

SUMMARY
Pyridoxal 5′-phosphate (PLP), the biologically active form of vitamin B6, is an essential cofactor in many biosynthetic pathways. The emergence of PLP-dependent enzymes as drug targets and biocatalysts, such as tryptophan synthase (TS), has underlined the demand to understand PLP-dependent catalysis and reaction specificity. The ability of neutron diffraction to resolve the positions of hydrogen atoms makes it an ideal technique to understand how the electrostatic environment and selective protonation of PLP regulates PLP-dependent activities. Facilitated by microgravity crystallization of TS with the Toledo Crystallization Box, we report the 2.1 Å joint X-ray/neutron (XN) structure of TS with PLP in the internal aldimine form. Positions of hydrogens were directly determined in both the α- and β-active sites, including PLP cofactor. The joint XN structure thus provides insight into the selective protonation of the internal aldimine and the electrostatic environment of TS necessary to understand the overall catalytic mechanism.
Graphical Abstract

Drago et al. visualize elusive hydrogen atoms in tryptophan synthase with neutron crystallography. The 2.1 Å joint X-ray/neutron structure provides insight into the enzyme electrostatics and selective protonation of the PLP cofactor that govern the catalytic mechanism.
INTRODUCTION
Pyridoxal 5′-phosphate (PLP) cofactor is the bioactive form of vitamin B6 found in all kingdoms of life and essential for a myriad of cellular processes (Figure 1A). The enzymes utilizing this ubiquitous cofactor belong to a highly versatile family of biocatalysts that primarily exist in metabolic pathways involving amino acids.1 The family of PLP-dependent enzymes is divided into five major fold-types, I through V, and facilitates a diverse range of chemical transformations such as transamination, β- and γ-elimination, decarboxylation, retro-aldol cleavage and aldol addition, racemization, replacement reactions, and glycogen phosphorylation.2,3 Many PLP-dependent enzymes, such as serine hydroxymethyltransferase, DOPA decarboxylase, and alanine racemase, have been implicated as pharmaceutical targets,4 while others, including aspartate aminotransferase, tyrosine decarboxylase, and tryptophan synthase (TS), are of great interest in enzyme engineering applications for the syntheses of unnatural amino acids and other value-added products.5–9 The question remains as to how the PLP cofactor is modulated by this large family of enzymes to achieve extremely diverse reaction specificity. The internal aldimine (Figure 1A) consists of the PLP cofactor covalently bound via a Schiff base linkage between the catalytic Lys ε-amine and the aldehyde moiety of PLP. Gem-diamine formation promotes the conversion of the enzyme-bound PLP internal aldimine into the substrate-coupled external aldimine (Figure 1B).10 Reaction specificity occurs when the electronics of the resultant PLP-substrate intermediate are modulated by the entire active site environment.11,12 To better understand PLP reaction specificities, catalytic mechanisms of PLP-dependent enzymes are deciphered at the atomic level using spectroscopic and structural analyses. Computational analysis is then used to determine the energy landscape based on these models. To acquire an accurate depiction of the catalytic process, knowledge of the positions of all atoms in the enzyme, including the mapping of proton movements throughout the Brønsted acid-base catalysis orchestrated by the PLP cofactor, is essential.13,14
Figure 1. Biochemistry of PLP.
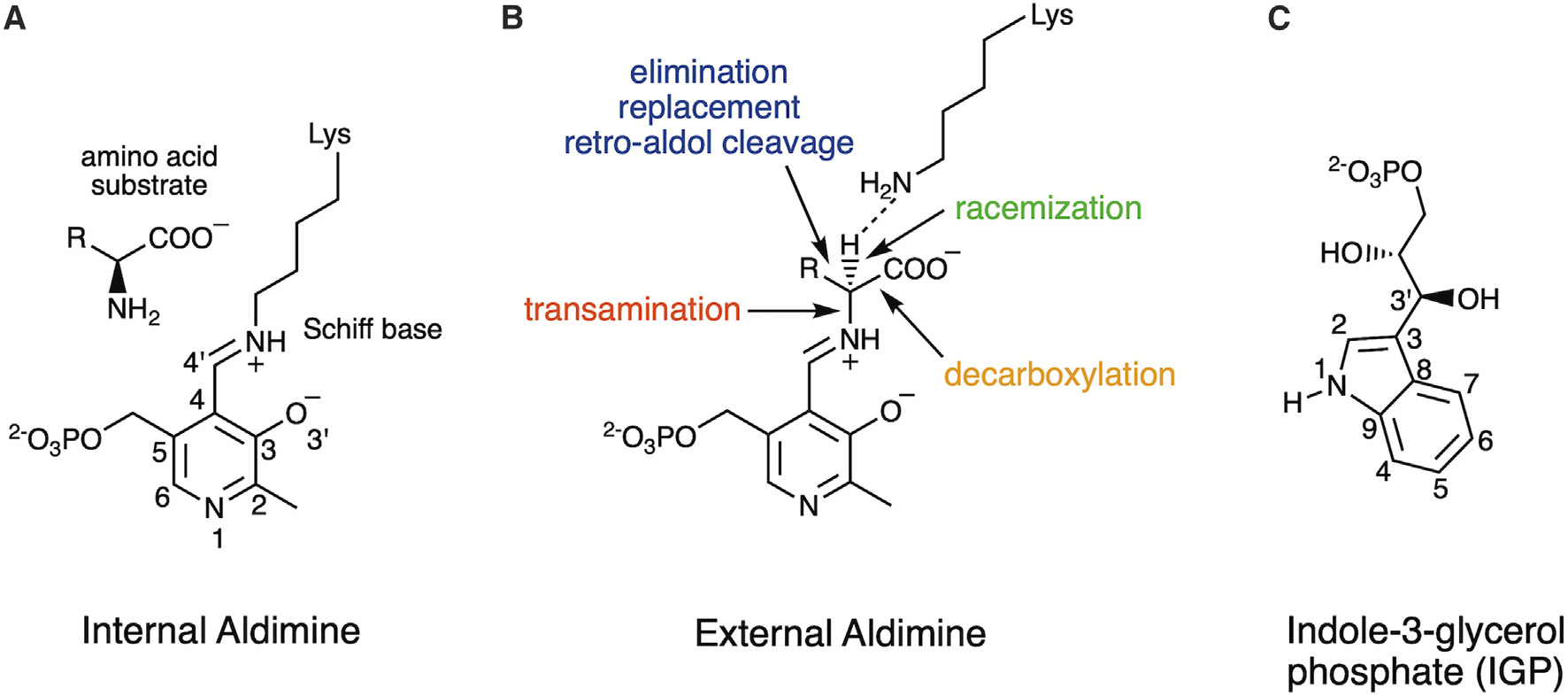
(A) Pyridoxal 5’-phosphate (PLP) forms an internal aldimine with the ε-amine of an active site lysine (Lys87 in TS) through a Schiff base linkage, and the amino acid substrate forms a Michaelis complex.
(B) Electronic overlap in the external aldimine promotes the deprotonation of Cα by the ε-amine of the released active site lysine. Indicated are the reaction specificities of PLP-dependent enzymes including transamination, decarboxylation, racemization, β- and γ-elimination (TS is β-elimination), replacement, and retro-aldol cleavage.
(C) The natural substrate for TS is indole-3-glycerol phosphate (IGP).
TS, the representative enzyme for fold-type-II PLP-dependent enzymes, catalyzes the final steps in the biosynthesis of L-Trp. TS has a rich history of research activity. It is well-established as a model for bienzyme assembly and intramolecular substrate channeling.15,16 Moreover, the lack of a human homolog makes TS an attractive target for specific inhibitor design against pathogenic microorganisms such as Salmonella enterica, Staphylococcus aureus, and Mycobacterium tuberculosis,17,18 which have become especially important considering the constant emergence of bacterial resistance to widely used antibiotics. More recently, TS has become the archetype for enantioselective synthesis of non-canonical Trp derivatives for pharmaceuticals including antibiotics, immunosuppressants, and cancer therapeutics.19 TS purifies as an αββα heterotetrameric bienzyme assembly in which the α- and β-chain active sites perform two separate reactions, and an internal channel connects the two active sites (Figure 2A). TS first releases indole from indole-3-glycerol phosphate (IGP) (Figures 1C and 2B) and then combines indole with a PLP-activated serine (Figure 2C) to form tryptophan. The spectral properties of the pyridoxal cofactor have established the many intermediate electronic states in the proposed mechanisms (Figure 2C).20–22 Acidic groups in the α-chain active site promote the retro-aldol cleavage of indole 3-glycerol phosphate to indole and glyceraldehyde 3-phosphate. The PLP cofactor in the β-chain active site promotes the condensation of L-Ser with indole to yield L-Trp through β-elimination and replacement steps.16 L-Ser displaces the catalytic lysine in the internal aldimine through a transimination reaction with the β-active site adopting an open conformation, yielding the external aldimine and the active site closing. Subsequent β-elimination releases the hydroxy group of L-Ser to form a sequestered, highly reactive α-aminoacrylate intermediate. Indole, a product of IGP cleavage occurring in the α-active site, traverses a 25 Å intramolecular tunnel to the β-active site where it reacts with the α-aminoacrylate intermediate to yield L-tryptophan. Inter-subunit communication is presumably relayed through residues in the channel and a monovalent cation (MVC) binding site near the β-active site (Figure 2A). Interestingly, in the absence of IGP to supply the β-active site with indole, TS catalyzes hydrolysis of L-Ser to ammonia and pyruvate,23 resulting in NH4+ displacing the Na+ occupying the MVC site.24
Figure 2. Salmonella typhimurium TS is an abba linearly arranged heterotetramer.
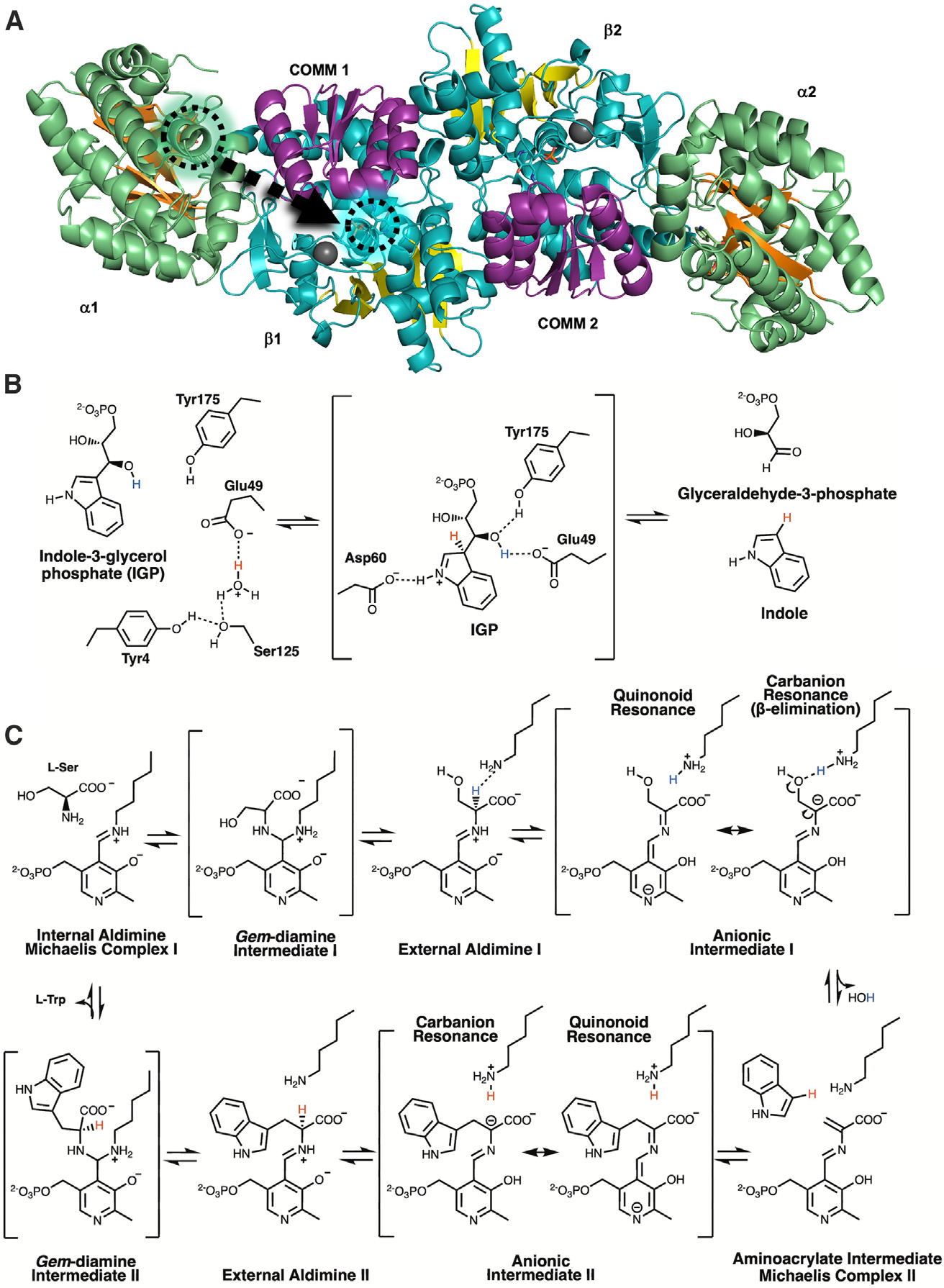
In the α-site reaction, indole-3-glycerol phosphate (IGP) is cleaved into indole and glyceraldehyde-3- phosphate. In the β-site reaction, indole is coupled to PLP-activated serine to form tryptophan.
