

Keywords: chondrocyte, kinesin, microtubules, motor proteins, skeletal dysplasia
Abstract
Skeletal dysplasias are group of rare genetic diseases resulting from mutations in genes encoding structural proteins of the cartilage extracellular matrix (ECM), signaling molecules, transcription factors, epigenetic modifiers, and several intracellular proteins. Cell division, organelle maintenance, and intracellular transport are all orchestrated by the cytoskeleton-associated proteins, and intracellular processes affected through microtubule-associated movement are important for the function of skeletal cells. Among microtubule-associated motor proteins, kinesins in particular have been shown to play a key role in cell cycle dynamics, including chromosome segregation, mitotic spindle formation, and ciliogenesis, in addition to cargo trafficking, receptor recycling, and endocytosis. Recent studies highlight the fundamental role of kinesins in embryonic development and morphogenesis and have shown that mutations in kinesin genes lead to several skeletal dysplasias. However, many questions concerning the specific functions of kinesins and their adaptor molecules as well as specific molecular mechanisms in which the kinesin proteins are involved during skeletal development remain unanswered. Here we present a review of the skeletal dysplasias resulting from defects in kinesins and discuss the involvement of kinesin proteins in the molecular mechanisms that are active during skeletal development.
INTRODUCTION
Skeletal dysplasias are a group of heterogeneous genetic disorders that affect skeletal growth and are characterized by often overlapping phenotypes such as dwarfism and long bone deformation and malformation (1, 2). Abnormalities in the early skeletal development, patterning, and maintenance that affect mainly bone and cartilage but often also tendons, ligaments, and skeletal muscles result in a variety of skeletal diseases with an estimated prevalence rate of 1/5,000 births worldwide (3, 4). Skeletal dysplasias are diagnosed based on radiographic, clinical, and molecular features and can be associated with further complications such as congenital cardiac defects, gastrointestinal abnormalities, endocranial, and neurological complications (2). Advances in molecular genetics and next generation sequencing have accelerated the identification of genetic variants in rare disorders of the skeleton and allowed the identification of 771 conditions associated with 552 genes (5) The molecular mechanisms underlying the skeletal dysplasias together with the identified genetic variants are detailed in the “Nosology of skeletal dysplasia,” which is regularly reviewed and updated (5). Research advances have shown that different genetic mutations acting through shared molecular pathways can cause similar skeletal phenotypes [e.g., osteogenesis imperfecta (OI) results from disruptions in genes encoding type I collagen (COL1A1 and COL1A2) and genes encoding molecular chaperones (e.g., SERPINH1 and FKBP10)], as well as that different skeletal dysplasias can result from mutations in the same gene (e.g., mutations in the FGFR3 cause achondroplasia, hypochondroplasia, and thanatophoric dysplasia) (6, 7). Genes associated with skeletal dysplasias are implicated in various biological processes and encode extracellular (matrix components, matrix remodeling enzymes, growth factors) (8–10), intracellular (chaperones, transcription factors, motor molecules) (6, 11), and cell surface molecules (receptors, channel proteins) (12, 13) that are essential for bone and cartilage formation and maintenance.
Kinesins Are Ubiquitously Expressed Microtubule-Associated Motor Proteins Involved in Multiple Cellular Processes
Kinesins are cytoskeletal motor proteins that carry cargo along the microtubules in an ATP-dependent manner (14). Kinesins were first discovered in 1985 as proteins involved in neuronal development and homeostasis, and together with dyneins they control the anterograde and retrograde axonal transport (14, 15). Since their discovery, 45 different kinesin (KIF) genes have been identified in mice and humans and were grouped into 15 classes based on sequence homology (Table 1) (78). Most kinesins function as a dimer, and their structure comprises a coiled-coil neck linker that mediates the dimerization of the two heavy chains and two light chains, which are associated by a heptad repeat regions near their N-terminus (79). The motor domain, also known as the head, binds to the microtubules and hydrolyses ATP to generate energy required for movement. The tail domain attaches to specific cargo, either directly or through association with adaptor proteins, and regulates the kinesin function (79).
