

Abstract
Flavin‐dependent monooxygenases (FMOs) constitute a diverse enzyme family that catalyzes crucial hydroxylation, epoxidation, and Baeyer–Villiger reactions across various metabolic pathways in all domains of life. Due to the intricate nature of this enzyme family's mechanisms, some aspects of their functioning remain unknown. Here, we present the results of molecular dynamics computations, supplemented by a bioinformatics analysis, that clarify the early stages of their catalytic cycle. We have elucidated the intricate binding mechanism of NADPH and L‐Orn to a class B monooxygenase, the ornithine hydroxylase from known as SidA. Our investigation involved a comprehensive characterization of the conformational changes associated with the FAD (Flavin Adenine Dinucleotide) cofactor, transitioning from the out to the in position. Furthermore, we explored the rotational dynamics of the nicotinamide ring of NADPH, shedding light on its role in facilitating FAD reduction, supported by experimental evidence. Finally, we also analyzed the extent of conservation of two Tyr‐loops that play critical roles in the process.
Keywords: flavin‐dependent monooxygenases, flavin dynamics, NADPH binding, NADPH dynamics, ornithine binding, uncoupling
1. INTRODUCTION
Flavin‐dependent monooxygenases (FMOs) are a large family of enzymes that catalyze hydroxylation, epoxidation, and Baeyer–Villiger reactions in primary and secondary metabolism in all kingdoms of life (Cashman, 1995; Mascotti et al., 2015; Mascotti et al., 2016; Van Berkel et al., 2006). The family has been divided into eight classes based on their fold and function (Huijbers et al., 2014; Mascotti et al., 2016). Structural, computational, and biochemical studies have provided significant insights into the mechanism of action of FMOs (Badieyan et al., 2015; Binda et al., 2015; Chocklett & Sobrado, 2010; Franceschini et al., 2012; Hissen et al., 2005; Romero, Fedkenheuer, et al., 2012; Sucharitakul et al., 2014). The overall reaction can be divided into reductive and oxidative half‐reactions. In the reductive half, the flavin cofactor is reduced by the co‐substrate, NAD(P)H. In the oxidative half, the reduced flavin reacts with molecular oxygen forming the C4a‐(hydro)peroxyflavin, which is the hydroxylating intermediate (Gatti et al., 1994; Reis et al., 2021). A crucial feature in the oxidative half‐reaction is that activation of molecular oxygen needs to be highly regulated such that the intended product is formed instead of hydrogen peroxide, which uncouples the reaction (e.g., no product formation per NAD(P)H oxidation).
Multiple mechanisms have been identified within the FMO classes to ensure effective coupling of the reaction. In class A FMOs, which possess a single Rossmann fold for flavin binding, reduction by NAD(P)H requires the presence of the substrate in the active site. Thus, after the reduced flavin reacts with molecular oxygen to produce the C4a‐hydroperoxyflavin, the subsequent reaction with the substrate is assured (Palfey & McDonald, 2010; Van Berkel et al., 2006). This mechanism has been shown to be facilitated by flavin motion (Palfey & McDonald, 2010). In the resting state, the flavin is in the active site (the in conformation), secluded from the solvent, where it cannot be reached by NAD(P)H. Upon substrate binding, conformational changes place the flavin closer to the protein surface (the out conformation), where it can more effectively react with NAD(P)H. This conformational change produces a 105‐fold enhancement in the reduction rate with respect to the in conformation (Gatti et al., 1994; Schreuder et al., 1989; Van Berkel et al., 1994). This mechanism has been described as the “cautious mechanism” because the enzyme is only active when the substrate is present (Palfey & McDonald, 2010; Van Berkel et al., 2006).
Within class B FMO members, it has also been established that flavin exists in either the out or in conformations (Campbell, Stiers, et al., 2020; Setser et al., 2014). However, despite this similarity with class A FMOs, they exhibit a distinct strategy to prevent uncoupling. In particular, class B FMOs do not necessitate substrate binding to achieve an effective reaction with NAD(P)H. Instead, after the reaction with molecular oxygen, the C4a‐hydroperoxyflavin is stabilized until the substrate is present and the catalytic cycle continues. This “bold mechanism” requires NAD(P)+ to remain bound in the active site. The role of NAD(P)H in this bold mechanism has been the focus of several studies (Chocklett & Sobrado, 2010; Mayfield et al., 2010; Romero, Fedkenheuer, et al., 2012). However, key features of NAD(P)H's reaction with oxidized flavin remain elusive. Several biochemical studies have shown that the hydride transfer step occurs with pro‐R stereoselectivity (Beaty & Ballou, 1981; Robinson, Rodriguez, & Sobrado, 2014; Romero, Robinson, & Sobrado, 2012; Torres Pazmiño et al., 2008; Van Berkel et al., 2006). However, all the structures solved with NADP+ bound into the active site suggest a pro‐S stereoselectivity (Campbell, Stiers, et al., 2020; Eswaramoorthy et al., 2006; Franceschini et al., 2012; Malito et al., 2004).
In this article, we report a computational study on the class B FMO Siderophore A (SidA) from Aspergillus fumigatus. We selected this enzyme because of the extensive array of mechanistic and structural examinations conducted on it (Campbell, Stiers, et al., 2020; Chocklett & Sobrado, 2010; Hissen et al., 2005; Martin del Campo et al., 2016; Robinson, Franceschini, et al., 2014). We focused on the binding mechanism of its substrate, L‐Ornithine (L‐Orn), and its co‐substrate, NADPH. In addition, we explored the FAD transition from the out () to the in () conformations, and an unprecedented nicotinamide ring flip that solves the pro‐R/pro‐S stereo‐selectivity conundrum observed in this class of FMOs. Analysis of these steps reveals that two Tyrosines, Tyr324 and Tyr407, actively participate in the catalytic mechanism. Accordingly, we conducted a bioinformatics investigation to assess their relevance within the broader FMO family. The results strongly indicate that short sequences containing both residues play a pivotal role in the function of the entire group of enzymes. We believe these new pieces of information address essential unanswered questions about the catalytic mechanism of class B FMOs, which could have implications in biotechnology and drug design.
2. METHODS
2.1. General procedures
In this section, we describe the methodology used to run the molecular dynamics (MD) simulations and analyze their results. First, we explain how we built the computational models of SidA in alternative states and then we provide the protocols we followed to run standard and umbrella sampling (US) simulations. Figure 1 depicts all the events studied with US. The computational models appearing in the figure, except those enclosed in a rectangle, were prepared from structures taken from the Protein Data Bank (PDB). Finally, we present the procedures we followed to investigate the conservation among FMOs of critical residues spotted by the simulations.
FIGURE 1.

Scheme of the putative events occurring at the initial stages in the catalytic mechanism of SidA. The computational models appearing in the figure, except for those enclosed in a rectangle, were prepared from PDB files as explained in the main text. All the events indicated with a green arrow were studied via US simulations. Those highlighted with a thick green line correspond to the most probable pathway according to the results of this work.