(A) Ribbon model of the αββα heterotetramer. In the α-domain (green), the core β-strands (orange) have a distinctive TIM barrel-like fold with IGP (green circle) bound in the active site (PDB: 1A5B). The b-domain (cyan) has two sub-domains. The large sub-domain forms contacts with the α-domain, the binding pocket for a monovalent cation (gray sphere), and the loops connecting the C termini of the core parallel b-strands (yellow) to the intervening a helices form the PLP binding site. A 25 Å hydrophobic, intramolecular tunnel connects the α- and β-active sites. The smaller, more flexible “COMM” β-subdomain (purple) forms the top of the channel and participates in interdomain communication.
(B) The TS α-active site catalyzes the cleavage of IGP into glyceraldehyde 3-phosphate and indole. When protonated, Glu49 forms hydrogen bonds with water molecules near Tyr173 (left). Shown is the assumed intermediate (center) when IGP binds, in which Glu49 delivers a proton to indole C3 (red) and aligns to deprotonate the 3′ hydroxy of IGP (blue). Asp60 is positioned to form a short hydrogen bond to the protonated indole N1. The ensuing retro-aldol cleavage involves adding a proton to indole C3 (red) and yielding glyceraldehyde-3-phosphate and indole. The presence of glyceraldehyde-3-phosphate in the active site and release of indole initiates conformational changes, primarily in loop αL6, allowing indole to travel through the hydrophobic channel, which is coordinated to the activation of serine in the β-site.
(C) In the β-reaction, L-Ser is first shown in the Michaelis complex near the PLP-Lys87 internal aldimine. Transimination through an intermediate gem-diamine forms the Ser-PLP external aldimine and a neutral Lys87. As the Ser-PLP external aldimine shifts into a more stable conformation, the neutral ε-amine of Lys87 is repositioned near the Ser-PLP Cα proton. Electronic overlap within the external aldimine coordinates the Cα for deprotonation by Lys87, acting as a general base, and the rearrangement of the transient carbanionic/quinonoid intermediate causes β-elimination of water (HOH). In the lower panel, from right to left, formation of the metastable α-aminoacrylate intermediate coincides with the arrival of indole transported through the intramolecular channel. Indole, positioned as a Michaelis complex, promotes coupling to the Cβ of the aminoacrylate through a second carbanionic intermediate. Lys87 then reprotonates the Cα position and the L-Trp external aldimine is produced. L-Trp is released through a gem-diamine intermediate regenerating the internal aldimine.
The chemical reactions occurring in the α- and β-active sites of TS proceed through a series of protonation and deprotonation events of the PLP cofactor and the active site residues. Consequently, the knowledge of the protonation states and therefore the locations of hydrogen atoms throughout the reaction stages is crucial to fully understand the catalytic mechanism of TS. While X-ray diffraction is considered the “gold standard” for macromolecular structure determination, X-ray data to sub-angstrom (Å) resolutions are typically required to resolve hydrogen atoms in biomacromolecules. However, even at such high resolutions, the positions of some key functional hydrogen atoms may remain undetermined.25 Neutrons, on the other hand, are superb at probing hydrogen atom locations in biological macromolecules.26 Neutrons interact with atomic nuclei rather than with the electron density, as X-rays do. Hydrogen (H) and its heavier isotope deuterium (D) scatter neutrons as well as carbon, oxygen, and nitrogen. Thus, neutron crystallography can provide positions of H and D atoms even at moderate resolutions,27–31 and the protonation states and hydrogen bonding networks can be visualized and accurately mapped in the protein neutron structures.32–36 Combining neutron and X-ray diffraction data in a joint X-ray/neutron (XN) refinement therefore results in complete protein structures where the positions of all atoms have been determined.37 The ability to directly visualize the positions of these otherwise elusive atoms makes neutron diffraction a preferred technique when trying to decipher how PLP-dependent enzymes achieve their reaction specificity. We have previously determined neutron structures of aspartate aminotransferase (AAT), the model PLP-dependent enzyme for fold-type-I and transamination, in the internal and external aldimine forms, and bound to the first half-reaction product, pyridoxamine 5′-phosphate.11,38,39 The neutron structures of AAT revealed how the interplay of hydrogen atoms on the cofactor and adjacent residues and their movements control the reaction specificity and pre-organize the active site to facilitate the second half-reaction.39
Here we report a 2.1 Å room temperature joint XN structure of TS from Salmonella typhimurium containing a substrate-free α-active site and the covalently linked PLP in the internal aldimine state in the β-active site, the latter of which adopts an open conformation. To overcome poor crystal packing and obtain TS crystals of magnitude and quality necessary for neutron diffraction data collection, microgravity capillary dialysis crystallization aboard the International Space Station (ISS) was performed using the Toledo Crystallization Box (TCB) hardware.40 In the β-active site, neutron scattering length density maps reveal the protonation states of the deuterated enzyme including the catalytically important atoms in the PLP cofactor and the active site residues. Quantum chemical calculations were performed on a cluster model derived from the neutron structure to characterize the hydrogen bond between the PLP Schiff base nitrogen, NSB, and the phenolic oxygen, O3′, revealing an asymmetric hydrogen bond favoring protonation of NSB, in agreement with solid-state nuclear magnetic resonance (NMR) measurements.41,42 In the α-active site, the catalytic Glu49, the proposed proton donor to IGP in the α-reaction, shows a dual conformation and is deprotonated. Using the neutron structure model and AlphaFold models to predict the positions of an unresolved 12-residue dynamic loop region covering the α-active site, the pKa values of Glu49 in the two experimentally observed conformations were calculated, and substantial differences in their acidities were obtained. Our structural analyses provide insight into how the protonation states of the cofactor and enzyme residues as well as side chain conformational changes promote catalysis in the active sites of TS.
RESULTS AND DISCUSSION
Neutron diffraction of TS
Neutron diffraction was used to visualize the protonation states of the ground-state TS enzyme: an empty α-active site and an internal aldimine-bound PLP cofactor in the β-active site (PDB: 8EYP). Salmonella typhimurium is a neutrophilic bacterium (typical pH = 7.6) that can tolerate low pH.43 Crystals were grown near neutral pH (pH = 7.8, pD = 8.2) to represent the normal state. Microgravity crystallization and preliminary data analysis were reported previously.40 Small (0.05 mm3) crystals are easily obtained for XRD analysis. However, low neutron flux necessitates large (≥ 0.5 mm3) defect-free crystals, which proved inaccessible at unit gravity. Experiments were delivered by SpaceX CRS-15 (CASIS PGC-8) and CRS-18 (CASIS PCG-15) to the ISS. For the PCG-15 project, a capillary diffusion device (TCB) was used to produce large crystals of perdeuterated TS. Several of the largest crystals were shipped to Institut Laue-Langevin (ILL), Grenoble, France, for data collection. Both X-ray and neutron diffraction data, 1.8 Å and 2.1 Å resolution respectively, were collected at ambient temperature to ensure comparable datasets for joint XN refinement.
The two-site catalytic mechanism of TS
TS combines IGP and L-Ser to synthesize L-Trp through a two-step process. TS is an αββα linear heterotetramer with a TIM-barrel fold α-subunit and an α/β fold with a twisted parallel β-sheet core in the β-subunit (Figure 2A). The larger β-subunits have a stable 2-fold symmetric interface with an additional linkage domain connecting the two distinct α- and β-active sites. TS crystallizes with the αβ dimer in the asymmetric unit, and the αββα heterotetrameric biological assembly is generated through the crystallographic 2-fold axis. The complex between the α- and β-subunits has a propensity to dissociate during purification.15 Within the heterodimer of TS, the two subunits are positioned side by side with the active sites facing the same direction. The β-linkage sub-domain creates the base of an internal channel. The flexibly tethered communication (β-COMM) sub-domain forms the top of the channel. The β-COMM sub-domain and a monovalent cation binding site in the β-linkage sub-domain assist in coordinating the two separate enzyme activities.15
The α-active site in TS catalyzes the retro-aldol cleavage of IGP to yield D-glyceraldehyde-3-phosphate and indole (Figure 2B).15,16 Like most enzyme active sites, the α-active site has two distinct conformations; open, with loop αL6 (α179–α193) disordered, and closed, with αL6 becoming ordered during the reaction.44–46 When IGP binds, Phe212 is located in a position that interferes with αL6 (PDB: 1A5B47 and PDB: 1QOQ48). The release of indole repositions Phe212, which allows ordering of αL6 that causes the movement of αL2(α52–α60), blocking the entry to the internal channel. The indole moiety of IGP is positioned at the top center of the α-TIM barrel adjacent to the blocked internal channel. The glycerol phosphate moiety fits across the active site with the phosphate oriented toward the surface. To promote the aldol cleavage in the closed conformation,Asp60 links to the N1 nitrogen of the indole, presumably through a short hydrogen bond, and Glu49 and Tyr175 bridge the 3′-OH of the glycerol phosphate (PDB: 1A5B).47 Glu49 is thought to act as the catalytic base that deprotonates the C3 hydroxy group of IGP, as well as the acid that protonates the indole leaving group. It is not clear if this happens by a stepwise or concerted mechanism.47,49,50 Electronic rearrangement results in the formation of the two products. In the D60N/IGP α-complex structure (PDB: 1A5B),47 αL6 remains disordered, αL2 appears to be partially ordered, and Phe212 points away from the binding site. When IGP is cleaved, indole enters the channel, and Phe212 moves ~8 Å toward the active site, covering the top of the channel. In concert with the large Phe212 repositioning, αL6 becomes ordered, D-glyceraldehyde-3-phosphate remains bound, and the β-COMM domain tilts toward the α-subunit(PDB:6XNC).51 Reversion to the open conformation releases D-glyceraldehyde-3-phosphate when indole couples to the activated L-Ser in the β-site.
