

Abstract
The cGAS-STING intracellular DNA sensing pathway has emerged as a key element of innate antiviral immunity and a promising therapeutic target. The existence of an innate immune sensor that can be activated by any double-stranded DNA of any origin raises fundamental questions about how cGAS is regulated and how it responds to “foreign” DNA while maintaining tolerance to ubiquitous self-DNA. In this review, we summarize recent evidence implicating important roles for cGAS in detection of foreign and self-DNA. We describe two recent and surprising insights into cGAS-STING biology: that cGAS is tightly tethered to the nucleosome, and that the cGAMP product of cGAS is an immunotransmitter acting at a distance to control innate immunity. We consider how these advances influence our understanding of the emerging roles of cGAS in the DNA damage response, senescence, aging, and cancer biology. Finally, we describe emerging approaches to harness cGAS-STING biology for therapeutic benefit.
Etoc blurb:
The cGAS-STING pathway detects intracellular DNA and activates a potent antiviral response. Dvorkin and colleagues summarize recent insights into cGAS-STING biology and its role in host defense, immunopathologies, and anti-tumor immunity.
Introduction
Our current understanding of innate antiviral immunity is rooted in elegant experiments conducted during the 1950s in which Isaacs and Lindenmann found that cells treated with inactivated influenza virus produced a soluble factor that could protect fresh cells from subsequent infection with live virus. They isolated this factor and termed it “interferon” owing to its ability to interfere with viral infection1,2. We now know that the type I interferons (IFNs) are a family of many closely related cytokines that are produced in response to virus infection3.
The induction of IFNs is, in most cases, under the control of innate immune sensors of nucleic acids. Because viruses can have a genome composed of either RNA or DNA, there are specific sensors that can detect either RNA or DNA to activate the antiviral IFN response. These include several Toll-like receptors (TLRs) expressed by sentinel immune cells that sample endosomal compartments for the presence of unusual nucleic acids4. In addition to the TLRs, there are broadly expressed sensors that detect intracellular nucleic acids and signal the presence of viral infection from within the infected cell itself5. The RIG-I-like receptors detect structural features of viral RNAs that are scarce within our own cells and signal through the adapter protein MAVS to activate the IFN-mediated antiviral response6. The principal sensor of intracellular DNA in vertebrates is cyclic GMP-AMP synthase (cGAS)7, a member of the nucleotidyltransferase family of enzymes that includes the oligoadenylate synthetases (OAS). cGAS binds the sugar-phosphate backbone of double-stranded DNA (dsDNA) in a sequence-independent fashion, resulting in structural rearrangements that activate the production of cyclic GMP-AMP (cGAMP) from GTP and ATP. cGAMP rapidly diffuses throughout the cell and binds to STING, a transmembrane protein that resides on the endoplasmic reticulum (ER). cGAMP binding to STING results in a conformational change in STING that exposes binding sites for the kinase TBK1 and the transcription factor IRF38,9. IRF3 undergoes TBK1-mediated phosphorylation, dimerization, and nuclear translocation to activate the IFN genes. In addition to activating IFNs, STING also engages the NF-kB transcription factor to induce the expression of proinflammatory cytokines and chemokines10.
Our understanding of the mechanisms and functions of the cGAS-STING DNA sensing pathway has grown exponentially since the first descriptions of intracellular DNA sensing in 200611,12, the discovery of STING in 200813,14, and the identification of cGAS and cGAMP in 20137,15. Among the many exciting developments in the field, we have learned that cGAS, cGAMP, and STING have deep evolutionary roots in prokaryote anti-phage immunity. Hundreds of bacterial cGAS-like enzymes have been identified that produce an array of cyclic dinucleotides and trinucleotides16. Even the ligand specificities and of eukaryotic cGAS enzymes have diverged over evolutionary time: cGAS-like receptors in Drosophila fruit flies can detect dsRNA, not dsDNA17–19.
In vertebrates, the activation of cGAS by dsDNA of any potential origin has created a conundrum of self/non-self discrimination that has intrigued the field since its inception: how does a sequence-independent sensor of dsDNA avoid constant activation by the billions of base pairs of self-DNA in all nucleated cells? Indeed, we have learned of important instances in which cGAS can be activated by self-DNA. An extreme example of this is the rare human “interferonopathy” Aicardi-Goutieres Syndrome, some cases of which are caused by mutations in enzymes that metabolize, modify, or package self-DNA, resulting in inappropriate cGAS activation and severe pathology20–22. Perhaps less extreme but more pervasive, accumulated damage of nuclear and mitochondrial self-DNA during cellular aging appears to activate cGAS and contribute to the processes of senescence and organismal “inflammaging,” both of which are typically viewed as pathological events that reduce healthy lifespan. In contrast, a growing body of evidence implicates these same mechanisms of self-DNA reactivity in protective immunity to cancer stimulated by radiation and chemotherapy. Thus, the high potential for activation of cGAS by self-DNA may have both pathological and protective functions.
In this review, we consider these pathological and protective functions of cGAS. We summarize two recent developments that offer unexpected insights into the regulation of the cGAS-STING pathway. The first involves a foundational concept in the field known as “cytosolic DNA sensing,” which was proposed in 2006 as a logical (but not evidence-based) explanation for how an intracellular DNA sensor could avoid reactivity to genomic DNA12. This proposal, which was made seven years before the discovery of cGAS as the key DNA sensor, suggested that sequestration of the sensor in the cytosol would avoid activation by nuclear DNA while enabling responses to DNA that appeared inappropriately in the cytosol. As described below, recent functional and structural studies have revealed that the majority of cGAS in most cell lines is tightly tethered to nuclear chromatin in a way that prevents activation by DNA. Thus, “cytosolic DNA sensing” may be an incomplete framework for considering cGAS biology. The second recent insight is the discovery that cGAMP is an “immunotransmitter” that is exported from and imported into cells, thus allowing for a mode of cell-to-cell transmission of cGAS-STING signaling23. Considered in the context of the emerging understanding of self-DNA detection by cGAS, these insights have important implications for therapeutic approaches to block cGAS signaling in the context of immunopathology, as well as for harnessing cGAS activation to improve anti-cancer immunity.
cGAS is tightly tethered to the nucleosome
Since its discovery7, cGAS has been considered a cytosolic PRR. Given cGAS has no sequence specificity, its localization in the cytosol was thought to be important to sequester it away from genomic DNA to prevent aberrant activation of the cGAS-STING pathway. However, several reports indicated a role for cGAS localized in the nucleus in several different functions including detection of nuclear-replicating viruses, regulation of DNA repair, and promotion of senescence24–27. These studies indicated that cGAS was only localized in the nucleus under certain conditions, such as viral infection or DNA damage, or during mitosis. In 2019, Manel and colleagues showed that cGAS accumulates on chromatin during mitosis and persists within the nucleus in daughter cells through the next interphase28. The N-terminus of cGAS was found to be important for its nuclear localization and enrichment on centromeric DNA and LINE elements28. Gekara and colleagues also demonstrated that cGAS was constitutively present in the nucleus, and found that this localization required cGAS binding to DNA29. In contrast, Funabiki and colleagues found that purified cGAS lacking the N-terminus bound to mononucleosomes with higher affinity than to naked dsDNA30. This interaction was not mediated by cGAS binding to nucleosomal DNA, as DNA-binding mutants of cGAS bound nucleosomes with similar affinity. Instead, direct interactions between cGAS and the H2A/H2B acidic patch of the nucleosome were required, as mutations of the acidic patch reduced binding of cGAS to the nucleosome30.
Volkman and colleagues demonstrated that in many cell human and mouse cell lines, most cGAS remains nuclear regardless of cell cycle31. In many cases, such as mouse macrophages and HeLa cells, the portion of cGAS that remains nuclear can be as high as 85–95%, indicating that nuclear cGAS represents a significant pool of cGAS within the cell. Nuclear cGAS is bound tightly to chromatin and is only liberated at very high salt concentrations. This binding is independent of the DNA-binding ability of cGAS and does not require the cGAS N-terminus. Instead, a set of evolutionarily conserved positively charged residues on the cGAS surface are required to tether cGAS to chromatin. Chief among these are two arginine residues, R236 and R255 in human cGAS. Mutation of each of these residues caused cGAS to become untethered from chromatin and highly autoreactive against self-DNA31, indicating that tethering of cGAS to chromatin inhibits cGAS activity, which explains how cGAS can remain nuclear at steady-state without inducing aberrant production of cGAMP.
Six independent groups32–37 published cryo-electron microscopy structures of both mouse and human cGAS bound to the nucleosome core particle (NCP), elucidating many key features of this interaction, and discovering how cGAS can be kept inactive while being held mere angstroms from its activating ligand (Figure 1). Confirming prior reports30, cGAS was found to bind to the nucleosome acidic patch, a charged pocket formed by a group of 8 residues from H2A and H2B that acts as a binding site for numerous chromatin-binding proteins38,39. The interaction between cGAS and the acidic patch was very strong, with a measured binding affinity (Kd) of 8.6nM for reconstituted nucleosomes and 6.3nM for cell-derived nucleosomes37. This makes the cGAS:nucleosome interaction over a hundred times stronger than the cGAS:DNA interaction. R236 and R255 of human cGAS were found to be the critical residues mediating binding of cGAS to the nucleosome acidic patch, acting as a canonical “arginine anchor” common to other acidic patch binding proteins, including regulator of chromatin condensation 1 (RCC1), high mobility group protein N2 (HMGN2), interleukin-33, and the latency-associated nuclear antigen (LANA) from Kaposi’s sarcoma herpesvirus (KSHV)38,39. In addition, residues K254 (which stabilizes the cGAS loop within the acidic patch pocket) and R353 (which binds to D51 of H2B), were also shown to strongly regulate cGAS tethering33,35–37. All of these studies used cGAS mutants lacking the N-terminus, confirming that it is completely dispensable for cGAS-chromatin tethering. Whether the N-terminus plays any role in further stabilizing cGAS on the nucleosome or participates in interactions that could untether cGAS from the nucleosome requires further investigation.
