinetobacter baumannii: emergence of a successful pathogen. Clin. Microbiol. Rev. 2008, 21, 538–582. 10.1128/CMR.00058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muduli S.; Karmakar S.; Mishra S. The coordinated action of the enzymes in the L-lysine biosynthetic pathway and how to inhibit it for antibiotic targets. Biochim. Biophys. Acta, General Subj. 2023, 1867, 130320 10.1016/j.bbagen.2023.130320. [DOI] [PubMed] [Google Scholar]
- Gillner D. M.; Becker D. P.; Holz R. C. Lysine biosynthesis in bacteria: a metallodesuccinylase as a potential antimicrobial target. JBIC, J. Biol. Inorg. Chem. 2013, 18, 155–163. 10.1007/s00775-012-0965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapin G.; Blanchard J. S.. Enzymology of Bacterial Lysine Biosynthesis. In Advances in Enzymologyand Related Areas of Molecular Biology; John Wiley & Sons, Inc., 2006; pp 279–324. [DOI] [PubMed] [Google Scholar]
- Heath T. K.; Lutz M. R. Jr; Reidl C. T.; Guzman E. R.; Herbert C. A.; Nocek B. P.; Holz R. C.; Olsen K. W.; Ballicora M. A.; Becker D. P. Practical spectrophotometric assay for the dapE-encoded N-succinyl-L, L-diaminopimelic acid desuccinylase, a potential antibiotic target. PLoS One 2018, 13, e0196010 10.1371/journal.pone.0196010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karita M.; Etterbeek M. L.; Forsyth M. H.; Tummuru M. K. R.; Blaser M. J. Characterization of Helicobacter pylori dapE and construction of a conditionally lethal dapE mutant. Infect. Immun. 1997, 65, 4158–4164. 10.1128/iai.65.10.4158-4164.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka M. S. Jr.; Jacobs W. R. Jr. Biosynthesis of diaminopimelate, the precursor of lysine and a component of peptidoglycan, is an essential function of Mycobacterium smegmatis. J. Bacteriol. 1996, 178, 6496–6507. 10.1128/jb.178.22.6496-6507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badger J.; Sauder J. M.; Adams J. M.; Antonysamy S.; Bain K.; Bergseid M. G.; Buchanan S. G.; Buchanan M. D.; Batiyenko Y.; Christopher J. A.; et al. Structural analysis of a set of proteins resulting from a bacterial genomics project. Proteins: Struct., Funct., Bioinf. 2005, 60, 787–796. 10.1002/prot.20541. [DOI] [PubMed] [Google Scholar]
- Nocek B. P.; Gillner D. M.; Fan Y.; Holz R. C.; Joachimiak A. Structural Basis for Catalysis by the Mono- and Dimetalated Forms of the dapE-Encoded N-succinyl-L,L-Diaminopimelic Acid Desuccinylase. J. Mol. Biol. 2010, 397, 617–626. 10.1016/j.jmb.2010.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starus A.; Nocek B.; Bennett B.; Larrabee J. A.; Shaw D. L.; Sae-Lee W.; Russo M. T.; Gillner D. M.; Makowska-Grzyska M.; Joachimiak A.; Holz R. C. Inhibition of the dapE-Encoded N-Succinyl-L,L-diaminopimelic Acid Desuccinylase from Neisseria meningitidis by L-Captopril. Biochemistry 2015, 54, 4834–4844. 10.1021/acs.biochem.5b00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillner D. M.; Bienvenue D. L.; Nocek B. P.; Joachimiak A.; Zachary V.; Bennett B.; Holz R. C. The dapE-encoded N-succinyl-L,L-diaminopimelic acid desuccinylase from Haemophilus influenzae contains two active-site histidine residues. JBIC, J. Biol. Inorg. Chem. 2009, 14, 1–10. 10.1007/s00775-008-0418-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocek B.; Reidl C.; Starus A.; Heath T.; Bienvenue D.; Osipiuk J.; Jedrzejczak R. P.; Joachimiak A.; Becker D. P.; Holz R. C. Structural Evidence for a Major Conformational Change Triggered by Substrate Binding in DapE Enzymes: Impact on the Catalytic Mechanism. Biochemistry 2018, 57, 574. 10.1021/acs.biochem.7b01151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D.; Mishra S. Structural and mechanistic insight into substrate binding from the conformational dynamics in apo and substrate-bound DapE enzyme. Phys. Chem. Chem. Phys. 2016, 18, 1671–1680. 10.1039/C5CP06024A. [DOI] [PubMed] [Google Scholar]