Table 1.
Classification of the kinesin proteins according to the motor domain position (16)
| N-Kinesins | ||
|---|---|---|
| Kinesin Class | Member Kinesins | Identified Roles/Processes |
| Kinesin 1 | KIF5A | RNA transport (17), mitochondria transport (18), autophagy (19), secretory vesicle transport (20) |
| KIF5B | RNA transport (17), cytokinesis (21), vesicle transport (22), peroxisome transport (23), nuclear positioning (24) | |
| KIF5C | RNA transport (17) | |
| Kinesin 2 | KIF3A | Embryonic cilia formation (25, 26), cilia function, anterograde IFT (27), hedgehog signaling (28) |
| KIF3B | Ciliogenesis (26), hedgehog signaling (29), vesicle transport (30) | |
| KIF3C | Ciliogenesis (31), axonal transport (32) | |
| KIF17 | Vesicle transport, receptor sorting (33) | |
| Kinesin 3 | KIF1A | Vesicle transport (34, 35) |
| KIF1B | Mitochondria transport (36), receptor transport (37) | |
| KIF1C | Retrograde transport of Golgi vesicles to the ER (38) | |
| KIF13A | Vesicle transport, Golgi to plasma membrane transport (39) | |
| KIF13B | VEGFR2 trafficking from the Golgi, angiogenesis (40), secretory vesicle transport (41) | |
| KIF14 | Cytokinesis (41) | |
| KIF16A | Cell division and spindle assembly (43), pericentriolar material stabilization (43) | |
| KIF16B | Vesicle transport, early to late endosomes (44), receptor recycling (45) | |
| Kinesin 4 | KIF4A | Chromokinesin (46), ciliogenesis (47, 48) |
| KIF4B | Spindle dynamics and cytokinesis (49) | |
| KIF7 | Primary cilium-associated hedgehog signaling (50) | |
| KIF21A | Microtubule dynamics (51) | |
| KIF21B | Microtubule dynamics, centrosome polarization (52) | |
| KIF27 | Construction of central pair in motile cilia, ciliogenesis, and hedgehog signaling (53) | |
| Kinesin 5 | KIF11 | Cytokinesis, spindle formation (54), ciliogenesis (55), translation, association of ribosomes with microtubules (56) |
| Kinesin 6 | KIF20A | Cytokinesis (57), Golgi-derived vesicle transport (58) |
| KIF20B | Cell division (59) | |
| KIF23 | Cytokinesis (60) | |
| Kinesin 7 | KIF10 | Cell division, kinetochore function (61) |
| Kinesin 8 | KIF18A | Mitotic chromosome positioning (62) |
| KIF18B | Microtubule depolymerization, cell division (63) | |
| KIF19A | Cilia length (64) | |
| Kinesin 9 | KIF6 | Ciliogenesis, cell division (65) |
| KIF9 | Motile cilia function (66), podosome function (67) | |
| Kinesin 10 | KIF22 | Chromokinesin (46) |
| Kinesin 11 | KIF26A | Microtubule stability, RET signaling (68) |
| KIF26B | MYH10 mediated cell-cell adhesion (69) | |
| Kinesin 12 | KIF12 | Cell polarity (70) |
| KIF15 | Cell division (71), integrin endocytosis (72) | |
| Kinesin 13 | KIF24 | Depolymerization of the centriolar microtubules (73) |
| Kinesin 14b | KIF25 | Centrosome operation and spindle orientation (74) |
| M-Kinesins | ||
|---|---|---|
| Kinesin class | Member kinesins | Identified roles/processes |
| Kinesin 13 | KIF2A | Microtubule depolymerization, spindle assembly, and chromosome movement (75) |
| KIF2B | Spindle assembly and chromosome movement (75) | |
| KIF2C | Microtubule depolymerization, cell division (63) | |
| C-Kinesins | ||
|---|---|---|
| Kinesin class | Member kinesins | Identified roles/processes |
| Kinesin 14a | KIFC1 | Retrograde transport from Golgi apparatus to the ER (38), retrograde transport of secretory vesicles (76) |