Figure 1. The cGAS-STING DNA sensing pathway.
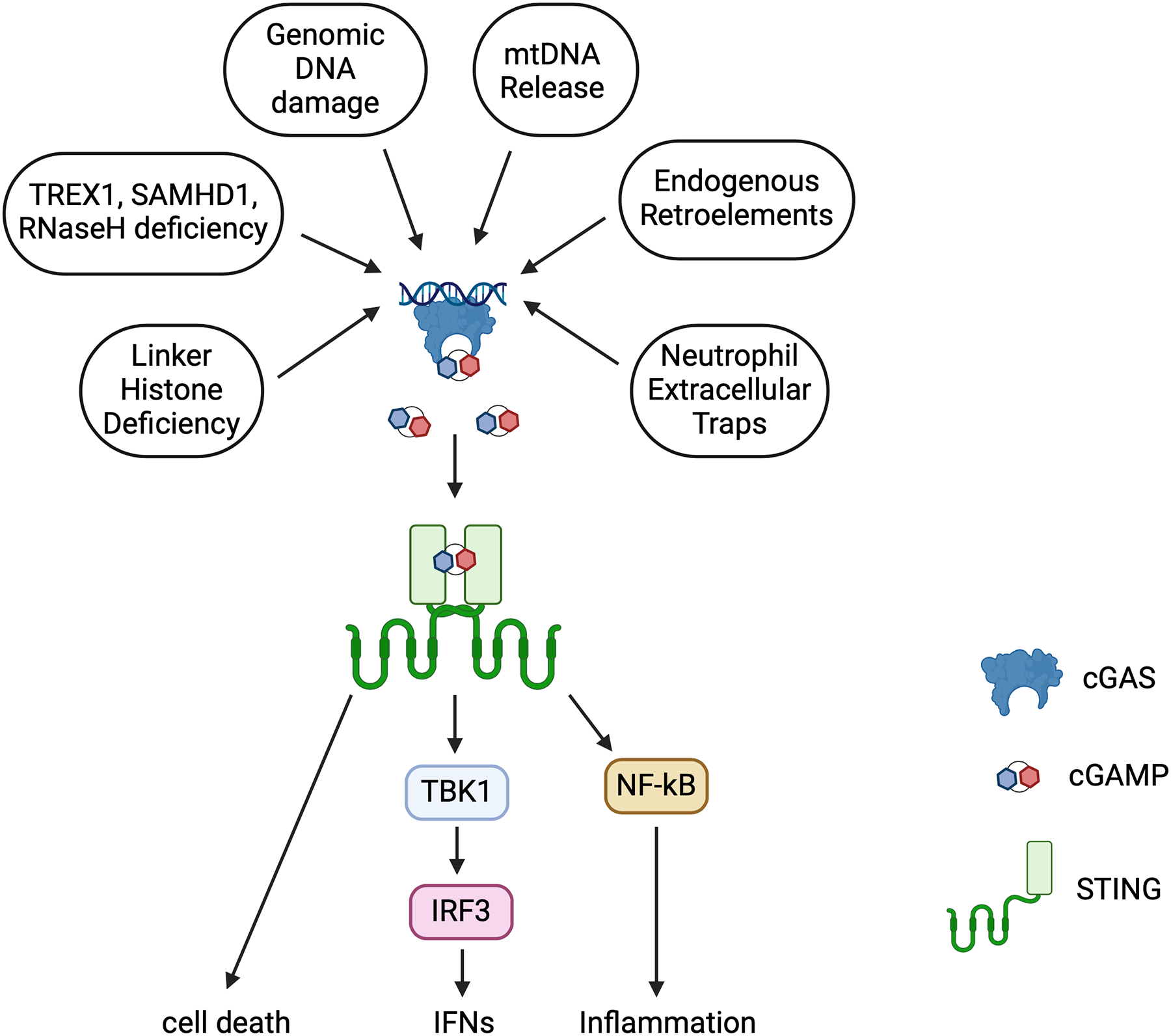
Multiple sources of self and foreign DNA converge on cGAS activation, which leads to STING-dependent signaling.
cGAS has 3 DNA binding sites, termed A, B, and C. Canonical activation of cGAS requires dimerization, forming a complex of two cGAS molecules and two molecules of double stranded DNA in which both site A and site B are occupied on both cGAS molecules40–42. Site C promotes further oligomerization and liquid phase separation to enhance cGAS signaling43,44. In the cGAS:NCP complex, the critical cGAS residues responsible for binding to the acidic patch originate in or are proximal to site B, such that nucleosome binding directly occludes site B. This includes key DNA-binding residues including R236 and K254, which are repurposed in the cGAS:NCP complex for protein-protein interactions. Site A remains solvent exposed and participates only minimally in nuclear tethering but cannot bind DNA due to steric hindrance from the NCP. Taken together, these data reveal that nucleosome binding inhibits cGAS in three ways: 1) key DNA-binding residues on site B are repurposed for acidic patch interactions, preventing DNA-binding to site B; 2) steric hindrance from the proximal NCP prevent dimerization of cGAS which is required for activation; 3) steric hindrance from the NCP prevents binding of DNA to site A.
In most cell types cGAS exists as both a cytosolic and a nuclear pool; however, the factors that regulate this distribution remain uncharacterized. Several studies have reported that cGAS distribution is affected by cell cycle29,45, whereas others have found that cGAS distribution is unaffected by cell cycle31,44. This may be due to cell type-specific differences, including the rate of cell division. Moreover, one study suggested that the N terminus of cGAS recruits it to the plasma membrane in the cytosol46.
Another mechanism that regulates protein distribution within the cell is the expression of nuclear localization sequences (NLS) and nuclear export sequences (NES). Two putative nuclear localization sequences have been described for cGAS, NLS1 and NLS2. NLS1 resides within the N-terminus and is therefore not required for nuclear localization of cGAS. Furthermore, when expressed alone, the N-terminus localizes primarily to the cytosol31,47, suggesting that NLS1 is non-functional. In contrast, deletion of NLS2 (residues 295DVIMKRKRGGS305) from human cGAS (cGASΔNLS2) has been shown to reduce the levels of cGAS within the nucleus27,48,49. NLS sequences target cells for nuclear import through importin. cGAS has been shown to interact with importin proteins, and treatment with the importin-α inhibitor importazole also reduces the level of nuclear cGAS27,49. Additionally, it should be noted that when expressed in cells, cGASΔNLS2 failed to activate an IFNβ luciferase reporter48. However, given that NLS2 resides proximally to the cGAS catalytic site, deletion of NLS2 may disrupt cGAS catalytic activity.
A putative NES sequence has also been described for cGAS (169LEKLKL174). Several studies have shown that mutation of the cGAS NES only modestly increases the level of nuclear cGAS47,50, suggesting that the NES plays a minor role in maintaining the cytosolic pool of cGAS at baseline. Proteins expressing an NES are exported from the nucleus by CRM1, which can be inhibited by the antifungal leptomycin B (LMB). One study observed that in HeLa cells after DNA transfection, a portion of cGAS translocated from the nucleus to the cytosol in a CRM1-dependent manner, as LMB treatment blocked this translocation50. This suggests that the cGAS NES is involved in cGAS translocation following activation in the nucleus. However, it should be noted that other studies have failed to see a shift in cGAS localization after DNA transfection in HeLa cells31, so the importance of the NES remains controversial.
Function of Nuclear cGAS
The function of nuclear cGAS is still an open question. Given that nuclear cGAS is held in an inactive state on the chromatin, nuclear tethering may merely be a method of sequestering excess cGAS. In this scenario, nuclear cGAS never becomes untethered or activated, and is held on the chromatin until otherwise degraded. We believe this is unlikely for several reasons. First, other acidic-patch binding proteins that bind with similar affinities, such as RCC1 and HMGN2, are known to be released from chromatin51,52. Therefore, there is precedent for regulated release of acidic patch-binding proteins that may also apply to cGAS. Second, in many cell lines, the majority of cGAS is tethered to chromatin31. It is difficult to imagine that a cell would maintain such a large pool of inactive protein as a “dead end”, rather than target it for degradation when in excess. In fact, given that mutations that untether cGAS from chromatin render it massively and constitutively active31, it seems dangerous for the cell to keep cGAS tethered on chromatin solely as a sequestration mechanism.
It is tempting then to speculate that cGAS can become untethered under certain conditions and participate in immune responses. Given that most DNA viruses, such as herpesviruses and adenoviruses, replicate within the nucleus, nuclear cGAS is uniquely positioned to detect incoming viral DNA. Perhaps during infection with nuclear replicating viruses, viral or host factors can cause cGAS to become released from the chromatin to detect replicating viral DNA. Indeed, one study found that during infection with herpes simplex virus 1 (HSV-1) or adenovirus (AdV), a portion of cGAS accumulates in the nuclear soluble fraction rather than the chromatin-bound fraction, suggesting that some portion of chromatin-bound cGAS can become untethered during DNA virus infection47. An alternative possibility is that cytosolic cGAS is recruited to the nucleus to detect replicating DNA virus.