The β-active site in TS catalyzes the synthesis of L-Trp by coupling PLP-activated L-Ser with the indole generated in the α-active site arriving through the internal channel (Figure 2A). In the first step, the PLP cofactor, sequestered as an internal aldimine to Lys87, is coupled to L-Ser substrate through transimination via a gem-diamine intermediate to form a PLP-Ser external aldimine (Figure 2C). The Schiff base form of the PLP-substrate complex is known to lower the pKa of the Cα proton.10,52 The neutral ε-amine of Lys87 abstracts the Cα proton from the substrate, prompting the formation of the first anionic intermediate, which has two dominant resonance forms, carbanion and quinonoid. Traditionally, proposed mechanisms of PLP-dependent enzymes assume a protonated PLP-N1; however, recent studies have suggested N1 is neutral in fold-type-II PLP-dependent enzymes and all PLP-dependent racemases.53 The current TS data (discussed in detail below) indicate that PLP-N1 is deprotonated as a hydrogen-bond acceptor with Ser377 as the donor, and the typical PLP quinonoid cannot form. Instead, the intermediate is shown with a protonated O3′ and an N1 anion in resonance with a carbanion forming at Cα. The Cα proton is abstracted by the ε-amine of Lys87 and then transfers to the hydroxy group of the serine. The resulting β-elimination releases H2O and forms the aminoacrylate intermediate. The ε-amine of Lys87 also participates in the second step of the reaction. Nucleophilic addition of indole to Cβ of the aminoacrylate produces the second anionic intermediate. Lys87 deprotonates indole C3 and reprotonates the carbanion formed at Cα, producing the L-Trp external aldimine. Through a second gem-diamine transimination, L-Trp is released as Lys87 displaces the product and regenerates the internal aldimine. When indole is not present for the subsequent nucleophilic attack on Cβ, Lys87 can displace dehydroalanine in the aminoacrylate Schiff base linkage, which will degrade to pyruvate and ammonia.54
Functional hydrogens visualized in the TS α-active site
When IGP is bound in the closed α-active site, Glu49 and Tyr175 are hydrogen bonded to the 3′-OH group of IGP, and Asp60 forms a hydrogen bond to the indole nitrogen (Figure 2B). During the retro-aldol cleavage of IGP, Glu49 appears to be responsible for protonating C3 of IGP and then accepting a proton from the 3′-OH of IGP to facilitate the cleavage of the C–C bond.16 Glu49 therefore must be protonated in the closed enzyme form to initiate catalysis. While bonded to the indole N1 nitrogen, Asp60 can also stabilize the positive charge on N1 during protonation of indole C3. This mechanism is proposed to occur either through stepwise proton transfers or a concerted mechanism that prevents the buildup of charged species. However, the hydrophobic microenvironment of the a-active site supports a concerted mechanism.44 Both Glu49 and Asp60 are conserved, and mutagenesis studies have determined that both are essential for the catalytic activity of TS.55,56
At neutral pH and in the absence of substrate, both loops αL2 (α52–α60) and αL6 (α179–α193) are expected to be disordered. X-ray refinement of microgravity-grown crystals yielded a mean overall B-factor of ~24 Å2. Residues α57–α62, including the catalytic Asp60, had refined B-factors >60 Å2. While most of the region is somewhat disordered, the joint refined map shows a short hydrogen bond (2.7 Å, D–O 1.7 Å) between the carboxyl group of Asp60 (H-bond acceptor) and the phenolic oxygen (H-bond donor) of Tyr102. In the room temperature X-ray model, used in the joint XN refinement, Glu49 appears in two alternate conformations (Figure 3A). Conformer A (Glu49a), with 60% refined occupancy (χ1 = 180°), adopts a position near Tyr175 found when IGP is bound.47 This is the active position in which Glu49 can deliver a proton to the indole C3 and remove a proton from IGP 3′-OH. Conformer B (Glu49b), with 40% refined occupancy (χ1 = −67°), is rotated to participate in a hydrogen bond network with two water molecules supported by residues Tyr4, Tyr173, and Ser125. Joint XN refinement limits the visualization of multiple conformations, where only the highest occupied position can be modeled accurately. The neutron scattering length density or “nuclear density map” suggests the carboxylic group of Glu49a is deprotonated, but the signal is reduced by the dual conformation (Figure 3B). Most well-defined waters have a boomerang shape in nuclear density maps, allowing correct modeling of hydrogen bond donor/acceptor pairs. The nuclear density of the water coupled to Tyr173/Ser125/Glu49b adopts a triangular shape, which may be interpreted as a dual-conformation D2O. This water molecule may be organized to donate a proton to Glu49 while in conformation B. Interestingly, in the cryo X-ray structure of TS (PDB: 8EZC), the populations of Glu49 switch dominance, with 40% refined occupancy for Glu49a and 60% refined occupancy for Glu49b (Figure 3C). The cryogenic X-ray data provided more completeness and higher resolution for model building. However, being slightly non-isomorphous with the ambient neutron data, the cryo data were not used in joint XN refinement.
Figure 3. Observed dual positions of a-active site residue Glu49.
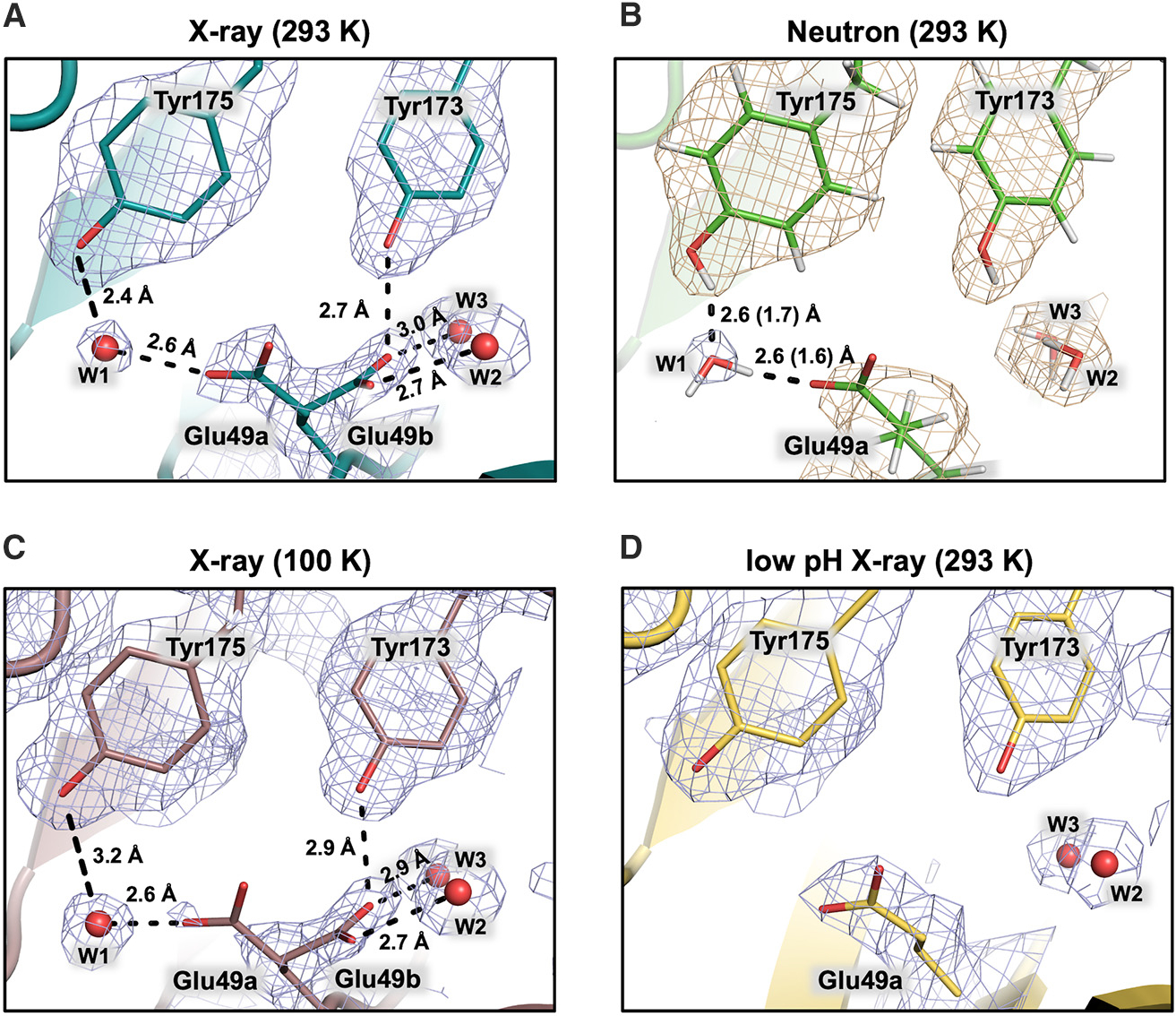
Glu49a is the active position oriented toward Tyr175 hydrogen bonded to water W1 in which Glu49 can deliver a proton to the indole C3 and abstract a proton from IGP 3′-OH. Glu49b is in a hydrogen bonding network with two water molecules (W2 and W3) reinforced by residues Tyr173 and Tyr4 and is the suspected reservoir for the proton delivered to indole C3. Not shown in this view, W2 is linked to Ser125, and W3 has a short 2.5 Å (D–O 1.7 Å) H-bond to the phenolic oxygen of Tyr4. The 2| FO|-|FC| neutron scattering length density is shown in wheat and the electron density in blue mesh.
(A) The room temperature X-ray model shows both Glu49a (60%) and b (40%) conformers.
(B) The room temperature neutron model shows only Glu49a, and deuteron-donor H-bonding distances are shown in parentheses.
(C) The cryo X-ray model shows Glu49b becoming the dominant conformer.
(D) The low-pH, room temperature X-ray model shows Glu49a becoming dominant, suggesting Glu49 is protonated.
The alternate conformations of Glu49 are likely to be important for the α-site mechanism with environmental factors affecting the pKas. For first estimations, we used the H++ webserver to estimate the pKa values from the neutron structure. We note that the α-chain of TS adopts an open conformation before the substrate binds. With Phe212 in the outward position, the αL6 loop residues covering the α-active site are disordered but may still influence the local pKa values. Therefore, we used a hybrid approach in which we provided the joint XN structure of TS as a custom template to generate complete TS models with five predicted conformations of the αL6 (α178–α189) loop. The structure of the loop region and the predicted local difference test (pLDDT) metrics are shown in Figures S2 and S3 and are compared to their positions in the structure, PDB: 6XNC, where Phe212 occupies the indole binding site.51 The pKa predictions were then performed on the joint XN structure and the five AlphaFold models (M1–M5) (Table S4). At an internal dielectric constant εint of 10, the pKa values of Glu49a and Glu49b using the joint XN structure model were predicted to be 6.4 and 7.9, respectively. Similarly, the pKa values for Glu49 in the top AlphaFold model, M1, were predicted to be 6.3 for Glu49a and 7.8 for Glu49b. Consequently, both the pKa values calculated using the experimental and AlphaFold models exhibited a 1.5 pK unit increase (ΔpKa) when the Glu49 conformation flips from the active Glu49a to the water-sequestered Glu49b position. Additional pKa predictions were performed at an εint. = 6 to determine the influence of the dielectric constant on the pKa. At εint = 6, the pKa values for Glu49a and Glu49b were estimated to be 8.2 and 10.7 (ΔpKa = 2.5) in the experimental model and 8.5 and 11.1 (ΔpKa = 2.6) in the best AlphaFold model. Lowering the dielectric constant both increased the predicted pKa values and increased the difference between the pKas of each conformation. While we anticipate errors in the absolute pKa values, the directionality of the pKa shift should be considered meaningful. The pKa predictions of the remaining AlphaFold models (M2, M3, M4, and M5) are also consistent with these findings. To add confidence to the pKa estimates, TS crystals were soaked in a pH 5.0 buffer, and X-ray diffraction data were collected at room temperature. At pH 7.8, the room temperature structure shows dual conformations for Glu49. TS crystals were acidified to investigate the effect of pH on the conformation of Glu49 (PDB: 8EYS). Under acidic conditions, however, the room temperature X-ray structure revealed Glu49a as the only conformation in which both the water cluster and Glu49 are likely protonated (Figure 3D).