| Kinesin 14b | KIFC2 | Correction of the improper microtubule attachments at kinetochores (75) |
| KIFC3 | Maintaining the integrity of the adherent junction (77) | |
The kinesin motor domain is located either in the amino (N)-terminus, middle of the protein, or in the carboxy (C)-terminus. Based on this, kinesins are subdivided into three classes: N-kinesins that are plus-end-directed motor protein, M-kinesins that act as microtubule depolymerizers, and C-kinesins that mediate minus-end-directed movement (Fig. 1) (81, 82). The motor domains present a high sequence homology in all kinesin superfamily and are highly conserved across a number of species (83). Kinesin microtubule binding and movement are regulated by various intramolecular and intermolecular interactions; for example, Kinesin-1 molecules are autoinhibited by the kinesin head and tail interactions, whereas Kinesin-3 molecules autoinhibition is via interaction of the coiled-coil domain with the motor head (79, 83, 84). The diversity of the kinesin light chains permits the recognition and transport of multiple cargos (85). There are four human kinesin light chains (KLCs, KLC1, KLC2, KLC3, and KLC4), and their complexity can be further enhanced by alternative splicing, with multiple splice variants allowing specific cargo binding (83). Cargos can bind directly to the kinesin heavy chains or attach to the tetratricopeptide (TRP) repeat domain in the kinesin light chain (KLC) (86). The TRP repeats are composed of multiple tandem repeats of 34 degenerate amino acids forming a helix-turn-helix structural motif able to mediate protein-protein interactions (80). Activation of kinesins is by effector proteins binding to the kinesin heavy chain or the TRPs on the light chain and breaking or weakening the intramolecular interaction (86–88).
Figure 1.

Molecular structure of kinesins. Kinesin dimers comprise two heavy chains (HCs) with a microtubule-binding site (motor domain) and a coiled-coil domain, and two light chains (LCs) comprising the tetratricopeptide repeat domain (KLCTPR) and a heptad repeat that associate to the coiled-coil domain of HCs (80). The position of the motor domain defines the kinesin classes: N-Kinesins have an N-terminus motor domain and drive plus-ended microtubule movement, M-Kinesins have their motor domain in the middle and are MT depolymerizers, and C-kinesin have a carboxy (C)-terminal motor domain and mediate minus-end-directed movement (81, 82). Created with BioRender.com.
Kinesin proteins are essential for neurodevelopment and have been implicated in cancer development and metastasis. They are involved in several key molecular processes, such as the transport of organelles, vesicles, membrane-bound organelles, nucleic acids, and protein complexes, and in the regulation of cell cycle progression, mitotic spindle formation, and movement of chromosomes during cell division (78). Kinesin family members also participate in the regulation of microtubule dynamics and were recently discovered to act as depolymerizers (KIF2A and KIF2C) and microtubule stabilizers (KIF26A and KIF21A), further emphasizing their importance for cellular morphogenesis, neuronal and axonal morphology, and ciliogenesis (Table 1) (89).