Because of the high affinity between cGAS and the nucleosome acidic patch, any mechanism to activate nuclear cGAS must overcome this high affinity. In addition to direct displacement from the nucleosome, cGAS tethering may be regulated by post-translational modifications (PTMs) on either cGAS or the histones, which could induce conformational changes or charge differences that could weaken the interaction between cGAS and the nucleosome. Other proteins that bind to the acidic patch are known to be regulated by PTMs. For example, binding of RCC1 is regulated by methylation, while binding of HMGN2 is regulated by phosphorylation51,52. cGAS, which is highly post-translationally modified53, may be regulated in a similar fashion. Several studies have suggested a role for cGAS PTMs in regulating cGAS distribution and chromatin-binding. Phosphorylation of tyrosine 215 by B-lymphoid tyrosine kinase (BLK) was associated with cytosolic retention of cGAS, and nuclear translocation of cGAS following DNA damage or VEGF-A signaling was accompanied by dephosphorylation of cGAS at that residue27,49. Overexpression of a Y215E mutant of cGAS impaired nuclear translocation following DNA damage27. Conversely, methylation of cGAS at K362 by the methyltransferase SUV39H1 was shown to promote nuclear localization of cGAS and tighter binding to the nucleosome54. Although no clear role has yet been ascribed to these PTMs during antiviral responses or autoimmunity, these examples suggest that post-translational modification of cGAS may be an important regulatory mechanism to control cGAS distribution and mobilization from the nucleosome. Nevertheless, many questions still remain about the role of PTMs in regulating cGAS localization. One such unexplored question is the role of histone PTMs in regulating cGAS tethering. Histones, especially histone tails, are also highly modified, and such modification may regulate the interaction between cGAS and the acidic patch. The H4 tail is known to bind to the acidic patch in a manner regulated by acetylation38, with acetylation of key lysines on H4 inhibiting the acidic patch interaction. Deacetylation of the H4 tail would promote acidic patch binding, which could compete with cGAS or decrease the number of available acidic patches for cGAS to bind. Further research should examine the role of the H4 tail acetylation, and other post-translational modifications of both cGAS and histones, in regulating the cGAS/acidic patch interaction. Other factors, such as histone turnover, variant histones, and chromatin condensation, could play a role in modulating cGAS nuclear tethering as well.
To summarize, cGAS is both a cytosolic and nuclear protein, and in many cells the nuclear pool of cGAS represents the majority of total cGAS within the cell. Structural analyses show elegant negative regulation of cGAS activation through tethering to the nucleosomal acidic patch, in which the DNA-binding residues of cGAS are repurposed for binding to the acidic patch and cGAS dimerization is impossible. Mutations within the tethering surface render cGAS massively autoreactive, highlighting the importance of this negative regulatory mechanism. Future research must consider this pool of cGAS and work to dissect the different contributions of the cytosolic and nuclear pool of cGAS under different conditions and in response to different sources of immunostimulatory DNA.
cGAMP is an immunotransmitter that can act at a distance
The generation of cGAMP by activated cGAS results in its rapid diffusion throughout the cell and engagement of STING on the ER membrane. Because cGAMP is such a potent immunostimulatory molecule, the question arose of whether and how cGAMP is metabolized. Cyclic AMP (cAMP) and cyclic GMP (cGMP), two cyclic mononucleotides that regulate cell signaling, are both metabolized by intracellular phosphodiesterases that cleave the intramolecular bond to yield AMP and GMP respectively55. It would make sense, therefore, if an analogous phosphodiesterase could cleave and inactivate cGAMP.
In 2014, Lingyin Li, Tim Mitchison, and colleagues identified the first enzyme that degrades cGAMP: ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1)56. ENPP1 was already known to metabolize ATP into AMP and pyrophosphate, a process that is important for regulation of skeletal and tissue mineralization57. Elegant biochemical and structural studies revealed that ENPP1 metabolizes cGAMP by cleaving both phosphodiester bonds, resulting in the release of GMP and AMP58,59. However, the identification of ENPP1 as the principal cGAMP degrading enzyme raised a conundrum that baffled the field for years and slowed acceptance of ENPP1 as a relevant regulator of cGAMP signaling: ENPP1 is an extracellular enzyme, whereas cGAMP is produced and signals inside of cells. Thus, ENPP1 was not truly analogous to the intracellular cAMP and cGMP phosphodiesterases. cGAMP transfer between certain cell types was known to occur via movement through gap junctions, and the incorporation of cGAMP into viral particles was later described as a means to transfer cGAMP from cell to cell. However, neither of these mechanisms results in exposure of cGAMP to extracellular space. The simple topological question remained: how could an extracellular phosphodiesterase metabolize an intracellular cyclic dinucleotide?
Early studies of cGAMP activity pointed to a potential solution that was not fully appreciated at the time: injection of “naked” cGAMP into mice caused potent STING activation, and the simple addition of cGAMP to cell culture media was sufficient to activate STING signaling60. Because nucleotides cannot passively cross the lipid bilayer of the cell membrane, these findings implied a mechanism of cGAMP import into cells. In 2019, two labs employed CRISPR screens to identify SLC19A1 as the first cGAMP importer61,62. Shortly thereafter, two additional cGAMP importers were identified, SLC46A2 and LRRC8, that mediate cell type specific cGAMP import63–65. Finally, another study found that the antimicrobial peptide LL-37 can mediate cGAMP transfer into cells66.
The question of how cGAMP exits cells remained. Two studies demonstrated that tumor cells produce and release cGAMP that activates STING signaling in neighboring cells to stimulate anti-tumor immunity67,68. In 2022, the ABCC1 transporter was identified as an ATP-dependent cGAMP exporter69. ABCC1, also known as multidrug resistance protein 1 (MRP1), was already well known for its ability to export antibiotics, chemotherapeutic drugs like doxorubicin, and glutathione-conjugated macromolecules70. Loss of ABCC1 potentiated STING signaling within cells and exacerbated disease in the Trex1 knockout mouse model of AGS, a disease that is caused by cGAS-STING signaling. Interestingly, ABCC1-deficient cells retained significant residual cGAMP export that was inhibited by the drug MK-571, which can block additional members of the ABCC exporter family69. This demonstrates the existence of multiple cGAMP exporters that are perhaps structurally related, the identification of which will broaden our understanding of how cGAMP exits cells (Figure 2).
Figure 2. Localization of cGAS.

At homeostasis, two pools of cGAS reside within the cell. The majority of cGAS is localized within the nucleus where it is sequestered and held inactive on the nucleosome acidic patch. A second, smaller pool of cGAS is located within the cytosol and is competent to signal. cGAS can be activated by DNA from pathogenic sources (e.g. viral DNA) or by self-DNA released after mitochondrial or genomic DNA damage. Activation of cGAS stimulates production of the secondary messenger molecule cGAMP. Many open questions remain in the regulation of cGAS distribution, including whether cGAS can be mobilized from the nucleosome, whether cGAS is dynamically trafficked between the nucleus and the cytosol, and how histone modifications impact cGAS tethering on the nucleosome.
Together, the definition of mechanisms of cGAMP export, import, and extracellular degradation have led to the description of cGAMP as an “immunotransmitter” that can signal to distal cells (Figure 2). Conceptually, the biology of the cGAS-cGAMP-STING pathway can now be expanded to include a three-cell model: one cell can produce and export cGAMP, another cell can import cGAMP and signal through STING, and a third cell can contribute the ENPP1 that degrades extracellular cGAMP. Very recently, another ENPP1 paralog, ENPP3, was identified that can also degrade extracellular cGAMP. Interestingly, ENPP1 and ENPP3 have distinct tissue dependent expression patterns, suggesting they are not redundant enzymes71. Whereas ENPP1 and ENPP3 are expressed at steady state, a third inducible cGAMP degrading enzyme, sphingomyelin phosphodiesterase acid-like 3A (SMPDL3A), was recently identified72. SMPDL3A expression is undetectable under steady state conditions, but it is upregulated through activation of Liver X receptor (LXR), which has a well-known role in lipid metabolism72. Thus, the regulation of cGAMP as an immunotransmitter will undoubtedly be further refined in the near future.
cGAS detection of self-DNA
In the initial description of intracellular DNA sensing12, it was postulated that the DNA sensor (identified later as cGAS) might aberrantly sense self-DNA, leading to autoimmunity. Potential sources of this self-DNA include multiple types of DNA damage, both endogenous and exogenous in origin73. These include chromosomal instability, which results from defects in chromosome cohesion, mitotic checkpoint function, centrosome copy number, kinetochoremicrotubule attachment dynamics, and cell-cycle regulation74. Another form of chromosome abnormality is micronuclei, which arise from chromosomes or chromosome fragments that fail to incorporate into daughter nuclei during telophase and recruit their own defective nuclear envelope75. An abundant source of extranuclear self-DNA is contained within mitochondria; this mitochondrial DNA can escape into the cytosol in the context of mitochondrial dysfunction and damage76,77. Additionally, neutrophil extracellular traps (NETs) are an abundant source of extracellular genomic DNA and have been show to activate cGAS after uptake by phagocytes78.
In 2011, damage of genomic DNA was shown to induce IFN production in HeLa cells and primary human monocytes treated with etoposide, thus linking DNA damage and the innate immune response79. It has since been shown that mitotic progression in cells following genotoxic cancer therapy leads to the formation of micronuclei80 and chromatin bridges81, with an ISG signature dependent on the cGAS-STING pathway. Moreover, the cGAS-STING pathway is activated by mitochondrial dysfunction due to loss of apoptotic caspases82,83, defective mitochondrial DNA condensation84, or defects in mitophagy85. Mitochondrial DNA damage and release have been implicated in cGAS responses in several infectious and hereditary diseases, including COVID1986 and ALS87. Z-DNA Binding Protein (ZBP1) was recently identified as a cooperative partner for cGAS sensing of mitochondrial DNA, sustaining and enhancing IFN production88. Together, these and many other studies highlight the ability for cGAS to detect both genomic and mitochondrial DNA.
