

Abstract
Experimental and computational studies illuminating the factors that guide metal-centered stereogenicity and, therefrom, selectivity in transfer hydrogenative carbonyl additions of alcohol proelectrophiles catalyzed by chiral-at-metal-and-ligand octahedral d6-metal ions, iridium(III) and ruthenium(II), are described. To augment or invert regio-, diastereo- and enantioselectivity, predominantly one from among as many as 15 diastereomeric-at-metal complexes is required. For iridium(III) catalysts, cyclometallation assists in defining the metal stereocenter, and for ruthenium(II) catalysts, iodide counterions play a key role. Whereas classical strategies to promote selectivity in metal catalysis aim for high-symmetry transition states, well-defined low-symmetry transition states can unlock selectivities that are otherwise difficult to achieve or inaccessible.
Keywords: Chirality, Octahedral Stereogenic Center, Iridium, Ruthenium, Enantioselective
Graphical Abstract
I. Introduction:
The development of atom-efficient catalytic methods for the conversion of abundant, renewable feedstocks to value-added products is an important objective of modern chemical research.1 As demonstrated by hydrogenation (e.g. the Haber-Bosch reaction),2–4 methane-steam reforming,5 hydroformylation6 and related Fischer-Tropsch process,7 and the cracking of ethane to ethylene,8 the atom-efficiency of transformations that occur through the addition, redistribution or removal of hydrogen is a key, enabling characteristic for implementation of any catalytic process at large volume. Guided and inspired by this characteristic, our laboratory has developed diverse catalytic carbonyl additions that operate through the reductive activation of π-unsaturated pronucleophiles under the conditions of hydrogenation or hydrogen auto-transfer from alcohol proelectrophiles (Figure 1).9 These processes represent an alternative to carbonyl additions mediated by stoichiometric organometallic reagents, which are often hazardous, functional group intolerant, and generate metal-containing waste. Hydrogen auto-transfer for carbonyl addition is mechanistically distinct from related “borrowing hydrogen” reactions that promote hydroxyl substitution of alcohol reactants.10
Figure 1.

Carbonyl addition beyond stoichiometric organometallic nucleophiles.
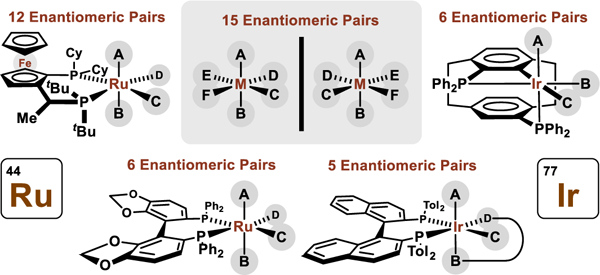
To date, nearly all asymmetric reactions of this type are catalyzed by iridium(III) and ruthenium(II) complexes.9 Such octahedral d6-metal ions possess unoccupied dz2 and dx2−y2 orbitals. This electronic configuration is especially conducive to alkoxide β-hydride elimination (alcohol dehydrogenation), an event that is likely preceded by an agostic interaction with the carbinol C-H bond. As shown, occupancy of the dx2−y2 orbital would place a relatively high-energy pair of electrons in the spatial domain of the migrating hydride (Figure 2). Alternatively, if the vacant coordination site of the pentacoordinate metal alkoxide is in the northern apical position, the unoccupied dz2 orbital would similarly facilitate alkoxide β-hydride elimination (not shown). We have found that the development of highly regio-, diastereo- and enantioselective carbonyl additions of alcohol proelectrophiles using iridium(III) and ruthenium(II) catalysts requires control of metal-centered stereogenicity,11 which is a daunting challenge given the multiplicity of stereoisomers associated with octahedral metal ions, as described in the tertiary literature.12 For an octahedral metal ion bearing six different monodentate ligands, there exist 15 enantiomeric pairs, meaning 30 stereoisomers are possible. Chiral nonracemic bidentate ligands can reduce the number of possible stereoisomers, but do not necessarily enforce formation of a single diastereomeric-at-metal complex. For example, when bound to a C1-symmetric ligand such as JOSIPHOS,13 12 diastereomeric-at-metal complexes remain possible. Similarly, when bound to a C2-symmetric ligand such as SEGPHOS,14 6 diastereomeric-at-metal complexes can exist. Naturally, to enforce optimal levels of regio-, diastereo- and enantioselectivity, intervention of predominantly one diastereomeric-at-metal complex is required. However, while chiral-at-metal-and-ligand catalysts are ubiquitous, metal-centered stereogenicity in such systems is often poorly understood or entirely ignored. Here, factors that guide metal-centered stereogenicity and, therefrom, regio-, diastereo- and enantioselectivity in C-C couplings of alcohols catalyzed by octahedral chiral-at-metal-and-ligand complexes are surveyed.
Figure 2:
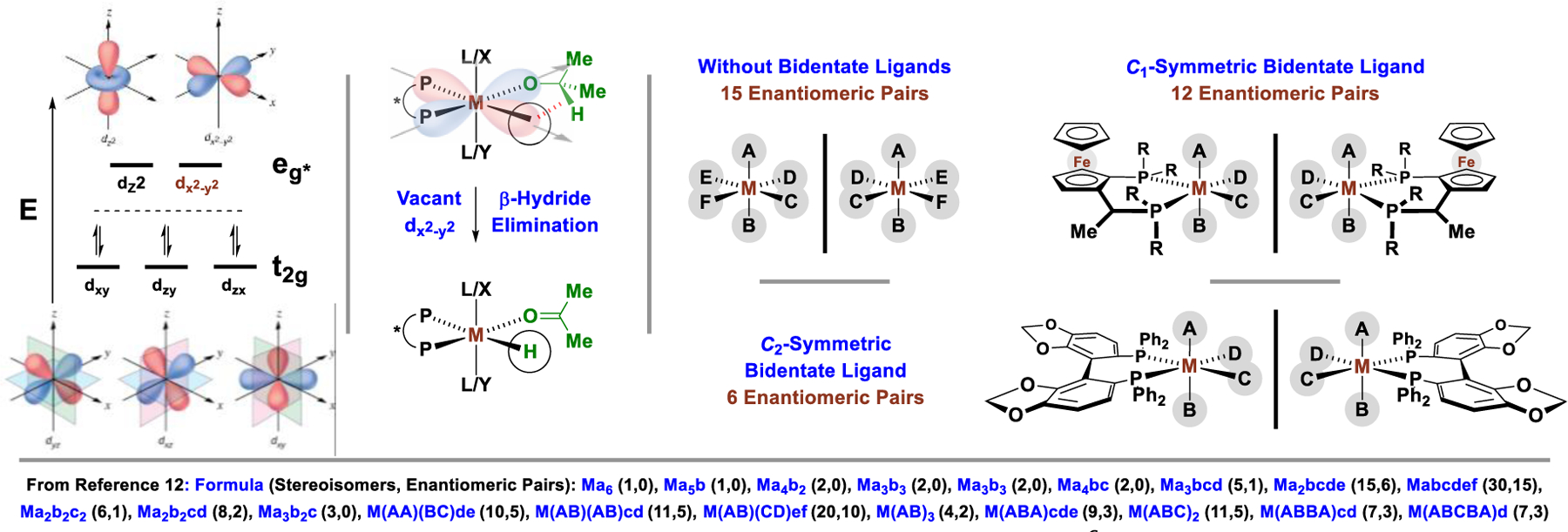
The unique reactivity and stereochemical complexity of octahedral d6-metal ions (an idealized octahedral splitting diagram is shown).