Several Kinesins Are Implicated in Skeletal Development and Disease
Kinesins play a pivotal role during early development and organogenesis. They are essential for neuronal development and axonal transport and play important roles in cell survival. Mutations in several kinesin genes affect neurodevelopment and result in secondary microcephaly, and in several mouse models the loss of kinesin function led to embryonic or perinatal lethality (KIF1A, KIF1B, KIF2A, KIF3A, KIF3B, KIF5A, KIF5B, KIF7, KIF10, KIF11, KIF18A, KIF20A, KIF20B, KIF21, and KIF26B) (90–92). Interestingly, mutations in several kinesin genes have also been specifically shown to have an impact on the development of the vertebral column and the appendicular skeleton and lead to multiple malformations, reduced bone formation, early onset joint degeneration, and multiple joint dislocations. Skeleton formation, maintenance, and homeostasis are tightly regulated by numerous molecular mechanisms in which kinesins play a key role; and mutations in four kinesin genes are associated with specific skeletal dysplasia (Table 2), through a variety of mechanisms (Fig. 2). Differential spatial and temporal gene expression was detected for Kif5b, Kif7, Kif10, and Kif22 in the murine tibial growth plate cartilage across the growth plate zones and with age (Fig. 3), highlighting their importance in the resting and proliferative zone homeostasis. Disruption of KIF5B has been shown to affect cytokinesis of proliferative chondrocytes and result in growth plate disorganization and skeletal abnormalities (21). HH signaling pathways are important for the maintenance of growth plate and skeletal patterning, and mutations in KIF7 impair HH signaling in the growth plate (50). In addition, KIF7 plays crucial role in cilia length regulation (94). Chondrocytes are highly mechanoresponsive and defects in primary cilia homeostasis have been shown to compromise cartilage mechanosensing and lead to skeletal dysplasia. Moreover, kinesin proteins are essential for cell division, and mutations in kinesins tend to strongly affect mitosis in skeletal cells. In the growth plate, chondrocytes undergo constant division and can be affected by mutations in the chromokinesin proteins (e.g., KID/KIF22 and kinetochore kinesin CENPE) through mechanisms such as aberrant chromosome attachment and movement on the spindle microtubules, chromosome misalignment on equatorial plate, and disrupted centrosome separation (95). Furthermore, kinesin proteins implicated in skeletal dysplasia are involved in organelle positioning and function and in vesicular transport, which can impact on ECM-related processes, and consequently lead to abnormal ECM organization and skeletal defects (Fig. 4) (20).
Table 2.
Skeletal dysplasias resulting from mutations in kinesin genes (5)
| Gene | Inheritance | Disorder | MIM No. | Skeletal Features | Complications | Disease Mechanism |
|---|---|---|---|---|---|---|
| KIF5B | AD | Kyphomelic dysplasia with facial dysmorphism (21, 107, 110) | 211350 | Short stature, vertebrae deformity, narrow chest, bowed legs, joint stiffness, micromelia, distinctive facial features, and micrognathia | Neonatal respiratory distress | Impaired mitochondria and lysosome trafficking, disruption of autophagy, cytokinesis defects, proliferative chondrocyte apoptosis, disrupted perichondral ossification |
| KIF7 | AR | Acrocallosal syndrome (ACLS) (115) | 200990 | Macrocephaly, facial dysmorphism, brain abnormalities | Mental retardation | Disruption of HH signaling, cilia length misregulation, proliferative chondrocyte apoptosis, and expansion of the hypertrophic zone |
| AR | Hydrolethalus syndrome (HLS2) (94) | 614120 | Hydrocephaly, postaxial polydactyly of the upper limbs, and pre- or postaxial polydactyly of the lower limbs and hallux duplication | Widened ventricles and midbrain-hindbrain malformation | ||
| AR | Al-Gazali-Bakalinova syndrome (AGBK) (114) | 607131 | Macrocephaly multiple epiphyseal dysplasia, and distinctive faces | Agenesis of the corpus callosum and frontotemporal brain atrophy | ||
| KIF10 | AR | Microcephalic osteodysplastic primordial dwarfism (61, 101, 102, 126) | 616051 | Microcephaly, short stature, metaphyseal sclerosis, structural brain malformations, mild micrognathia, short metacarpals, and osteopenia | Partial agenesis of the corpus callosum and cerebellar hypoplasia, cardio-myopathy | Chromosome mis-alignment, spindle defects, and arrest of G2-M phase |
| KIF22 | AD | SEMD with joint laxity (Hall type or leptodactylic type) (103, 129, 131) | 603546 | Short stature, progressive knee malalignment, slender metacarpals/metatarsals, mild spinal deformity, and early onset osteoarthritis | Ligamentous laxity, multiple dislocations | Chromosome segregation defects, lack in chromosome compaction, and disrupted cytokinesis |
Figure 2.