Perhaps the most compelling demonstrations of the pathological consequences of cGAS detection of self-DNA are the human type I interferonopathies, of which Aicardi-Goutieres Syndrome (AGS) is the most well studied20,89. AGS is a monogenic disorder caused by mutations in any of nine genes that have been defined to date, seven of which have been demonstrated in mouse models to result in activation of the cGAS-STING pathway. First, the DNA exonuclease Trex1 degrades single stranded DNA and is particularly adept at resecting DNA from a nick or discontinuity in dsDNA90. Trex1 was found to exhibit potent anti-retroviral activity against both endogenous retroelements and infectious HIV91,92. Trex1 has also been shown to limit cGAS activity in micronuclei (MN) by resection of micronuclear DNA45. The dNTP phosphohydrolase SAMHD1 limits cellular dNTP pools and is a potent restriction factor for HIV that is antagonized by the virus-encoded Vpx factor93,94. The RNaseH2 enzyme, which is composed of three subunits, each of which can be mutated in AGS, is essential for the removal of misincorporated ribonucleotides that appear with a surprisingly high frequency during genomic DNA replication95,96. The two most recently identified AGS genes, RNU7-1 and LSM11, form part of a ribonucleoprotein complex that is essential for the processing of mRNAs encoding replication-dependent histones97. Fibroblasts from AGS patients harboring these mutations had normal levels of core histones, but the abundance of linker histone H1.4 was significantly reduced, which led to IFN production in a cGAS-STING-dependent manner. Intriguingly, nuclear cGAS in patient fibroblasts was enriched in the nuclear periphery and patient fibroblasts also had more misshapen nuclei, including the presence of nuclear herniations enriched for cGAS97. These data suggest linker histones may regulate cGAS by either “shielding” genomic DNA from cGAS or by maintaining nuclear integrity.
Together, the AGS genes suggest potential sources of endogenous DNA that may contribute to cGAS activation. Two AGS genes encode potent antiretroviral enzymes (TREX1 and SAMHD1), implicating reverse-transcribed endogenous retroelements as a potential source of immunostimulatory DNA. Interestingly, treatment of Trex1−/− mice with reverse transcriptase inhibitors (RTIs) ameliorates disease98, and treatment of TREX1-mutant human AGS patients with RTIs reduces the interferon signature in peripheral blood99. Four of the AGS genes encode enzymes that act to prevent the formation of micronuclei (RNaseH2)100 or metabolize damaged DNA within micronuclei (Trex1)45. Finally, two AGS genes (LSM11, RNU7) contribute to the proper expression of histones, most notably linker histones97. It is worth considering these processes in light of a predominant pool of nucleosome-tethered cGAS. Whereas the cytosolic DNA detection model would require the leakage or ejection of DNA fragments from the nucleus to engage cytosolic cGAS, the existence of a substantial pool of nuclear cGAS raises the strong possibility that each of these DNA abnormalities could be detected in situ, in the nucleus (or micronucleus) itself.
In addition to AGS, other diseases similarly provide insight into cGAS-STING biology. Bloom Syndrome is a rare genetic disease characterized by marked genomic instability resulting from a deficiency in the BLM RecQ-like helicase, a DNA helicase essential for maintaining genome integrity. Bloom Syndrome patient fibroblasts show an increase in micronuclei and an ISG signature dependent on the cGAS-STING pathway101. cGAS activation has also been found in cells from Ataxia-Telangectasia (A-T) patients that have mutations in the ATM kinase102, and cGAS activation has been implicated in neurodegeneration in a rat model of A-T103.Together, the study of human diseases that result in inappropriate cGAS-STING activation, along with new insights into how cGAS can respond to genotoxic stress, reveal an emerging paradigm that places cGAS as a self-referential innate immune sensor positioned to detect perturbations in nuclear and mitochondrial DNA.
In addition to being activated downstream of DNA damage, the cGAS-STING pathway has also recently been implicated in regulating and maintaining DNA damage response (DDR) pathways. However, both the outcome of cGAS-STING signaling on DDR pathways and the mechanism by which these outcomes are produced remain controversial. Several studies have shown that signaling through the cGAS-STING pathway is required to prevent chromosomal instability104–106. Conversely, several studies have pointed to a role for the cGAS-STING pathway in promoting, rather than suppressing, genomic instability through inhibition of homology-directed repair (HDR)27,29,107. Further work is needed in order to elucidate the multifaceted role of cGAS in modulating DDR and future studies should examine more rigorously which pool of cGAS contributes to regulation of DDR. One possibility is that the nuclear pool and the cytosolic pool of cGAS differ in their regulation of DDR.
cGAS-STING in Senescence
Many recent studies have demonstrated a critical role for cGAS in promoting senescence and regulating inflammation in senescent and aging cells25,108–118. Loss of cGAS led to enhanced proliferation and spontaneous immortalization in MEFs25,108, as well as loss of senescence markers such as production of β-galactosidase and cytokines representative of the secretory-associated-senescence phenotype (SASP), suggesting cGAS is important for senescence induced by serial passage. cGAS was also required for senescence induced by DNA damage and oxidative stress, as well as oncogene-induced senescence25,108,109. Initial reports suggested that STING was dispensable for senescence25, however most studies have found that STING is required for cGAS-mediated senescence by activating IRF3 and NF-kB to upregulate the SASP108–114,116.
Senescence is a hallmark of aging, and the cGAS-STING pathway plays a key role in regulating age-induced senescence and inflammation through several different mechanisms. Senescence is accompanied by a loss of Lamin B1 and a corresponding loss of nuclear integrity119,120, potentially leading to activation of cGAS25,108,109. Indeed, cells from patients with aging-related diseases including ataxia telangiectasia, Hutchinson-Gilford progeria (HGPS), and Werner syndrome have evidence for elevated cGAS-STING activation25,108,109,121,122. Senescent cells also downregulate TREX1 and DNase2, leading to enhanced accumulation of cytosolic DNA in senescent cells121. In stromal cells and professional contractile cells, aging was also associated with a loss of YAP/TAZ activity and nuclear localization113. YAP and TAZ are transcriptional coactivators that regulate gene expression downstream of mechanical signaling123. Loss of YAP/TAZ function led to a loss in nuclear integrity that promoted senescence and inflammation primarily through activation of the cGAS-STING pathway, with cGAS shown to accumulate at the nuclear border in complex with DNA113.
Aging is also associated with de-repression of endogenous retroviral elements. Two of these endogenous retroviral elements, Line elements and human endogenous retrovirus K (HERVK) have been shown to induce senescence and inflammaging in both human cells and mice in part by activation of the cGAS-STING pathway and SASP induction110–112,114,116. In human cells harboring mutations that induce progeroid diseases including HGPS and Werner syndrome, upregulation of HERVK activated cGAS-STING signaling to drive senescence and inflammation116.
Mitochondrial dysfunction has also been linked to aging, with two recent studies showing that mitochondrial DNA (mtDNA) released into the cytosol in aged cells activates the cGAS-STING pathway to promote senescence and neurodegeneration117,118. Mitochondrial outer membrane permeabilization (MOMP), a key feature of apoptosis, was found to occur only in a minority of mitochondria in senescent cells, leading to inflammation but not cell death118. This process, termed “minority MOMP” (miMOMP), required BAX and BAK, and drove expression of the SASP in senescent cells through activation of the cGAS-STING pathway. Treatment of senescent fibroblasts with the BAX inhibitor BAI1 reduced mtDNA release and IL-6 secretion, and treatment of aged mice with BAI1 reduced brain inflammation, ameliorated the age-related decline in neuromuscular coordination, improved bone density, and reduced SASP expression118. Similarly, aged mice treated with the STING inhibitor H-151 performed better in tests of strength and memory117. This was accompanied by reduced microgliosis and increased neuronal density in the hippocampus. cGAS-STING activation was suggested to be driven by mtDNA, as aged microglia had misshapen mitochondria and increased amounts of cytosolic mtDNA.
Together, these studies indicate a critical role for cGAS in senescence and inflammaging and point to the cGAS-STING pathway as a potential therapeutic target for progeroid and aging-related diseases. Future studies should investigate in greater depth whether cGAS−/− and STING−/− mice are protected from aging-related conditions and assess the translational potential of cGAS and STING inhibitors in treating progeroid and aging-related disorders. Furthermore, it remains unclear whether this function is unique to cGAS, or whether other PRRs may also contribute to senescence and inflammaging. One study found that senescent cells upregulated RIG-I, and that knockout of RIG-I or MAVS reduced expression and secretion of IL-6 and IL-8, two key SASP genes, in senescent cells124. The intracellular form of Klotho, a protein identified as an anti-aging factor125, was shown to block RIG-I signaling and suppress IL-6 and IL-8 secretion124. Still, the contribution of RIG-I or MDA5 in promoting senescence in vivo remains unclear, and future studies using MAVS−/− and MDA5−/− mice will be needed to establish their relative importance. Nevertheless, it is intriguing to speculate that detection of a variety of mislocalized or aberrantly expressed nucleic acids by diverse PRRs may be a general feature of aging-related senescence and inflammation.
cGAS in cancer biology
Recent advances in immuno-oncology highlight the importance of the immune system in the prevention, control, and elimination of cancer. Many immunotherapies function by helping the body’s own immune system attack and eliminate tumor cells. Immune checkpoint blockade of PD-1/PD-L1 and CTLA4 has revolutionized cancer therapy by unleashing T cell responses to tumors. However, since activation of most adaptive immunity relies on innate immune responses126, it is not surprising that a growing body of evidence highlights the critical role of the innate immune system in these anti-tumor responses.