Based on our experimental observations and pKa calculations, we argue that the Glu49b conformation stabilizes the proton required for indole C3 activation. We thus suggest that the conformational flexibility of the Glu49 side chain plays a crucial role in catalysis. We propose that when Glu49 is in the flipped Glu49b conformation exhibiting the elevated pKa value, it obtains a proton, most likely from the water (W3, Figures 3A, 3C, and 3D) stabilized by Tyr173 and Ser125, to become the protonated carboxylic acid. During IGP cleavage, the C3′-OH is deprotonated by Glu49a, which rotates back to conformation b in a concerted motion as indole departs into the channel, Phe212 rotates, and αL6 becomes ordered. The observation that Glu49 adopts the active, likely protonated, Glu49a conformation at low pH is consistent with its role as the general acid catalyst in the α-active site of TS. A similar mechanism for a proton transfer to a substrate was previously proposed for a family 11 glycoside hydrolase where the catalytic glutamate also alternated between “upward” and “downward” conformations having different acidities to obtain a proton from the bulk solvent and then to deliver it to the glycosidic oxygen of the substrate to initiate catalysis.33
Functional hydrogens visualized in the TS β-active site
The planarity of the PLP Schiff base affects orbital overlap, which modulates the cofactor effect as an electron sink. In the internal aldimine form, the Schiff base nitrogen (NSB) is in plane with the PLP ring and prepared for gem-diamine formation with serine (Figure 2C). During catalysis, the PLP ring and NSB external aldimine become coplanar, and the bond perpendicular to the plane is selectively cleaved, a mechanism known as the Dunathan alignment.57 When the cofactor is in the active planar structure, two tautomeric forms exist: the zwitterionic ketoenamine with protonated positively charged NSB and deprotonated negatively charged phenolic O3′ and the neutral enolimine where the phenolic O30 is protonated and NSB is deprotonated. The Schiff base tautomers have different UV-vis absorption spectra, with the ketoenamine and enolimine absorbing at 420 nm and 330 nm, respectively.58,59 The internal aldimine form of TS has two distinct absorption peaks at 410 nm and 336 nm, typically favoring protonation of NSB and the ketoenamine tautomer.20,60 In our joint XN structure, a deuterium atom is visible in both the 2|FO|-|FC| (wheat mesh) and |FO|-|FC| (purple mesh) neutron scattering length density maps 1.0 Å from the Schiff base NSB and 1.8 Å from O3′ (Figure 4A). These observations support the presence of the ketoenamine tautomer reported using NMR crystallography.41,42 In addition to the hydrogen bond with NSB, the accumulation of negative charge on O3′ (phenoxy-anion) is stabilized through partial double bond character of the C3–O3′ bond (i.e., π-conjugation with the pyridine ring) and by a short hydrogen bond of 2.6 Å (D–O 1.7 Å) with a nearby water molecule.
Figure 4. β-Active site perspectives from a TS neutron structure.
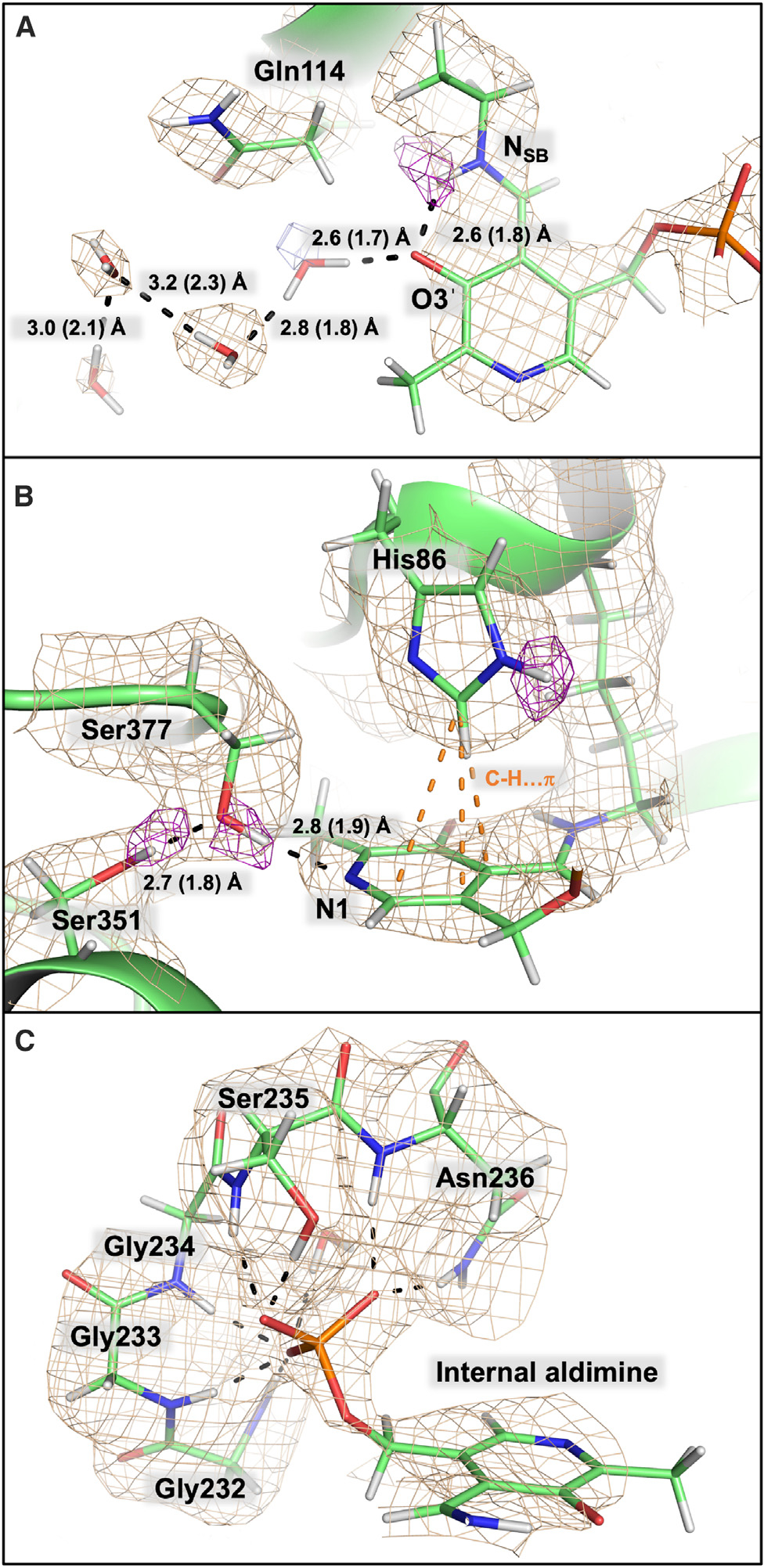
The 2|FO|-|FC| neutron scattering length density map is depicted in wheat mesh contoured at 1 σ, and the omit |FO|-|FC| neutron scattering length density is shown in purple mesh contoured at 2.2 σ. Hydrogen bonding distances between heavy atoms are shown with the deuteron-donor distances in parentheses.
(A) The Schiff base nitrogen, NSB, is protonated and hydrogen bonded to the phenolic oxygen,O3′. The 2|FO|-|FC| electron density is shown in blue mesh.
(B) Ser377 prevents protonation of pyridine nitrogen, N1, and is stabilized by an additional hydrogen bond with Ser351. His86 is neutral and monoprotonated on the ε-nitrogen, positioned above the cofactor.
(C) The glycine-rich phosphate binding loop is composed of Gly232, Gly233, Gly234, Ser235, Asn236, and two crystallographic waters, as well as His86 and Thr109 (not shown for clarity).
During PLP-dependent catalysis, the cofactor withdraws electrons from the substrate, lowering the pKa of Cα allowing a weak base, the deprotonated ε-amine of the active site lysine, to extract a proton from a C–H bond (blue H atom, Figure 2C). The pyridine nitrogen, N1, of the PLP cofactor is speculated to increase the efficacy of the pyridine ring to act as an electron sink, stabilizing the formation of a carbanionic intermediate after Cα deprotonation.10 The formation of a transient carbanion is stabilized by a quinonoid intermediate, a resonance structure with a distinct absorbance at ~490–580 nm.10,61–64 In AAT, N1 is protonated during catalysis, and a chain of histidine and structural water molecules promote proton hopping.11 For TS, N1 was observed to be deprotonated with no protonation network present (Figure 4B). The side chain hydroxy group of Ser377 is a hydrogen bond donor to N1 at a distance of 2.8 Å (D–N 1.9 Å) and a hydrogen bond acceptor from the Ser351 hydroxy group at a distance of 2.7 Å (D–O 1.8 Å). This N1–Ser377–Ser351 hydrogen bond linkage would presumably prevent protonation of the pyridine nitrogen by the bulk solvent. The nearby Glu350 is salt-bridged to a protonated Lys382 on the re face of the cofactor and is unlikely to contribute to N1 protonation. In TS, it is more favorable to retain negative charge on Cα so that –OH β-elimination from the L-Ser substrate in the external aldimine can proceed. In PLP-dependent transaminases, it is important to withdraw electrons from Cα, where a stabilized quinonoid intermediate is required for the characteristic 1,3-proton shift.10,11,65 During TS catalysis, a quinonoid-like resonance must exist to allow formation of a Cα carbanion. However, the requisite quinonoid is less stable and forces electrons toward the L-Ser hydroxy group for β-elimination. In AAT, the stabilized quinonoid appears in kinetic studies with an absorption peak at 495 nm. In TS, the quinonoid appears in the spectra with a maximum at 475 nm, blue shifted compared to AAT. To explain this quinonoid blue shift, two anionic external aldimine intermediates are shown, an N− anionic quinonoid in which the Cα proton is abstracted and a carbanion, which promotes β-elimination (anionic intermediate I, Figure 2C).
Our observation that NSB is protonated in the internal aldimine of TS agrees with previously reported solid-state NMR data.41 Later, 4D and 5D solid-state NMR studies of TS revealed equilibrium between the ketoenamine and enolimine tautomers in the internal aldimine, with a fast tautomeric exchange and equilibrium populations of 61% and 39% for the protonated Schiff base and phenolic oxygen at 30°C, respectively.42 NMR studies have also demonstrated the temperature dependence of the tautomeric equilibrium, with a significant increase of the ketoenamine population as temperature decreases. At the temperature of the neutron diffraction data collection, ~16°C, there would be 70% of protonated NSB present, which agrees well with our observation of a protonated Schiff base. At the resolution of the neutron diffraction data in this study, the deuteron appears at the major NSB position but is not well defined at the minor O3′ position.