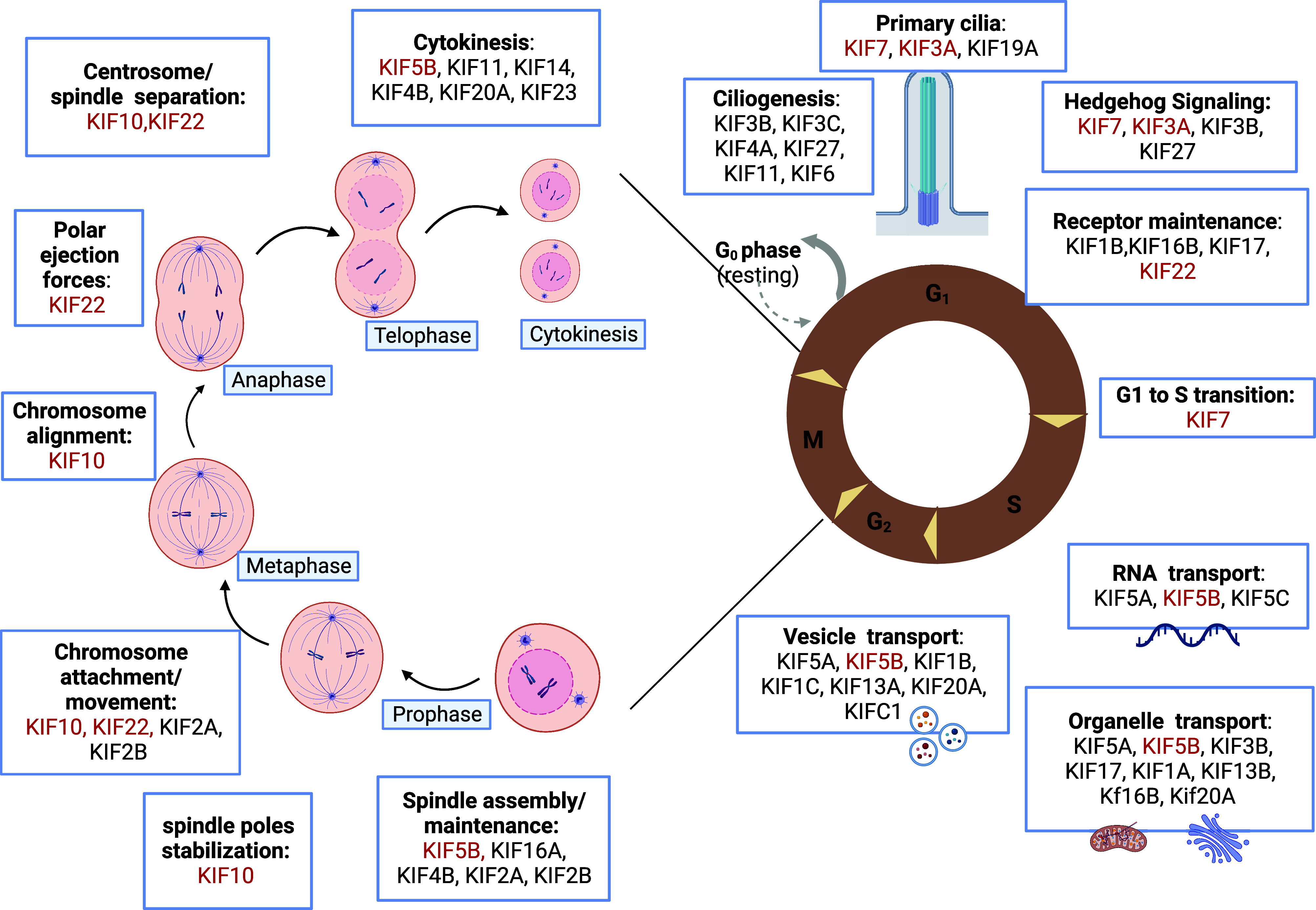
Kinesins play a role in multiple cell processes. Kinesins ensure specific functions during different phases of cell division, mediating chromosome movement and alignment, spindle formation, and regulating microtubule-kinetochore interaction. They are also involved in the maintenance of the primary cilia structure and function, and drive intracellular transport of organelles, vesicles, receptors, and RNA along microtubules. The kinesins implicated in skeletal dysplasia have been highlighted in red. Created with BioRender.com.