2.2. Model construction and initial settings
The computational models of the enzyme in alternative states were built from the crystal structures of SidA recently determined by Campbell, Stiers, et al. (2020). For the resting form of the enzyme, which contains FAD in the out conformation and no ligands in the binding pocket, we used structure 6X0H from PDB. This model will be denoted as SidA(). This structure contains an acetate ion in the L‐Orn pocket that was removed to build model SidA() and was replaced by L‐Orn to create model SidA(, L‐Orn). We utilized the PDB structure 6X0I to generate a model of SidA with FAD in the in conformation and NADPH bound into the active site, SidA(, NADPH). A model of SidA with NADPH bound to the active site but FAD in the out conformation was produced by combining models SidA(, NADPH) and SidA(). First, we aligned SidA() with SidA(, NADPH). Then, we replaced the FAD molecule of SidA(, NADPH) with that of SidA(). By a similar procedure, but using models SidA(, L‐Orn) and SidA(, NADPH), we created model SidA(, NADPH, L‐Orn). The relative positions of FAD, NADPH, L‐Orn in both, SidA(, NADPH) and SidA(, NADPH, L‐Orn), are similar to those found in the corresponding structures of KtzI, with PDB codes 4TLZ and 4TM0. Also, the active site structure observed in SidA(, NADPH) agrees with that of the M101A SidA's mutant, which stabilizes FAD in the out conformation (PDB code 7JVL) (Campbell, Robinson, et al., 2020). Finally, we took structure 6X0J to build a model that contains NADPH and L‐Orn docked into the active site, with FAD in the in conformation. This model will be called SidA(, NADPH, L‐Orn). Figure 1 presents a sketch of the alternative steps in the mechanism of the reaction catalyzed by SidA. A drawing for each of the models described above is also provided, indicating their place in the mechanism. We remark that all the steps indicated in Figure 1 occur before FAD reduction by NADPH (i.e., they contain oxidized FAD). For future references, Table 1 presents the names of the computational models created for this work along with the IDs of the PDB structures used to build them.
TABLE 1.
Names of the computational models of Aspergillus fumigatus SidA created for this work.
| Model | PBD IDs |
|---|---|
| SidA() | 6X0H |
| SidA(, L‐Orn) | 6X0H |
| SidA(, NADPH) | 6X0H and 6X0I |
| SidA(, NADPH, L‐Orn) | 6X0H and 6X0J |
| SidA(, NADPH) | 6X0I |
| SidA(, NADPH, L‐Orn) | 6X0J |
Note: The subscripts indicates whether the cofactor is in the in or out conformation. In all cases, FAD is oxidized. The PDB codes of the structures utilized to build each model are also provided.
SidA is a homo‐tetramer. However, in the PDB files, the chains are not exactly the same. In general, all chains lack several residues, but they differ in the identity of the missing parts. Chain B of structure 6X0J presents the highest degree of completeness. It possesses only one gap of just four residues (Glu386‐Gly387‐Ala388‐Ala389). The lacking fragment is not present in any of the crystal structures of SidA, but the related enzyme KtzI (PDB 4TM0) contains an equivalent segment (Asp338‐Val339‐Leu340‐Val341). Accordingly, we first completed chain B of 6X0J with the corresponding fragment taken from 4TM0. Subsequently, we utilized the structure of this newly formed chain B as a template. Missing residues in chains A, C, and D were incorporated according to this template. All the simulations presented in this article were carried out with the tetrameric form of SidA.
The initial PDB structures were fed into the LEAP module of AMBER18 (Case et al., 2018). Water molecules were then introduced to fill an octahedral cell whose walls were 15 Å from the nearest protein atom. The models were neutralized by adding ions. The ff14SB force field (Maier et al., 2015) was used for the protein, the TIP3P for the water molecules (Joung & Cheatham, 2008) and the frcmod.ionsjc_tip3p parameters for the ions (Jorgensen et al., 1983). The PMEMD module of AMBER18 was used to run the simulations. After model building, all the systems were first minimized. Then, they were heated at constant volume for 2 ns to reach 303K using the Langevin thermostat with a collision frequency of . The SHAKE algorithm was used to constrain the bonds involving hydrogen atoms (Kräutler et al., 2001). The Particle Mesh Ewald method, with a cutoff radius of 10.0 Å, was implemented to evaluate the electrostatic interactions. Accordingly, these interactions were computed in the direct space for Å and otherwise in the reciprocal space (Darden et al., 1993; Essmann et al., 1995). This setting was based on efficiency considerations (Salomon‐Ferrer et al., 2013). A cutoff of 10.0 Å was also applied to the rest of the non‐bonded interactions. After the heating, we changed from NVT to NPT conditions and ran another 20 ns to allow density to relax and equilibrate. Pressure was controlled with a Berendsen barostat with a coupling constant of 2.0 ps. The time step was set to 2.0 fs in all the simulations described in this article.
2.3. Standard MD simulations
Standard MD simulations were performed to scrutinize the structural and dynamical characteristics of models SidA(), SidA(, NADPH), and SidA(, NADPH). In all cases, we used the last snapshot of the equilibration stage as the initial structure of the production runs. For each model, we performed 20 MD simulations of 50 ns, adding up a total time of for each of them. The initial velocities of these trajectories were randomly chosen from a Maxwellian distribution corresponding to T = 303K. Snapshots were collected every 0.25 ns.
2.4. Umbrella sampling
The US technique was employed to simulate the unbinding of NADPH from models SidA(, NADPH) and SidA(, NADPH, L‐Orn) and the unbinding of L‐Orn from models SidA(, L‐Orn), SidA(, NADPH, L‐Orn) and SidA(, NADPH, L‐Orn). We recall that the US simulations are carried out under quasi‐equilibrium conditions. Therefore, when they are analyzed in reverse order (as we did in this work), simulations of an unbinding process provide information about the corresponding binding event.
The US technique was also applied to simulate the rotation of the isoalloxazine moiety of FAD and the nicotinamide ring of NADPH. The rotation of the isoalloxazine group takes the FAD cofactor from the out to the in conformations. This movement was analyzed in models SidA(, NADPH) and SidA(, NADPH, L‐Orn). The rotation of the nicotinamide ring of NADPH was evaluated to see if it could produce conformations more appropriate for the H‐transfer between NADPH and FAD. This rotation was simulated in models SidA(, NADPH) and SidA(, NADPH, L‐Orn).
The reaction coordinate (RC) for the unbinding of NADPH was defined as the distance between two centers of mass (COMs). One of them is the COM of the heteroatoms of NADPH. The other is the COM of the backbone atoms of residues Val96, Ser99, Lys100, and Ser230. The position of this COM lies within the active site and remains almost invariant during the NADPH unbinding. The RC for the unbinding of L‐Orn was defined as the distance between the COM of the heteroatoms of L‐Orn and the COM of the backbone atoms of residues Ser104, Phe105, Asp288, Pro290, and Asn293.