II.A. Cyclometallated π-Allyliridium-C,O-Benzoates:
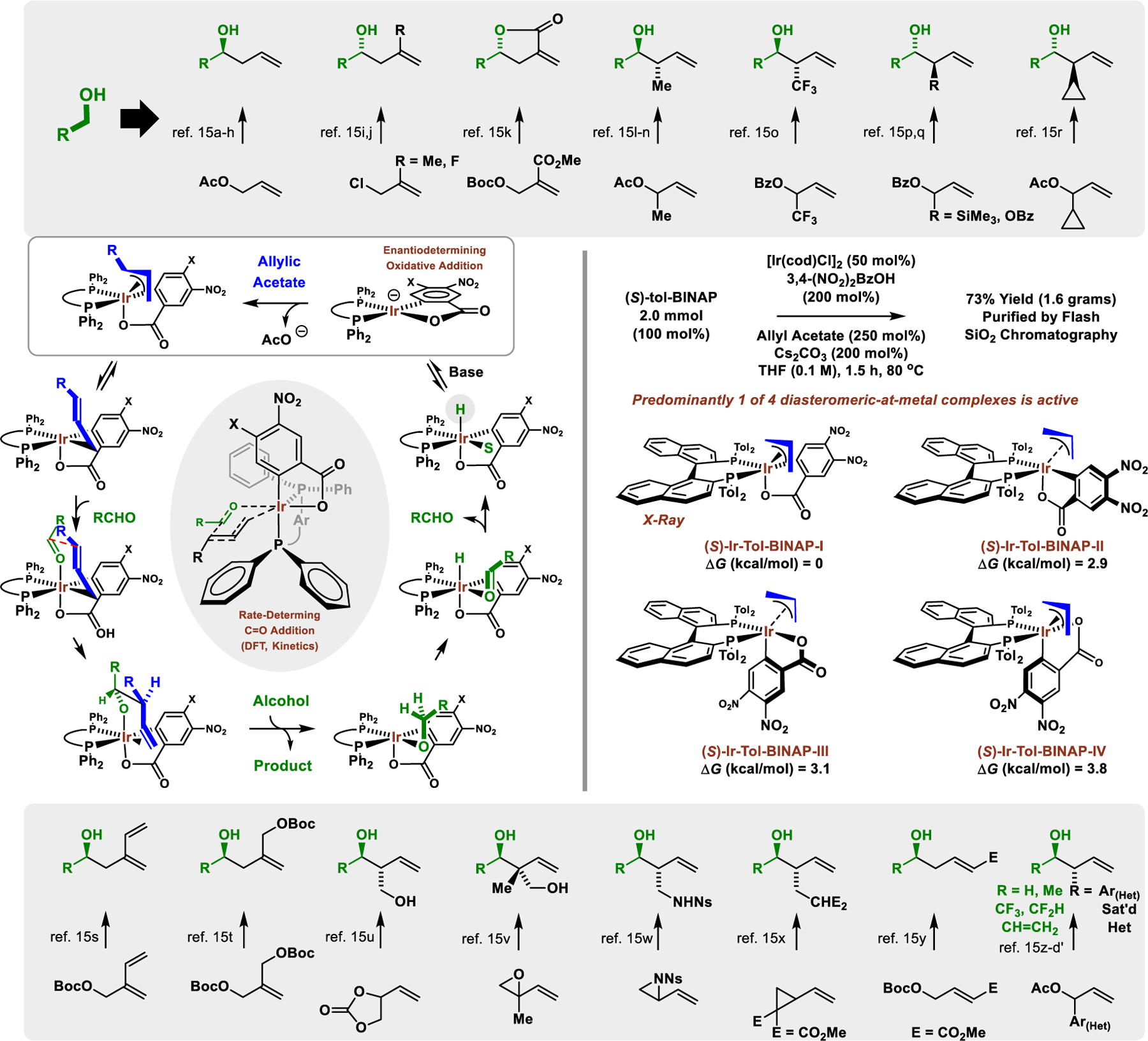
π-Allyliridium-C,O-benzoate complexes bound by various chelating axially chiral C2-symmetric phosphine ligands (BINAP, SEGPHOS, etc) catalyze an exceptionally diverse array of highly enantioselective carbonyl allylations from alcohol proelectrophiles (Figure 3).15,16 The π-allyliridium-C,O-benzoate complex, which is stable to conventional silica gel chromatography, can exist as four possible diastereomeric-at-metal complexes. The relative energies of these complexes have been estimated by DFT calculations.17 The three most stable diastereomers are observed when the complex is formed and can be separated by HPLC, but they slowly equilibrate upon standing. The most stable diastereomer has been characterized by single crystal X-ray diffraction.17 A catalytic cycle corroborated by DFT calculations17 reveals that the initially formed diastereomeric mixture of π-allyliridium complexes is inconsequential, as the stereocenter at iridium is ablated upon deprotonation of the iridium hydride18 to form a transient square planar iridium(I) complex. Thus, the enantiodetermining step of the catalytic cycle is not carbonyl addition, but oxidative addition of the allylic acetate, which defines the stereochemistry at iridium. The carbonyl addition, which occurs in a stereospecific manner through a closed Zimmerman-Traxler-type transition structure,19 simply transfers stereochemical information from the stereogenic center at iridium to carbon. Due to full occupancy of coordination sites in the XY-plane, the vacant dz2 orbital facilitates β-hydride elimination in a manner akin to that shown for the vacant dx2−y2 orbital in Figure 2.
Figure 3.
Metal-centered stereogenicity in enantioselective π-allyliridium-C,O-benzoate-catalyzed carbonyl allylations of alcohol proelectrophiles.
This interpretation of the reaction mechanism suggests that stereochemistry at iridium might be more important in directing the enantiofacial selectivity of carbonyl addition than the chiral phosphine ligand. Indeed, using the same antipode of chiral ligand, (S)-tol-BINAP, roughly equal yet opposite enantioselectivities are observed in carbonyl allylations mediated by allyl acetate versus allene, H2C=C=CH2 (Figure 4).17 Our collective experimental and computational studies demonstrate that the observed inversion in enantioselectivity stems from an inversion in stereochemistry at iridium. Specifically, while the square planar iridium(I) complex oxidatively adds allyl acetate to form the most stable diastereomer of the π-allyliridium complex, allene hydrometallation from the octahedral iridium(III) hydride delivers the less stable π-allyliridium complex in which the stereocenter at iridium is inverted. Thus, the inversion of enantioselectivity observed upon aldehyde addition for diastereomeric π-allyliridium complexes, each bearing (S)-BINAP, demonstrates that the stereocenter at iridium overrides the influence of the chiral ligand in defining the enantioselectivity of carbonyl addition.
Figure 4:
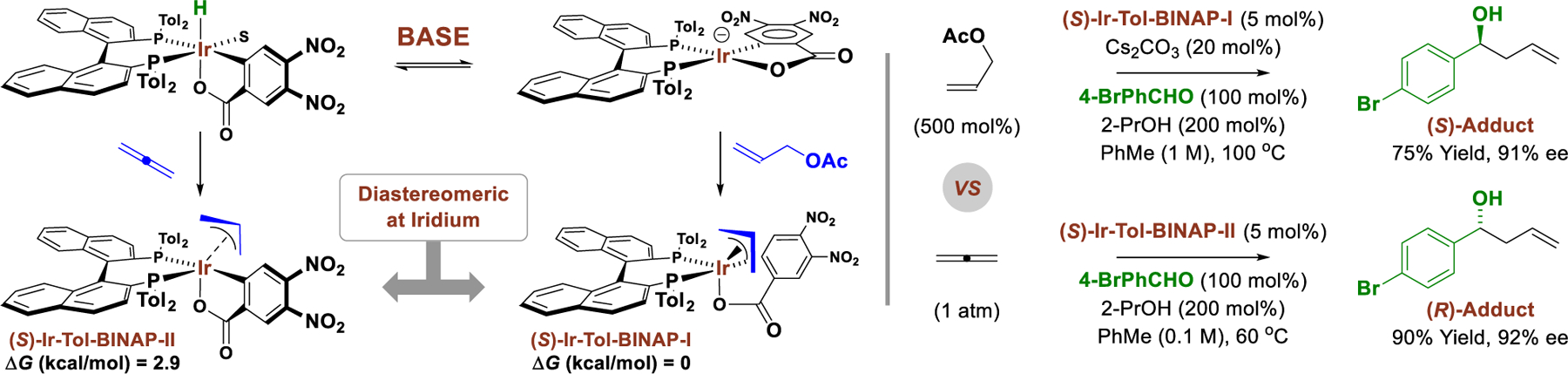
Inversion of enantioselectivity in carbonyl allylations catalyzed by diastereomeric-at-iridium complexes.
II.B. Cyclometallated π-Allyliridium-PhanePhos Complexes:
Another distinct class of diastereomeric-at-metal catalysts for alcohol-mediated C-C coupling are ortho-cyclometallated iridium-PhanePhos complexes.20 These catalysts are generated in situ from [Ir(cod)Cl]2 and (R)-PhanePhos and were initially identified in connection with their ability to catalyze enantioselective C-C couplings of methanol with dienes or CF3-bearing allenes to form primary neopentyl alcohols via formaldehyde allylation (Figure 5, eq. 1 and 2).9k,20a,b Notably, the latter process enables enantioselective formation of CF3-bearing quaternary carbon stereocenters (Figure 5, eq. 2).20b At first, it was not recognized that the active catalysts were ortho-cyclometallated. However, in subsequent work, a chromatographically stable ortho-cyclometalated iridium-(R)-PhanePhos complex was isolated, characterized by single crystal X-ray diffraction and demonstrated to be catalytically competent in the redox-neutral C-C couplings of methanol and related 2-propanol mediated allene-fluoral reductive couplings (Figure 5, eq. 3).20c This ortho-cyclometallated iridium-PhanePhos complex is chiral-at-metal-and-ligand and, given the tridentate nature of ortho-cyclometallated ligand, 6 diastereomeric-at-metal complexes are potentially active. Only one diastereomeric-at-metal complex is observed in the solid state in which the chloride and C-aryl ligands are trans, and DFT calculations of the possible stereoisomeric transition states for carbonyl addition suggest the analogous diastereomeric-at-metal transition structure is most stable. Mechanistic studies involving a combination of deuterium labelling, reaction progress kinetic analysis (RPKA), and DFT calculations were applied to the allene-fluoral reductive couplings. The collective data corroborate a catalytic mechanism in which rapid, reversible allene hydrometalation precedes turnover-limiting carbonyl addition.20c As shown in the indicated stereochemical model, interactions involving the ortho-CH2 group of the ethano-bridge that lies proximal to iridium play a key role in directing relative and absolute stereochemistry. More recently, the ortho-cyclometallated iridium-PhanePhos complexes were found to catalyze enantioselective 2-propanol-mediated reductive couplings of 2-substituted dienes with oxetanones and N-acyl-azetidinones (Figure 5, eq. 4).20d Here, in contrast to reactions of methanol/formaldehyde, which result in coupling of the diene at the 2-position to form quaternary carbon stereocenters, C-C bond formation occurs at the diene 3-position to form tertiary carbon stereocenters. These data suggest a Curtin-Hammet scenario in which the steric demand of the carbonyl electrophile influences the relative energies of competing transition states for rate-determining carbonyl addition from a rapidly equilibrating pool of isomeric allyliridium haptomers (not shown).