Innate immune cells can directly detect and eliminate tumor cells, as well as induce and amplify adaptive immune cell responses. Activation and/or manipulation of the innate immune system has unlocked potential new avenues for anti-tumor immunotherapies. The cGAS-STING pathway is particularly well positioned to contribute to anti-tumor immunity: as described above, cGAS can be activated during key processes of oncogenesis by endogenous DNA damage, chromosomal instability, and micronuclei formation; and by DNA damage resulting from therapeutics including chemotherapy and radiation. Gajewski and colleagues demonstrated in 2014 that STING-deficient mice support accelerated tumor growth in numerous transplantable tumor models; this defective anti-tumor immunity was accompanied by weaker tumor-specific cytotoxic T cell responses127. In other work, dendritic cells (DCs) were shown to present tumor antigens and generate anti-tumor CD8 + T cell responses in both humans and mice128. It was further demonstrated that STING signaling in DCs themselves was required for effective tumor antigen cross-presentation and subsequent CD8+ T cell activation129. Together, these findings highlight how the activation of the STING pathway in antigen presenting cells (APCs) contributes to activation of anti-tumor CD8+ T cells.
Natural Killer (NK) cells are another type of innate immune cell that can detect and kill tumor cells directly. Recently it was shown that their activation and effector function also depend on the activation of the STING pathway. Intratumoral injection of STING agonists in the form of cyclic-di-nucleotides (CDNs) causes NK cell-mediated killing of tumor cells that is independent of CD8+ T cell responses. DCs also contributed to NK anti-tumoral activity in vivo through their production of type I IFNs130. Mice depleted of NK cells before tumor implantation were not able to control tumor growth, highlighting the importance of these cells in tumor rejection. Furthermore, in human melanoma patients there is a positive correlation between cGAS expression, NK cell receptor expression, and survival67.
Interestingly, it has been shown that cGAMP produced by the tumor cells themselves is required to activate STING in non-tumor cells and to drive anti-tumor responses. cGAS-deficient mice control growth of implanted tumors as well as wild type mice. However, when tumor cells were deleted for cGAS before implantation into WT mice, tumor growth was no longer restricted67. Further investigation found that many human and mouse cancer cells produce and export cGAMP at steady state, but this exported cGAMP could only be detected when ENPP1 was either genetically deleted or chemically inhibited56,68. Mice deficient for ENPP1 or treated with a chemical ENPP1 inhibitor, showed greater immune activation and control of tumor burdens68. Furthermore, ENPP3 activity can be tumor promoting in some settings, and mice expressing a hydrolase-deficient ENPP3 have fewer lung metastases in a model of melanoma. However, because both tumor cells and the surrounding TME can express different levels of ENPP1 and ENPP3, the contribution of either enzyme in anti-tumor immunity may depend on the tumor type71. cGAMP therefore serves as a potent immunotransmitter from tumor cells to the surrounding non-tumor cells. The recent identification of specific mechanisms of cGAMP export and import raises new questions about how these transporters contribute to anti-tumor immunity. It should also be noted that increased chromosomal instability is also associated with greater metastatic potential, and in some settings, chronic activation of the cGAS-STING pathway in tumors is associated with metastasis131. This suggests that outcomes of STING activation are more nuanced, potentially leading to both tumor restrictive and tumor promoting outcomes depending on context.
Innate immune activation through the cGAS-STING pathway is not only important for cancer therapies that cause DNA damage, but also plays a role in the anti-tumor effects of checkpoint blockade. In mice it was shown that cGAS and STING were required for the antitumor effects of anti-PD-L1 therapy, both in restricting tumor growth and recruitment of tumor specific CD8+ T cells129. This suggests a direct interplay between these two pathways that could be leveraged to improve existing anti-tumor therapies.
Harnessing cGAS biology for therapeutic benefit
Ample human genetic evidence and mouse models have connected the cGAS-STING pathway to numerous human immune diseases, including AGS, familial chilblain lupus (FCL), STING-associated vasculopathy with onset in infancy (SAVI), and some cases of systemic lupus erythematosus (SLE). STING signaling has also been implicated in mouse models of numerous diseases, including Parkinson’s disease, Amyotrophic Lateral Sclerosis (ALS), and Niemann-Pick disease. Conversely, our emerging understand of the anti-cancer functions of cGAS-STING has motivated interest in triggering this axis for cancer therapy. Immense resources in biotech and pharma have been invested in the development of two classes of drugs: cGAS-STING inhibitors, and STING agonists. These efforts have been reviewed recently132; we will summarize them here and consider how our emerging understanding of the biology of this pathway may point to new avenues and applications.
cGAS and STING inhibitors have been reported and are advancing in clinical development132. Obvious applications of such inhibitors include AGS, FCL, SLE, and SAVI. Importantly, the disease mechanisms might warrant the selective use of such antagonists. In particular, mouse models of Niemann-Pick disease have demonstrated that STING activation can occur in the absence of cGAS133; such mutants would benefit from STING inhibitors but not cGAS inhibitors.
More broadly, the emerging contributions of cGAS-STING to chronic conditions associated with aging warrant consideration of the possibility that low-dose, long term STING inhibition might contribute to preserving healthy life span. Just as low dose aspirin is widely used to reduce risk of cardiovascular disease, could STING inhibition broadly improve tissue function and ameliorate inflammaging? At this point, this is more of a rhetorical question, and only the development of safe, inexpensive, orally bioavailable STING inhibitors would enable such a possibility.
In immuno-oncology, STING agonists have shown considerable promise in mouse models for their ability to stimulate inflammation in tumors and enhance antigen presentation and cytotoxic T cell responses134,135. Clinical trials of these STING agonists have been performed in human cancer patients, based on the idea that introduction of a STING agonist might turn immunologically “cold” tumors “hot.” So far, these clinical trials have yielded disappointing results, with a general failure of efficacy either as a monotherapy or in combination with checkpoint blockade136. Although the reasons underlying these outcomes remain unclear, three possibilities may contribute. First, dose-limiting toxicity might prevent the administration of a therapeutic dose of STING agonist. Second, because the efficacy of STING agonists in mouse models requires STING signaling in the host and not in the tumor cells, it is possible that treating “cold” tumors might result in failure to activate sufficient immune cells because they are not present in the tumor at the time of treatment. Third, STING signaling causes toxicity in T cells, which may cripple the very cells that are important for immunotherapy.
Based on the emerging paradigms of nuclear cGAS and the immunotransmitter function of cGAMP, an alternative approach to boost cGAS-STING signaling in tumors could be explored in which the endogenous signaling pathway can be optimized and amplified. Such an approach would require four elements. First, optimizing the activation of cGAS in tumor cells to maximize intratumoral cGAMP production. This will require an understanding of how cGAS activation is restrained in the context of chemotherapy and radiation. A simple way to bypass such restraint would be to introduce constitutively active cGAS into tumors137. Second, enhancing cGAMP export from tumor cells, in part by identifying those tumors with high ABCC1 expression that closely correlates with efficiency of cGAMP export69. Third, promoting cGAMP import into the relevant immune cells that contribute to anti-tumor immunity while perhaps avoiding cGAMP import into T cells that may restrain their function23. Finally, preventing degradation of extracellular cGAMP by inhibiting ENPP1138, ENPP3, and perhaps SMPDL3A. While this approach is currently aspirational, a first step would be to leverage the deep data sets and single cell atlases of gene expression within tumors to identify those with tumor cells that express the most cGAS and ABCC1, those that contain the highest frequency of immune cells that are important for the response to cGAMP, and those that would be most responsive to inhibition of ENPP1, ENPP3, and SMPDL3A. Just as tumor genomics and expression profiles now contribute to the decision of whether to administer checkpoint blockade, we predict that an analogous set of criteria focusing on cGAS-STING biology might inform how to enhance endogenous activation of this pathway in tumors.
Concluding thoughts
The cGAS-STING DNA sensing pathway has emerged as a central mediator of host defense, a cause of numerous human immune diseases, a contributor to fundamental pathological processes of senescence and aging, and a new avenue for triggering inflammation in tumors to improve anti-cancer immunity. All of these myriad roles for cGAS can be attributed to its ability to be activated by any double stranded DNA of any origin. The study of the high potential for self-reactivity of cGAS has revealed elegant new regulatory mechanisms that restrain cGAS and touch on broad swaths cell biology. Thus, a new frontier of cGAS-STING biology will be to leverage our understanding of this cGAS regulation to develop new ways to treat a variety of human diseases, from autoimmunity to inflammaging to cancer.
Figure 3. cGAMP signaling and regulation.

cGAMP can act in both an autocrine and paracrine manner when activating STING. cGAMP is exported from cells by ABCC1 and other unknown transporter(s). Export can be chemically inhibited by MK-571, which acts on several ATP-binding cassette (ABC) transporters including ABCC1. cGAMP import then occurs through several known importers, (SLC19A1, SLC46A2, and LRRC8), thus allowing cGAMP to act as an immunotransmitter. Concentrations of extracellular cGAMP can be regulated via degradation by both membrane-bound ENPP1 and ENPP3, or soluble ENPP1. SMPDL3A, another cGAMP degrading enzyme, may act both intra and extracellularly.
