

Abstract
Objectives
Interstitial lung disease (ILD) in CTDs has highly variable morphology. We aimed to identify imaging features and their impact on ILD progression, mortality, and immunosuppression response.
Methods
Patients with CTD-ILD had high-resolution chest CT (HRCT) reviewed by expert radiologists blinded to clinical data for overall imaging pattern [usual interstitial pneumonia (UIP); non-specific interstitial pneumonia (NSIP); organizing pneumonia (OP); fibrotic hypersensitivity pneumonitis (fHP); and other]. Transplant-free survival and change in percent-predicted forced vital capacity (FVC) were compared using Cox and linear mixed-effects models adjusted for age, sex, smoking, and baseline FVC. FVC decline after immunosuppression was compared with pre-treatment.
Results
Among 645 CTD-ILD patients, the most frequent CTDs were SSc (n = 215), RA (n = 127), and inflammatory myopathies (n = 100). NSIP was the most common pattern (54%), followed by UIP (20%), fHP (9%), and OP (5%). Compared with the case for patients with UIP, FVC decline was slower in patients with NSIP (by 1.1%/year, 95% CI 0.2, 1.9) or OP (by 3.5%/year, 95% CI 2.0, 4.9), and mortality was lower in patients with NSIP [hazard ratio (HR) 0.65, 95% CI 0.45, 0.93] or OP (HR 0.18, 95% CI 0.05, 0.57), but higher in fHP (HR 1.58, 95% CI 1.01, 2.40). The extent of fibrosis also predicted FVC decline and mortality. After immunosuppression, FVC decline was slower compared with pre-treatment in NSIP (by 2.1%/year, 95% CI 1.4, 2.8), with no change for UIP or fHP.
Conclusion
Multiple radiologic patterns are possible in CTD-ILD, including a fHP pattern. NSIP and OP were associated with better outcomes and response to immunosuppression, while fHP had worse survival compared with UIP.
Keywords: interstitial lung disease, connective tissue disease, radiologic patterns, prognosis
Rheumatology key messages.
A fibrotic hypersensitivity pneumonitis pattern was associated with the worst survival in patients with interstitial lung disease associated with a CTD, followed by usual interstitial pneumonia.
Non-specific interstitial pneumonia and organizing pneumonia patterns were associated with better outcomes and immunosuppression response compared with usual interstitial pneumonia.
Increased extent of fibrosis, measured by honeycombing and reticulation, was associated with progression and mortality.
Introduction
CTDs are a common cause of interstitial lung disease (ILD), particularly in RA, SSc and idiopathic inflammatory myositis (IIM). Radiologic patterns of usual interstitial pneumonia (UIP), non-specific interstitial pneumonia (NSIP), and less commonly a fibrotic hypersensitivity pneumonitis (fHP) pattern, are found in all CTD-ILD subtypes, although the prevalence of ILD and the radiologic morphology can vary significantly across CTD-ILD subtypes [1]. These imaging patterns may impact disease prognosis and treatment decisions regarding the use of antifibrotic vs immunosuppressive medication; however, this remains unclear, especially across the different CTD-ILD subtypes.
Our goal was to identify the different lung imaging patterns found in various CTD-ILD subtypes in a large prospectively enrolled cohort and to examine the association of imaging pattern with CTD-ILD progression, mortality, and immunosuppression response. We hypothesized that the UIP pattern would be associated with more rapid progression, lower transplant-free survival, and a reduced immunosuppression response compared with the NSIP pattern.
Methods
Seven of eight sites from the CAnadian REgistry for Pulmonary Fibrosis (CARE-PF) participated in this study. CARE-PF prospectively enrolls adults with fibrotic ILD from specialized ILD centres across Canada. Patients provided written informed consent and completed standardized questionnaires and testing [2]. This study included a subgroup of patients with CTD-ILD who underwent structured re-evaluation in multidisciplinary conference, focused on documentation and measurement of clinical characteristics and radiologic features and patterns [3]. The patients with CTD-ILD included in this study had rheumatologist-confirmed diagnoses and were enrolled between 2001 and 2021. Research ethics board approval was obtained for this study at each site (coordinating centre: University of British Columbia H20-02619).
Radiologic review
High-resolution CT of the chest performed within 1 year prior to or up to 3 months after the initial ILD clinical evaluation were reviewed for all patients. Imaging patterns and specific features were evaluated by expert chest radiologists blinded to the underlying diagnosis and any clinical information beyond the patient’s age and sex. The radiologists ranked the most likely imaging pattern by providing their radiologic confidence, based on published guidelines, for idiopathic pulmonary fibrosis (IPF) and hypersensitivity pneumonitis (HP) [4, 5], as well as their own interpretation and gestalt. Patients without any radiologist-defined pattern reaching >50% confidence were labelled as unclassifiable. Patients with NSIP who also displayed elements of organizing pneumonia (OP) present were classified as NSIP. Patients were analysed based on the main imaging patterns: UIP, NSIP, fHP, OP, lymphoid interstitial pneumonia (LIP), unclassifiable, or ‘other’ pattern.