Figure 5.
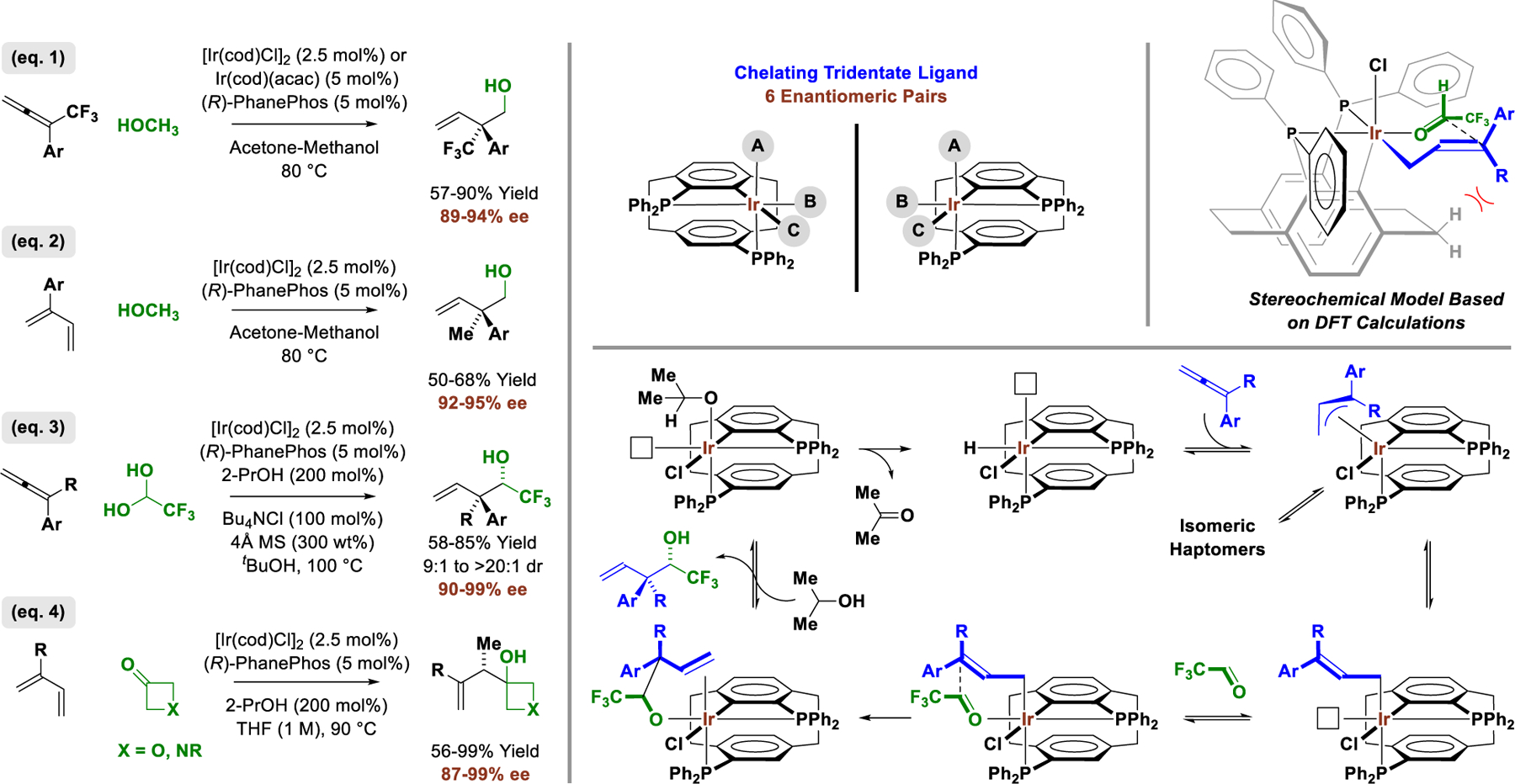
Metal-centered stereogenicity in enantioselective ortho-cyclometallated π-allyliridium-PhanePhos-catalyzed carbonyl allylations.
III.A. Iodide-Bound π-Allylruthenium(JOSIPHOS) Complexes:
The formation of racemic homoallylic alcohols via ruthenium-catalyzed reductive or redox neutral couplings of dienes or allenes with aldehydes or alcohol proelectrophiles was first described in 2008.21 The development of related enantioselective processes proved to be quite challenging due to the inability to control stereochemistry at ruthenium in the requisite chiral-at-ligand-and-metal catalysts. Catalysts modified by chiral phosphate counterions displayed good levels of diastereo- and enantioselectivity in certain cases,22 but the chiral phosphate counterions are not commercially available, which impelled efforts toward a more practical solution. Eventually, it was found that ruthenium-JOSIPHOS catalysts modified by iodide counterions are highly efficient catalysts for the enantioselective carbonyl allylation of alcohol proelectrophiles (Figure 6).23,24 These processes exploit alkynes as precursors to nucleophilic allylruthenium species.25 At first, Bu4NI was exploited as an exogenous iodide source (Figure 6, eq. 5, 6),23a-c however, the iodide-containing precatalyst RuI(CO)(JOSIPHOS)(η3-C3H5) is also effective (Figure 6, eq. 7).23d As the corresponding chloride- or bromide-modified catalysts displayed lower enantioselectivities and diminished turnover, experimental and computational studies were undertaken to understand the origins of iodide’s uniquely beneficial effects. Analysis of X-ray crystallographic data for the halide-bound π-allylruthenium complexes, RuX(CO)(JOSIPHOS)(η3-C3H5), where X = Cl, Br, I, revealed a trans-relationship between the π-acidic carbonyl ligand and π-donating halide in each case. However, occupancy of the diastereotopic northern and southern apical coordination sites varied. For the chloride and bromide complexes, mixtures of diastereomeric-at-metal complexes were evident, but for the iodide complex a single diastereomer was observed (Figure 7). Additionally, for the iodide complex, QTAIM analysis26 implicated the presence of a formyl CH···I hydrogen bond that stabilizes the preferred transition state for carbonyl addition.27 The collective data are consistent with the indicated catalytic cycle where, from a plurality of stereoisomeric structures, predominantly one diastereomeric-at-metal complex intervenes in the enantiodetermining transition state for carbonyl addition due to the enhanced size and polarizability of iodide versus chloride or bromide.
Figure 6.

Metal-centered stereogenicity in enantioselective π-allylruthenium-JOSIPHOS-catalyzed carbonyl allylations.
Figure 7.

Structures of RuX(CO)(JOSIPHOS)(?3-C3H5), where X = Cl, Br, I, determined by X-ray diffraction.
The development of iodide-bound ruthenium-JOSIPHOS catalysts unlocked numerous enantioselective alcohol-mediated C-C couplings that occur with complete atom-efficiency (Figure 8).28 This includes the redox-neutral C−C couplings of primary alcohols with methylallene (not shown) or 1,3-butadiene to form products of anti-crotylation (Figure 8, eq. 8).28a Similarly, related C-C couplings of primary alcohols with gaseous allene (propadiene) deliver highly enantiomerically enriched secondary homoallylic alcohols (Figure 8, eq. 9).28b Ruthenium hydrides obtained via primary alcohol dehydrogenation catalyze the isomerization of skipped dienes to form conjugated dienes, which, in turn, participate in transfer hydrogenative carbonyl addition. Thus, 1,4-pentadiene and 1,5-hexadiene provide entry to products of (α-ethyl)allylation and (α-propyl)allylation, respectively (Figure 8, eq. 10).28c Finally, primary alcohols react with 2-butyne to form chiral allylic alcohols (Figure 8, eq. 11).28d Distinct from other methods for asymmetric aldehyde allylations16 and vinylations,29 a kinetic preference for primary alcohol dehydrogenation enables primary-secondary diols to participate in highly chemoselective C-C coupling at the primary alcohol, meaning unprotected secondary alcohols are tolerated (not shown). For each process, use of the corresponding chloride- and bromide-bound ruthenium catalysts led to lower yields and enantioselectivities, corroborating iodide’s uniquely capacity to define stereochemistry at ruthenium and stabilize the preferred transition state for carbonyl addition via formyl CH···I hydrogen bonding.
Figure 8.