The RCs employed to simulate the transition, and the rotation of the nicotinamide ring of NADPH, are torsional angles. The comparison between FAD conformations in the 6X0H and 6X0I crystal structures reveals that the out to in transition involves rotations around the C2 ‐C1
‐C1 and N10‐C1
and N10‐C1 bonds (Campbell, Stiers, et al., 2020). However, in trial calculations aimed to determine the best parameters for these simulations, we noticed that when enforcing the rotation of FAD around the N10‐C1
bonds (Campbell, Stiers, et al., 2020). However, in trial calculations aimed to determine the best parameters for these simulations, we noticed that when enforcing the rotation of FAD around the N10‐C1 bond, the dihedral angle around C2
bond, the dihedral angle around C2 ‐C1
‐C1 relaxes spontaneously. The opposite does not occur. Based on these observations, we defined the RC for this process as the C2
relaxes spontaneously. The opposite does not occur. Based on these observations, we defined the RC for this process as the C2 ‐C1
‐C1 ‐N10‐C10 dihedral angle. The RC used to analyze the rotation of the nicotinamide ring of NADPH was the C2D‐C1D‐N1N–C6N dihedral.
‐N10‐C10 dihedral angle. The RC used to analyze the rotation of the nicotinamide ring of NADPH was the C2D‐C1D‐N1N–C6N dihedral.
In all US simulations, the last structure of a given simulation was employed as the initial conformation of the next one. Each window was sampled during 10 ns. However, the first 5 ns were considered part of an equilibration period. Accordingly, only the last 5 ns of each window were included to compute the Potential of Mean Force (PMF). Snapshots were recorded every 150 ps. Other numerical details related to the sampling of each RC, the algorithms employed to compute the PMFs as well as the assessments of their consistency and statistical uncertainty, are provided in the Supporting Information section. Snapshots taken from the US simulations were used to create pictures and movies of the processes they represent. All movies and images were generated with visual molecular dynamics (Humphrey et al., 1996).
2.5. Bioinformatic analysis
After examining the results of the standard and US simulations, we decided to carry out a bioinformatic analysis to evaluate the extent of conservation of the Tyr324‐loop (Thr‐322‐Asn323‐Tyr324) and the Tyr407‐loop (Thr405‐Gly406‐Tyr407), among FMOs. We first analyzed the degree of conservation of these loops within class‐B FMOs, using the group of 24 enzymes described in Mascotti et al. (2015). However, as this number is somewhat limited, we decided to investigate the conservation in larger datasets. Consequently, we used BlastP and Jackhmmer to identify two distinct clusters of SidA homologous sequences. BlastP was executed on the Uniprot website employing UniprotKB as the target database and default settings except for the maximum number of hits (set to 1000). The results underwent subsequent processing through MMSeq2 on the MPI server (https://toolkit.tuebingen.mpg.de/), to select representative sequences according to the following parameters: Minimum sequence identity, 0.7; Minimum alignment coverage, 0.8 and Clustering mode, normalslow_sensitive. The final set contained 173 sequences. Jackhmmer, on the other side, was performed locally based on the Uniref50 database, downloaded from UniprotKB in October 2022. After three iterations, the results were filtered by E‐value (<0.0001) and query coverage (>70%). Then, we excluded sequences that either were not mapping in UniprotKB or possessed lengths below 368 or above 698. This final set included 437 sequences. The sequences derived from BlastP and Jackhmmer were subjected to a Multiple Sequence Alignment (MSA) utilizing Muscle (Gabler et al., 2020) with default parameters. The alignments were then visualized in the alignment viewer web (alignmentviewer.org).
Two additional analyses were exploited to examine the relevance of the Tyr‐loops. One of them employed the Consurf server (Yariv et al., 2023) to detect important functional sites of closest and distant SidA‐homologs. To this end, we employed chain B of SidA (PDB ID 6X0J). The parameters were adjusted as described below. Homologs were collected from UNIREF90 database using HMMER, with an E‐value cutoff of 0.0001 and three iterations (Jackhmmer). A maximum number of 150 final homologs were selected with CD‐HIT and a cutoff of 95%. The maximal overlap between homologs permitted was 10%. Homologs should cover at least 60% of the query and share a minimal sequence identity of 15%. For Alignment, Phylogeny and Conservation Scores parameters were unchanged. The default settings include alignment by MAFFT, phylogenetic tree built using Neighbor‐Joining with ML distance, conservation scores calculated with the Bayesian method and amino acid substitution model chosen by best fit. The last examination was an Evolutionary trace analysis (Lichtarge et al., 1996) with default parameters using 6X0JB from PDB as query in the Universal Evolutionary Trace server (http://evolution.lichtargelab.org/).
3. RESULTS AND DISCUSSION
In this section, we initially present the simulation results related to the binding process of the substrate (L‐Orn) and the co‐substrate (NADPH) to the resting form of the enzyme, analyzing the alternative orders these processes could have. Then, we show the results of the calculations focused on the FAD to transition, as well as the rotation of the nicotinamide ring of NADPH that generates a conformation appropriate for FAD reduction. All the assessments on the accuracy and consistency of these computations are presented in the Supporting Information section. Finally, we provide the outcome of the bioinformatics analysis that sheds light on the significance of the Tyr324 and Tyr407 loops on FMOs.
3.1. Binding of NADPH and L‐Orn to SidA()
We start the analysis by considering the apo form of the enzyme, SidA(). This stage can be followed by either the binding of NADPH or L‐Orn (events a b1 and a b2 of Figure 1, respectively). We present in Figure 2 the PMFs for both events. They reveal that NADPH is held much more tightly than L‐Orn, as expected. Thus, the bound state of L‐Orn is more labile and shorter in duration than that of NADPH. The free energy differences between the bound and unbound states of L‐Orn and NADPH observed in Figure 2 can be used as an approximation of their binding free energies. From that, we estimate that at 303K, the ratio between the probability of finding SidA(, L‐Orn) and that of SidA(, NADPH) is . Although this analysis does not consider the effect of the concentrations, it is clear that the probability of observing SidA(, L‐Orn) is completely negligible in comparison with that of SidA(, NADPH).
FIGURE 2.

PMF for the binding of NADPH and L‐Orn to the resting form of SidA (SidA()). The calculations we carried out to assess their consistency and the procedure we followed to compute the error bars are presented in the Supporting Information section.