The percentage of normal lung and parenchyma affected by honeycombing, isolated reticulation, reticulation with ground glass, isolated ground-glass opacities, hypoattenuating lung, consolidation, and emphysema was visually quantified on the inspiratory scan. The sum of these features was required to add to 100%. Total fibrosis was defined as the amount of honeycombing plus reticulation with or without admixed ground-glass opacities. The ratio of total ground-glass opacities to total fibrosis was calculated by the sum of ground glass admixed with reticulation with pure ground-glass opacities divided by the total fibrosis. Traction bronchiectasis was quantified as the percentage of lung parenchyma fed by an airway exhibiting this feature. Presence of nodules, cysts, axillary or mediastinal lymphadenopathy (one or more lymph nodes ≥1 cm), and dilated oesophagus (defined as the largest linear measurement of the oesophageal air column >1.5 cm on transverse section below the ventricles and above the diaphragm) were also recorded [6, 7]. The cases were split among a total of 18 chest radiologists who participated in the review, each of whom were trained by the same core group of investigators overseeing the study to maximize inter-observer consistency [3].
Other measurements
Baseline demographic data and smoking history were obtained from patient-completed questionnaires. ILD progression was assessed by longitudinal change in percent predicted FVC on serial pulmonary function tests, which are typically performed every 3–6 months as part of standard clinical practice. ANA patterns with a titre of ≥1/80 were collected and included centromere, cytoplasmic, homogeneous, nucleolar, speckled, or negative ANA. The association between imaging pattern and immunosuppression response was assessed in a subset of patients who received immunosuppressive treatment (defined as ≥3 months of MMF, AZA, CYC, tocilizumab, and/or ≥1 dose of rituximab) with at least 6 months of pulmonary function test follow-ups before and after immunosuppression. Medication start and stop dates were recorded based on a detailed and standardized chart review as previously described [8].
Statistical analyses
Data are presented as mean ± S.D., median (interquartile range), or number (percentage). Longitudinal change in percent-predicted FVC was compared between imaging patterns, using linear mixed-effect models (LMMs) with a random intercept and random slope to account for between-patient variability. The occurrence of either death or lung transplant on follow-up was assessed between groups using Kaplan Meier and Cox proportional hazards analyses. UIP served as the reference group for all models. Among patients who were immunosuppressed, the rate of FVC decline after treatment was compared with FVC decline before treatment in a LMM whereby treatment was considered as a binary intervention. All multivariable models were adjusted for age, sex, smoking pack-years, and baseline FVC. Sensitivity analysis included the year of cohort entry in each multivariable model, and an additional sensitivity analysis was performed in SSc-ILD examining the association between ANA pattern type with FVC decline and mortality.
Exploratory analyses examined the association of specific imaging features with FVC decline and with transplant-free survival. This included the total percentage of lung fibrosis, ratio of ground-glass opacities to fibrosis, emphysema, consolidation, mosaic attenuation, traction bronchiectasis, presence of cysts, presence of pleuroparenchymal fibroelastosis (PPFE), and presence of a dilated oesophagus. These models were also adjusted for age, sex, smoking pack-years, and baseline FVC, except for analyses involving total percentage of lung fibrosis where multivariable models did not include baseline FVC due to collinearity. All statistical analyses were performed using R version 4.2.2 and ‘nlme’ statistical package version 3.1–162.
Results
Patient characteristics and imaging findings
A total of 645 patients underwent blinded radiological review and had a mean follow-up of 5.2 ± 3.2 years with a mean of 8.9 ± 6.8 FVC measurements per person. The most frequent CTD diagnoses were SSc (33%), RA (20%) and IIM (16%). The baseline demographics are shown in Table 1. Among SSc patients, 70% had available ANA status with patterns. UIP was found most frequently in RA (47%) and SS (33%). NSIP and/or OP were the predominant patterns in SSc (77%), IIM (78%), and MCTD (73%) (Fig. 1A). A fHP pattern was present across all CTD-ILD diagnoses (range 4–20%). LIP was present in a minority of SS patients (13%), and more rarely in MCTD (2%) and RA (1%). Fig. 1B shows the extent of specific imaging features across CTD subtypes.
Table 1.