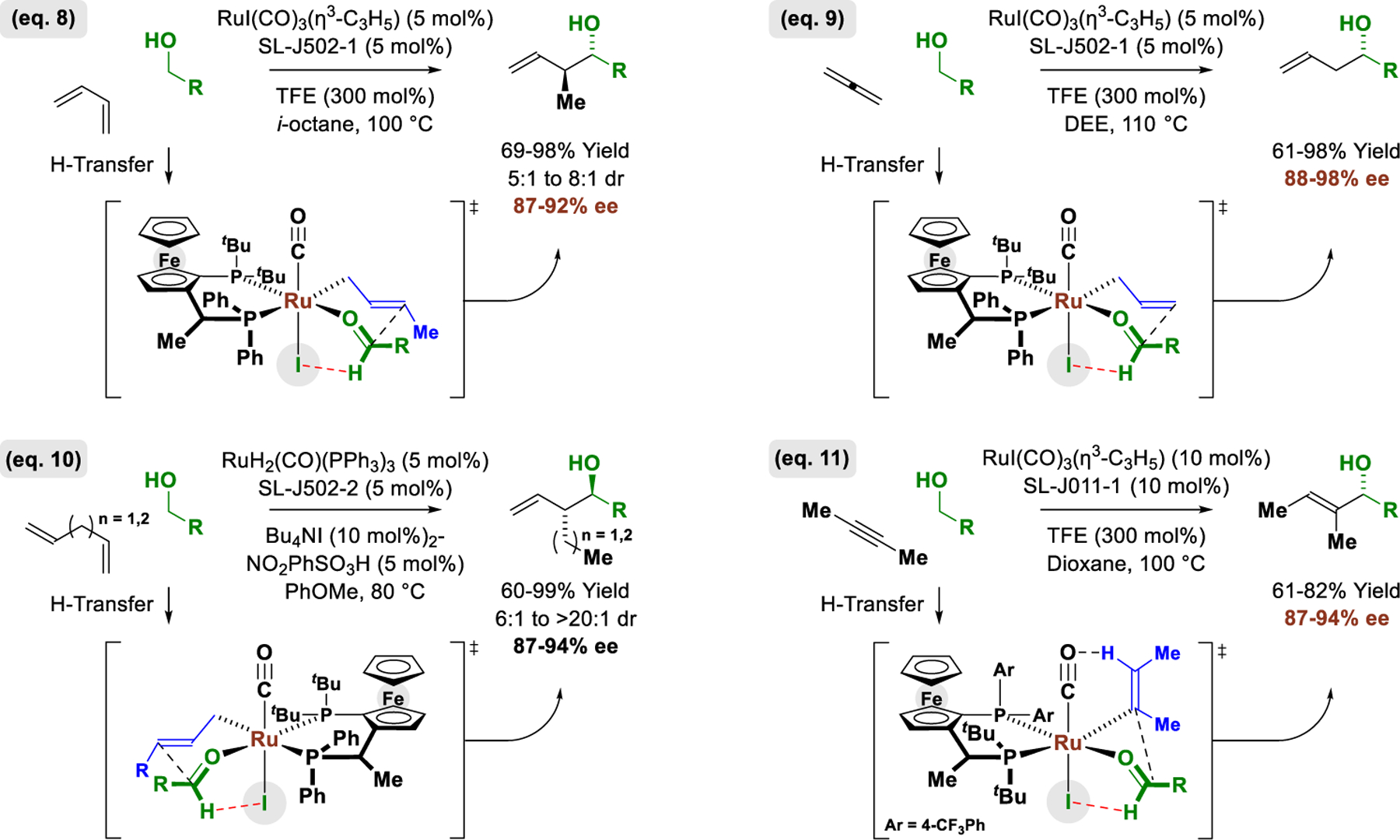
The control of metal-centered stereogenicity unlocks diverse enantioselective ruthenium-JOSIPHOS-catalyzed carbonyl additions.
III.B. Iodide-Bound π-Allylruthenium(SEGPHOS) Complexes:
An especially compelling illustration of the effect of metal-centered stereogenicity on selectivity is found in the C-C coupling of primary alcohols with isoprene (Figure 9).30 Here, pseudo-diastereomeric chiral-at-ruthenium complexes RuX(CO)[η3-prenyl][(S)-SEGPHOS], where X = Cl and I, deliver products of sec-prenylation and tert-prenylation, respectively. To determine the origins of regiodivergence,31 the parent chloride- and iodide-bound π-allylruthenium complexes modified by SEGPHOS were analyzed via single crystal X-ray diffraction. For the chloride-bound complex, the normal trans-diapical relationship between the π-acidic carbonyl ligand and π-donating chloride ligand was retained. This is a preference that persists across diverse phosphine-modified ruthenium carbonyl complexes bearing π-allyl,33a π-propargyl,33b hydride,33c,d,e vinyl,33f,g and acetylide ligands.33h In contrast, for the iodide complex, a highly uncommon cis-relationship between an apical carbonyl ligand and equatorial iodide was observed. This anomalous preference is due to nonbonded interactions that would be incurred between the equatorial diphenylphosphino moiety of SEGPHOS with an apical iodide. Solid and solution phase 31P NMR analysis revealed that the observed diastereomeric preferences persist in solution and are not an artifact of crystal packing forces. Furthermore, DFT calculations suggest the trans-halide/carbonyl diastereomers of both the π-allyl and π- prenyl complexes are more stable in the case of the chloride complex and, conversely, the cis-halide/carbonyl diastereomers are more stable in the case of the iodide complex. DFT calculations of the iodide-bound catalyst point to a Curtin-Hammett scenario in which the relative energies of competing transition states for aldehyde coordination from an equilibrating mixture of sec- and n-prenylruthenium complexes is rate- and product-determining. Whereas the influence of metal-centered stereogenicity on enantioselectivity is well-established, to our knowledge, these data represent the first correlation between metal-centered stereogenicity and regioselectivity in metal catalysis.
Figure 9.
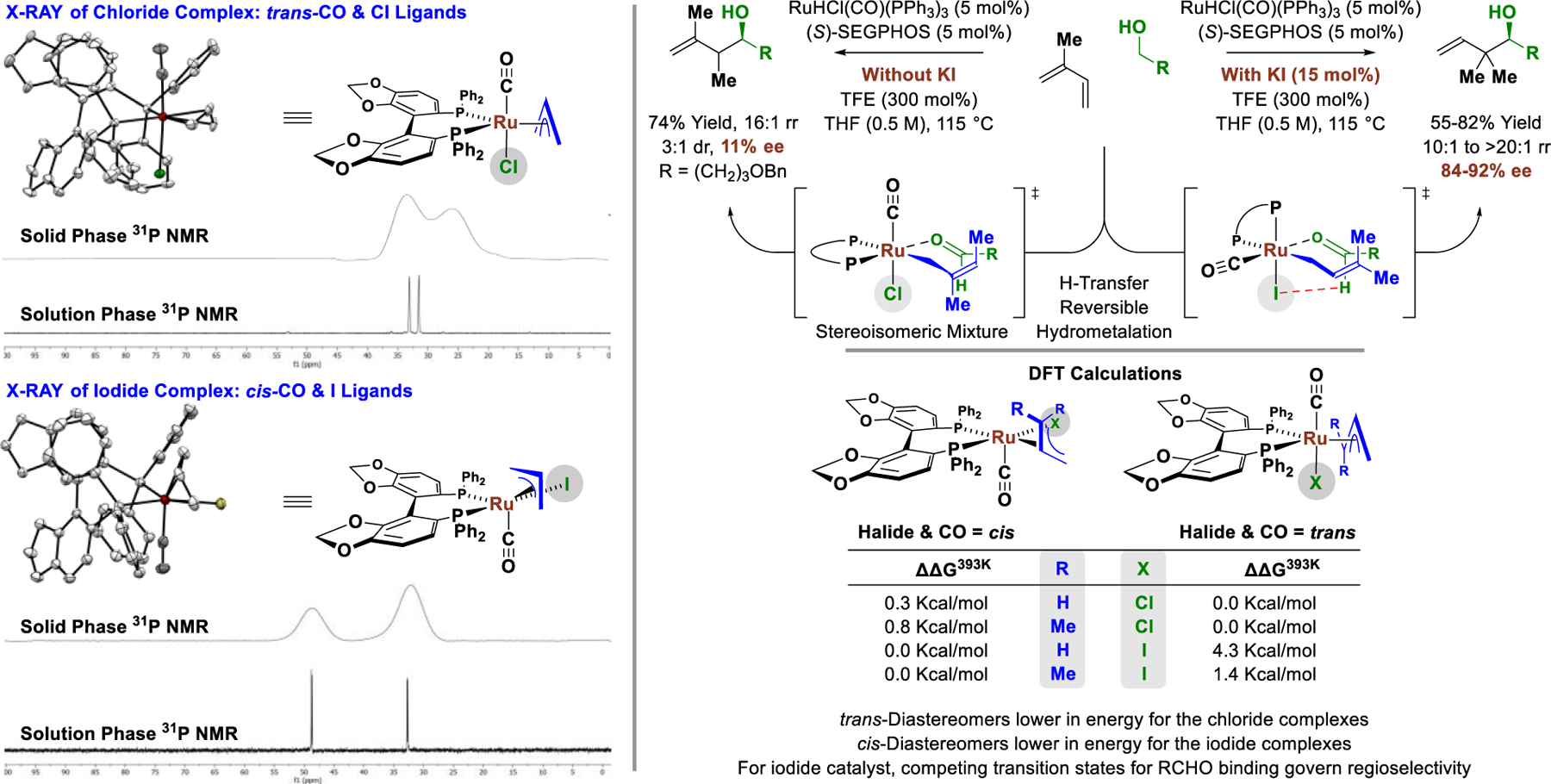
Divergent regioselectivity guided by metal-centered stereogenicity in enantioselective π-allylruthenium-SEGPHOS-catalyzed C-C couplings of primary alcohols with isoprene.
IV. Conclusion and Outlook:
Classical strategies for optimizing enantioselectivity in metal catalysis have emphasized minimization of the number of competing transition structures, for example, through use of square planar d8-metal ions bound by chelating C2-symmetric ligands.32 However, with reliable strategies for the stereocontrolled formation of octahedral chiral-at-metal-and-ligand complexes, one may craft low-symmetry environments for catalysis that are topologically distinct and may unlock selectivities that are otherwise difficult to achieve or inaccessible. In this monograph, strategies for the generation of diastereomeric-at-metal catalysts based on the octahedral d6-metal ions, iridium(III) and ruthenium(II), were described and correlations of metal-centered stereochemistry to regio, diastereo- and enantioselectivity were made using experimental and computational methods. Surprising outcomes include the fact that metal-stereochemistry can override ligand-stereochemistry to invert enantioselectivity (Figure 4) or invert regioselectivity (Figure 9). It is our hope that the present approaches to generating stereochemically-defined chiral-at-metal-and-ligand octahedral complexes will inform and accelerate advances in the broader field of regio- and stereoselective metal catalysis.