More detailed information about these processes can be gained by analyzing the conformations and interactions of these molecules along the paths to their binding sites. Movies S1 and S2, available in the Supporting Information section, illustrate both processes while Figures 3 and 4 show typical structures of NADPH and L‐Orn, at different values of the RC. At the first stages of the NADPH binding, its phosphate groups make contact with the enzyme via a triad of positive residues: Arg364, Lys379, and Lys393. These amino acids are located in a three‐stranded ‐sheet sited at the border of the enzyme, away from the active site. Figure 3 panel (a) illustrates this situation, highlighting the SidA residues involved in this initial recognition. Subsequently, for 25.0 < RC < 35.0, NADPH loses the contacts with this initial triad, but establishes new contacts with another three residues: Lys361, Arg279, and Ser218. We recall that the involvement of Arg279 in facilitating the binding of NADPH has been experimentally verified (Robinson, Franceschini, et al., 2014). The adenine moiety of NADPH fits in a pocket sited between Arg279 and Ser218 (see Figure 3 panel b). Its location, except for minor aspects, remains unchanged until the end of the process. Later on, the nicotinamide portion of NADPH rotates using the adenine moiety as a pivot. While this happens, the interactions of the phosphate groups of NADPH with Lys361, Arg279, and Ser218 get weaker, but those with another group of positive residues, Lys215 and Lys320, gain strength. Once the nicotinamide ring of NADPH is close to FAD, the Tyr407‐loop moves away from the cofactor. Figure 3 panel (c) presents a comparison of the locations of the Tyr407‐loop, before and after this motion. It is interesting to note the similarities between the Tyr407‐loop and the much well characterized Tyr324‐loop. Both are part of larger flexible structures located close to FAD. They bear similar sequences (TSY and TGY) and present a high degree of conservation within SidA analogs (Campbell, Stiers, et al., 2020). The motion of the Tyr407‐loop away from FAD enables the entrance of the nicotinamide ring of NADPH which forms a sandwich interacting with the si‐face of FAD, on the one side, and the phenyl group of Tyr324, on the other side (see Figure 3 panel d). Once NADPH has reached its final position, Tyr324 moves apart from FAD. At the end of this movement, its phenyl ring lies almost perpendicular to the isoalloxazine moiety of (Figure 3 panel e). The conformation of the Tyr324‐loop, at this stage, is almost the same as that observed in structures 4TLZ and 4TM0 of KtzI, and in the M101A SidA's mutant (Campbell, Robinson, et al., 2020).
FIGURE 3.

Representative structures observed during the binding of NADPH to SidA(). NADPH is highlighted in yellow. Structures were selected by visual inspection. The values of the RC at which the snapshots were taken are shown below each panel. Residues that bear significant interactions with NADPH are represented in . Panel (a): NADPH interacts via its phosphate groups with positive residues (Arg364, Lys379, and Lys393) located at the border of the enzyme. Panel (b): The adenine moiety of NADPH reaches its final position and interacts with Lys361, Arg279, and Ser218. Panel (c): Two alternative snapshots that illustrate the displacement of the Tyr407‐loop as the nicotinamide ring of NADPH gets close to the FAD cofactor. Panel (d): The nicotinamide ring of NADPH is sandwiched between the si‐face of FAD and the phenyl group of Tyr324. Panel (e): At the end of the process, Tyr324 is displaced out and NADPH reaches its final conformation.
FIGURE 4.

Representative conformations observed during the binding of L‐Orn to SidA(). L‐Orn is highlighted in yellow. Structures were selected by visual inspection. The values of the reaction coordinate at which the snapshots were taken are shown below each panel. Residues Arg279 and Asp280, together with Tyr324 and are represented in . Panel (a) describes the initial interaction between L‐Orn and Arg279/Asp280. Panel (b) represents a conformation in which L‐Orn is entering the active site. It is flanked by the phenyl ring of Tyr324, on one side, and by the si‐face of on the other. Panel (c): L‐Orn docked in its active site pocket.
Relevant conformations for the binding event of L‐Orn to SidA() are presented in Figure 4. Panel (a) of Figure 4 depicts the initial contacts between the molecular partners. The main interactions are established with Arg279 and Asp280. Then, L‐Orn leaves this site and approaches the FAD cofactor. When this occurs, Tyr324 has to move out to make room for the substrate, which binds between its phenyl moiety and the si‐face of FAD, as shown in panel (b). In this regard, the entrance of L‐Orn is similar to that of the nicotinamide ring of NADPH. Both use the same “FAD gate” with an analogous displacement of the Tyr324‐loop. Finally, L‐Orn moves further into the active site to reach its binding pocket, as can be seen in panel (c). Interestingly, when this happens, the Tyr324‐loop returns to its original location reestablishing the stacking interaction with the isoalloxazine group of FAD. This is an important difference between the two processes: only the entrance of NADPH triggers the conformational change of the Tyr324‐loop that distinguishes between the out and in conformations of FAD in the crystal structures.
3.2. Binding of NADPH to SidA(, L‐Orn) and L‐Orn to SidA(, NADPH)
The entrance of NADPH to SidA(, L‐Orn) is analogous to its entrance to SidA(). The PMF presents a similar depth and the interactions of NADPH with the active site residues are also the same. The result is not surprising considering that the L‐Orn pocket is buried in the active site so that it does not pose any obstacle to the entrance of NADPH. The situation is the opposite for the entrance of L‐Orn to SidA(, NADPH). We recall that we studied this event by enforcing the unbinding of L‐Orn from SidA(, NADPH, L‐Orn) via US simulations. In all our trial calculations, we found that the bound NADPH molecule blocked the way out to L‐Orn. To overcome the obstacle, we applied increasingly larger bias potentials. However, the procedure did not uncover any alternative way out for L‐Orn. Instead, it broke the active site of the protein. We, therefore, conclude that L‐Orn cannot use this gate to bind SidA when NADPH is already attached to the active site and the FAD cofactor is in the out conformation. Accordingly, there are two potential routes to form the SidA(, NADPH, L‐Orn) complex. One of them entails the attachment of NADPH to SidA(, L‐Orn), already discussed, followed by the transition from FAD to FAD
to FAD (event c2 d2 of Figure 1). Alternatively, once SidA(, NADPH) is formed, the transition could occur, followed by the binding of L‐Orn (events b1 c1 d2 of Figure 1). We analyze these possibilities in the following sections.
(event c2 d2 of Figure 1). Alternatively, once SidA(, NADPH) is formed, the transition could occur, followed by the binding of L‐Orn (events b1 c1 d2 of Figure 1). We analyze these possibilities in the following sections.
3.3. Transition between and
We evaluated this transition from both, SidA(, NADPH) (Figure 1 b1 c1) and SidA(, NADPH, L‐Orn) (Figure 1 c2 d2). The results are similar, as can be seen in Figure 5 which depicts the corresponding PMF profiles. In both cases, the most stable state is the one with FAD in the out conformation. The free energy difference of the transition is somewhat smaller when L‐Orn is not present and the barrier for the conformational change is ~2.0 kcal/mol lower. Therefore, the most likely path among those indicated in Figure 1 is a b1 c1 d2. Movie S3, available in the Supporting Information section, illustrates the dynamics of the active site of SidA(, NADPH) during this event.
FIGURE 5.

PMFs for the out to in transition of the FAD cofactor. The calculations we carried out to assess their consistency and the procedure we followed to compute the error bars are presented in the Supporting Information section. The red curve represents the profile corresponding to the SidA(, NADPH) SidA(, NADPH) transition, while the blue one indicates the SidA(, NADPH, L‐Orn) SidA(, NADPH, L‐Orn) transition.