Baseline characteristics
| All CTD (n = 645) | SSc (n = 215) | RA (n = 127) | IIM (n = 100) | MCTD (n = 61) | SS (n = 40) | SLE (n = 19) | UCTD (n = 83) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Age | 60 ± 12 | 57 ± 12 | 66 ± 9 | 57 ± 13 | 54 ± 13 | 64 ± 10 | 56 ± 12 | 63 ± 10 | ||
| Male | 198 (31) | 45 (21) | 59 (47) | 44 (44) | 9 (15) | 14 (35) | 3 (16) | 24 (29) | ||
| Baseline FVC (L) | 2.8 (3.7) | 2.9 (5.0) | 3.3 (5.1) | 2.6 (0.8) | 2.4 (0.7) | 2.6 (0.8) | 2.3 (0.8) | 2.4 (0.7) | ||
| Baseline FVC % | 75 ± 20 | 73 ± 19 | 82 ± 21 | 73 ± 20 | 74 ± 20 | 78 ± 19 | 68 ± 18 | 74 ± 19 | ||
| Ever smoked | 335 (52) | 88 (41) | 87 (69) | 54 (54) | 26 (43) | 20 (50) | 7 (37) | 53 (64) | ||
| Pack years | 17 [5-30] | 15 [2, 30] | 21 [8, 35] | 10 [4, 22] | 16 [6, 30] | 12 [5, 21] | 8 [6, 37] | 20 [6, 31] | ||
| Years of follow-up | 5.2 ± 3.2 | 5.7 ± 3.6 | 4.5 ± 2.6 | 5.6 ± 2.6 | 5.7 ± 3.9 | 5.7 ± 3.6 | 4.5 ± 2.6 | 5.6 ± 2.6 | ||
| Death | 185 (29) | 70 (33) | 44 (35) | 12 (12) | 14 (23) | 9 (23) | 5 (26) | 31 (37) | ||
| Transplant | 30 (5) | 10 (5) | 8 (6) | 1 (1) | 5 (8) | 1 (3) | 0 0 | 5 (6) | ||
| Antifibrotic use | 6 (1) | 1 (0.5) | 4 (3) | 0 | 1 (2) | 0 | 0 | 0 | ||
| Radiologic pattern (%) | ||||||||||
| UIP | 126 (20) | 18 (8) | 59 (47) | 9 (9) | 7 (12) | 13 (33) | 3 (16) | 17 (21) | ||
| NSIP | 351 (54) | 163 (76) | 24 (19) | 62 (62) | 40 (66) | 12 (30) | 9 (47) | 41 (49) | ||
| OP | 32 (5) | 2 (1) | 3 (2) | 16 (16) | 4 (7) | 5 (13) | 0 (0.0) | 2 (2) | ||
| fHP | 60 (9) | 12 (6) | 19 (15) | 4 (4) | 7 (12) | 3 (8) | 4 (21) | 11 (13) | ||
| LIP | 7 (1) | 0 0 | 1 (1) | 0 0 | 1 (2) | 5 (13) | 0 0 | 0 0 | ||
| Unclassifiable | 64 (10) | 19 (9) | 19 (15) | 9 (9) | 2 (3) | 1 (3) | 3 (15) | 11 (13) | ||
| Other | 5 (1) | 1 (1) | 2 (2) | 0 0 | 0 0 | 1 (3) | 0 0 | 1 (1) | ||
Data are shown as mean ± S.D., number (percentage), or median [interquartile range]. fHP: fibrotic hypersensitivity pneumonitis; FVC (L) : absolute forced vital capacity in litres; FVC %: percent-predicted forced vital capacity; IIM: idiopathic inflammatory myositis; LIP: lymphoid interstitial pneumonia; MCTD: mixed CTD; NSIP: non-specific interstitial pneumonia; OP: organizing pneumonia; UCTD: undifferentiated CTD; antifibrotic use: defined by the use of nintedanib or pirfenidone for at least 3 months; UIP: usual interstitial pneumonia. Other patterns include appearances of predominant pleuroparenchymal fibroelastosis, desquamative interstitial pneumonia, respiratory bronchiolitis-associated interstitial lung disease, pulmonary Langerhans’ cell histiocytosis.
Figure 1.