ACKNOWLEDGMENTS
The Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM069445, R35 GM128779), JSPS (KAKENHI 20H05671) and KAUST are acknowledged for partial support of this research.
Footnotes
The authors declare no competing financial interest.
References
- (1).For reviews, see: (a) Trost BM The Atom Economy—A Search for Synthetic Efficiency. Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]; (b) Doerksen RS; Meyer CC; Krische MJ Feedstock Reagents in Metal-Catalyzed Carbonyl Reductive Coupling: Minimizing Preactivation for Efficiency in Target-Oriented Synthesis. Angew. Chem. Int. Ed 2019, 58, 14055–14064 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).For selected reviews on enantioselective hydrogenation and transfer hydrogenation in the synthesis of pharmaceutical ingredients, see: (a) Hawkins JM; Watson TJN Asymmetric Catalysis in the Pharmaceutical Industry. Angew. Chem. Int. Ed 2004, 43, 3224–3228. [DOI] [PubMed] [Google Scholar]; (b) Thommen M Homogeneous Asymmetric Hydrogenation: Mature and Fit for Early Stage Drug Development. Spec. Chem. Mag 2005, 25, 26–28. [Google Scholar]; (c) Thayer AM Chiral Catalysis. Chem. Eng. News 2005, 83, 40–58. [Google Scholar]; (d) Farina V; Reeves JT; Senanayake CH; Song JJ Asymmetric Synthesis of Active Pharmaceutical Ingredients. Chem. Rev 2006, 106, 2734–2793. [DOI] [PubMed] [Google Scholar]; (e) Carey JS; Laffan D; Thomson C; Williams MT Analysis of The Reactions Used for The Preparation of Drug Candidate Molecules. Org. Biomol. Chem 2006, 4, 2337–2347. [DOI] [PubMed] [Google Scholar]; (f) Ager DJ; de Vries AHM; de Vries JG Asymmetric Homogeneous Hydrogenations at Scale. Chem. Soc. Rev 2012, 41, 3340–3380. [DOI] [PubMed] [Google Scholar]; (g) Etayo P; Vidal-Ferran A Rhodium-Catalyzed Asymmetric Hydrogenation as a Valuable Synthetic Tool for The Preparation of Chiral Drugs. Chem. Soc. Rev 2013, 42, 728–754. [DOI] [PubMed] [Google Scholar]; (h) Hayler JD; Leahy DK; Simmons EM A Pharmaceutical Industry Perspective on Sustainable Metal Catalysis. Organometallics 2019, 38, 36–46. [Google Scholar]
- (3).Stoffels MA; Klauck FJR; Hamadi T; Glorius F; Leker J Technology Trends of Catalysts in Hydrogenation Reactions: A Patent Landscape Analysis. Adv. Synth. Catal 2020, 362, 1258–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For a review on the Haber-Bosch process, see: Smil V Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, MIT Press: Cambridge, MA, 2004; pp 68–107. [Google Scholar]
- (5).For a review on methane-steam reforming, see: Hook van J. P. Methane-Steam Reforming. Catal. Rev 1980, 21, 1–51. [Google Scholar]
- (6).For a review on alkene hydroformylation, see: P.W.N.M. van Leeuwen, Claver C Rhodium Catalyzed Hydroformylation. Catalysis by Metal Complexes, Vol. 22; Springer Dordrecht, 2000. DOI: 10.1007/0-306-46947-2 [DOI] [Google Scholar]
- (7).For a review on the Fischer-Tropsch process, see: Dry ME The Fischer–Tropsch Process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar]
- (8).Kniel L; Winter O; Stork K Ethylene: Keystone to the Petrochemical Industry; Vol. 2; Marcel Dekker: New York, 1980. [Google Scholar]
- (9).For selected reviews on carbonyl addition via addition or redistribution of hydrogen, see: (a) Ngai M-Y; Kong J-R; Krische MJ Hydrogen-Mediated C-C Bond Formation: A Broad New Concept in Catalytic C-C Coupling. J. Org. Chem 2007, 72, 1063–1072. [DOI] [PubMed] [Google Scholar]; (b) Iida H; Krische MJ Catalytic Reductive Coupling of Alkenes and Alkynes to Carbonyl Compounds and Imines Mediated by Hydrogen. Top. Curr. Chem 2007, 279, 77–104. [Google Scholar]; (c) Hassan A; Krische MJ Unlocking Hydrogenation for C-C Bond Formation: A Brief Overview of Enantioselective Methods. Org. Proc. Res. Devel 2011, 15, 1236–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Moran J; Krische MJ Formation of C-C Bonds via Ruthenium-Catalyzed Transfer Hydrogenation. Pure Appl. Chem 2012, 84, 1729–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ketcham JM; Shin I; Montgomery TP; Krische MJ Catalytic Enantioselective C-H Functionalization of Alcohols by Redox-Triggered Carbonyl Addition: Borrowing Hydrogen, Returning Carbon. Angew. Chem. Int. Ed 2014, 53, 9142–9150. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Perez F; Oda S; Geary LM; Krische MJ Ruthenium-Catalyzed Transfer Hydrogenation for C-C Bond Formation: Hydrohydroxyalkylation and Hydroaminoalkylation via Reactant Redox Pairs. Top. Curr. Chem 2016, 374, 365–387. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Nguyen KD; Park BY; Luong T; Sato H; Garza VJ; Krische MJ Metal-Catalyzed Reductive Coupling of Olefin-Derived Nucleophiles: Reinventing Carbonyl Addition. Science 2016, 354, aah5133. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Kim SW; Zhang W; Krische MJ Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res 2017, 50, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Santana CG; Krische MJ From Hydrogenation to Transfer Hydrogenation to Hydrogen Auto-Transfer in Enantioselective Metal-Catalyzed Carbonyl Reductive Coupling: Past, Present and Future. ACS Catal 2021, 11, 5572–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Ortiz E; Shezaf JZ; Shen W; Krische MJ Historical Perspective on Ruthenium-Catalyzed Hydrogen Transfer and Survey of Enantioselective Hydrogen Auto-Transfer Processes for the Conversion of Lower Alcohols to Higher Alcohols. Chem. Sci 2022, 13, 12625–12633. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Meyer CC; Krische MJ Iridium-, Ruthenium- and Nickel-Catalyzed C-C Couplings of Methanol, Formaldehyde and Ethanol with π-Unsaturated Pronucleophiles via Hydrogen Transfer. J. Org. Chem 2023, 88, 4965–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Ortiz E; Saludares C; Wu J; Cho Y; Santana CG; Krische MJ Carbonyl Allylation and Crotylation: Historical Perspective, Relevance to Polyketide Synthesis and Evolution of Enantioselective Ruthenium-Catalyzed Hydrogen Auto-Transfer Processes. Synthesis 2023, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For selected reviews on “borrowing hydrogen” processes for hydroxyl substitution, see: (a) Hamid MHSA; Slatford PA; Williams JMJ Borrowing Hydrogen in The Activation of Alcohols. Adv. Synth. Catal 2007, 349, 1555–1575. [Google Scholar]; (b) Guillena G; Ramón DJ; Yus M Alcohols as Electrophiles in C-C Bond-Forming Reactions: The Hydrogen Autotransfer Process. Angew. Chem. Int. Ed 2007, 46, 2358–2364. [DOI] [PubMed] [Google Scholar]; (c) Dobereiner GE; Crabtree RH Dehydrogenation as a Substrate-Activating Strategy in Homogeneous Transition Metal Catalysis. Chem. Rev 2010, 110, 681–703. [DOI] [PubMed] [Google Scholar]; (d) Bähn S; Imm S; Neubert L; Zhang M; Neumann H; Beller M The Catalytic Amination of Alcohols. ChemCatChem 2011, 3, 1853–1864. [Google Scholar]; (e) Yang Q; Wang Q; Yu Z Substitution of Alcohols by N-Nucleophiles via Transition Metal-Catalyzed Dehydrogenation. Chem. Soc. Rev 2015, 44, 2305–2329. [DOI] [PubMed] [Google Scholar]; (f) Aitchison H; Wingad RL; Wass DF Homogeneous Ethanol to Butanol Catalysis - Guerbet Renewed. ACS Catal 2016, 6, 7125–7132. [Google Scholar]; (g) Quintard A; Rodriguez J Catalytic Enantioselective OFF ↔ ON Activation Processes Initiated by Hydrogen Transfer: Concepts and Challenges. Chem. Commun 2016, 52, 10456–10473. [DOI] [PubMed] [Google Scholar]; (h) Reed-Berendt BG; Polidano K; Morrill LC Recent Advances in Homogeneous Borrowing Hydrogen Catalysis Using Earth-Abundant First Row Transition Metals. Org. Biomol. Chem 2019, 17, 1595–1607. [DOI] [PubMed] [Google Scholar]; (i) Kwok T; Hoff O; Armstrong RJ; Donohoe TJ Control of Absolute Stereochemistry in Transition-Metal-Catalysed Hydrogen-Borrowing Reactions. Chem. Eur. J 2020, 26, 12912–12926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For selected reviews on enantioselective catalysis via chiral stereogenic-at-metal complexes, see: (a) Knight PD; Scott P Predetermination of Chirality at Octahedral Centres with Tetradentate Ligands: Prospects for Enantioselective Catalysis. Coord. Chem. Rev 2003, 242, 125–143. [Google Scholar]; (b) Bauer EB Chiral-at-Metal Complexes and Their Catalytic Applications in Organic Synthesis. Chem. Soc. Rev 2012, 41, 3153–3167. [DOI] [PubMed] [Google Scholar]; (c) Gong L; Chen L-A; Meggers E Asymmetric Catalysis Mediated by the Ligand Sphere of Octahedral Chiral-at-Metal Complexes. Angew. Chem. Int. Ed 2014, 53, 10868–10874. [DOI] [PubMed] [Google Scholar]; (d) Steinlandt PS; Zhang L; Meggers E Metal Stereogenicity in Asymmetric Transition Metal Catalysis. Chem. Rev 2023, 123, 4764–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Miessler GL; Fischer PJ; Tarr DA Inorganic Chemistry, 5th ed.; Pearson: Upper Saddle River, NJ, 2014; pp 326. [Google Scholar]