- Kochert M.; Nocek B. P.; Habeeb Mohammad T. S.; Gild E.; Lovato K.; Heath T. K.; Holz R. C.; Olsen K. W.; Becker D. P. Atomic-Resolution 1.3 Å Crystal Structure, Inhibition by Sulfate, and Molecular Dynamics of the Bacterial Enzyme DapE. Biochemistry 2021, 60, 908–917. 10.1021/acs.biochem.0c00926. [DOI] [PubMed] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 859–866. 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Langer G.; Cohen S. X.; Lamzin V. S.; Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Davis I. W.; Richardson D. C. KING (Kinemage, Next Generation): a versatile interactive molecular and scientific visualization program. Protein Sci. 2009, 18, 2403–2409. 10.1002/pro.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C. J.; Headd J. J.; Moriarty N. W.; Prisant M. G.; Videau L. L.; Deis L. N.; Verma V.; Keedy D. A.; Hintze B. J.; Chen V. B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayannis M. I.; Samios D. N.; Gousetis C. A study of the molar absorptivity of ascorbic acid at different wavelengths and pH values. Anal. Chim. Acta 1977, 93, 275–279. 10.1016/0003-2670(77)80032-9. [DOI] [PubMed] [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Soding J.; Thompson J. D.; Higgins D. G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet P. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003, 31, 3320–3323. 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhayani J. A.; Ballicora M. A. Determination of dissociation constants of protein ligands by thermal shift assay. Biochem. Biophys. Res. Commun. 2022, 590, 1–6. 10.1016/j.bbrc.2021.12.041. [DOI] [PubMed] [Google Scholar]
- Gillner D.; Armoush N.; Holz R. C.; Becker D. P. Inhibitors of bacterial N-succinyl-L,L-diaminopimelic acid desuccinylase (DapE) and demonstration of in vitro antimicrobial activity. Bioorg. Med. Chem. Lett. 2009, 19, 6350–6352. 10.1016/j.bmcl.2009.09.077. [DOI] [PubMed] [Google Scholar]
- Reidl C. T.; Heath T. K.; Darwish I.; Torrez R. M.; Moore M.; Gild E.; Nocek B. P.; Starus A.; Holz R. C.; Becker D. P. Indoline-6-Sulfonamide Inhibitors of the Bacterial Enzyme DapE. Antibiotics 2020, 9, 595 10.3390/antibiotics9090595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassner N. C.; Baase W. A.; Hausrath A. C.; Matthews B. W. Substitution with selenomethionine can enhance the stability of methionine-rich proteins. J. Mol. Biol. 1999, 294, 17–20. 10.1006/jmbi.1999.3220. [DOI] [PubMed] [Google Scholar]
- Burmeister W. P. Structural changes in a cryo-cooled protein crystal owing to radiation damage. Acta Crystallogr., D: Biol. Crystallogr. 2000, 56, 328–341. 10.1107/S0907444999016261. [DOI] [PubMed] [Google Scholar]
- Díaz-Sánchez Á. G.; Terrazas-López M.; Aguirre-Reyes L. G.; Lobo-Galo N.; Álvarez-Parrilla E.; Martínez-Martínez A.. Aspectos estructurales y funcionales de la N-Succinil-L, L-diaminopimelato desuccinilasa, una enzima clave para el crecimiento bacteriano y un blanco para el control antimicrobiano. TIP, Rev. Espec. Cienc. Quim.-Biol. 2019, 22. 10.22201/fesz.23958723e.2019.0.191. [DOI] [Google Scholar]
- Muduli S.; Mishra S. Ligands-Induced Open-Close Conformational Change during DapE Catalysis: Insights from Molecular Dynamics Simulations. Proteins: Struct., Funct., Bioinf. 2023, 91, 781–797. 10.1002/prot.26466. [DOI] [PubMed] [Google Scholar]
- Terrazas-López M.; Lobo-Galo N.; Aguirre-Reyes L. G.; Bustos-Jaimes I.; Marcos-Víquez J. Á.; González-Segura L.; Díaz-Sánchez Á. G. Interaction of N-succinyl diaminopimelate desuccinylase with orphenadrine and disulfiram. J. Mol. Struct. 2020, 1222, 128928 10.1016/j.molstruc.2020.128928. [DOI] [Google Scholar]
- Bhusal R. P.; Patel K.; Kwai B. X.; Swartjes A.; Bashiri G.; Reynisson J.; Sperry J.; Leung I. K. Development of NMR and thermal shift assays for the evaluation of Mycobacterium tuberculosis isocitrate lyase inhibitors. MedChemComm 2017, 8, 2155–2163. 10.1039/C7MD00456G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmperman P.; Baranauskienė L.; Jachimovičiu̅tė S.; Jachno J.; Torresan J.; Michailovienė V.; Matulienė J.; Sereikaitė J.; Bumelis V.; Matulis D. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys. J. 2008, 95, 3222–3231. 10.1529/biophysj.108.134973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-García R.; Zaldívar-Machorro V. J.; Mújica-Jiménez C.; González-Segura L.; Muñoz-Clares R. A. Disulfiram irreversibly aggregates betaine aldehyde dehydrogenase—a potential target for antimicrobial agents against Pseudomonas aeruginosa. Biochem. Biophys. Res. Commun. 2006, 341, 408–415. 10.1016/j.bbrc.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Douse C. H.; Vrielink N.; Wenlin Z.; Cota E.; Tate E. W. Targeting a dynamic protein–protein interaction: fragment screening against the malaria myosin A motor complex. ChemMedChem 2015, 10, 134–143. 10.1002/cmdc.201402357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir A.; Honda R. P.; Kamatari Y. O.; Endo S.; Fukuoka M.; Kuwata K. Effects of ligand binding on the stability of aldo–keto reductases: Implications for stabilizer or destabilizer chaperones. Protein Sci. 2016, 25, 2132–2141. 10.1002/pro.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.