Radiologist-defined imaging patterns (a) and individual features (b) present in CTD-ILD. n: number of patients with each CTD diagnosis; fHP: fibrotic hypersensitivity pneumonitis; GGO: ground-glass opacities; IIM: idiopathic inflammatory myositis; LIP: lymphoid interstitial pneumonia; MCTD: mixed CTD; NSIP: non-specific interstitial pneumonia; OP: organizing pneumonia; UCTD: undifferentiated CTD; UIP: usual interstitial pneumonia
Association of imaging pattern with FVC decline
On unadjusted analyses, FVC declined most rapidly in CTD-ILD patients with the fHP pattern (annualized rate of FVC percentage change of –2.3%, 95% CI –3.5, –1.0), followed by UIP (–1.7%, 95% CI –2.4, –1.0), other patterns (–1.3%, 95% CI –2.5, –0.1) and NSIP (–0.6%, 95% CI –1.4, 0.15). Patients with isolated OP had improved FVC over time by 1.8% (95% CI 0.4, 3.1). Compared with UIP, the rate of FVC decline was slower in patients with NSIP (by 1.1%, 95% CI 0.2, 1.9) and OP (by 3.5%, 95% CI 2.0, 4.9) on multivariable adjusted analyses, whereas fHP was associated with a trend towards faster FVC decline (by –0.7%, 95% CI –2.0, 0.6) (Fig. 2). There was no difference in the rate of progression between radiologic patterns in either RA or SSc-ILD. There were insufficient SSc patients with a fHP pattern to analyse. The addition of the year of cohort entry in the multivariable model did not change any of the findings, although a later year of cohort entry was associated with slower FVC decline for each year of diagnosis (by 0.17%, 95% CI 0.08, 0.25). Additional sensitivity analyses by including ANA antibody patterns in patients with SSc-ILD did not show any association with FVC decline in SSc-ILD.
Figure 2.

Linear mixed-model of FVC decline in all CTD-ILD. Multivariable analyses adjusted for age, sex, smoking pack years, and baseline FVC. CTD-ILD: CTD-associated interstitial lung disease; FVC: forced vital capacity; FVC%: percent-predicted forced vital capacity; UIP: usual interstitial pneumonia; NSIP: non-specific interstitial pneumonia; OP: organizing pneumonia; fHP: fibrotic hypersensitivity pneumonitis
On exploratory analyses, the total amount of lung fibrosis (honeycombing and reticulation) was associated with subsequent FVC decline in CTD-ILD and SSc-ILD, whereas the amount of consolidation was associated with subsequent FVC improvement in CTD-ILD (Supplementary Table S1, available at Rheumatology online).
Association of imaging pattern with mortality or lung transplant
Among all CTD-ILD patients, NSIP and OP patterns were associated with better mortality compared with UIP [hazard ratio (HR) 0.65, 95% CI 0.45, 0.93; and 0.18, 95% CI 0.05, 0.57, respectively], whereas an fHP pattern was associated with increased mortality compared with UIP (HR of 1.58, 95% CI 1.01, 2.48; Fig. 3). In RA, there was no mortality difference between NSIP and UIP, but a fHP pattern was also associated with increased mortality compared with UIP (HR 2.53, 95% CI 1.13–5.62). In SSc, NSIP was not statistically different compared with UIP (HR 0.55, 95% CI 0.27, 1.17). Additional sensitivity analyses in SSc patients by including ANA antibody patterns did not show any association with mortality.
Figure 3.
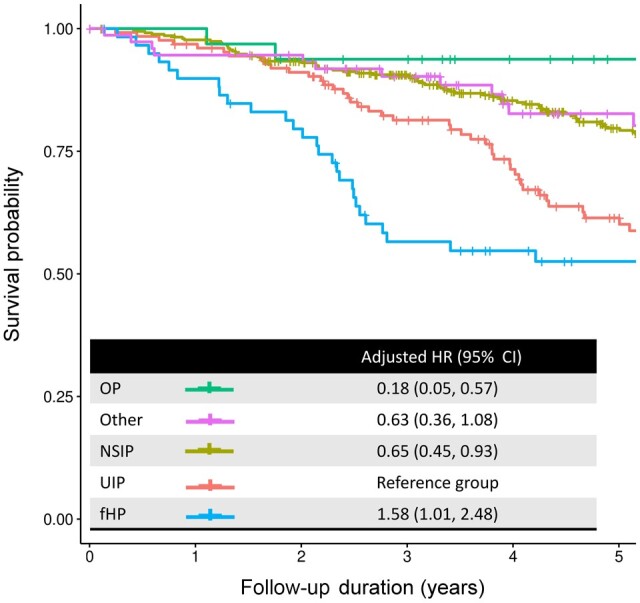
Mortality in CTD-ILD stratified by imaging pattern. Adjusted HR, hazard ratio adjusted for age, sex, smoking pack years, and baseline FVC using Cox regression analyses. CTD-ILD: CTD-associated interstitial lung disease; FVC: forced vital capacity; UIP: usual interstitial pneumonia; NSIP: non-specific interstitial pneumonia; OP: organizing pneumonia; fHP: fibrotic hypersensitivity pneumonitis
On exploratory analyses in all CTD-ILD, increased baseline fibrosis, traction bronchiectasis, emphysema, presence of PPFE, and dilated oesophagus were associated with mortality, whereas a higher ratio of ground-glass opacities to fibrosis was associated with decreased mortality on adjusted analysis. Similar findings for fibrosis and traction bronchiectasis were present in both RA and SSc-ILD. Emphysema and dilated oesophagus were additionally associated with mortality in RA-ILD. Increased ratio of ground-glass opacities to fibrosis was also associated with decreased mortality in SSc-ILD (Supplementary Table S2, available at Rheumatology online).