The analysis of the structures achieved after the transition (c1 and d2 in Figure 1) shows that the relative positions of FAD and NADPH are not appropriate for the H‐transfer required for FAD reduction. This observation agrees with those directly obtained from the inspection of the crystal structures of SidA with FAD in the in conformation (see, e.g., Franceschini et al., 2012). This reaction consists of the transfer of a hydride from C4(NADPH) to N5(FAD). However, the distances separating N5(FAD) from any of the H‐atoms attached to C4(NADPH) are too long. Figure 6 shows the probability distribution functions for these distances (blue and light blue curves), calculated from the snapshots collected from MD simulations of models SidA(, NADPH) and SidA(, NADPH, L‐Orn). It can be seen that the pro‐ hydrogen reaches shorter distances than the pro‐R, but they are never smaller than 4.0 Å. Clearly, the transfer cannot occur under such conditions. Besides, the fact that the pro‐ hydrogen is closer to N5 than the pro‐ hydrogen contradicts kinetic studies which indicate that the transfer has a pro‐ stereochemistry (Franceschini et al., 2012; Robinson, Franceschini, et al., 2014; Romero, Fedkenheuer, et al., 2012).
FIGURE 6.

Probability density function for the distances between the H‐atoms bound to C4(NADPH) and N5(FAD). The blue and light‐blue distributions were computed before the rotation of NADPH around the C1D‐N1N bond, while the red and orange functions were obtained after the rotation.
3.4. Binding of L‐Orn to SidA(, NADPH)
As explained in Section 3.2, US simulations demonstrated that L‐Orn cannot enter the active site of SidA through the “FAD gate” if the FAD cofactor is in the out conformation and NADPH is already bound to the enzyme. However, the simulations also revealed that the binding of NADPH destabilizes the out conformation by displacing the Tyr324 ring which otherwise forms stacking interactions with the isoalloxazine group of FAD. This event would trigger the out to in conformational change of FAD and could provide a new opportunity for the binding of L‐Orn. Therefore, we studied the binding of L‐Orn to SidA(, NADPH) by enforcing its unbinding from the SidA(, NADPH, L‐Orn) model via US simulations. The PMF for this process is presented in Figure S6 of the Supporting Information section, where it is compared with the binding of L‐Orn to the resting state of the enzyme. The results demonstrate that, despite the presence of NADPH, L‐Orn can enter the active site of the enzyme if FAD is in the in conformation. Moreover, the bound state of the ligand is significantly more stable in this situation than in the resting state. Movie S4, available in the Supporting Information, outlines this event. It can be observed that the transition of FAD from the out to the in conformation makes room for the passage of L‐Orn to its binding pocket. We also note that the Tyr324‐loop plays a different role in this event than in the entrance of L‐Orn to SidA(). In the present case, the phenyl ring of Tyr324 remains almost perpendicular to the isoalloxazine moiety of FAD during the whole process. In this position, it rotates, acting as a gate and enabling the L‐Orn passage to the active site. In Figure S7 of the Supporting Information section, this process is described.
during the whole process. In this position, it rotates, acting as a gate and enabling the L‐Orn passage to the active site. In Figure S7 of the Supporting Information section, this process is described.
3.5. Rotation of the nicotinamide ring of NADPH
As explained in Section 3.3, the relative positioning of the isoalloxazine group of FAD and the nicotinamide ring of NADPH is not appropriate for the hydride transfer required for FAD reduction. The structures show that the pro‐ hydrogen is the closest to the N5 acceptor atom, while all kinetic studies indicate that the process has a pro‐ stereochemistry (Franceschini et al., 2012; Robinson, Franceschini, et al., 2014; Romero, Fedkenheuer, et al., 2012).
It has been suggested that the positioning detected in the crystal structures is required to stabilize the C4a hydroperoxyflavin intermediate. Accordingly, NADPH should carry out a conformational change to shift from a location appropriate for the protection of the intermediate to one consistent with the hydride transfer. Such a conformational change has been observed in other monooxygenases (Manenda et al., 2020). In L‐Orn N‐hydroxylase from Pseudomonas aeruginosa (PvdA), a close homolog of SidA, high flexibility of the nicotinamide ring of NADPH has been observed in the oxidized state of the enzyme (Olucha et al., 2011). In addition, for SidA, there is indirect evidence that NADPH modifies its conformation to allow FAD reduction. In particular, it was noticed that the rate of reduction of the cofactor is significantly larger in the S257A mutant than in the native enzyme (Shirey et al., 2013), while the uncoupling between the reductive and oxidative halves of the reaction is also much more prominent. Residue Ser257 stabilizes the NADPH conformation that protects the hydroperoxyflavin intermediate via an H‐bond interaction. The lack of this interaction in the S257A mutant facilitates the movement of NADPH to alternative positions where it can reduce FAD. But, at the same time, it reduces the stability of the hydroperoxyflavin intermediate enhancing the uncoupling.
The analysis of the relative positioning of FAD and NADPH, in models SidA(, NADPH) and SidA(, NADPH, L‐Orn), reveals that a rotation of the nicotinamide ring around the C1D‐N1N bond would cause the pro‐ hydrogen to get closer to the N5 acceptor atom than the pro‐ hydrogen. In other words, the rotation would take NADPH from a position where it can protect the hydroperoxyflavin intermediate to one appropriate for FAD reduction. We evaluated the feasibility of this rotation in models SidA(, NADPH) and SidA(, NADPH, L‐Orn) via US simulations. The PMFs are presented in Figure 7 panel (c). Movie S5, available in the Supporting Information section, illustrates this process in SidA(, NADPH, L‐Orn).
FIGURE 7.
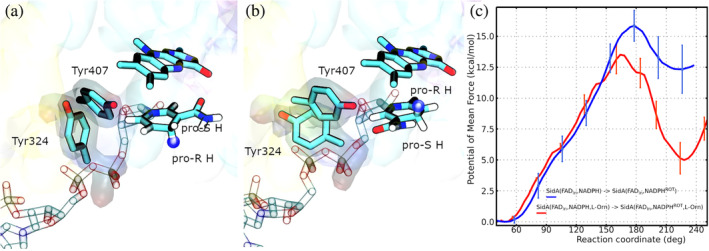
Panels (a and b) illustrate the active site of SidA(, NADPH) with the nicotinamide moiety of NADPH before and after rotation, respectively. Tyr324, Tyr407, FAD, and NADPH are drawn as . The two Tyrosine residues are shadowed. The pro‐R hydrogen atom is highlighted with a blue sphere. Panel (c) shows the PMFs for the rotation of the nicotinamide ring of NADPH in models SidA(, NADPH) (blue line) and SidA(, NADPH, L‐Orn) (red line). The calculations we carried out to assess the consistency of their and the procedure we followed to compute the error bars are presented in the Supporting information section.
Before the rotation, the hydroxyl group of Tyr324 is H‐bonded to the amino group of the backbone of Tyr407 hindering their relative movements (see panel (a) of Figure 7). This is the conformation seen in the structures with PDB codes 6X0J and 6X0I. However, as the rotation progresses, the Tyr407‐ and Tyr324‐ loops separate from each other, and then Tyr324 establishes new interactions with Glu458. This new conformation (panel (b) of Figure 7) makes room for the nicotinamide moiety of NADPH, which can then reach the location appropriate for FAD reduction.