- (13).Blaser H-U; Brieden W; Pugin B; Spindler F; Studer M; Togni A Solvias Josiphos Ligands: From Discovery to Technical Applications. Top. Catal 2002, 19, 3–16. [Google Scholar]
- (14).Saito T; Yokozawa T; Ishizaki T; Moroi T; Sayo N; Miura T; Kumobayashi H New Chiral Diphosphine Ligands Designed to have a Narrow Dihedral Angle in the Biaryl Backbone. Adv. Synth. Catal 2001, 343, 264–267. [Google Scholar]
- (15).(a) Kim IS; Ngai M-Y; Krische MJ Enantioselective Iridium Catalyzed Carbonyl Allylation from the Alcohol or Aldehyde Oxidation Level Using Allyl Acetate as an Allyl Metal Surrogate. J. Am. Chem. Soc 2008, 130, 6340–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim IS; Ngai M-Y; Krische MJ Enantioselective Iridium Catalyzed Carbonyl Allylation from the Alcohol or Aldehyde Oxidation Level via Transfer Hydrogenative Coupling of Allyl Acetate: Departure from Chirally Modified Allyl Metal Reagents in Carbonyl Addition. J. Am. Chem. Soc 2008, 130, 14891–14899. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu Y; Kim IS; Hassan A; Del Valle DJ; Krische MJ 1,n-Glycols as Dialdehyde Equivalents in Iridium Catalyzed Enantioselective Carbonyl Allylation and Iterative Two-Directional Assembly of 1,3-Polyols. Angew. Chem. Int. Ed 2009, 48, 5018–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hassan A; Lu Y; Krische MJ Elongation of 1,3-Polyols via Iterative Catalyst-Directed Carbonyl Allylation from the Alcohol Oxidation Level. Org. Lett 2009, 11, 3112–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Schmitt DC; Dechert-Schmitt A-MR; Krische MJ Iridium-Catalyzed Allylation of Chiral β-Stereogenic Alcohols: Bypassing Discrete Formation of Epimerizable Aldehydes. Org. Lett 2012, 14, 6302–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dechert-Schmitt A-MR; Schmitt DC; Krische MJ Protecting-Group-Free Diastereoselective C-C Coupling of 1,3-Glycols and Allyl Acetate through Site-Selective Primary Alcohol Dehydrogenation. Angew. Chem. Int. Ed 2013, 52, 3195–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Shin I; Wang G; Krische MJ Catalyst-Directed Diastereo- and Site-Selectivity in Successive Nucleophilic and Electrophilic Allylations of Chiral 1,3-Diols: Protecting-Group-Free Synthesis of Substituted Pyrans. Chem. Eur. J 2014, 20, 13382–13389. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Kim SW; Lee W; Krische MJ Asymmetric Allylation of Glycidols Mediated by Allyl Acetate via Iridium-Catalyzed Hydrogen Transfer. Org. Lett 2017, 19, 1252–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Hassan A; Townsend IA; Krische MJ Catalytic Enantioselective Grignard Nozaki-Hiyama Methallylation from the Alcohol Oxidation Level: Chloride Compensates for π-Complex Instability. Chem. Comm 2011, 47, 10028–10030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Hassan A; Montgomery TP; Krische MJ Consecutive Iridium Catalyzed C-C and C-H Bond Forming Hydrogenations for the Diastereo- and Enantioselective Synthesis of syn-3-Fluoro-1-Alcohols: C-H (2-Fluoro)allylation of Primary Alcohols. Chem. Comm 2012, 48, 4692–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Montgomery TP; Hassan A; Park BY; Krische MJ Enantioselective Conversion of Primary Alcohols to α-exo-Methylene Butyrolactones via Iridium Catalyzed C-C Bond Forming Transfer Hydrogenation: 2-(Alkoxycarbonyl)allylation. J. Am. Chem. Soc 2012, 134, 11100–11103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Kim IS; Han SB; Krische MJ anti-Diastereo- and Enantioselective Carbonyl Crotylation from the Alcohol or Aldehyde Oxidation Level Employing a Cyclometallated Iridium Catalyst: α-Methyl Allyl Acetate as a Surrogate to Preformed Crotylmetal Reagents. J. Am. Chem. Soc 2009, 131, 2514–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Gao X; Townsend IA; Krische MJ Enhanced anti-Diastereo- and Enantioselectivity in Alcohol-Mediated Carbonyl Crotylation Using an Isolable Single Component Iridium Catalyst. J. Org. Chem 2011, 76, 2350–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Gao X; Han H; Krische MJ Direct Generation of Acyclic Polypropionate Stereopolyads via Double Diastereo- and Enantioselective Iridium-Catalyzed Crotylation of 1,3-Diols: Beyond Stepwise Carbonyl Addition in Polyketide Construction. J. Am. Chem. Soc 2011, 133, 12795–12800. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Gao X; Zhang YJ; Krische MJ Iridium-Catalyzed anti-Diastereo- and Enantioselective Carbonyl (α-Trifluoromethyl)allylation from the Alcohol or Aldehyde Oxidation Level. Angew. Chem. Int. Ed 2011, 50, 4173–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Han SB; Gao X; Krische MJ Iridium-Catalyzed anti-Diastereo- and Enantioselective Carbonyl (Trimethylsilyl)allylation from the Alcohol or Aldehyde Oxidation Level. J. Am. Chem. Soc 2010, 132, 9153–9156. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Han SB; Han H; Krische MJ Diastereo- and Enantioselective anti-Alkoxyallylation Employing Allylic gem-Dicarboxylates as Ally Donors via Iridium Catalyzed Transfer Hydrogenation. J. Am. Chem. Soc 2010, 132, 1760–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Tsutsumi R; Hong S; Krische MJ Diastereo- and Enantioselective Iridium Catalyzed Carbonyl (α-Cyclopropyl)allylation via Transfer Hydrogenation. Chem. Eur. J 2015, 21, 12903–12907. [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Xiang M; Luo G; Wang Y; Krische MJ Enantioselective Iridium-Catalyzed Carbonyl Isoprenylation via Alcohol-Mediated Hydrogen Transfer. Chem. Comm 2019, 55, 981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]; (t) Luo G; Xiang M; Krische MJ Successive Nucleophilic and Electrophilic Allylation for The Catalytic Enantioselective Synthesis of 2,4-Disubstituted Pyrrolidines. Org. Lett 2019, 21, 2493–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]; (u) Zhang YJ; Yang JH; Kim SH; Krische MJ anti-Diastereo- and Enantioselective Carbonyl (Hydroxymethyl)allylation from The Alcohol or Aldehyde Oxidation Level: Allyl Carbonates as Allylmetal Surrogates. J. Am. Chem. Soc 2010, 132, 4562–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]; (v) Feng J; Garza VJ; Krische MJ Redox-Triggered C-C Coupling of Alcohols and Vinyl Epoxides: Diastereo- and Enantioselective Formation of All-Carbon Quaternary Centers via tert-(Hydroxy)-Prenylation. J. Am. Chem. Soc 2014, 136, 8911–8914. [DOI] [PMC free article] [PubMed] [Google Scholar]; (w) Wang G; Franke J; Ngo CQ; Krische MJ Diastereo- and Enantioselective Iridium Catalyzed Coupling of Vinyl Aziridines and Alcohols: Site-Selective Modification of Unprotected Diols and Synthesis of Substituted Piperidines. J. Am. Chem. Soc 2015, 137, 7915–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]; (x) Moran J; Smith AG; Carris RM; Johnson JS; Krische MJ Polarity Inversion of Donor–Acceptor Cyclopropanes: Disubstituted δ-Lactones via Enantioselective Iridium Catalysis. J. Am. Chem. Soc 2011, 133, 18618–18621. [DOI] [PMC free article] [PubMed] [Google Scholar]; (y) Enantioselective Iridium Catalyzed Vinylogous Reformatsky-Aldol Reaction from the Alcohol Oxidation Level: Linear Regioselectivity by Way of Carbon-Bound Enolates. Angew. Chem. Int. Ed 2011, 50, 3493–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]; (z) Garza VJ; Krische MJ Hydroxymethylation beyond Carbonylation: Enantioselective Iridium Catalyzed Reductive Coupling of Formaldehyde with Allylic Acetates via Enantiotopic π-Facial Discrimination. J. Am. Chem. Soc 2016, 138, 3655–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]; (a′) Cabrera JM; Tauber J; Zhang W; Xiang M; Krische MJ Selection between Diastereomeric Kinetic vs Thermodynamic Carbonyl Binding Modes Enables Enantioselective Iridium-Catalyzed anti-(α-Aryl)allylation of Aqueous Fluoral Hydrate and Difluoroacetaldehyde Ethyl Hemiacetal. J. Am. Chem. Soc 2018, 140, 9392–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b′) Meyer CC; Stafford NP; Cheng MJ; Krische MJ Ethanol: Unlocking an Abundant Renewable C2-Feedstock for Catalytic Enantioselective C-C Coupling. Angew. Chem. Int. Ed 2021, 60, 10542–10546. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c′) Meyer CC; Verboom KL; Evarts MM; Jung W-O; Krische MJ Allyl Alcohol as an Acrolein Equivalent in Enantioselective C–C Coupling: Total Synthesis of Amphidinolides R, J, and S. J. Am. Chem. Soc 2023, 145, 8242–8247. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d′) Verboom KL; Meyer CC; Evarts MM; Jung W-O; Krische MJ O-Acetyl 1,3-Propanediol as an Acrolein Proelectrophile in Enantioselective Iridium-Catalyzed Carbonyl Allylation. Org. Lett 2023, 25, 3659–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).For selected reviews on catalytic enantioselective carbonyl allylation, see: (a) Denmark SE; Fu J Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones. Chem. Rev 2003, 103, 2763–2794. [DOI] [PubMed] [Google Scholar]; (b) Hall DG Lewis and Brønsted Acid Catalyzed Allylboration of Carbonyl Compounds: From Discovery to Mechanism and Applications. Synlett 2007, 1644–1655. [Google Scholar]; (c) Yus M; González-Gómez JC; Foubelo F Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev 2011, 111, 7774–7854. [DOI] [PubMed] [Google Scholar]; (d) Huo H-X; Duvall JR; Huang M-Y; Hong R Catalytic Asymmetric Allylation of Carbonyl Compounds and Imines with Allylic Boronates. Org. Chem. Front 2014, 1, 303–320. [Google Scholar]; (e) Spielmann K; Niel G; de Figueiredo RM; Campagne J-M Catalytic Nucleophilic ‘Umpoled’ π-Allyl Reagents. Chem. Soc. Rev 2018, 47, 1159–1173. [DOI] [PubMed] [Google Scholar]
- (17).Kim SW; Meyer CC; Mai BK; Liu P; Krische MJ Inversion of Enantioselectivity in Allene Gas versus Allyl Acetate Reductive Aldehyde Allylation Guided by Metal-Centered Stereogenicity: An Experimental and Computational Study. ACS Catal 2019, 9, 9158–9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kristjánsdóttir SS; Norton JR Acidity of Hydrido Transition Metal Complexes in Solution. In Transition Metal Hydrides; Dedieu A, Ed.; VCH: New York, 1992; pp 309–359. [Google Scholar]
- (19).Zimmerman HE; Traxler MD The Stereochemistry of the Ivanov and Reformatsky Reactions. I. J. Am. Chem. Soc 1957, 79, 1920–1923. [Google Scholar]
- (20).(a) Nguyen KD; Herkommer D; Krische MJ Enantioselective Formation of All-Carbon Quaternary Centers via C−H Functionalization of Methanol: Iridium-Catalyzed Diene Hydrohydroxymethylation. J. Am. Chem. Soc 2016, 138, 14210–14213. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Holmes M; Nguyen KD; Schwartz LA; Luong T; Krische MK Enantioselective Formation of CF3‑Bearing All-Carbon QuaternaryStereocenters via C−H Functionalization of Methanol: Iridium Catalyzed Allene Hydrohydroxymethylation. J. Am. Chem. Soc 2017, 139, 8114–8117. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schwartz LA; Holmes M; Brito GA; Gonçalves TP; Richardson J; Ruble JC; Huang K-W; Krische MJ Cyclometalated Iridium−PhanePhos Complexes Are Active Catalysts in Enantioselective Allene−Fluoral Reductive Coupling and Related Alcohol-Mediated Carbonyl Additions That Form Acyclic Quaternary Carbon Stereocenters. J. Am. Chem. Soc 2019, 141, 2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Meyer CC; Dubey ZJ; Krische MJ Enantioselective Iridium-Catalyzed Reductive Coupling of Dienes with Oxetanones and N-Acyl-Azetidinones Mediated by 2-Propanol. Angew. Chem. Int. Ed 2022, 61, No. e202115959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).For ruthenium-catalyzed reductive (or redox-neutral) couplings of dienes or allenes with aldehydes (or alcohols) to form racemic homoallylic alcohols, see: (a) Shibahara F; Bower JF; Krische MJ Ruthenium Catalyzed C-C Bond Forming Transfer Hydrogenation: Carbonyl Allylation from the Alcohol or Aldehyde Oxidation Level Employing Acyclic 1,3-Dienes as Surrogates to Preformed Allyl Metal Reagents. J. Am. Chem. Soc 2008, 130, 6338–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ngai M-Y; Skucas E; Krische MJ Ruthenium Catalyzed C-C Bond Formation via Transfer Hydrogenation: Branch-Selective Reductive Coupling of Allenes to Paraformaldehyde and Higher Aldehydes. Org. Lett 2008, 10, 2705–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Skucas E; Zbieg JR; Krische MJ anti-Aminoallylation of Aldehydes via Ruthenium Catalyzed Transfer Hydrogenative Coupling of Sulfonamido-Allenes: 1,2-Aminoalcohols. J. Am. Chem. Soc 2009, 131, 5054–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smejkal T; Han H; Breit B; Krische MJ All Carbon Quaternary Centers via Ruthenium Catalyzed Hydroxymethylation of 2-Substituted Butadienes Mediated by Formaldehyde: Beyond Hydroformylation. J. Am. Chem. Soc 2009, 131, 10366–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zbieg JR; McInturff EL; Krische MJ Allenamide Hydro-Hydroxyalkylation: 1,2-Aminoalcohols via Ruthenium Catalyzed Carbonyl anti-Aminoallylation. Org. Lett 2010, 12, 2514–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Han H; Krische MJ Direct Ruthenium Catalyzed C-C Coupling of Ethanol: Diene Hydro-Hydroxyethylation to Form All Carbon Quaternary Centers. Org. Lett 2010, 12, 2844–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zbieg JR; McInturff EL; Leung JC; Krische MJ Amplification of anti-Diastereoselectivity via Curtin-Hammett Effects in Ruthenium Catalyzed Hydrohydroxyalkylation of 1,1-Disubstituted Allenes: Diastereoselective Formation of All-Carbon Quaternary Centers. J. Am. Chem. Soc 2011, 133, 1141–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Leung JC; Geary LM; Chen T-Y; Zbieg JR; Krische MJ Direct, Redox Neutral Prenylation and Geranylation of Secondary Carbinol C-H Bonds: C4 Regioselectivity in Ruthenium Catalyzed C-C Couplings of Dienes to α-Hydroxy Esters. J. Am. Chem. Soc 2012, 134, 15700–15703. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Köpfer A; Sam B; Breit B; Krische MJ Regiodivergent Reductive Coupling of 2-Substituted Dienes to Formaldehyde Employing Ruthenium or Nickel Catalysts: Hydrohydroxymethylation via Transfer Hydrogenation. Chem. Sci 2013, 4, 1876–1880. [Google Scholar]; (j) Chen T-Y; Krische MJ Regioselective Ruthenium Catalyzed Hydrohydroxyalkylation of Dienes with 3-Hydroxy-2-Oxindoles: Prenylation, Geranylation and Beyond. Org. Lett 2013, 15, 2994–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Park BY; Montgomery TP; Garza VJ; Krische MJ Ruthenium Catalyzed Hydrohydroxyalkylation of Isoprene Employing Heteroaromatic Secondary Alcohols: Isolation and Reversible Formation of the Putative Metallacycle Intermediate. J. Am. Chem. Soc 2013, 135, 16320–16323. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Sam B; Luong T; Krische MJ Ruthenium-Catalyzed C-C Coupling of Fluorinated Alcohols with Allenes: Dehydrogenation at the Energetic Limit of β-Hydride Elimination. Angew. Chem. Int. Ed 2015, 54, 5465–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Zbieg JR; Yamaguchi E; McInturff EL; Krische MJ Enantioselective C-H Crotylation of Primary Alcohols via Hydrohydroxyalkylation of Butadiene. Science 2012, 336, 324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McInturff EL; Yamaguchi E; Krische MJ Chiral Anion Dependent Inversion of Diastereo- and Enantioselectivity in Carbonyl Crotylation via Ruthenium Catalyzed Butadiene Hydrohydroxyalkylation. J. Am. Chem. Soc 2012, 134, 20628–20631. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Grayson MN; Krische MJ; Houk KN Ruthenium-Catalyzed Asymmetric Hydrohydroxyalkylation of Butadiene: The Role of the Formyl Hydrogen Bond in Stereochemical Control. J. Am. Chem. Soc 2015, 137, 8838–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).For enantioselective ruthenium-JOSIPHOS-catalyzed C-C couplings of aldehydes from alcohols proelectrophiles, see: (a) Liang T; Nguyen KD; Zhang W; Krische MJ Enantioselective Ruthenium Catalyzed Carbonyl Allylation via Alkyne-Alcohol C-C Bond Forming Transfer Hydrogenation: Allene Hydrometallation vs. Oxidative Coupling. J. Am. Chem. Soc 2015, 137, 3161–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liang T; Zhang W; Chen T-Y; Nguyen KD; Krische MJ Ruthenium Catalyzed Diastereo- and Enantioselective Coupling of Propargyl Ethers with Alcohols: Siloxy-Crotylation via Hydride Shift Enabled Conversion of Alkynes to π-Allyls. J. Am. Chem. Soc 2015, 137, 13066–13071. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xiang M; Ghosh A; Krische MJ Diastereo- and Enantioselective Ruthenium-Catalyzed C-C Coupling of 1-Arylpropynes and Alcohols: Alkynes as Chiral Allylmetal Precursors in Carbonyl anti-(α-Aryl)allylation. J. Am. Chem. Soc 2021, 143, 2838–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ortiz E; Shezaf JZ; Chang Y-H; Gonçalves TP; Huang K-W; Krische MJ Understanding Halide Counterion Effects in Enantioselective Ruthenium-Catalyzed Carbonyl (α-Aryl)allylation: Alkynes as Latent Allenes and Trifluoroethanol-Enhanced Turnover in The Conversion of Ethanol to Higher Alcohols via Hydrogen Auto-Transfer. J. Am. Chem. Soc 2021, 143, 16709–16717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).For selected reviews on halide counterion effects in metal catalysis, see: (a) Maitlis PM; Haynes A; James BR; Catellani M; Chiusoli GP Iodide Effects in Transition Metal Catalyzed Reactions. Dalton Trans 2004, 3409–3419. [DOI] [PubMed] [Google Scholar]; (b) Fagnou K; Lautens M Halide Effects in Transition Metal Catalysis. Angew. Chem. Int. Ed 2002, 41, 26–47. [DOI] [PubMed] [Google Scholar]
- (25).For a review, see: Haydl AM; Breit B; Liang T; Krische MJ Alkynes as Electrophilic or Nucleophilic Allylmetal Precursors in Transition Metal Catalysis. Angew. Chem. Int. Ed 2017, 56, 11312–11325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lu T; Chen F Bond Order Analysis Based on the Laplacian of Electron Density in Fuzzy Overlap Space. J. Phys. Chem. A 2013, 117, 3100–3108. [DOI] [PubMed] [Google Scholar]
- (27).For selected studies of formyl CH hydrogen bonding, see reference 22c and the following: (a) Corey EJ; Lee TW. The Formyl C–H···O Hydrogen Bond as a Critical Factor in Enantioselective Lewis-Acid Catalyzed Reactions of Aldehydes. Chem. Commun 2001, 1321–1329. [Google Scholar]; (b) Thakur TS; Kirchner MT; Bläser D; Boese R; Desiraju GR Nature and Strength of C–H⋯O Interactions Involving Formyl Hydrogen Atoms: Computational and Experimental Studies of Small Aldehydes. Phys. Chem. Chem. Phys 2011, 13, 14076–14091. [DOI] [PubMed] [Google Scholar]
- (28).(a) Ortiz E; Spinello BJ; Cho Y; Wu J; Krische MJ Stereo- and Site-Selective Crotylation of Alcohol Proelectrophiles via Ruthenium-Catalyzed Hydrogen Auto-Transfer Mediated by Methylallene and Butadiene. Angew. Chem. Int. Ed 2022, 61, e202212814. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saludares C; Ortiz E; Santana CG; Spinello BJ Krische, M. J. Asymmetric Ruthenium-Catalyzed Carbonyl Allylations by Gaseous Allene via Hydrogen Auto-Transfer: 1º vs 2º Alcohol Dehydrogenation for Streamlined Polyketide Construction. ACS Catal 2023, 13, 1662–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dubey ZJ; Shen W; Little JA; Krische MJ Dual Ruthenium-Catalyzed Alkene Isomerization-Hydrogen Auto-Transfer Unlocks Skipped Dienes as Pronucleophiles for Enantioselective Carbonyl Allylation. J. Am. Chem. Soc 2023, 145, 8576–8582. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ortiz E; Chang Y-H; Shezaf JZ; Shen W; Krische MJ Stereo- and Site-Selective Conversion of Primary Alcohols to Allylic Alcohols via Ruthenium-Catalyzed Hydrogen Auto-Transfer Mediated by 2-Butyne. J. Am. Chem. Soc 2022, 144, 8861–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).For selected reviews encompassing enantioselective carbonyl vinylation, see: (a) Hargaden GC; Guiry PJ The Development of the Asymmetric Nozaki−Hiyama−Kishi Reaction. Adv. Synth. Catal 2007, 349, 2407–2424. [Google Scholar]; (b) Lumbroso A; Cooke ML; Breit B Catalytic Asymmetric Synthesis of Allylic Alcohols and Derivatives and their Applications in Organic Synthesis. Angew. Chem. Int. Ed 2013, 52, 1890–1932. [DOI] [PubMed] [Google Scholar]; (c) Bauer T Enantioselective Dialkylzinc-Mediated Alkynylation, Arylation and Alkenylation of Carbonyl Groups. Coord. Chem. Rev 2015, 299, 83–150. [Google Scholar]; (d) Tian Q; Zhang G Recent Advances in the Asymmetric Nozaki−Hiyama−Kishi Reaction. Synthesis 2016, 48, 4038–4049. [Google Scholar]; (e) Gil A; Albericio F; Álvarez, M. Role of the Nozaki−Hiyama−Takai−Kishi Reaction in the Synthesis of Natural Products. Chem. Rev 2017, 117, 8420–8446. [DOI] [PubMed] [Google Scholar]; (f) Ortiz E; Shezaf JZ; Chang Y-H; Krische MJ Enantioselective Metal-Catalyzed Reductive Coupling of Alkynes with Carbonyl Compounds and Imines: Convergent Construction of Allylic Alcohols and Amines. ACS Catal 2022, 12, 8164–8174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shezaf JZ; Santana CG; Saludares C; Briceno ES; Sakata K; Krische MJ Chiral-at-Ruthenium-SEGPHOS Catalysts Display Diastereomer-Dependent Regioselectivity: Enantioselective Isoprene-Mediated Carbonyl tert-Prenylation via Halide Counterion Effects. J. Am. Chem. Soc 2023, 145, 18676–18683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).For selected reviews highlighting regiodivergence in metal-catalysis, see: (a) Funken N; Zhang Y-Q; Gansäuer A Regiodivergent Catalysis: A Powerful Tool for Selective Catalysis. Chem. Eur. J 2017, 23, 19–32. [DOI] [PubMed] [Google Scholar]; (b) Najera C; Beletskaya IP; Yus M Metal-Catalyzed Regiodivergent Organic Reactions Chem. Soc. Rev 2019, 48, 4515–4618. [DOI] [PubMed] [Google Scholar]
- (32).Knowles WS; Sabacky MJ Catalytic Asymmetric Hydrogenation Employing a Soluble, Optically Active, Rhodium Complex Chem. Commun 1968, 1445–1446. [Google Scholar]
- (33).For single crystal x-ray diffraction data on chloride- and bromide-bound ruthenium carbonyl complexes, see: (a) Xue P; Bi S; Sung HHY; Williams ID; Lin Z; Jia G Isomerism of [Ru(η3-allyl)Cl(CO)(PPh3)2] Organometallics 2004, 23, 4735–4743. [Google Scholar]; (b) Wakatsuki Y; Yamazaki H; Maruyama Y; Shimizu I Novel Regioselection in Insertion of a 1,4-Disubstituted-1,3-Enyne into Ruthenium–Hydrogen Bonds. J. Chem. Soc. Chem. Comm 1991, 261–263. [Google Scholar]; (c) Santos A; López J; Montoya J; Noheda P; Romero A; Echavarren AM Synthesis of New Ruthenium(II) Carbonyl Hydrido, Alkenyl, and Alkynyl Complexes with Chelating Diphosphines. Organometallics 1994, 13, 3605–3615. [Google Scholar]; (d) Pingen D; Lebl T; Lutz M; Nichol GS; Kamer PCJ; Vogt D Catalytic Activity and Fluxional Behavior of Complexes Based on RuHCl(CO)(PPh3)3 and Xantphos-Type Ligands. Organometallics 2014, 33, 2798–2805. [Google Scholar]; (e) Kunihiro K; Heyte S; Paul S; Roisnel T; Carpentier J -F.; Kirillov, E. Ruthenium-Catalyzed Coupling Reactions of CO2 with C2H4 and Hydrosilanes towards Silyl Esters. Chem. Eur. J 2021, 27, 3997–4003. [DOI] [PubMed] [Google Scholar]; (f) Hill AF; McQueen CM A. Dihydroperimidine-Derived PNP Pincer Complexes as Intermediates en Route to N-Heterocyclic Carbene Pincer Complexes. Organometallics 2014, 33, 1909–1912. [Google Scholar]; (g) Jago D; Walkey MC; Gaschk EE; Spackman PR; Piggott MJ; Moggach SA; Koutsantonis GA Multistate Switching of Some Ruthenium Alkynyl and Vinyl Spiropyran Complexes. Inorg. Chem 2023, 62, 12283–12297. [DOI] [PubMed] [Google Scholar]; (h) Bartlett MJ; Frogley BJ; Hill AF; Sharma M; Smith MK; Ward JS Hydrogenating an Organometallic Carbon Chain: Buten-yn-diyl (CH=CHC≡C) as a Missing Link. Dalton Trans 2019, 48, 16534–16554. [DOI] [PubMed] [Google Scholar]