Figure 3.
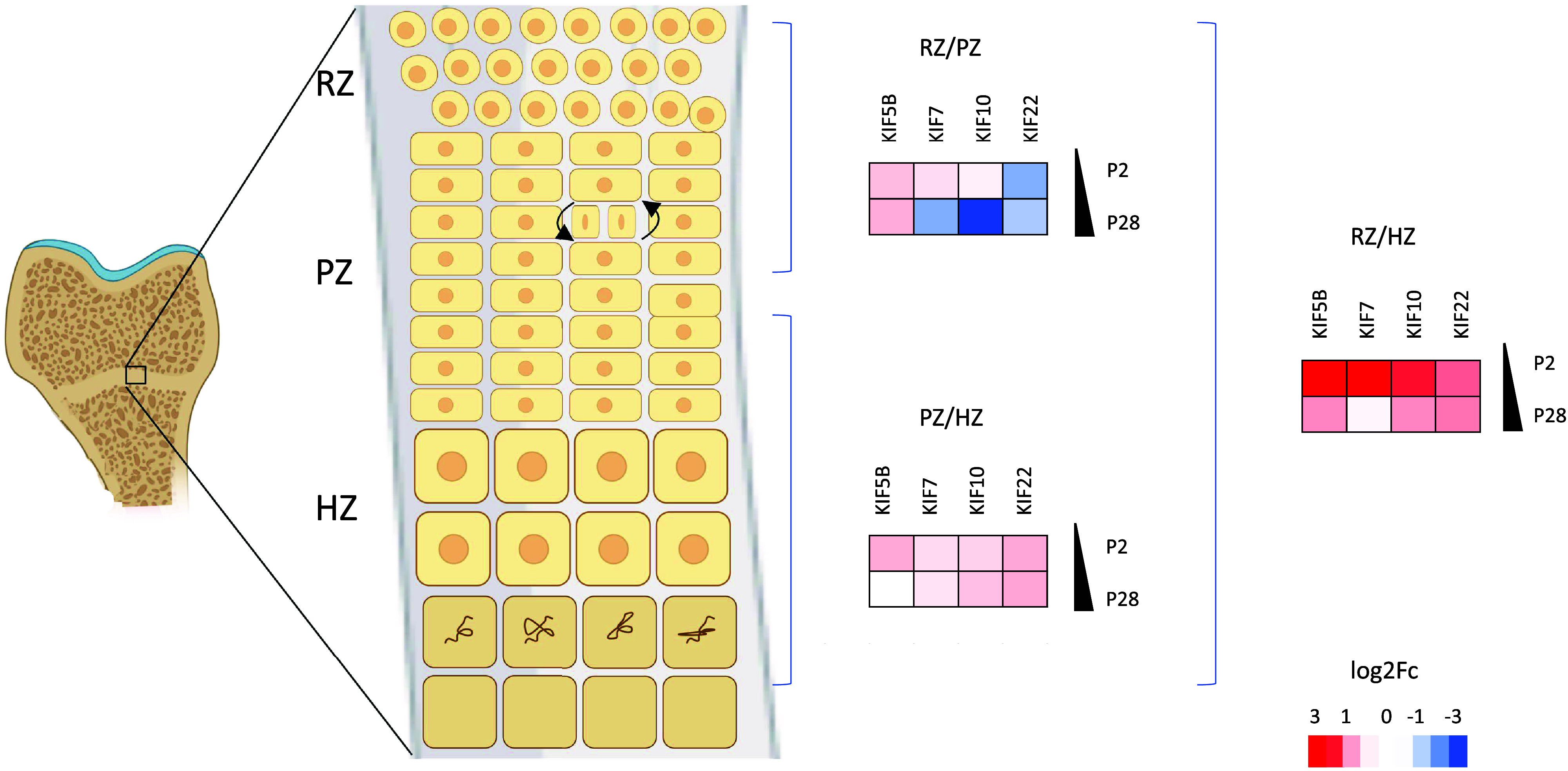
KIF5B, KIF7, KIF10, and KIF22 are differentially expressed in the zones of murine epiphyseal cartilage during development. Fold change data were obtained from SkeletalVis (106), and heat maps showing differential expression (log2FC) were generated. Key: HZ, hypertrophic zone; PZ, proliferative zone; P2, postnatal day 2; P28, postnatal day 28; RZ, resting zone. Created with BioRender.com.
Figure 4.

Kinesin motor proteins mediate ECM-related processes. Kinesin molecules implicated in skeletal dysplasia play roles in organelle positioning (including the endoplasmic reticulum and Golgi apparatus) and receptor recycling, and contribute to the intracellular transport of vesicles containing ECM components. Kinesins also play roles in ciliogenesis and mechanosensing. ECM, extracellular matrix. Created with BioRender.com.
Loss of Function of KIF5B Affects Cell Polarity, Cytokinesis, and Lysosomal Function and Results in Kyphomelic Dysplasia with Facial Dysmorphism
Kyphomelic dysplasia with facial dysmorphism is an autosomal recessive skeletal disorder characterized by a disproportionately short stature with a short narrow chest, shortening and bending (bowing) of the long bones, flared irregular metaphyses, and characteristic facial features (depressed nasal bridge, hypertelorism, micrognathia, and mid face hypoplasia) (107). Three de novo heterozygous variants in KIF5B encoding kinesin heavy chain have recently been reported, affecting the highly conserved amino acids near the ATP binding site in the catalytic motor domain of KIF5B (107). KIF5B is a member of the Kinesin-1 family, widely expressed in the human body with relative abundance in the retina, skeletal muscle, and central nervous system (108, 109).
In zebrafish, mutations in Kif5B (Kif5Blof) impaired lysosomal function and consequently led to a deregulation of autophagy (110). Moreover, Kif5B loss of function induced chondrocyte apoptosis and led to a loss of flattened appearance of the proliferative chondrocytes, accumulation of abnormally rounded chondrocytes with impaired polarization, loss of hypertrophic chondrocytes, and disrupted perichondral ossification (110). Centrosomes in the mutant cells were abnormally orientated, suggesting the loss of polarization of the microtubule-organizing center (MTOC). A chondrocyte-specific knockout mouse model of Kif5b (Col2cre; Kif5bfl/− mice) provided further insights into the