The probability distribution functions for the distance between the N5 atom of FAD and the H‐atoms bound to C4 of NADPH, at the end of the nicotinamide ring rotation, are presented in Figure 6. It can be observed that both hydrogen atoms are substantially closer to N5 than in the original structures. Furthermore, the pro‐ hydrogen (red curve) is within an H‐bond distance to the acceptor, and it is in a position more appropriate for the H‐transfer than the pro‐ hydrogen (orange curve). The PMF profiles of Figure 7 panel (c), on the other hand, confirm that NADPH rotation is possible for both models, SidA(, NADPH) and SidA(, NADPH, L‐Orn), although it is not thermodynamically favorable in either of them. However, the free energy barrier is lower and the rotated state is significantly more stable in the presence of the substrate than in its absence. From the free energy difference of these rotations, we estimate that in the presence of L‐Orn, the probability of finding the rotated state of NADPH is ~4000 times smaller than the one of the unrotated state, while it is times smaller in L‐Orn absence. Since the reduction of FAD can only occur from the rotated NADPH, its stabilization because of the presence of L‐Orn could be a strategy developed by the enzyme to mitigate the uncoupling.
We could try using the barriers of Figure 7 to compute the absolute values of the rate constants of NADPH rotations. However, two concerns prevent us from doing that. First, the barriers determined with empirical classical potentials do not have the accuracy required to make meaningful comparisons with experiments. Besides, we ignore the pre‐exponential factor we should use. Nevertheless, interesting conclusions can be drawn by comparing the rotation of NADPH (Figure 7c) with that of FAD (Figure 5). The pre‐exponential factors of both transitions should be comparable, and the inaccuracies of the force field should affect the two barriers similarly. Under these assumptions, the ratio between the rate constants for these two rotations is,
| (1) |
Considering the most likely events (red curves of Figure 5 and 7c) along with the uncertainties in their barriers, the maximum and minimum estimates for are ~2.48 and −2.35 kcal/mol, respectively. These figures indicate the barriers of the two rotations are the same, within the statistical uncertainty of the results. More precisely, at 303K, the ratio would rank between 0.016 and 50.0.
After determining the putative existence of a rotated form of the nicotinamide ring of NADPH, we examined the possibility that NADPH could bind SidA() being already in the alternative (rotated) conformation. We computed the PMF for this process and found it is similar to the one shown in the right panel of Figure 2. However, we also noted that, in this scenario, the location of the nicotinamide ring prevents the FAD out to in transitioning. We, therefore, conclude that this pathway does not lead to the activation of SidA.
3.6. Conservation of Tyr324 and Tyr407 loops in FMOs
Figure 8 depicts sequence logos showing the degree of conservation of the Tyr324 and Tyr407 loops within class B FMOs (24 sequences), as well as in the sets of homology sequences obtained by BlastP (173 representatives from 1000 original sequences) and Jackhmmer (437 sequences). The alignments are provided in the Supporting Information section.
FIGURE 8.

Sequence logos built using data derived from class‐B FMOs and SidA homologs obtained from BlastP and Jackhmmer. The number of sequences for each case is 24 (class‐B), 173 (BlastP), and 437 (Jackhmmer). The percentage of conservation of the sequence motifs considered in each case is indicated below. FMO, flavin‐dependent monooxygenase.
In either class B FMOs or the Jackhmmer sets, there is a moderate level of conservation in the Tyr324 loop (16.6% and 33.1%, respectively). However, within the BlastP sequences, this triad exhibits a notably high conservation (98.8%). The scenario contrasts with that of the Tyr407‐loop, which presents significantly high conservation across all datasets (91.9% in class B FMOs, 95.3% in BlastP, and 67.0% in Jackhmmer). When examining the aligned sequences, we noticed the occasional presence of a phenylalanine(F) residue within the Tyr407‐loop. As this residue might potentially contribute to the enzyme mechanism in a similar way as Tyrosine(Y), the computed percentages for class B FMOs and Jackhmmer include both TGY and TGF triads. Additionally, we also detected a considerable level of conservation of SGY motifs in the BlastP set. Consequently, when calculating the percentage of conservation of the Tyr407‐loop within this set, we took into account both TGY and SGY.
Consurf results are presented in Figure 9 panel (a). This tool identifies sites of functional significance by assigning conservation scores based on the evolutionary rate of each site (Yariv et al., 2023). Regions exposed to the solvent that exhibit high levels of conservation are assumed to play a role in the protein's function. This approach predicts the Tyr324 and Tyr407 loops are key elements in the enzyme's functionality, with scores similar to those of active site residues (residues from ~100 to ~150). Panel (b) of Figure 9 shows the results of the Evolutionary Trace analysis. This method consists of classifying the residues in a protein sequence according to their relative evolutionary importance, aiming to pinpoint sites that bear functional significance (Lichtarge et al., 1996). Notably, although this algorithm assigns the “relevance” to each residue with distinct criteria than Consurf, it also puts the Tyr324/Tyr407 loops at the top of the ranking along with the active site residues. Finally, we must mention that after examining the complete sequence in the MSAs, we also found that Arg279, which according to the simulations, plays important roles in the L‐Orn/NADPH binding mechanism, also exhibits significant levels of conservation (see Jackhmmer.afa, BlastP.afa and ClassB.afa files available in the Supporting Information section). Moreover, it is identified as an “important” position in both the Consurf and Evolutionary Trace examinations.
FIGURE 9.

Relevance of SidA residues according to Consurf (panel a) and the Evolutionary Trace algorithms (panel b). Both Tyr324 and Tyr407 loops are circled in both panels. Arg279 is underlined in green. As ET server depicts the results on the structure residues, regions with missing residues are indicated with an asterisk. These are at the beginning of the sequence, in positions 379–383, and at the end.
4. CONCLUSIONS
We have studied the binding of NADPH and L‐Orn to SidA via MD simulations. Also, we analyzed the conformational changes that take the FAD cofactor from the to the position, as well as a rotation of the nicotinamide ring of NADPH that would allow the reduction of FAD according to the available experimental evidence. The study was complemented with a bioinformatics analysis focused on the conservation of residues which, according to the simulations, plays significant roles in the mechanism. The main findings of this work are the following:
The most likely sequence of events taking SidA from the resting state to the state just before FAD reduction is a b1 c1 d2 e of Figure 1.
The last step of this sequence is a rotation of the nicotinamide ring of NADPH that brings together the donor and acceptor of the H atom involved in FAD reduction. It also resolves the controversy over which of the two hydrogen atoms (pro‐ or pro‐) is transferred.
The simulations reveal that the loop formed by Tyr405 Gly406 and Tyr407 actively participates in the catalytic mechanism. A bioinformatic analysis revealed these residues are conserved within class B FMOs and SidA homologs and also predicted they are functionally important.