Acknowledgements
We thank members of the Stetson lab for insightful discussions. DBS is an advisor for Related Sciences and a co-founder of DangerBio. This work was supported by a National Science Foundation Graduate Research Fellowship to S.D., and by RO1 awards AI150716 and AI084914.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Isaacs A, and Lindenmann J (1957). Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147, 258–267. [PubMed] [Google Scholar]
- 2.Isaacs A, Lindenmann J, and Valentine RC (1957). Virus interference. II. Some properties of interferon. Proc R Soc Lond B Biol Sci 147, 268–273. [PubMed] [Google Scholar]
- 3.Stark GR, Kerr IM, Williams BR, Silverman RH, and Schreiber RD (1998). How cells respond to interferons. Annu Rev Biochem 67, 227–264. [DOI] [PubMed] [Google Scholar]
- 4.Lind NA, Rael VE, Pestal K, Liu B, and Barton GM (2022). Regulation of the nucleic acid-sensing Toll-like receptors. Nat Rev Immunol 22, 224–235. 10.1038/s41577-021-00577-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goubau D, Deddouche S, and Reis ESC (2013). Cytosolic sensing of viruses. Immunity 38, 855–869. 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kato H, Takahasi K, and Fujita T (2011). RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol Rev 243, 91–98. 10.1111/j.1600-065X.2011.01052.x. [DOI] [PubMed] [Google Scholar]
- 7.Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ergun SL, Fernandez D, Weiss TM, and Li L (2019). STING Polymer Structure Reveals Mechanisms for Activation, Hyperactivation, and Inhibition. Cell 178, 290–301 e210. 10.1016/j.cell.2019.05.036. [DOI] [PubMed] [Google Scholar]
- 9.Shang G, Zhang C, Chen ZJ, Bai XC, and Zhang X (2019). Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 567, 389–393. 10.1038/s41586-019-0998-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Bai XC, and Chen ZJ (2020). Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 53, 43–53. 10.1016/j.immuni.2020.05.013. [DOI] [PubMed] [Google Scholar]
- 11.Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, et al. (2006). A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol 7, 40–48. [DOI] [PubMed] [Google Scholar]
- 12.Stetson DB, and Medzhitov R (2006). Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103. [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. nature07317 [pii] 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, and Shu HB (2008). The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550. 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 15.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slavik KM, and Kranzusch PJ (2023). CBASS to cGAS-STING: The Origins and Mechanisms of Nucleotide Second Messenger Immune Signaling. Annu Rev Virol 10, 423–453. 10.1146/annurev-virology-111821-115636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slavik KM, Morehouse BR, Ragucci AE, Zhou W, Ai X, Chen Y, Li L, Wei Z, Bahre H, Konig M, et al. (2021). cGAS-like receptors sense RNA and control 3’2’-cGAMP signalling in Drosophila. Nature 597, 109–113. 10.1038/s41586-021-03743-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holleufer A, Winther KG, Gad HH, Ai X, Chen Y, Li L, Wei Z, Deng H, Liu J, Frederiksen NA, et al. (2021). Two cGAS-like receptors induce antiviral immunity in Drosophila. Nature 597, 114–118. 10.1038/s41586-021-03800-z. [DOI] [PubMed] [Google Scholar]
- 19.Cai H, Li L, Slavik KM, Huang J, Yin T, Ai X, Hedelin L, Haas G, Xiang Z, Yang Y, et al. (2023). The virus-induced cyclic dinucleotide 2’3’-c-di-GMP mediates STING-dependent antiviral immunity in Drosophila. Immunity 56, 1991–2005 e1999. 10.1016/j.immuni.2023.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crow YJ, and Manel N (2015). Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15, 429–440. 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 21.Uggenti C, Lepelley A, and Crow YJ (2019). Self-Awareness: Nucleic Acid-Driven Inflammation and the Type I Interferonopathies. Annu Rev Immunol 37, 247–267. 10.1146/annurev-immunol-042718-041257. [DOI] [PubMed] [Google Scholar]
- 22.Crowl JT, Gray EE, Pestal K, Volkman HE, and Stetson DB (2017). Intracellular Nucleic Acid Detection in Autoimmunity. Annu Rev Immunol 35, 313–336. 10.1146/annurev-immunol-051116-052331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ritchie C, Carozza JA, and Li L (2022). Biochemistry, Cell Biology, and Pathophysiology of the Innate Immune cGAS-cGAMP-STING Pathway. Annu Rev Biochem. 10.1146/annurev-biochem-040320-101629. [DOI] [PubMed] [Google Scholar]
- 24.Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, and Knipe DM (2015). cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112, E1773–1781. 10.1073/pnas.1424637112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Wang H, Ren J, Chen Q, and Chen ZJ (2017). cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A 114, E4612–E4620. 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lahaye X, Gentili M, Silvin A, Conrad C, Picard L, Jouve M, Zueva E, Maurin M, Nadalin F, Knott GJ, et al. (2018). NONO Detects the Nuclear HIV Capsid to Promote cGAS-Mediated Innate Immune Activation. Cell 175, 488–501 e422. 10.1016/j.cell.2018.08.062. [DOI] [PubMed] [Google Scholar]
- 27.Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, Jiang Y, Fei Y, Zhu C, Tan R, et al. (2018). Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136. 10.1038/s41586-018-0629-6. [DOI] [PubMed] [Google Scholar]
- 28.Gentili M, Lahaye X, Nadalin F, Nader GPF, Lombardi EP, Herve S, De Silva NS, Rookhuizen DC, Zueva E, Goudot C, et al. (2019). The N-Terminal Domain of cGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep 26, 3798. 10.1016/j.celrep.2019.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang H, Xue X, Panda S, Kawale A, Hooy RM, Liang F, Sohn J, Sung P, and Gekara NO (2019). Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J, e102718. 10.15252/embj.2019102718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, and Funabiki H (2019). The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 178, 302–315 e323. 10.1016/j.cell.2019.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volkman HE, Cambier S, Gray EE, and Stetson DB (2019). Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife 8. 10.7554/eLife.47491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyer JA, Spangler CJ, Strauss JD, Cesmat AP, Liu P, McGinty RK, and Zhang Q (2020). Structural basis of nucleosome-dependent cGAS inhibition. Science 370, 450–454. 10.1126/science.abd0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao D, Han X, Fan X, Xu RM, and Zhang X (2020). Structural basis for nucleosome-mediated inhibition of cGAS activity. Cell Res 30, 1088–1097. 10.1038/s41422-020-00422-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, Hirai S, Funabiki H, and Kurumizaka H (2020). Structural basis for the inhibition of cGAS by nucleosomes. Science 370, 455–458. 10.1126/science.abd0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michalski S, de Oliveira Mann CC, Stafford CA, Witte G, Bartho J, Lammens K, Hornung V, and Hopfner KP (2020). Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 587, 678–682. 10.1038/s41586-020-2748-0. [DOI] [PubMed] [Google Scholar]
- 36.Pathare GR, Decout A, Gluck S, Cavadini S, Makasheva K, Hovius R, Kempf G, Weiss J, Kozicka Z, Guey B, et al. (2020). Structural mechanism of cGAS inhibition by the nucleosome. Nature 587, 668–672. 10.1038/s41586-020-2750-6. [DOI] [PubMed] [Google Scholar]
- 37.Zhao B, Xu P, Rowlett CM, Jing T, Shinde O, Lei Y, West AP, Liu WR, and Li P (2020). The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature 587, 673–677. 10.1038/s41586-020-2749-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalashnikova AA, Porter-Goff ME, Muthurajan UM, Luger K, and Hansen JC (2013). The role of the nucleosome acidic patch in modulating higher order chromatin structure. J R Soc Interface 10, 20121022. 10.1098/rsif.2012.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGinty RK, and Tan S (2021). Principles of nucleosome recognition by chromatin factors and enzymes. Curr Opin Struct Biol 71, 16–26. 10.1016/j.sbi.2021.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, Brautigam CA, Zhang X, and Chen ZJ (2014). The Cytosolic DNA Sensor cGAS Forms an Oligomeric Complex with DNA and Undergoes Switch-like Conformational Changes in the Activation Loop. Cell Rep 6, 421–430. 10.1016/j.celrep.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andreeva L, Hiller B, Kostrewa D, Lassig C, de Oliveira Mann CC, Jan Drexler D, Maiser A, Gaidt M, Leonhardt H, Hornung V, and Hopfner KP (2017). cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 549, 394–398. 10.1038/nature23890. [DOI] [PubMed] [Google Scholar]
- 42.Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, Gray NS, Mekalanos JJ, and Kranzusch PJ (2018). Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 174, 300–311 e311. 10.1016/j.cell.2018.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie W, Lama L, Adura C, Tomita D, Glickman JF, Tuschl T, and Patel DJ (2019). Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc Natl Acad Sci U S A 116, 11946–11955. 10.1073/pnas.1905013116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li T, Huang T, Du M, Chen X, Du F, Ren J, and Chen ZJ (2021). Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science 371. 10.1126/science.abc5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohr L, Toufektchan E, von Morgen P, Chu K, Kapoor A, and Maciejowski J (2021). ER-directed TREX1 limits cGAS activation at micronuclei. Mol Cell 81, 724–738 e729. 10.1016/j.molcel.2020.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barnett KC, Coronas-Serna JM, Zhou W, Ernandes MJ, Cao A, Kranzusch PJ, and Kagan JC (2019). Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 176, 1432–1446. 10.1016/j.cell.2019.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu Y, Song K, Hao W, Li J, Wang L, and Li S (2022). Nuclear soluble cGAS senses double-stranded DNA virus infection. Commun Biol 5, 433. 10.1038/s42003-022-03400-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cui S, Yu Q, Chu L, Cui Y, Ding M, Wang Q, Wang H, Chen Y, Liu X, and Wang C (2020). Nuclear cGAS Functions Non-canonically to Enhance Antiviral Immunity via Recruiting Methyltransferase Prmt5. Cell Rep 33, 108490. 10.1016/j.celrep.2020.108490. [DOI] [PubMed] [Google Scholar]