intracellular role of KIF5B in chondrocytes. Kif5b mutant mice displayed short stature with restricted growth of spine vertebrae and long bones (21). Mutant growth plate showed disrupted cartilage characterized by disorganized columnar structure, cellular misorientation, abnormal apoptosis of proliferative chondrocytes, and a decrease in the number of hypertrophic chondrocytes (24). Monitoring cell division of isolated mutant chondrocytes demonstrated defects in cytokinesis characterized by a delay in cell abscission or failure in forming daughter cells due to back fusion during abscission of the primary chondrocytes. KIF5B has been shown to concentrate in the central spindle of primary chondrocytes, and the disruption of the mitotic spindle in the KIF5B null chondrocytes outlines its role in the organization and maintenance of the central spindle during chondrocyte cytokinesis. Deletion of KIF5B in all mouse tissues results in embryonic lethality and severe growth retardation (111). Interestingly, the extraembryonic cells used to study the intracellular mechanisms affected by the global Kif5B deletion showed abnormally clustered mitochondria in the perinuclear region of cells and impaired lysosomal dispersion. The mitochondria phenotype was rescued by an exogenous expression of Kif5B, highlighting a role of KIF5B in both mitochondria and lysosome trafficking and positioning (111). Kif5blof zebrafish mutants also showed abnormal mitochondria and ER localization (110). The impaired lysosomal function reported in the Kif5Blof zebrafish mutants correlated with decreased mTOR pathway activation that may have contributed to the decrease in the number of hypertrophic chondrocytes and suggested a role for KIF5B in regulating autophagy. Moreover, a heterozygous mutation in KIF5B was identified in four patients with osteogenesis imperfecta (OI) and analysis of primary patient fibroblasts showed an impaired intracellular transport of mitochondria and abnormal Golgi positioning and resulted in a downregulation of mTOR signaling supporting the implication of KIF5B in regulation of autophagy (98). In addition, Kinesin-1 and -3 proteins have been shown to drive lysosome movement along different microtubule tracks and particularly KIF5B selectively recruits lysosomes to the microtubules (97). KIF5B has also been shown to be important for the initial formation of the autophagosome in the HeLa cells (97). Interestingly, KIF5B has been implicated