AUTHOR CONTRIBUTIONS
Pablo Sobrado: Writing – review and editing; visualization; validation; investigation; funding acquisition; conceptualization; project administration; resources. Gustavo Pierdominici‐Sottile: Conceptualization; investigation; funding acquisition; writing – original draft; methodology; validation; data curation. Juliana Palma: Conceptualization; validation; visualization; writing – review and editing; formal analysis; software; methodology; investigation; funding acquisition; writing – original draft. María Leticia Ferrelli: Data curation; formal analysis; writing – review and editing; investigation; conceptualization; writing – original draft.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
Movie S1. Binding mechanism of NADPH to SidA in its resting state (SidA(FADout)). The main enzyme residues that are involved during this process are highlighted. The backbone of both Tyr324 and Tyr407 loops are colored in blue and red, respectively. The distance between the center of mass of the backbone atoms of these two loops is outlined throughout the process. Colored shadows surround different types of residues/molecules: blue (positive residues), green (polar residues), and orange (NADPH and FAD).
Movie S2. Binding mechanism of L‐Orn to SidA in its resting state (SidA(FADout)). Residues involved in this process are highlighted. The backbones of the Tyr324 and Tyr407 loops are colored in blue and red, respectively.
Movie S3. FADout → FADin transition highlighting the dynamics of NADPH and active site residues of SidA. Model SidA(FADout, NADPH) was employed to simulate this event.
Movie S4. Binding mechanism of L‐Orn to SidA when NADPH is already docked and FAD is in its conformation (model SidA(FADin, NADPH)). The phenyl ring of Tyr324 remains almost perpendicular to the FAD ring during the whole process, acting as a top that spins as L‐Orn enters the active site. The distance between the center of mass of the backbone atoms of Tyr324 and Tyr497 loops is indicated throughout the process.
Movie S5. Nicotinamide ring rotation in the active site of SidA(FADin, NADPH, L‐Orn). At the beginning of the process, Tyr324 is H‐bonded to the Tyr407 backbone. This interaction limits the motion of Tyr324 and Tyr407 loops. When this H‐bond is broken, the mobility of nicotinamide moiety increases, so that it can then flip to a new conformation in which its pro‐R C4 hydrogen is within an h‐bond distance from N5(FAD).
Data S1. Proteins_SI.
Data S2. ClassB.afa Multiple sequence alignment.
Data S3. BlastP.afa Multiple sequence alignment.
Data S4. Jackhmmer.afa Multiple sequence alignment.
ACKNOWLEDGMENTS
The authors thank Sydney Johnson for her kind help, CONICET, Fulbright, UNQ, and ANPCyT for their financial support. This research was supported in part by the National Science Foundation grants CHE‐2003658 (to PS), Universidad Nacional de Quilmes (UNQ) (ID: 1292/19), ANPCyT (PICT 2020‐SerieA‐00192), CONICET (ID: 11220130100260CO), and Fulbright.
Pierdominici‐Sottile G, Palma J, Ferrelli ML, Sobrado P. The dynamics of the flavin, NADPH, and active site loops determine the mechanism of activation of class B flavin‐dependent monooxygenases. Protein Science. 2024;33(4):e4935. 10.1002/pro.4935
Review Editor: Lynn Kamerlin
Contributor Information
Gustavo Pierdominici‐Sottile, Email: gsottile@unq.edu.ar.
Pablo Sobrado, Email: psobrado@vt.edu.
REFERENCES
- Badieyan S, Bach RD, Sobrado P. Mechanism of N‐hydroxylation catalyzed by flavin‐dependent monooxygenases. J Org Chem. 2015;80:2139–2147. [DOI] [PubMed] [Google Scholar]
- Beaty NB, Ballou DP. The reductive half‐reaction of liver microsomal FAD‐containing monooxygenase. J Biol Chem. 1981;256:4611–4618. [PubMed] [Google Scholar]
- Binda C, Robinson RM, Del Campo JSM, Keul ND, Rodriguez PJ, Robinson HH, et al. An unprecedented NADPH domain conformation in lysine monooxygenase NBTG provides insights into uncoupling of oxygen consumption from substrate hydroxylation. J Biol Chem. 2015;290:12676–12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell AC, Robinson R, Mena‐Aguilar D, Sobrado P, Tanner JJ. Structural determinants of flavin dynamics in a class B monooxygenase. Biochemistry. 2020;59:4609–4616. [DOI] [PubMed] [Google Scholar]
- Campbell AC, Stiers KM, Del Campo JSM, Mehra‐Chaudhary R, Sobrado P, Tanner JJ. Trapping conformational states of a flavin‐dependent N‐monooxygenase in crystallo reveals protein and flavin dynamics. J Biol Chem. 2020;295:13239–13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D, Ben‐Shalom I, Brozell S, Cerutti D, Cheatham T, Cruzeiro V, et al. Amber 18. San Francisco: University of California; 2018. [Google Scholar]
- Cashman JR. Structural and catalytic properties of the mammalian flavin‐containing monooxygenase. Chem Res Toxicol. 1995;8:165–181. [DOI] [PubMed] [Google Scholar]
- Chocklett SW, Sobrado P. Aspergillus fumigatus SidA is a highly specific ornithine hydroxylase with bound flavin cofactor. Biochemistry. 2010;49:6777–6783. [DOI] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- Eswaramoorthy S, Bonanno JB, Burley SK, Swaminathan S. Mechanism of action of a flavin‐containing monooxygenase. Proc Natl Acad Sci U S A. 2006;103:9832–9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini S, Fedkenheuer M, Vogelaar NJ, Robinson HH, Sobrado P, Mattevi A. Structural insight into the mechanism of oxygen activation and substrate selectivity of flavin‐dependent N‐hydroxylating monooxygenases. Biochemistry. 2012;51:7043–7045. [DOI] [PubMed] [Google Scholar]
- Gabler F, Nam SZ, Till S, Mirdita M, Steinegger M, Söding J, et al. Protein sequence analysis using the MPI bioinformatics toolkit. Curr Protoc Bioinformatics. 2020;72:e108. [DOI] [PubMed] [Google Scholar]
- Gatti DL, Palfey BA, Lah MS, Entsch B, Massey V, Ballou DP, et al. The mobile flavin of 4‐OH benzoate hydroxylase. Science. 1994;266:110–114. [DOI] [PubMed] [Google Scholar]
- Hissen AH, Wan AN, Warwas ML, Pinto LJ, Moore MM. The aspergillus fumigatus siderophore biosynthetic gene sidA, encoding L‐ornithine N5‐oxygenase, is required for virulence. Infect Immun. 2005;73:5493–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers MM, Montersino S, Westphal AH, Tischler D, Van Berkel WJ. Flavin dependent monooxygenases. Arch Biochem Biophys. 2014;544:2–17. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- Joung IS, Cheatham TE. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B. 2008;112:9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kräutler V, Van Gunsteren WF, Hünenberger PH. A fast shake algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J Comput Chem. 2001;22:501–508. [Google Scholar]
- Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257:342–358. [DOI] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malito E, Alfieri A, Fraaije MW, Mattevi A. Crystal structure of a Baeyer–Villiger monooxygenase. Proc Natl Acad Sci U S A. 2004;101:13157–13162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manenda MS, Picard ME, Zhang L, Cyr N, Zhu X, Barma J, et al. Structural analyses of the Group A flavin‐dependent monooxygenase PieE reveal a sliding FAD cofactor conformation bridging OUT and IN conformations. J Biol Chem. 2020;295:4709–4722. 10.1074/jbc.RA119.011212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin del Campo JS, Vogelaar N, Tolani K, Kizjakina K, Harich K, Sobrado P. Inhibition of the flavin‐dependent monooxygenase siderophore A (SidA) blocks siderophore biosynthesis and Aspergillus fumigatus growth. ACS Chem Biol. 2016;11:3035–3042. [DOI] [PubMed] [Google Scholar]
- Mascotti ML, Ayub MJ, Furnham N, Thornton JM, Laskowski RA. Chopping and changing: the evolution of the flavin‐dependent monooxygenases. J Mol Biol. 2016;428:3131–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascotti ML, Lapadula WJ, Juri AM. The origin and evolution of Baeyer–Villiger monooxygenases (BVMOs): an ancestral family of flavin monooxygenases. PLoS One. 2015;10:e0132689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield JA, Frederick RE, Streit BR, Wencewicz TA, Ballou DP, DuBois JL. Comprehensive spectroscopic, steady state, and transient kinetic studies of a representative siderophore‐associated flavin monooxygenase. J Biol Chem. 2010;285:30375–30388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olucha J, Meneely KM, Chilton AS, Lamb AL. Two structures of an N‐hydroxylating flavoprotein monooxygenase: ornithine hydroxylase from Pseudomonas aeruginosa . J Biol Chem. 2011;286:31789–31798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfey BA, McDonald CA. Control of catalysis in flavin‐dependent monooxygenases. Arch Biochem Biophys. 2010;493:26–36. [DOI] [PubMed] [Google Scholar]
- Reis RA, Li H, Johnson M, Sobrado P. New frontiers in flavin‐dependent monooxygenases. Arch Biochem Biophys. 2021;699:108765. [DOI] [PubMed] [Google Scholar]
- Robinson R, Franceschini S, Fedkenheuer M, Rodriguez PJ, Ellerbrock J, Romero E, et al. Arg279 is the key regulator of coenzyme selectivity in the flavin‐dependent ornithine monooxygenase SidA. Biochim Biophys Acta. 2014;1844:778–784. [DOI] [PubMed] [Google Scholar]
- Robinson RM, Rodriguez PJ, Sobrado P. Mechanistic studies on the flavin‐dependent N6‐lysine monooxygenase mbsg reveal an unusual control for catalysis. Arch Biochem Biophys. 2014;550:58–66. [DOI] [PubMed] [Google Scholar]
- Romero E, Fedkenheuer M, Chocklett SW, Qi J, Oppenheimer M, Sobrado P. Dual role of NADP (H) in the reaction of a flavin dependent N‐hydroxylating monooxygenase. Biochim Biophys Acta. 2012;1824:850–857. [DOI] [PubMed] [Google Scholar]
- Romero E, Robinson R, Sobrado P. Monitoring the reductive and oxidative half‐reactions of a flavin‐dependent monooxygenase using stopped‐flow spectrophotometry. J Vis Exp. 2012;e3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon‐Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J Chem Theory Comput. 2013;9:3878–3888. [DOI] [PubMed] [Google Scholar]
- Schreuder HA, Prick PA, Wierenga RK, Vriend G, Wilson KS, Hol WG, et al. Crystal structure of the p‐hydroxybenzoate hydroxylase‐substrate complex refined at 1.9 Å resolution: analysis of the enzyme‐substrate and enzyme‐product complexes. J Mol Biol. 1989;208:679–696. [DOI] [PubMed] [Google Scholar]
- Setser JW, Heemstra JR Jr, Walsh CT, Drennan CL. Crystallographic evidence of drastic conformational changes in the active site of a flavin‐dependent N‐hydroxylase. Biochemistry. 2014;53:6063–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey C, Badieyan S, Sobrado P. Role of ser‐257 in the sliding mechanism of NADP(H) in the reaction catalyzed by the Aspergillus fumigatus flavin‐dependent ornithine N5‐monooxygenase SidA . J Biol Chem. 2013;288:32440–32448. 10.1074/jbc.M113.487181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucharitakul J, Tinikul R, Chaiyen P. Mechanisms of reduced flavin transfer in the two‐component flavin‐dependent monooxygenases. Arch Biochem Biophys. 2014;555:33–46. [DOI] [PubMed] [Google Scholar]
- Torres Pazmiño DE, Baas BJ, Janssen DB, Fraaije MW. Kinetic mechanism of phenylacetone monooxygenase from Thermobifida fusca . Biochemistry. 2008;47:4082–4093. [DOI] [PubMed] [Google Scholar]
- Van Berkel W, Kamerbeek N, Fraaije M. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J Biotechnol. 2006;124:670–689. [DOI] [PubMed] [Google Scholar]
- Van Berkel WJ, Eppink MH, Schreuder HA. Crystal structure of p‐hydroxybenzoate hydroxylase reconstituted with the modified FAD present in alcohol oxidase from methylotrophic yeasts: evidence for an arabinoflavin. Protein Sci. 1994;3:2245–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yariv B, Yariv E, Kessel A, Masrati G, Chorin AB, Martz E, et al. Using evolutionary data to make sense of macromolecules with a “face‐lifted” consurf. Protein Sci. 2023;32:e4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Binding mechanism of NADPH to SidA in its resting state (SidA(FADout)). The main enzyme residues that are involved during this process are highlighted. The backbone of both Tyr324 and Tyr407 loops are colored in blue and red, respectively. The distance between the center of mass of the backbone atoms of these two loops is outlined throughout the process. Colored shadows surround different types of residues/molecules: blue (positive residues), green (polar residues), and orange (NADPH and FAD).
Movie S2. Binding mechanism of L‐Orn to SidA in its resting state (SidA(FADout)). Residues involved in this process are highlighted. The backbones of the Tyr324 and Tyr407 loops are colored in blue and red, respectively.
Movie S3. FADout → FADin transition highlighting the dynamics of NADPH and active site residues of SidA. Model SidA(FADout, NADPH) was employed to simulate this event.
Movie S4. Binding mechanism of L‐Orn to SidA when NADPH is already docked and FAD is in its conformation (model SidA(FADin, NADPH)). The phenyl ring of Tyr324 remains almost perpendicular to the FAD ring during the whole process, acting as a top that spins as L‐Orn enters the active site. The distance between the center of mass of the backbone atoms of Tyr324 and Tyr497 loops is indicated throughout the process.
Movie S5. Nicotinamide ring rotation in the active site of SidA(FADin, NADPH, L‐Orn). At the beginning of the process, Tyr324 is H‐bonded to the Tyr407 backbone. This interaction limits the motion of Tyr324 and Tyr407 loops. When this H‐bond is broken, the mobility of nicotinamide moiety increases, so that it can then flip to a new conformation in which its pro‐R C4 hydrogen is within an h‐bond distance from N5(FAD).
Data S1. Proteins_SI.
Data S2. ClassB.afa Multiple sequence alignment.
Data S3. BlastP.afa Multiple sequence alignment.
Data S4. Jackhmmer.afa Multiple sequence alignment.