Association with immunosuppression
Among the 153 CTD-ILD patients with sufficient data to assess for immunosuppression response, 31 had UIP, 85 had NSIP and 18 had fHP. Only 1 person had concurrent antifibrotic medication treatment for at least 3 months before and after immunosuppression initiation. FVC decline was slower by 2.1% (1.4, 2.8) after treatment compared with pre-treatment in CTD-ILD with an NSIP pattern, after adjusting for age, sex, smoking, and baseline FVC (Fig. 4). In UIP there was no significant difference between pre- and post-treatment rates of FVC decline. The sample size was limited for analyses in the other ILD pattern groups, for subset analyses in individual CTD diagnoses, and to adequately assess interaction between treatment response and specific imaging findings.
Figure 4.

FVC decline before and after immunosuppression in CTD-ILD. There were too few patients with relevant treatment data to perform a similar analysis for patients with a CT pattern of fHP. CTD-ILD: CTD-associated interstitial lung disease; UIP: usual interstitial pneumonia; NSIP: non-specific interstitial pneumonia; Tx: treatment, P adj, P value for multivariable analyses adjusted for age, sex, smoking pack years, and baseline FVC
Discussion
We used a large well-characterized, prospectively enrolled cohort of patients to assess the radiologic patterns frequently present across CTD-ILD subtypes, notably including the fHP or airway-centric pattern that has historically been underappreciated in this population. We show that CTD subtypes have a propensity for different radiologic patterns, with these patterns predicting prognosis and treatment response. Patients with a UIP pattern have accelerated FVC decline, worse transplant-free survival, and attenuated response, if any, to immunosuppression, compared with patients with NSIP or OP patterns. Patients with a fHP pattern had worse survival than those with UIP, which is a novel and clinically relevant finding. Taken together, these findings emphasize the importance of radiologic patterns in CTD-ILD and sets the stage for further research into the prognosis and treatment of this population.
It is likely that the worse prognosis of UIP compared with NSIP and OP could in part be explained by the irreversible nature of fibrosis and the lack of prominent inflammation that might be responsive to immunosuppressive agents. Current pathophysiological mechanisms identify pro-fibrotic pathways driving the progression of fibrosis from existing fibrosis [9, 10]. Indeed, the extent of fibrotic changes, defined by the amount of honeycombing and reticulations, was also consistently associated with worse progression and mortality outcomes. These changes are a key element in the classification of UIP, and it is possible that these fibrotic changes may be a driver behind the worse prognosis associated with the UIP pattern. Patients with NSIP, which has more ground-glass opacities, and those with OP pattern, characterized by multifocal consolidation, may be more likely to have an inflammatory component that will respond to immunosuppression, such as is seen in IIM that has a predominance of OP and NSIP patterns [11, 12]. Consistent with this, we found that patients with an OP pattern had increasing FVC over time, likely because of reversibility of lung involvement with treatment.
The frequent presence of a ‘fHP’ pattern in ILD diagnoses other than clinical HP (range 4–20% across the CTD-ILD subtypes in our cohort) indicates the need for an alternative term to describe a pattern that is not specific to the clinical diagnosis of fHP. The term ‘bronchiolocentric interstitial pneumonia’ has been proposed for this purpose, appropriately highlighting the presence of significant airway involvement alongside common features of other interstitial pneumonias [3]. The clinical relevance of this imaging pattern in CTD-ILD is confirmed by the worse prognosis of these patients compared with patients with all other imaging patterns, including UIP. It remains unclear why prognosis is worse with a fHP pattern; however, this poor prognosis is also consistent with other data from CARE-PF showing that patients with a clinical diagnosis of fHP had a rate of progression and mortality that was similar to IPF [13]. Additional studies are urgently needed to determine the correlation between radiologic and pathologic findings, as well as the biological underpinnings of this poor prognosis and whether there are management implications—most notably whether immunosuppression is appropriate, as suggested in some CTD-ILD subtypes [14], or whether this is potentially harmful, as has been demonstrated in IPF [15].