The binding pocket for the PLP phosphate group is composed of the side chains of His86, Thr190, Ser235, and Asn236, two crystallographic waters, and the backbone of Gly232, Gly233, Gly234, and Ser235, which create a peculiar turn that forms a charge-stabilizing pocket with six amide ND groups directed toward the phosphate oxygens (Figure 4C). The His86 imidazole positioned perpendicular to PLP on the si face is neutral, protonated only on Nε2, whereas Nδ1 faces this residue’s own main chain amide ND preventing Nd1 protonation. The position of the imidazole group of His86 is locked in place by close C–H• • •π interactions with the PLP pyridine ring with distances between heavy atoms of 3.7–3.9 Å and with Cβ of Asn236, whose side chain contributes to the PLP phosphate binding pocket, with distances of 3.3–3.6 Å (Figure S5). Mutation of the histidine residue to leucine (H86L) reduces the ability of PLP to bind to the enzyme by 20-fold,66 indicating that His86 plays an important role in binding PLP. On the re face of the cofactor, Glu350 sits parallel to the pyridine ring, forming hydrogen bonds with the side chain of Lys382 and the main chain of Gly303 (Figure S6). Site-directed mutagenesis revealed that, similar to His86, Glu350 is not essential for catalysis but rather plays a role in PLP binding.67
Quantum chemical cluster calculations on the TS β active site
The position of the proton in the NSB–O3′ hydrogen bond affects the resonance structures of the PLP cofactor. Density functional theory (DFT) calculations were carried out on a 195-atom model of the β-active site taken from the neutron structure of TS at the SMD68/ωB97X-D69/Def2-SVP70,71 level of theory. The deuterium atoms from the structure were modeled as hydrogen in the cluster model but the protonation states were kept identical to those observed in the neutron structure. The geometries of the reactant (NSB-protonated state) and product (O3′-protonated state) were optimized independently, and vibrational frequencies were calculated to confirm that each structure was at an energy minimum. A relaxed potential energy scan (PES) was performed to map the energy of intramolecular proton transfer between NSB and O3′. The maximum point on the relaxed PES was used as an initial guess in a transition state optimization. The transition state was confirmed to be a first-order saddle point based on vibrational frequency calculations. We estimated that the product state, enolimine tautomer, is 4.4 kcal/mol higher in energy than the reactant state, ketoenamine tautomer (Figure 5A). This result suugests that the hydrogen bond between NSB and O30 is an asymmetrical hydrogen bond with the proton favoring the nitrogen atom. The transition state is only 0.7 kcal/mol greater in energy than the O3′-protonated product, but the barrier for hydrogen transfer from NSB to O3′ is significantly higher at 5.1 kcal/mol. The effective energy profile of the tautomeric exchange at 30°C was calculated from the solid-state NMR data to give a free energy barrier of 8.8 kcal/mol for proton transfer from NSB to O3′ and the enolimine tautomer enthalpy higher by 2.4 kcal/mol compared to the ketoenamine tautomer. Our quantum chemical calculations performed at 0 K, with no zero-point energy corrections applied and treating hydrogen nuclei as classical particles, agree with solid-state NMR studies. Additionally, preference for NSB protonation in the internal aldimine is supported by molecular dynamics simulations performed on TS.72 The partial charges for both DFT optimized clusters were calculated with the NBO program (Figure S8).73 In the lower-energy, NSB-protonated state, C4′ has a larger partial positive charge, making it better suited for nucleophilic attack by an incoming L-Ser to form the substrate-bound external aldimine.
Figure 5. Potential energy profile for proton transfer between PLP-NSB and PLP-O3′.
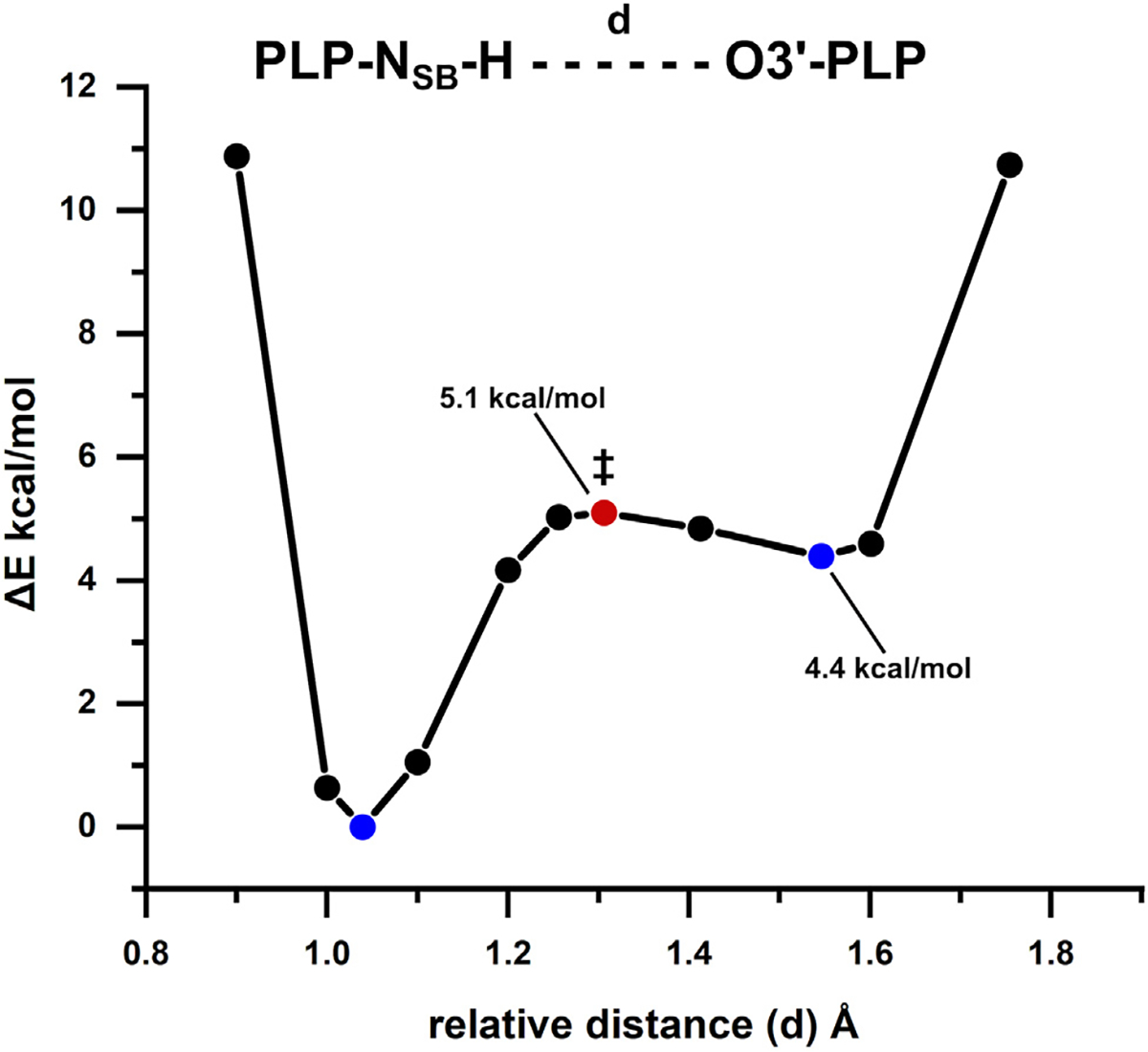
The calculated energy barrier height for the intramolecular proton transfer from the Schiff base to the phenolic oxygen is reported relative to the NSB-protonated (reactant) structure. The energies for the reactant and product are denoted with blue circles, while the calculated transition state energy (‡) is signified with a red circle.
Comparison of the TS β-active site to AAT
Fold-type-I enzymes are the most prevalent in PLP-dependent enzyme family and contain transaminases (except type IV transaminases), decarboxylases, and some enzymes that catalyze α-, β-, or γ-eliminations.1 Aspartate aminotransferase (AAT), the fold-type-I representative enzyme, is the most studied PLP-dependent enzyme and has been the focus of our previous neutron diffraction experiments.38,74 AAT catalyzes the reversible transamination of L-Asp and L-Glu to a-ketoglutarate and oxaloacetate, respectively.74 We have previously assigned protonation states to AAT for the internal and external aldimines and most recently for the pyridoxamine 5′-phosphate (PMP) intermediate, which has an N–H–N low-barrier hydrogen bond.11,38,39 TS is the representative enzyme for fold-type-II, and we can now directly compare the all-atom models of the PLP internal aldimine forms of TS and AAT (Figure S9).
In AAT, the pyridine nitrogen N1 is protonated and hydrogen bonded to a conserved aspartate, Asp222. As a model for transamination, the hallmark step in the reaction mechanism is the formation of a stabilized carbanionic intermediate, the quinonoid intermediate, which facilitates the transfer of the Cα proton to C4′ through the acid-base reaction assisted by the catalytic Lys257. The quinonoid form proposed to be utilized by AAT is not possible in the absence of N1 protonation, indicating the dominant quinonoid resonance form influences the reaction specificity. The significance of this observation was demonstrated in AAT with a D222A mutant, forcing deprotonation of N1 and reducing the turnover rate by over 1000-fold.75,76 In a similar study using TS, N1 protonation was forced in the S377D mutant, and the formation and accumulation of the quinonoid intermediate, as well as slowing of the reaction rate by over 100-fold, were observed.61 As mentioned above, it is more favorable in TS to retain the negative charge on Cα to promote β-elimination, whereas AAT needs to delocalize the charge to complete the 1,3-proton shift.
It was proposed long ago that the protonated Schiff base is more reactive in the formation of the external aldimine.77 In the AAT internal aldimine, neutron diffraction revealed that both the Schiff base NSB and phenolic oxygen O3′ are deprotonated.11 Protonation of the Schiff base also influences the rotation of the NSB–C4′ bond and the planarity of PLP-NSB.11 In the AAT internal aldimine, the NSB–C4′ bond is rotated 46° above the plane of the pyridine ring on the si face, but when protonation of NSB is forced with a low-pH environment, the torsion angle is reduced to 22°, with the same effect observed in DFT optimizations of AAT active site models.11 In the joint XN structure of TS, NSB is protonated, and the NSB–C4′ dihedral angle is positioned 17° above the plane of the pyridine ring, which agrees with the previously observed values for protonated NSB from experiments and calculations.11,14,78,79
Microgravity crystallization using the TCB was used to grow large crystals (1 mm3) for neutron crystallography experiments with perdeuterated Salmonella typhimurium TS. The resulting 2.1 Å neutron diffraction data were jointly refined with room temperature X-ray diffraction data to give an all-atom structure of TS with accurate hydrogen positions. Additional X-ray diffraction data, pKa predictions, and DFT calculations provide a more complete view of the overall TS mechanism. In the α-active site, the proposed proton donor to IGP in the cleavage of glycerol phosphate, Glu49, adopts two conformations with different pKas that appear to be functionally relevant to the α-reaction mechanism. We propose that Glu49 adopts conformation B (higher pKa) to acquire a proton and flips to the conformation A (lower pKa) to donate a proton to IGP-C3. In the β-active site, we observed the PLP cofactor with a non-protonated (neutral) N1, protonated (positively charged) NSB, and deprotonated (negatively charged) O3′ using the positions of hydrogen atoms revealed through neutron diffraction. The NSB–O3′ interaction was characterized as an asymmetric hydrogen bond favoring NSB protonation by mapping the potential energy of proton transfer and agrees with both our XN model and previously published solid-state NMR data.41 Thorough understanding of the TS α- and β-reaction mechanisms will require additional neutron diffraction experiments on different reaction intermediates to delineate the various protonation and deprotonation states in addition to thorough kinetic studies and biophysical techniques such as NMR.
EXPERIMENTAL PROCEDURES
Resource availability
Lead contact
Requests for further information should be directed to the lead contact, Timothy Mueser (timothy.mueser@utoledo.edu).
Materials availability
This study did not generate new unique materials.
Data and code availability
The accession numbers for the joint XN and X-ray structures reported in this paper are PDB: 8EYP, 8EZC, and 8EYS.