- 49.Luo J, Lu C, Chen Y, Wu X, Zhu C, Cui W, Yu S, Li N, Pan Y, Zhao W, et al. (2023). Nuclear translocation of cGAS orchestrates VEGF-A-mediated angiogenesis. Cell Rep 42, 112328. 10.1016/j.celrep.2023.112328. [DOI] [PubMed] [Google Scholar]
- 50.Sun H, Huang Y, Mei S, Xu F, Liu X, Zhao F, Yin L, Zhang D, Wei L, Wu C, et al. (2021). A Nuclear Export Signal Is Required for cGAS to Sense Cytosolic DNA. Cell Rep 34, 108586. 10.1016/j.celrep.2020.108586. [DOI] [PubMed] [Google Scholar]
- 51.Hao Y, and Macara IG (2008). Regulation of chromatin binding by a conformational switch in the tail of the Ran exchange factor RCC1. J Cell Biol 182, 827–836. 10.1083/jcb.200803110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prymakowska-Bosak M, Misteli T, Herrera JE, Shirakawa H, Birger Y, Garfield S, and Bustin M (2001). Mitotic phosphorylation prevents the binding of HMGN proteins to chromatin. Mol Cell Biol 21, 5169–5178. 10.1128/MCB.21.15.5169-5178.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu Y, and Li S (2020). Role of Post-Translational Modifications of cGAS in Innate Immunity. Int J Mol Sci 21. 10.3390/ijms21217842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fang L, Hao Y, Yu H, Gu X, Peng Q, Zhuo H, Li Y, Liu Z, Wang J, Chen Y, et al. (2023). Methionine restriction promotes cGAS activation and chromatin untethering through demethylation to enhance antitumor immunity. Cancer Cell 41, 1118–1133 e1112. 10.1016/j.ccell.2023.05.005. [DOI] [PubMed] [Google Scholar]
- 55.Bender AT, and Beavo JA (2006). Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58, 488–520. 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 56.Li L, Yin Q, Kuss P, Maliga Z, Millan JL, Wu H, and Mitchison TJ (2014). Hydrolysis of 2’3’-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat Chem Biol 10, 1043–1048. 10.1038/nchembio.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kato K, Nishimasu H, Okudaira S, Mihara E, Ishitani R, Takagi J, Aoki J, and Nureki O (2012). Crystal structure of Enpp1, an extracellular glycoprotein involved in bone mineralization and insulin signaling. Proc Natl Acad Sci U S A 109, 16876–16881. 10.1073/pnas.1208017109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kato K, Nishimasu H, Oikawa D, Hirano S, Hirano H, Kasuya G, Ishitani R, Tokunaga F, and Nureki O (2018). Structural insights into cGAMP degradation by Ecto-nucleotide pyrophosphatase phosphodiesterase 1. Nat Commun 9, 4424. 10.1038/s41467-018-06922-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carozza JA, Brown JA, Bohnert V, Fernandez D, AlSaif Y, Mardjuki RE, Smith M, and Li L (2020). Structure-Aided Development of Small-Molecule Inhibitors of ENPP1, the Extracellular Phosphodiesterase of the Immunotransmitter cGAMP. Cell Chem Biol 27, 1347–1358 e1345. 10.1016/j.chembiol.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li XD, Wu J, Gao D, Wang H, Sun L, and Chen ZJ (2013). Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394. 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ritchie C, Cordova AF, Hess GT, Bassik MC, and Li L (2019). SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol Cell 75, 372–381 e375. 10.1016/j.molcel.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luteijn RD, Zaver SA, Gowen BG, Wyman SK, Garelis NE, Onia L, McWhirter SM, Katibah GE, Corn JE, Woodward JJ, and Raulet DH (2019). SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438. 10.1038/s41586-019-1553-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou C, Chen X, Planells-Cases R, Chu J, Wang L, Cao L, Li Z, Lopez-Cayuqueo KI, Xie Y, Ye S, et al. (2020). Transfer of cGAMP into Bystander Cells via LRRC8 Volume-Regulated Anion Channels Augments STING-Mediated Interferon Responses and Anti-viral Immunity. Immunity 52, 767–781 e766. 10.1016/j.immuni.2020.03.016. [DOI] [PubMed] [Google Scholar]
- 64.Lahey LJ, Mardjuki RE, Wen X, Hess GT, Ritchie C, Carozza JA, Bohnert V, Maduke M, Bassik MC, and Li L (2020). LRRC8A:C/E Heteromeric Channels Are Ubiquitous Transporters of cGAMP. Mol Cell 80, 578–591 e575. 10.1016/j.molcel.2020.10.021. [DOI] [PubMed] [Google Scholar]
- 65.Cordova AF, Ritchie C, Bohnert V, and Li L (2021). Human SLC46A2 Is the Dominant cGAMP Importer in Extracellular cGAMP-Sensing Macrophages and Monocytes. ACS Cent Sci 7, 1073–1088. 10.1021/acscentsci.1c00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wei X, Zhang L, Yang Y, Hou Y, Xu Y, Wang Z, Su H, Han F, Han J, Liu P, et al. (2022). LL-37 transports immunoreactive cGAMP to activate STING signaling and enhance interferon-mediated host antiviral immunity. Cell Rep 39, 110880. 10.1016/j.celrep.2022.110880. [DOI] [PubMed] [Google Scholar]
- 67.Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, and Raulet DH (2018). Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763 e754. 10.1016/j.immuni.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carozza JA, Bohnert V, Nguyen KC, Skariah G, Shaw KE, Brown JA, Rafat M, von Eyben R, Graves EE, Glenn JS, et al. (2020). Extracellular cGAMP is a cancer cell-produced immunotransmitter involved in radiation-induced anti-cancer immunity. Nat Cancer 1, 184–196. 10.1038/s43018-020-0028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maltbaek JH, Cambier S, Snyder JM, and Stetson DB (2022). ABCC1 transporter exports the immunostimulatory cyclic dinucleotide cGAMP. Immunity 55, 1799–1812 e1794. 10.1016/j.immuni.2022.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cole SP (2014). Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu Rev Pharmacol Toxicol 54, 95–117. 10.1146/annurevpharmtox-011613-135959. [DOI] [PubMed] [Google Scholar]
- 71.Mardjuki R, Wang S, Carozza JA, Abhiraman GC, Lyu X, and Li L (2024). Identification of extracellular membrane protein ENPP3 as a major cGAMP hydrolase, cementing cGAMP’s role as an immunotransmitter. bioRxiv. 10.1101/2024.01.12.575449. [DOI] [PubMed] [Google Scholar]
- 72.Hou Y, Wang Z, Liu P, Wei X, Zhang Z, Fan S, Zhang L, Han F, Song Y, Chu L, and Zhang C (2023). SMPDL3A is a cGAMP-degrading enzyme induced by LXR-mediated lipid metabolism to restrict cGAS-STING DNA sensing. Immunity 56, 2492–2507 e2410. 10.1016/j.immuni.2023.10.001. [DOI] [PubMed] [Google Scholar]
- 73.Chatterjee N, and Walker GC (2017). Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 58, 235–263. 10.1002/em.22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thompson SL, Bakhoum SF, and Compton DA (2010). Mechanisms of chromosomal instability. Curr Biol 20, R285–295. 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Krupina K, Goginashvili A, and Cleveland DW (2021). Causes and consequences of micronuclei. Curr Opin Cell Biol 70, 91–99. 10.1016/j.ceb.2021.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.West AP, and Shadel GS (2017). Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17, 363–375. 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rongvaux A (2018). Innate immunity and tolerance toward mitochondria. Mitochondrion 41, 14–20. 10.1016/j.mito.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 78.Apel F, Andreeva L, Knackstedt LS, Streeck R, Frese CK, Goosmann C, Hopfner KP, and Zychlinsky A (2021). The cytosolic DNA sensor cGAS recognizes neutrophil extracellular traps. Sci Signal 14. 10.1126/scisignal.aax7942. [DOI] [PubMed] [Google Scholar]
- 79.Brzostek-Racine S, Gordon C, Van Scoy S, and Reich NC (2011). The DNA damage response induces IFN. J Immunol 187, 5336–5345. 10.4049/jimmunol.1100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Flynn PJ, Koch PD, and Mitchison TJ (2021). Chromatin bridges, not micronuclei, activate cGAS after drug-induced mitotic errors in human cells. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2103585118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, et al. (2014). Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577. 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME, et al. (2014). Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562. 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, et al. (2018). Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262. 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 86.Domizio JD, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, Nass T, Guenova E, Schaller M, Conrad C, et al. (2022). The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature 603, 145–151. 10.1038/s41586-022-04421-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, Louis C, Low RRJ, Moecking J, De Nardo D, et al. (2020). TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 183, 636–649 e618. 10.1016/j.cell.2020.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lei Y, VanPortfliet JJ, Chen YF, Bryant JD, Li Y, Fails D, Torres-Odio S, Ragan KB, Deng J, Mohan A, et al. (2023). Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. Cell 186, 3013–3032 e3022. 10.1016/j.cell.2023.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Crow YJ, and Stetson DB (2022). The type I interferonopathies: 10 years on. Nat Rev Immunol 22, 471–483. 10.1038/s41577-021-00633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, and Perrino FW (2008). The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. J Biol Chem 283, 31649–31656. M806155200 [pii] 10.1074/jbc.M806155200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stetson DB, Ko JS, Heidmann T, and Medzhitov R (2008). Trex1 prevents cellintrinsic initiation of autoimmunity. Cell 134, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, and Lieberman J (2010). The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol 11, 1005–1013. ni.1941 [pii] 10.1038/ni.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, et al. (2009). Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41, 829–832. ng.373 [pii] 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, and Benkirane M (2011). SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657. 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, et al. (2012). Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149, 1008–1022. 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B, et al. (2016). Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J 35, 831–844. 10.15252/embj.201593339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, Rice GI, Dhir S, Wheeler AP, Dhir A, et al. (2020). cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet 52, 1364–1372. 10.1038/s41588-020-00737-3. [DOI] [PubMed] [Google Scholar]