Exploratory findings showed that the amount of fibrosis (honeycombing and reticulation) on imaging was the most consistent factor in predicting future FVC decline and mortality. Traction bronchiectasis, a sign of architectural distortion in fibrotic disease, was also associated with mortality. Other poor prognostic factors included the presence of PPFE, extent of emphysema, and a dilated oesophagus. These are all features generally considered irreversible and not expected to improve with immunosuppression in CTD. Conversely, features conventionally thought to represent inflammation (ground-glass opacities and consolidation) were associated with a better prognosis and relative improvement in FVC with treatment. These findings highlight the fact that the extent of baseline fibrotic changes is an important factor for subsequent progression, and that early initiation of treatment is key. It is well documented that a lower baseline FVC is associated with worse prognosis in ILD [16, 17], which is why this was adjusted for in all analyses. These findings also lend support to the concept of a morphological approach to treating progressive ILD, especially with antifibrotics showing benefit in treating IPF and progressive fibrosing ILD of non-IPF etiologies, with the latter finding most notable in patients with features suggestive of a UIP pattern [18–21].
Subgroup analyses of specific CTD diagnoses and assessment of immunosuppression response was limited by the low prevalence of some patterns and particularly the number of patients who had sufficient data for treatment response analyses. Exploratory analyses were performed without a predefined hypothesis and without histologic confirmation, but the findings are concordant with findings based on imaging features and support the inclusion of a morphological approach to treatment, while also emphasizing the importance of the extent of fibrosis as a key feature. While patients in the medication analyses acted as their own pre-treatment controls, these findings also have limitations inherent in this retrospective uncontrolled analysis.
In summary, radiologic morphology remains an essential part of individualized patient management decisions in CTD-ILD, with different imaging patterns impacting prognosis. Notably, patterns of UIP and fHP predict worse outcomes and relatively poor response to immunosuppressive medication, while NSIP and OP patterns are associated with better outcomes and a more positive response to immunosuppressive medication. These findings should be further explored in randomized controlled trials. This study confirms the importance of radiologic patterns in CTD-ILD and highlights priorities for future research.
Supplementary Material
Contributor Information
Boyang Zheng, Department of Medicine, University of British Columbia, Vancouver, BC, Canada; Department of Medicine, McGill University, Montreal, QC, Canada.
Daniel-Costin Marinescu, Department of Medicine, University of British Columbia, Vancouver, BC, Canada; Centre for Heart Lung Innovation, St Paul’s Hospital, Vancouver, BC, Canada.
Cameron J Hague, Department of Radiology, University of British Columbia, Vancouver, BC, Canada.
Nestor L Muller, Department of Radiology, University of British Columbia, Vancouver, BC, Canada.
Darra Murphy, Department of Radiology, St James’ Hospital, Dublin 8, Ireland.
Andrew Churg, Department of Pathology, University of British Columbia, Vancouver, BC, Canada.
Joanne L Wright, Department of Pathology, University of British Columbia, Vancouver, BC, Canada.
Amna Al-Arnawoot, Department of Radiology, McMaster University, Hamilton, ON, Canada.
Ana-Maria Bilawich, Department of Radiology, University of British Columbia, Vancouver, BC, Canada.
Patrick Bourgouin, Department of Radiology, University of Montreal, Montreal, QC, Canada.
Gerard Cox, Department of Medicine, McMaster University, Hamilton, ON, Canada.
Celine Durand, Département de Médecine, Centre de recherche du Centre hospitalier de l, ’Université de Montréal, Montréal, QC, Canada.
Tracy Elliot, Department of Radiology, University of Calgary, Calgary, AB, Canada.
Jennifer Ellis, Department of Radiology, University of British Columbia, Vancouver, BC, Canada.
Jolene H Fisher, Department of Medicine, University of Toronto, Toronto, ON, Canada.
Derek Fladeland, Department of Medical Imaging, University of Saskatchewan, Saskatoon, SK, Canada.
Amanda Grant-Orser, Department of Medicine, University of Calgary, Calgary, AB, Canada.
Gillian C Goobie, Department of Medicine, University of British Columbia, Vancouver, BC, Canada; Centre for Heart Lung Innovation, St Paul’s Hospital, Vancouver, BC, Canada; Division of Pulmonary, Allergy and Critical Care Medicine, Department of Medicine, University of Pittsburgh, Pittsburgh, PA, USA.
Zachary Guenther, Department of Radiology, University of Calgary, Calgary, AB, Canada.
Ehsan Haider, Department of Radiology, McMaster University, Hamilton, ON, Canada.
Nathan Hambly, Department of Medicine, McMaster University, Hamilton, ON, Canada.
James Huynh, Department of Radiology, McMaster University, Hamilton, ON, Canada.