Protein perdeuteration, purification, and crystallization
The perdeuteration of TS was carried out in the Deuteration Laboratory (D-Lab) of the Life Sciences Group at ILL. The procedures for the perdeuteration, expression, and purification of TS have been previously described.40 Purified perdeuterated TS was crystallized in microgravity using the TCB aboard the ISS as part of experiment Protein Crystal Growth (PCG)-15 in July 2019. The TCB crystallization experiments remained aboard the ISS for approximately 6 months, returning in January 2020. The crystallization conditions for perdeuterated TS in the TCB apparatus were 50 mM Bicine (pH 7.8), 1 mM EDTA, 0.2 mM PLP, 2 mM spermine, and 6%–8% PEG 8000. To prepare crystals for neutron data collection, the polypropylene bag of an individual TCB experiment was opened, and the sealed quartz capillary was removed. The beeswax on the top of the capillary was gently pulled off, and a blunt-end needle and syringe were used to draw liquid away from the crystal in the capillary. The Tygon tube and dialysis membrane were then detached from the opposite end of the capillary, and the blunt-end needle and syringe were used to remove the remaining solution. A paper wick was used to absorb any excess liquid in the capillary, particularly around the crystal. H/D vapor exchange was performed in the capillary by placing plugs of deuterated mother liquor inside the quartz capillary and resealing both ends with capillary wax.
Neutron diffraction data collection
Neutron diffraction was tested on microgravity-grown TS crystals at room temperature on the IMAGINE80–83 instrument located at the High Flux Isotope Reactor (Oak Ridge National Laboratory, ORNL) using the broad-bandpass functionality with neutron wavelengths between 2.8 and 10 Å. Neutron diffraction data were collected on a microgravity-grown TS crystal on the quasi-Laue diffractometer, LADI-III,84 at the ILL, Grenoble, France. A neutron wavelength range of (Δλ/λ ~30%) of 2.85–3.80 Å was used in data collection with neutron diffraction extending to a resolution of 2.1 Å. Four different ϕ orientations (vertical rotation axis) were sampled during data collection, and a total of 57 images were collected. The neutron diffraction images were processed with a modified version of the program LAUEGEN85,86 of the Daresbury Laboratory Laue Software Suite to account for the cylindrical geometry of the area detector. LSCALE,87 of the same software suite, was used to establish a wavelength normalization curve from symmetry-equivalent intensities measured at different wavelengths. The neutron data were scaled and merged using SCALA.88 Data collection statistics can be found in Table S1.
Room temperature X-ray data collection and structure refinement
Room temperature X-ray diffraction data were collected on a microgravity-grown TS crystal on a Rigaku HighFlux HomeLab instrument at ORNL equipped with a MicroMax-007 HF X-ray generator, Osmic VariMax optics, and a Dectris EIGER R 4M detector. The X-ray data were integrated with CrysalisPro from Rigaku. The data were subsequently scaled with SCALA, and structure factors were calculated with CTRUNCATE, both in CCP4.89 The TS X-ray model was refined against ambient X-ray diffraction data in PHENIX90 to establish the positions of non-hydrogen atoms and to create an appropriate input model for joint XN refinement. X-ray data collection and refinement statistics are given in Table S1.
Joint X-ray/neutron structure refinement
The patch, nCNS,91 in the structure solution program CNS37,92 was used to perform the joint XN refinement of TS. Isomorphous, room temperature X-ray and neutron data were utilized for joint X/N refinement. Following a single rigid-body refinement, the atomic positions, individual atomic displacement parameters, and occupancies were refined until satisfactory. The graphics program Coot93 was used to view the model and neutron scattering length density maps, 2|FO|-|FC| and |FO|-|FC|. Hydrogen/deuterium atoms were rotated in residue side chains and waters to demonstrate correct hydrogen bonding. All hydrogens in the protein structure were initially modeled as deuterium, as TS was perdeuterated. Because the PLP used in the experiment was hydrogenous, the portions of the covalently linked internal aldimine cofactor originating from PLP were modeled with hydrogens, except at exchangeable sites (N1 and NSB).
Quantum chemical calculations
Quantum chemical calculations were performed on a 195-atom cluster extracted from the neutron structure of TS. The model of the β-active site consists of twelve active site residues (Ala85, His86, Lys87, Thr88, Gln114, Thr190, Ser235, Asn236, Ser351, Lys382, Ser377, and Glu350), the internal aldimine (PLP covalently linked to Lys87), and six crystallographic waters (Figure S7). The peptide backbone between consecutive residues, Ala85, His86, Lys87, and Thr88, was retained in the model. Geometry optimizations were performed at the SMD68/ωB97X-D69/Def2-SVP70,71 level of theory. A dielectric constant of 10 was used for the SMD solvent model to mimic the enzyme active site environment. Energy minima in the product and reactant structures and the maximum in the transition state structure were confirmed with vibrational frequency calculations in which the two minima had zero imaginary frequencies and the transition state had one. The potential energy path of proton transfer between NSB and O3′ was mapped with a relaxed PES. The geometry of the energy maximum of the resulting curve was used as an initial guess for a transition state optimization. Natural population analysis charges of the product and reactant structures were calculated with the NBO program73 in Gaussian 16, along with all other calculations.94
AlphaFold2 predictions
Complete structural models of the TS heterodimer were generated by using a hybrid approach in which residues that were unresolved in the experimentally determined structure (i.e., residues 178–191 of β-chain) were modeled with AlphaFold-multimer-v2,95 which is a version of AlphaFold296 trained specifically to model protein complexes. The MMSeqs2 web server97 was used to generate paired multiple sequence alignment by searching the UniRef database. The neutron structure determined in this work was used as a single custom template. Models were ranked by their average pLDDT, predicted TM-score (pTMscore), and interface predicted TM-score (ipTM), the latter of which scores interactions between residues of different chains. The metrics for the top model were as follows: pLDDT score = 95.2, pTMscore = 0.94, and iPTM = 0.93. The top model was selected based on a weighted combination of the ipTM and pTM (confidence = 0.8 · ipTM + 0.2 · pTM). The per-residue pLDDT scores are shown in Figure S3. All models were generated with the ColabFold98 implementation of AlphaFold2 (AF2).
H++ pKa predictions of αGlu49
The H++ (version 4.0) web server99 was used to compared predicted pKas of two observed conformations of the catalytic αGlu49 in TS. H++ uses a finite-difference Poisson-Boltzmann continuum electrostatics approach to compute pKas and titration curves for each residue in a protein.100 The predictions were carried out at 0.15 M salinity with internal and external dielectric constant of 10 and 80, respectively. Both α- and β-chains of the experimental TS structure missing residues α178–α189 in the α-chain and five full-length AF2 models were submitted to the server. For the experimental structure, the cofactor was removed. Predicted pKas for each model are shown in Table S4.
Low-pH X-ray data collection
TS crystals grown in a 9-well glass plate and sandwich box were equilibrated against 50 mM NaOAc (pH 5.0) for 5 days prior to data collection to lower the pH of the crystals. The low pH, room temperature X-ray diffraction data were collected and processed with the same procedure described above. PHENIX was used for structure refinement.90
Low-temperature X-ray data collection
X-ray diffraction data at 100 K were collected on a microgravity-grown crystal of TS using the LS-CAT beamline 21-ID-F equipped with a Rayonix MX300 detector at the Advanced Photon Source (APS). In the CCP4 program suite,89 the data were processed with Mosflm, then scaled and merged with SCALA. Structure refinement was carried out in PHENIX.90
Supplementary Material
Highlights.
2.1 Å joint XN structure of tryptophan synthase from a microgravity-grown crystal
Hydrogen atom positions are accurately determined in both α- and β-active sites
Schiff base is protonated, but O3′ and pyridine N1 are not, making PLP zwitterionic
Catalytic Glu49 in α-active site adopts two conformations with different pKa values
ACKNOWLEDGMENTS
We are especially grateful for the assistance of April Spinale, Ray Polniak, and Marc Giulianotti, International Space Station (ISS) National Laboratory, for flight preparation. We thank SpaceX for access to CRS-15 and CRS-18 and ESA Astronaut Alexander Gerst and NASA astronauts Christina Koch and Nicklaus Hague for sample handling at the International Space Station. For beamline access, we thank the Life Sciences Collaborative Team (LS-CAT) for access to beamline 21-ID-F at APS.
We also thank Lisa Keefe and Kevin Battaile for access and assistance at APS IMCA-CAT 17-ID-B used in early stages of this study. Additionally, thank you to Rob Phillips, Len Mueller, and Mike Toney for helpful discussion and suggestions. V.T.F. acknowledges the UK Engineering and Physical Sciences Research Council for grants EP/C015452/1 and GR/R99393/01 under which the Deuteration Laboratory was created within ILL’s Life Sciences Group. Funding was provided by the Center for the Advancement of Science in Space (contract GA2017–251, T.C.M.) and the National Institutes of Health (1R01GM137008–01A1, T.C.M. and A.K.). The research at ORNL’s High Flux Isotope Reactor (IMAGINE beamline) was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, US Department of Energy. The authors thank the Institut Laue–Langevin for provision of neutron beam time on the LADI-III beamline.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.xcrp.2024.101827.