- 98.Beck-Engeser GB, Eilat D, and Wabl M (2011). An autoimmune disease prevented by anti-retroviral drugs. Retrovirology 8, 91. 10.1186/1742-4690-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rice GI, Meyzer C, Bouazza N, Hully M, Boddaert N, Semeraro M, Zeef LAH, Rozenberg F, Bondet V, Duffy D, et al. (2018). Reverse-Transcriptase Inhibitors in the Aicardi-Goutieres Syndrome. N Engl J Med 379, 2275–2277. 10.1056/NEJMc1810983. [DOI] [PubMed] [Google Scholar]
- 100.Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gratia M, Rodero MP, Conrad C, Bou Samra E, Maurin M, Rice GI, Duffy D, Revy P, Petit F, Dale RC, et al. (2019). Bloom syndrome protein restrains innate immune sensing of micronuclei by cGAS. J Exp Med 216, 1199–1213. 10.1084/jem.20181329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, Lienenklaus S, Nilsson LM, Kroger A, Nilsson JA, et al. (2015). DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote antimicrobial innate immunity. Immunity 42, 332–343. 10.1016/j.immuni.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 103.Quek H, Luff J, Cheung K, Kozlov S, Gatei M, Lee CS, Bellingham MC, Noakes PG, Lim YC, Barnett NL, et al. (2017). A rat model of ataxia-telangiectasia: evidence for a neurodegenerative phenotype. Hum Mol Genet 26, 109–123. 10.1093/hmg/ddw371. [DOI] [PubMed] [Google Scholar]
- 104.Ranoa DRE, Widau RC, Mallon S, Parekh AD, Nicolae CM, Huang X, Bolt MJ, Arina A, Parry R, Kron SJ, et al. (2019). STING Promotes Homeostasis via Regulation of Cell Proliferation and Chromosomal Stability. Cancer Res 79, 1465–1479. 10.1158/0008-5472.CAN-18-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Basit A, Cho MG, Kim EY, Kwon D, Kang SJ, and Lee JH (2020). The cGAS/STING/TBK1/IRF3 innate immunity pathway maintains chromosomal stability through regulation of p21 levels. Exp Mol Med 52, 643–657. 10.1038/s12276-020-0416-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen H, Chen H, Zhang J, Wang Y, Simoneau A, Yang H, Levine AS, Zou L, Chen Z, and Lan L (2020). cGAS suppresses genomic instability as a decelerator of replication forks. Sci Adv 6. 10.1126/sciadv.abb8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Banerjee D, Langberg K, Abbas S, Odermatt E, Yerramothu P, Volaric M, Reidenbach MA, Krentz KJ, Rubinstein CD, Brautigan DL, et al. (2021). A non-canonical, interferon-independent signaling activity of cGAMP triggers DNA damage response signaling. Nat Commun 12, 6207. 10.1038/s41467-021-26240-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, and Ablasser A (2017). Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19, 1061–1070. 10.1038/ncb3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, et al. (2019). L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78. 10.1038/s41586-018-0784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Simon M, Van Meter M, Ablaeva J, Ke Z, Gonzalez RS, Taguchi T, De Cecco M, Leonova KI, Kogan V, Helfand SL, et al. (2019). LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab 29, 871–885 e875. 10.1016/j.cmet.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bi S, Liu Z, Wu Z, Wang Z, Liu X, Wang S, Ren J, Yao Y, Zhang W, Song M, et al. (2020). SIRT7 antagonizes human stem cell aging as a heterochromatin stabilizer. Protein Cell 11, 483–504. 10.1007/s13238-020-00728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sladitschek-Martens HL, Guarnieri A, Brumana G, Zanconato F, Battilana G, Xiccato RL, Panciera T, Forcato M, Bicciato S, Guzzardo V, et al. (2022). YAP/TAZ activity in stromal cells prevents ageing by controlling cGAS-STING. Nature 607, 790–798. 10.1038/s41586-022-04924-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liang C, Ke Q, Liu Z, Ren J, Zhang W, Hu J, Wang Z, Chen H, Xia K, Lai X, et al. (2022). BMAL1 moonlighting as a gatekeeper for LINE1 repression and cellular senescence in primates. Nucleic Acids Res 50, 3323–3347. 10.1093/nar/gkac146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang D, Liu Y, Zhu Y, Zhang Q, Guan H, Liu S, Chen S, Mei C, Chen C, Liao Z, et al. (2022). A non-canonical cGAS-STING-PERK pathway facilitates the translational program critical for senescence and organ fibrosis. Nat Cell Biol 24, 766–782. 10.1038/s41556-022-00894-z. [DOI] [PubMed] [Google Scholar]
- 116.Liu X, Liu Z, Wu Z, Ren J, Fan Y, Sun L, Cao G, Niu Y, Zhang B, Ji Q, et al. (2023). Resurrection of endogenous retroviruses during aging reinforces senescence. Cell 186, 287–304 e226. 10.1016/j.cell.2022.12.017. [DOI] [PubMed] [Google Scholar]
- 117.Gulen MF, Samson N, Keller A, Schwabenland M, Liu C, Gluck S, Thacker VV, Favre L, Mangeat B, Kroese LJ, et al. (2023). cGAS-STING drives ageing-related inflammation and neurodegeneration. Nature 620, 374–380. 10.1038/s41586-023-06373-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Victorelli S, Salmonowicz H, Chapman J, Martini H, Vizioli MG, Riley JS, Cloix C, Hall-Younger E, Machado Espindola-Netto J, Jurk D, et al. (2023). Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature 622, 627–636. 10.1038/s41586-023-06621-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Freund A, Laberge RM, Demaria M, and Campisi J (2012). Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 23, 2066–2075. 10.1091/mbc.E11-10-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, et al. (2013). Lysosome-mediated processing of chromatin in senescence. J Cell Biol 202, 129–143. 10.1083/jcb.201212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, Watanabe S, Kawamoto S, Miyata K, Barber GN, et al. (2018). Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 9, 1249. 10.1038/s41467-018-03555-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lan YY, Heather JM, Eisenhaure T, Garris CS, Lieb D, Raychowdhury R, and Hacohen N (2019). Extranuclear DNA accumulates in aged cells and contributes to senescence and inflammation. Aging Cell 18, e12901. 10.1111/acel.12901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kechagia JZ, Ivaska J, and Roca-Cusachs P (2019). Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol 20, 457–473. 10.1038/s41580-019-0134-2. [DOI] [PubMed] [Google Scholar]
- 124.Liu F, Wu S, Ren H, and Gu J (2011). Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat Cell Biol 13, 254–262. 10.1038/ncb2167. [DOI] [PubMed] [Google Scholar]
- 125.Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, et al. (2005). Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833. 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Iwasaki A, and Medzhitov R (2015). Control of adaptive immunity by the innate immune system. Nat Immunol 16, 343–353. 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. (2014). Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 26, 938. 10.1016/j.ccell.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 129.Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, and Chen ZJ (2017). cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A 114, 1637–1642. 10.1073/pnas.1621363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nicolai CJ, Wolf N, Chang IC, Kirn G, Marcus A, Ndubaku CO, McWhirter SM, and Raulet DH (2020). NK cells mediate clearance of CD8(+) T cell-resistant tumors in response to STING agonists. Sci Immunol 5. 10.1126/sciimmunol.aaz2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. 10.1038/nature25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.McWhirter SM, and Jefferies CA (2020). Nucleic Acid Sensors as Therapeutic Targets for Human Disease. Immunity 53, 78–97. 10.1016/j.immuni.2020.04.004. [DOI] [PubMed] [Google Scholar]
- 133.Chu TT, Tu X, Yang K, Wu J, Repa JJ, and Yan N (2021). Tonic prime-boost of STING signalling mediates Niemann-Pick disease type C. Nature 596, 570–575. 10.1038/s41586-021-03762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, Hudson TE, Vu UT, Francica BJ, Banda T, et al. (2018). Magnitude of Therapeutic STING Activation Determines CD8(+) T Cell-Mediated Anti-tumor Immunity. Cell Rep 25, 3074–3085 e3075. 10.1016/j.celrep.2018.11.047. [DOI] [PubMed] [Google Scholar]
- 135.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al. (2015). Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 11, 1018–1030. 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Meric-Bernstam F, Sweis RF, Kasper S, Hamid O, Bhatia S, Dummer R, Stradella A, Long GV, Spreafico A, Shimizu T, et al. (2023). Combination of the STING Agonist MIW815 (ADU-S100) and PD-1 Inhibitor Spartalizumab in Advanced/Metastatic Solid Tumors or Lymphomas: An Open-Label, Multicenter, Phase Ib Study. Clin Cancer Res 29, 110–121. 10.1158/1078-0432.CCR-22-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dowling QM, Volkman HE, Gray EE, Ovchinnikov S, Cambier S, Bera AK, Sankaran B, Johnson MR, Bick MJ, Kang A, et al. (2023). Computational design of constitutively active cGAS. Nat Struct Mol Biol 30, 72–80. 10.1038/s41594-022-00862-z. [DOI] [PubMed] [Google Scholar]
- 138.Carozza JA, Cordova AF, Brown JA, AlSaif Y, Bohnert V, Cao X, Mardjuki RE, Skariah G, Fernandez D, and Li L (2022). ENPP1’s regulation of extracellular cGAMP is a ubiquitous mechanism of attenuating STING signaling. Proc Natl Acad Sci U S A 119, e2119189119. 10.1073/pnas.2119189119. [DOI] [PMC free article] [PubMed] [Google Scholar]