Kerri A Johannson, Department of Medicine, University of Calgary, Calgary, AB, Canada.
Geoffrey Karjala, Department of Medical Imaging, University of Saskatchewan, Saskatoon, SK, Canada.
Nasreen Khalil, Department of Medicine, University of British Columbia, Vancouver, BC, Canada.
Martin Kolb, Department of Medicine, McMaster University, Hamilton, ON, Canada.
Jonathon Leipsic, Department of Radiology, University of British Columbia, Vancouver, BC, Canada.
Stacey D Lok, Department of Medicine, University of Saskatchewan, Saskatoon, SK, Canada.
Sarah MacIsaac, Department of Medicine, McMaster University, Hamilton, ON, Canada.
Micheal McInnis, Department of Medical Imaging, University of Toronto, Toronto, ON, Canada.
Helene Manganas, Département de Médecine, Centre de recherche du Centre hospitalier de l, ’Université de Montréal, Montréal, QC, Canada.
Veronica Marcoux, Department of Medicine, University of Saskatchewan, Saskatoon, SK, Canada.
John Mayo, Department of Radiology, University of British Columbia, Vancouver, BC, Canada.
Julie Morisset, Département de Médecine, Centre de recherche du Centre hospitalier de l, ’Université de Montréal, Montréal, QC, Canada.
Ciaran Scallan, Department of Medicine, McMaster University, Hamilton, ON, Canada.
Tony Sedlic, Department of Radiology, University of British Columbia, Vancouver, BC, Canada.
Shane Shapera, Department of Medicine, University of Toronto, Toronto, ON, Canada.
Kelly Sun, Department of Medicine, University of Toronto, Toronto, ON, Canada.
Victoria Tan, Department of Radiology, McMaster University, Hamilton, ON, Canada.
Alyson W Wong, Department of Medicine, University of British Columbia, Vancouver, BC, Canada; Centre for Heart Lung Innovation, St Paul’s Hospital, Vancouver, BC, Canada.
Christopher J Ryerson, Department of Medicine, University of British Columbia, Vancouver, BC, Canada; Centre for Heart Lung Innovation, St Paul’s Hospital, Vancouver, BC, Canada.
Supplementary material
Supplementary material is available at Rheumatology online.
Data availability
Data are not available for ethical/privacy reasons, and data sharing has not been approved by the research ethics board.
Funding
The Canadian Registry for Pulmonary Fibrosis (CARE-PF) CARE-PF is sponsored by Boehringer Ingelheim. The sponsor had no input over the study design, data analysis, presentation of findings, or interpretation of results.
Disclosure statement: B.Z. reports consulting fees and honoraria from Boehringer-Ingelheim, Abbvie, Novartis. D.-C.M. reports honoraria from Boehringer-Ingelheim. S.D.L. reports consulting/personal fees and moderator honoraria from Boehringer-Ingelheim, honoraria from and Hoffman-La Roche Ltd, and grants from AstraZeneca and the University of Saskatchewan. J.H.F. reports grants from the Canadian Pulmonary Fibrosis Foundation and the University of Toronto, and personal fees from Boehringer-Ingelheim and AstraZeneca. G.C.G. reports research funding through the Canadian Pulmonary Fibrosis Foundation, the University of British Columbia, the Canadian Institutes of Health Research, and the Canadian Lung Association, and honoraria from Boehringer-Ingelheim. K.A.J. reports grants from University Hospital Foundation, the Three Lakes Foundation and the University of Calgary, and personal fees from Boehringer-Ingelheim, Hoffman-La Roche, Pliant Therapeutics, Thyron, Brainomix and the Three Lakes Foundation. M.K. reports grants from Boehringer-Ingelheim, Hoffman-La Roche and Pieris, personal fees from Boehringer Ingelheim, Hoffman-La Roche, Horizon, Cipla, Abbvie, Bellerophon, Algernon, CSL Behring, United Therapeutics Novartis and LabCorp, participation on the advisory board of United Therapeutics Novartis and LabCorp, and an allowance for serving as Chief Editor of the European Respiratory Journal. H.M. reports research grants from Boehringer-Ingelheim, Galapagos, BMS and Hoffman-La Roche, and personal fees from Boehringer-Ingelheim. V.M. reports personal fees from Boehringer Ingelheim Canada, Hoffman-La Roche Ltd and Astra Zeneca, and grants from the University of Saskatchewan, the Royal University Hospital Foundation, Boehringer Ingelheim, Astra Zeneca and Hoffman-La Roche. J.M. reports personal fees from Boehringer-Ingelheim and Hoffman-La Roche. S.S. reports advisory board participation and personal fees from Boehringer-Ingelheim, Hoffman-La Roche and AstraZeneca. A.W.W. reports speaker’s honoraria from Boehringer-Ingelheim and AstraZeneca. C.J.R. reports a relationship with Boehringer Ingelheim Canada Ltd that includes: consulting or advisory fees, funding grants, and speaking and lecturing fees, and reports consultancy fees/honoraria from Hoffman-La Roche, Veracyte, AstraZeneca, Pliant Therapeutics, Trevi Therapeutics, Cipla Ltd and Ensho Health. The other authors have declared no conflicts of interest.