REFERENCES
- 1.Percudani R, and Peracchi A (2003). A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 4, 850–854. 10.1038/sj.embor.embor914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jansonius JN (1998). Structure, evolution and action of vitamin B6-dependent enzymes. Curr. Opin. Struct. Biol. 8, 759–769. 10.1016/s0959-440x(98)80096-1. [DOI] [PubMed] [Google Scholar]
- 3.Eliot AC, and Kirsch JF (2004). Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 73, 383–415. 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 4.Amadasi A, Bertoldi M, Contestabile R, Bettati S, Cellini B, di Salvo ML, Borri-Voltattorni C, Bossa F, and Mozzarelli A (2007). Pyridoxal 5’-phosphate enzymes as targets for therapeutic agents. Curr. Med. Chem. 14, 1291–1324. 10.2174/092986707780597899. [DOI] [PubMed] [Google Scholar]
- 5.Graber R, Kasper P, Malashkevich VN, Strop P, Gehring H, Jansonius JN, and Christen P (1999). Conversion of aspartate aminotransferase into an L-aspartate beta-decarboxylase by a triple active-site mutation. J. Biol. Chem. 274, 31203–31208. 10.1074/jbc.274.44.31203. [DOI] [PubMed] [Google Scholar]
- 6.Zhu H, Xu G, Zhang K, Kong X, Han R, Zhou J, and Ni Y (2016). Crystal structure of tyrosine decarboxylase and identification of key residues involved in conformational swing and substrate binding. Sci. Rep. 6, 27779. 10.1038/srep27779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steffen-Munsberg F, Vickers C, Kohls H, L and H, Mallin H, Nobili A, Skalden L, van den Bergh T, Joosten HJ, Berglund P, et al. (2015). Bioinformatic analysis of a PLP-dependent enzyme superfamily suitable for biocatalytic applications. Biotechnol. Adv. 33, 566–604. 10.1016/j.biotechadv.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 8.Dick M, Sarai NS, Martynowycz MW, Gonen T, and Arnold FH (2019). Tailoring Tryptophan Synthase TrpB for Selective Quaternary Carbon Bond Formation. J. Am. Chem. Soc. 141, 19817–19822. 10.1021/jacs.9b09864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Novick SJ, Dellas N, Garcia R, Ching C, Bautista A, Homan D, Alvizo O, Entwistle D, Kleinbeck F, Schlama T, and Ruch T (2021). Engineering an Amine Transaminase for the Efficient Production of a Chiral Sacubitril Precursor. ACS Catal. 11, 3762–3770. 10.1021/acscatal.0c05450. [DOI] [Google Scholar]
- 10.Toney MD (2011). Controlling reaction specificity in pyridoxal phosphate enzymes. Biochim. Biophys. Acta 1814, 1407–1418. 10.1016/j.bbapap.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dajnowicz S, Johnston RC, Parks JM, Blakeley MP, Keen DA, Weiss KL, Gerlits O, Kovalevsky A, and Mueser TC (2017). Direct visualization of critical hydrogen atoms in a pyridoxal 5’-phosphate enzyme. Nat. Commun. 8, 955. 10.1038/s41467-017-01060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dajnowicz S, Parks JM, Hu X, Johnston RC, Kovalevsky AY, and Mueser TC (2018). Hyperconjugation Promotes Catalysis in a Pyridoxal 5 ‘-Phosphate-Dependent Enzyme. ACS Catal. 8, 6733–6737. 10.1021/acscatal.8b01911. [DOI] [Google Scholar]
- 13.Phillips RS (2015). Chemistry and diversity of pyridoxal-5’-phosphate dependent enzymes. Biochim. Biophys. Acta 1854, 1167–1174. 10.1016/j.bbapap.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 14.Richard JP, Amyes TL, Crugeiras J, and Rios A (2009). Pyridoxal 5 ‘-phosphate: electrophilic catalyst extraordinaire. Curr. Opin. Chem. Biol. 13, 475–483. 10.1016/j.cbpa.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hyde CC, Ahmed SA, Padlan EA, Miles EW, and Davies DR (1988). 3-Dimensional Structure of the Tryptophan Synthase Alpha-2-Beta-2 Multienzyme Complex from Salmonella-Typhimurium. J. Biol. Chem. 263, 17857–17871. [PubMed] [Google Scholar]
- 16.Raboni S, Bettati S, and Mozzarelli A (2009). Tryptophan synthase: a mine for enzymologists. Cell. Mol. Life Sci. 66, 2391–2403. 10.1007/s00018-009-0028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Michalska K, Chang C, Maltseva NI, Jedrzejczak R, Robertson GT, Gusovsky F, McCarren P, Schreiber SL, Nag PP, and Joachimiak A (2020). Allosteric inhibitors of Mycobacterium tuberculosis tryptophan synthase. Protein Sci. 29, 779–788. 10.1002/pro.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bosken YK, Ai R, Hilario E, Ghosh RK, Dunn MF, Kan SH, Niks D, Zhou H, Ma W, Mueller LJ, et al. (2022). Discovery of antimicrobial agent targeting tryptophan synthase. Protein Sci. 31, 432–442. 10.1002/pro.4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watkins-Dulaney E, Straathof S, and Arnold F (2021). Tryptophan Synthase: Biocatalyst Extraordinaire. Chembiochem 22, 5–16. 10.1002/cbic.202000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faeder EJ, and Hammes GG (1970). Kinetic studies of tryptophan synthetase. Interaction of substrates with the B subunit. Biochemistry 9, 4043–4049. 10.1021/bi00823a003. [DOI] [PubMed] [Google Scholar]
- 21.Faeder EJ, and Hammes GG (1971). Kinetic studies of tryptophan synthetase. Interaction of L-serine, indole, and tryptophan with the native enzyme. Biochemistry 10, 1041–1045. 10.1021/bi00782a016. [DOI] [PubMed] [Google Scholar]
- 22.Hur O, Niks D, Casino P, and Dunn MF (2002). Proton transfers in the beta-reaction catalyzed by tryptophan synthase. Biochemistry 41, 9991–10001. 10.1021/bi025568u. [DOI] [PubMed] [Google Scholar]
- 23.Anderson KS, Kim AY, Quillen JM, Sayers E, Yang XJ, and Miles EW (1995). Kinetic characterization of channel impaired mutants of tryptophan synthase. J. Biol. Chem. 270, 29936–29944. 10.1074/jbc.270.50.29936. [DOI] [PubMed] [Google Scholar]
- 24.Holmes JB, Liu V, Caulkins BG, Hilario E, Ghosh RK, Drago VN, Young RP, Romero JA, Gill AD, Bogie PM, et al. (2022). Imaging active site chemistry and protonation states: NMR crystallography of the tryptophan synthase alpha-aminoacrylate intermediate. Proc. Natl. Acad. Sci. USA 119, e2109235119. 10.1073/pnas.2109235119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gardberg AS, Del Castillo AR, Weiss KL, Meilleur F, Blakeley MP, and Myles DAA (2010). Unambiguous determination of H-atom positions: comparing results from neutron and high-resolution X-ray crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 558–567. 10.1107/S0907444910005494. [DOI] [PubMed] [Google Scholar]
- 26.Blakeley MP, and Podjarny AD (2018). Neutron macromolecular crystallography. Emerg. Top. Life Sci. 2, 39–55. 10.1042/Etls20170083. [DOI] [PubMed] [Google Scholar]
- 27.Niimura N, and Podjarny AD (2011). Neutron Protein Crystallography : Hydrogen, Protons, and Hydration in BioMacromolecules (Oxford University Press; ). [Google Scholar]
- 28.Banco MT, Mishra V, Ostermann A, Schrader TE, Evans GB, Kovalevsky A, and Ronning DR (2016). Neutron structures of the Helicobacter pylori 5’-methylthioadenosine nucleosidase highlight proton sharing and protonation states. Proc. Natl. Acad. Sci. USA 113, 13756–13761. 10.1073/pnas.1609718113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerlits OO, Coates L, Woods RJ, and Kovalevsky A (2017). Mannobiose Binding Induces Changes in Hydrogen Bonding and Protonation States of Acidic Residues in Concanavalin A As Revealed by Neutron Crystallography. Biochemistry 56, 4747–4750. 10.1021/acs.biochem.7b00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerlits O, Weiss KL, Blakeley MP, Veglia G, Taylor SS, and Kovalevsky A (2019). Zooming in on protons: Neutron structure of protein kinase A trapped in a product complex. Sci. Adv. 5, eaav0482. 10.1126/sciadv.aav0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kneller DW, Phillips G, Weiss KL, Pant S, Zhang Q, O’Neill HM, Coates L, and Kovalevsky A (2020). Unusual zwitterionic catalytic site of SARS-CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 295, 17365–17373. 10.1074/jbc.AC120.016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golden EA, and Vrielink A (2014). Looking for Hydrogen Atoms: Neutron Crystallography Provides Novel Insights Into Protein Structure and Function. Aust. J. Chem. 67, 1751–1762. 10.1071/CH14337. [DOI] [Google Scholar]
- 33.Wan Q, Parks JM, Hanson BL, Fisher SZ, Ostermann A, Schrader TE, Graham DE, Coates L, Langan P, and Kovalevsky A (2015). Direct determination of protonation states and visualization of hydrogen bonding in a glycoside hydrolase with neutron crystallography. Proc. Natl. Acad. Sci. USA 112, 12384–12389. 10.1073/pnas.1504986112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oksanen E, Chen JCH, and Fisher SZ (2017). Neutron Crystallography for the Study of Hydrogen Bonds in Macromolecules. Molecules 22, 596. 10.3390/molecules22040596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Correy GJ, Kneller DW, Phillips G, Pant S, Russi S, Cohen AE, Meigs G, Holton JM, Gahbauer S, Thompson MC, et al. (2022). The mechanisms of catalysis and ligand binding for the SARS-CoV-2 NSP3 macrodomain from neutron and x-ray diffraction at room temperature. Sci. Adv. 8, eabo5083. 10.1126/sciadv.abo5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gajdos L, Blakeley MP, Haertlein M, Forsyth VT, Devos JM, and Imberty A (2022). Neutron crystallography reveals mechanisms used by Pseudomonas aeruginosa for host-cell binding. Nat. Commun. 13, 194. 10.1038/s41467-021-27871-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams PD, Mustyakimov M, Afonine PV, and Langan P (2009). Generalized X-ray and neutron crystallographic analysis: more accurate and complete structures for biological macromolecules. Acta Crystallogr. D Biol. Crystallogr 65, 567–573. 10.1107/S0907444909011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mueser TC, Drago V, Kovalevsky A, and Dajnowicz S (2020). Pyridoxal 5’-phosphate dependent reactions: Analyzing the mechanism of aspartate aminotransferase. Methods Enzymol. 634, 333–359. 10.1016/bs.mie.2020.01.009. [DOI] [PubMed] [Google Scholar]
- 39.Drago VN, Dajnowicz S, Parks JM, Blakeley MP, Keen DA, Coquelle N, Weiss KL, Gerlits O, Kovalevsky A, and Mueser TC (2022). An N/ H/ N low-barrier hydrogen bond preorganizes the catalytic site of aspartate aminotransferase to facilitate the second half-reaction. Chem. Sci. 13, 10057–10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drago VN, Devos JM, Blakeley MP, Forsyth VT, Kovalevsky AY, Schall CA, and Mueser TC (2022). Microgravity crystallization of perdeuterated tryptophan synthase for neutron diffraction. NPJ Microgravity 8, 13. 10.1038/s41526-022-00199-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caulkins BG, Bastin B, Yang C, Neubauer TJ, Young RP, Hilario E, Huang Y.m.M., Chang C.e.A., Fan L, Dunn MF, et al. (2014). Protonation states of the tryptophan synthase internal aldimine active site from solid-state NMR spectroscopy: direct observation of the protonated Schiff base linkage to pyridoxal-5’-phosphate. J. Am. Chem. Soc. 136, 12824–12827. 10.1021/ja506267d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klein A, Rovó P, Sakhrani VV, Wang Y, Holmes JB, Liu V, Skowronek P, Kukuk L, Vasa SK, Güntert P, et al. (2022). Atomicresolution chemical characterization of (2x)72-kDa tryptophan synthase via four- and five-dimensional (1)H-detected solid-state NMR. Proc. Natl. Acad. Sci. USA 119, e2114690119. 10.1073/pnas.2114690119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foster JW, and Spector MP (1995). How Salmonella Survive against the Odds. Annu. Rev. Microbiol. 49, 145–174. 10.1146/annurev.mi.49.100195.001045. [DOI] [PubMed] [Google Scholar]
- 44.Dunn MF, Niks D, Ngo H, Barends TRM, and Schlichting I (2008). Tryptophan synthase: the workings of a channeling nanomachine. Trends Biochem. Sci. 33, 254–264. 10.1016/j.tibs.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 45.Sakhrani VV, Hilario E, Caulkins BG, Hatcher-Skeers ME, Fan L, Dunn MF, and Mueller LJ (2020). Backbone assignments and conformational dynamics in the S. typhimurium tryptophan synthase alpha-subunit from solution-state NMR. J. Biomol. NMR 74, 341–354. 10.1007/s10858-020-00320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ito S, Yagi K, and Sugita Y (2022). Computational Analysis on the Allostery of Tryptophan Synthase: Relationship between α/β-Ligand Binding and Distal Domain Closure. J. Phys. Chem. B 126, 3300–3308. 10.1021/acs.jpcb.2c01556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rhee S, Miles EW, and Davies DR (1998). Cryo-crystallography of a true substrate, indole-3-glycerol phosphate, bound to a mutant (alphaD60N) tryptophan synthase alpha2beta2 complex reveals the correct orientation of active site alphaGlu49. J. Biol. Chem. 273, 8553–8555. [PubMed] [Google Scholar]