References
- 1. Joy GM, Arbiv OA, Wong CK et al. Prevalence, imaging patterns and risk factors of interstitial lung disease in connective tissue disease: a systematic review and meta-analysis. Eur Respir Rev 2023;32:220210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ryerson CJ, Tan B, Fell CD et al. The Canadian registry for pulmonary fibrosis: design and rationale of a national pulmonary fibrosis registry. Can Respir J 2016;2016:3562923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marinescu D-C, Hague CJ, Muller NL et al. Integration and application of radiologic patterns from clinical practice guidelines on idiopathic pulmonary fibrosis and fibrotic hypersensitivity pneumonitis. CHEST 2023;164:1466–75. [DOI] [PubMed] [Google Scholar]
- 4. Raghu G, Remy-Jardin M, Richeldi L et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2022;205:e18–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Raghu G, Remy-Jardin M, Ryerson CJ et al. Diagnosis of hypersensitivity pneumonitis in adults: an official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2020;202:e36–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schraufnagel DE, Michel JC, Sheppard TJ, Saffold PC, Kondos GT. CT of the normal esophagus to define the normal air column and its extent and distribution. AJR Am J Roentgenol 2008;191:748–52. [DOI] [PubMed] [Google Scholar]
- 7. Takekoshi D, Arami S, Sheppard TJ et al. Computed tomography of the esophagus in scleroderma and lung disease. Tohoku J Exp Med 2015;237:345–52. [DOI] [PubMed] [Google Scholar]
- 8. Khor YH, Goh NSL, Wong AW et al. ; CARE-PF Investigators. Impact of concomitant medication burden on tolerability of disease-targeted therapy and survival in interstitial lung disease. Ann Am Thorac Soc 2022;19:962–70. [DOI] [PubMed] [Google Scholar]
- 9. Selman M, Pardo A. When things go wrong: exploring possible mechanisms driving the progressive fibrosis phenotype in interstitial lung diseases. Eur Respir J 2021;58:2004507. [DOI] [PubMed] [Google Scholar]
- 10. Renzoni EA, Poletti V, Mackintosh JA. Disease pathology in fibrotic interstitial lung disease: is it all about usual interstitial pneumonia? Lancet 2021;398:1437–49. [DOI] [PubMed] [Google Scholar]
- 11. Lega J-C, Reynaud Q, Belot A et al. Idiopathic inflammatory myopathies and the lung. Eur Respir Rev 2015;24:216–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tashkin DP, Roth MD, Clements PJ et al. ; Sclerodema Lung Study II Investigators. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med 2016;4:708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hambly N, Farooqi MM, Dvorkin-Gheva A et al. Prevalence and characteristics of progressive fibrosing interstitial lung disease in a prospective registry. Eur Respir J 2022;60:2102571. [DOI] [PubMed] [Google Scholar]
- 14. Rahaghi FF, Hsu VM, Kaner RJ et al. Expert consensus on the management of systemic sclerosis-associated interstitial lung disease. Respir Res 2023;24:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ et al. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. New Engl J Med 2012;366:1968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wong AW, Ryerson CJ, Guler SA. Progression of fibrosing interstitial lung disease. Respir Res 2020;21:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brown KK, Inoue Y, Flaherty KR et al. Predictors of mortality in subjects with progressive fibrosing interstitial lung diseases. Respirology 2022;27:294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Behr J, Prasse A, Kreuter M et al. ; RELIEF Investigators. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir Med 2021;9:476–86. [DOI] [PubMed] [Google Scholar]
- 19. Flaherty KR, Wells AU, Cottin V et al. ; INBUILD Trial Investigators. Nintedanib in progressive fibrosing interstitial lung diseases. New Engl J Med 2019;381:1718–27. [DOI] [PubMed] [Google Scholar]
- 20. King TE, Bradford WZ, Castro-Bernardini S et al. ; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. New Engl J Med 2014;370:2083–92. [DOI] [PubMed] [Google Scholar]
- 21. Richeldi L, du Bois RM, Raghu G et al. ; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. New Engl J Med 2014;370:2071–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are not available for ethical/privacy reasons, and data sharing has not been approved by the research ethics board.