- 48.Weyand M, and Schlichting I (1999). Crystal Structure of Wild-Type Tryptophan Synthase Complexed with the Natural Substrate Indole-3-glycerol Phosphate. Biochemistry 38, 16469–16480. 10.1021/bi9920533. [DOI] [PubMed] [Google Scholar]
- 49.Brzović PS, Hyde CC, Miles EW, and Dunn MF (1993). Characterization of the functional role of a flexible loop in the alpha-subunit of tryptophan synthase from Salmonella typhimurium by rapid-scanning, stopped-flow spectroscopy and site-directed mutagenesis. Biochemistry 32, 10404–10413. 10.1021/bi00090a016. [DOI] [PubMed] [Google Scholar]
- 50.Dunn MF (2012). Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch. Biochem. Biophys. 519, 154–166. 10.1016/j.abb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phillips RS, and Harris AP (2021). Structural Basis of the Stereochemistry of Inhibition of Tryptophan Synthase by Tryptophan and Derivatives. Biochemistry 60, 231–244. 10.1021/acs.biochem.0c00635. [DOI] [PubMed] [Google Scholar]
- 52.Liang J, Han Q, Tan Y, Ding H, and Li J (2019). Current Advances on Structure-Function Relationships of Pyridoxal 5’-Phosphate-Dependent Enzymes. Front. Mol. Biosci. 6, 4. 10.3389/fmolb.2019.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Griswold WR, and Toney MD (2011). Role of the pyridine nitrogen in pyridoxal 5’-phosphate catalysis: activity of three classes of PLP enzymes reconstituted with deazapyridoxal 5’-phosphate. J. Am. Chem. Soc. 133, 14823–14830. 10.1021/ja2061006. [DOI] [PubMed] [Google Scholar]
- 54.Miles EW, and McPhie P (1974). Evidence for a rate-determining proton abstraction in the serine deaminase reaction of the beta 2 subunit of tryptophan synthetase. J. Biol. Chem. 249, 2852–2857. [PubMed] [Google Scholar]
- 55.Yutani K, Ogasahara K, Tsujita T, Kanemoto K, Matsumoto M, Tanaka S, Miyashita T, Matsushiro A, Sugino Y, and Miles EW (1987). Tryptophan Synthase Alpha-Subunit Glutamic-Acid 49 Is Essential for Activity - Studies with 19 Mutants at Position-49. J. Biol. Chem. 262, 13429–13433. [PubMed] [Google Scholar]
- 56.Nagata S, Hyde CC, and Miles EW (1989). The alpha subunit of tryptophan synthase. Evidence that aspartic acid 60 is a catalytic residue and that the double alteration of residues 175 and 211 in a second-site revertant restores the proper geometry of the substrate binding site. J. Biol. Chem. 264, 6288–6296. [PubMed] [Google Scholar]
- 57.Dunathan HC (1966). Conformation and Reaction Specificity in Pyridoxal Phosphate Enzymes. P Natl Acad Sci USA 55, 712–716. 10.1073/pnas.55.4.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heinert D, and Martell AE (1963). Pyridoxine and Pyridoxal Analogs. VII. Acid-Base Equilibria of Schiff Bases. J. Am. Chem. Soc. 85, 188–193. 10.1021/ja00885a018. [DOI] [Google Scholar]
- 59.Metzler CM, Cahill A, and Metzler DE (1980). Equilibriums and absorption spectra of Schiff bases. J. Am. Chem. Soc. 102, 6075–6082. 10.1021/ja00539a017. [DOI] [Google Scholar]
- 60.Ahmed SA, McPhie P, and Miles EW (1996). A thermally induced reversible conformational transition of the tryptophan synthase beta2 subunit probed by the spectroscopic properties of pyridoxal phosphate and by enzymatic activity. J. Biol. Chem. 271, 8612–8617. 10.1074/jbc.271.15.8612. [DOI] [PubMed] [Google Scholar]
- 61.Jhee KH, Yang LH, Ahmed SA, McPhie P, Rowlett R, and Miles EW (1998). Mutation of an active site residue of tryptophan synthase (beta-serine 377) alters cofactor chemistry. J. Biol. Chem. 273, 11417–11422. 10.1074/jbc.273.19.11417. [DOI] [PubMed] [Google Scholar]
- 62.Phillips RS, Sundararaju B, and Koushik SV (1998). The catalytic mechanism of kynureninase from Pseudomonas fluorescens: evidence for transient quinonoid and ketimine intermediates from rapid-scanning stopped-flow spectrophotometry. Biochemistry 37, 8783–8789. 10.1021/bi980066v. [DOI] [PubMed] [Google Scholar]
- 63.Toney MD, and Kirsch JF (1991). The K258R mutant of aspartate aminotransferase stabilizes the quinonoid intermediate. J. Biol. Chem. 266, 23900–23903. [PubMed] [Google Scholar]
- 64.Phillips RS, Demidkina TV, Zakomirdina LN, Bruno S, Ronda L, and Mozzarelli A (2002). Crystals of tryptophan indole-lyase and tyrosine phenol-lyase form stable quinonoid complexes. J. Biol. Chem. 277, 21592–21597. 10.1074/jbc.M200216200. [DOI] [PubMed] [Google Scholar]
- 65.Casasnovas R, Salvá A, Frau J, Donoso J, and Muñoz F (2009). Theoretical study on the distribution of atomic charges in the Schiff bases of 3-hydroxypyridine-4-aldehyde and alanine. The effect of the protonation state of the pyridine and imine nitrogen atoms. Chem. Phys. 355, 149–156. [Google Scholar]
- 66.Ro HS, and Miles EW (1999). Structure and function of the tryptophan synthase alpha(2) beta(2) complex. Roles of beta subunit histidine 86. J. Biol. Chem. 274, 36439–36445. 10.1074/jbc.274.51.36439. [DOI] [PubMed] [Google Scholar]
- 67.Yang L.h., Ahmed SA, and Miles EW (1996). PCR mutagenesis and overexpression of tryptophan synthase from Salmonella typhimurium: on the roles of beta2 subunit Lys-382. Protein Expr. Purif. 8, 126–136. 10.1006/prep.1996.0082. [DOI] [PubMed] [Google Scholar]
- 68.Marenich AV, Cramer CJ, and Truhlar DG (2009). Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 69.Chai JD, and Head-Gordon M (2008). Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- 70.Weigend F, and Ahlrichs R (2005). Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 71.Weigend F (2006). Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 8, 1057–1065. 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- 72.Huang YMM, You W, Caulkins BG, Dunn MF, Mueller LJ, and Chang CEA (2016). Protonation states and catalysis: Molecular dynamics studies of intermediates in tryptophan synthase. Protein Sci. 25, 166–183. 10.1002/pro.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Glendening E, Reed A, Carpenter J, and Weinhold F (2001). Gaussian NBO Version 3.1 (Pittsburgh PA: Gaussian Inc.). [Google Scholar]
- 74.Toney MD (2014). Aspartate aminotransferase: an old dog teaches new tricks. Arch. Biochem. Biophys. 544, 119–127. 10.1016/j.abb.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yano T, Kuramitsu S, Tanase S, Morino Y, and Kagamiyama H (1992). Role of Asp222 in the catalytic mechanism of Escherichia coli aspartate aminotransferase: the amino acid residue which enhances the function of the enzyme-bound coenzyme pyridoxal 5’-phosphate. Biochemistry 31, 5878–5887. 10.1021/bi00140a025. [DOI] [PubMed] [Google Scholar]
- 76.Yano T, Mizuno T, and Kagamiyama H (1993). A hydrogen-bonding network modulating enzyme function: asparagine-194 and tyrosine-225 of Escherichia coli aspartate aminotransferase. Biochemistry 32, 1810–1815. 10.1021/bi00058a015. [DOI] [PubMed] [Google Scholar]
- 77.Cordes EH, and Jencks WP (1962). Semicarbazone formation from pyridoxal, pyridoxal phosphate, and their Schiff bases. Biochemistry 1, 773–778. 10.1021/bi00911a007. [DOI] [PubMed] [Google Scholar]
- 78.Hayashi H, Mizuguchi H, and Kagamiyama H (1999). The imine-pyridine torsion of the pyridoxal 5’-phosphate schiff base of aspartate aminotransferase lowers its pKa in the unliganded enzyme and is crucial for the successive increase in the pKa during catalysis. Biochemistry 38, 854. 10.1021/bi985061c. [DOI] [PubMed] [Google Scholar]
- 79.Casasnovas R, Adrover M, Ortega-Castro J, Frau J, Donoso J, and Muñoz F (2012). C-H Activation in Pyridoxal-5 ‘-phosphate Schiff Bases: The Role of the Imine Nitrogen. A Combined Experimental and Computational Study. J. Phys. Chem. B 116, 10665–10675. 10.1021/jp303678n. [DOI] [PubMed] [Google Scholar]
- 80.Meilleur F, Kovalevsky A, and Myles DAA (2020). IMAGINE: The neutron protein crystallography beamline at the high flux isotope reactor. Methods Enzymol. 634, 69–85. 10.1016/bs.mie.2019.11.016. [DOI] [PubMed] [Google Scholar]
- 81.Coates L, Cao HB, Chakoumakos BC, Frontzek MD, Hoffmann C, Kovalevsky AY, Liu Y, Meilleur F, Dos Santos AM, Myles DAA, et al. (2018). A suite-level review of the neutron single-crystal diffraction instruments at Oak Ridge National Laboratory. Rev. Sci. Instrum. 89, 092802. 10.1063/1.5030896. [DOI] [PubMed] [Google Scholar]
- 82.Meilleur F, Coates L, Cuneo M, Kovalevsky A, and Myles D (2018). The Neutron Macromolecular Crystallography Instruments at Oak Ridge National Laboratory: Advances, Challenges, and Opportunities. Crystals 8, 388. 10.3390/cryst8100388. [DOI] [Google Scholar]
- 83.Meilleur F, Munshi P, Robertson L, Stoica AD, Crow L, Kovalevsky A, Koritsanszky T, Chakoumakos BC, Blessing R, and Myles DAA (2013). The IMAGINE instrument: first neutron protein structure and new capabilities for neutron macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 69, 2157–2160. 10.1107/S0907444913019604. [DOI] [PubMed] [Google Scholar]
- 84.Blakeley MP, Teixeira SCM, Petit-Haertlein I, Hazemann I, Mitschler A, Haertlein M, Howard E, and Podjarny AD (2010). Neutron macromolecular crystallography with LADI-III. Acta Crystallogr. D 66, 1198–1205. 10.1107/S0907444910019797. [DOI] [PubMed] [Google Scholar]
- 85.Campbell JW (1995). Lauegen, an X-Windows-Based Program for the Processing of Laue X-Ray-Diffraction Data. J. Appl. Crystallogr. 28, 228–236. 10.1107/S002188989400991x. [DOI] [Google Scholar]
- 86.Campbell JW, Hao Q, Harding MM, Nguti ND, and Wilkinson C (1998). LAUEGEN version 6.0 and INTLDM. J. Appl. Crystallogr. 31, 496–502. 10.1107/S0021889897016683. [DOI] [Google Scholar]
- 87.Arzt S, Campbell JW, Harding MM, Hao Q, and Helliwell JR (1999). LSCALE - the new normalization, scaling and absorption correction program in the Daresbury Laue software suite. J. Appl. Crystallogr. 32, 554–562. 10.1107/S0021889898015350. [DOI] [Google Scholar]
- 88.Weiss MS (2001). Global indicators of X-ray data quality. J. Appl. Crystallogr. 34, 130–135. 10.1107/S0021889800018227. [DOI] [Google Scholar]
- 89.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877. 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mustyakimov M, and Langan P (2007). nCNS: An Open Source Distribution Patch for CNS for Macromolecular Structure Refinement (Los Alamos, NM, USA: Los Alamos National Security; ). [Google Scholar]
- 92.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. (1998). Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921. 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- 93.Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 94.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, et al. (2016). Gaussian 16 Rev. C.01.
- 95.Evans R, O’Neill M, Pritzel A, Antropova N, Senior A, Green T, Zídek A, Bates R, Blackwell S, Yim J, et al. (2021). Protein Complex Prediction with AlphaFold-Multimer. Preprint at bioRxiv. 10.1101/2021.10.04.463034. [DOI]
- 96.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mirdita M, Steinegger M, and Söding J (2019). MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics 35, 2856–2858. 10.1093/bioinformatics/bty1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, and Steinegger M (2022). ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682. 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Anandakrishnan R, Aguilar B, and Onufriev AV (2012). H++3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40, W537–W541. 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bashford D, and Karplus M (1990). pKa’s of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry 29, 10219–10225. 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the joint XN and X-ray structures reported in this paper are PDB: 8EYP, 8EZC, and 8EYS.