in the delivery of matrix metalloproteinases to the cell surface in macrophages and cancer cells (96, 112), highlighting the diversity of individual kinesin functions and potential additional roles for this kinesin in skeletal development and homeostasis. For example, it has been shown that KIF5B is important for MMP14 (also implicated in cartilage resorption and ossification) trafficking in macrophages (113). KIF5B has also been shown to be involved in the intracellular trafficking of secretory vesicles containing extracellular matrix components and colocalized with collagen I-containing vesicles in the human pleural mesothelial cells, with KIF5B deletion resulting in reduced type I collagen secretion (20). Type II collagen secretion was also affected in Kif5blof zebrafish mutants (110). KIF5B may therefore contribute to the extracellular matrix production and organization by playing a role in transporting secretory vesicles. Taken together, KIF5B is a major contributor to the intracellular trafficking of various organelles (mitochondria, lysosomes, and secretory vesicles) that are implicated in extracellular matrix synthesis and skeletal development, as well as playing a role in cell division where it contributes to the organization and maintenance of the central spindle during cytokinesis.
Allelic Series of KIF7 Mutations Lead to Ciliopathy-Related Skeletal Disorders
Genome-wide linkage analysis and gene sequencing of individuals manifesting with several skeletal disorders identified homozygous mutations in KIF7 in three autosomal recessive lethal disorders commonly characterized by dysmorphic features (macrocephaly, hydrocephaly, or anencephaly) and skeletal dysplasia (114). Hydrolethalus syndrome manifests with hydrocephaly, micrognathia, postaxial polydactyly of the upper limbs, and pre- or postaxial polydactyly of the lower limbs and hallux duplication and is associated with a homozygous deletion in the KIF7 coiled-coil domain (94). Al-Gazali-Bakalinova syndrome resulting from the missense mutations in KIF7 is characterized by macrocephaly, multiple epiphyseal dysplasia, genu valgum (knock knees), clinodactyly, short neck, pectus excavatum, and distinctive facial features including hypertelorism and frontal bossing (114). Acrocallosal syndrome (ACLS) results from nonsense or frameshift KIF7 mutations affecting the motor domain or the Gli binding site and presents with macrocephaly, mental retardation, polydactyly, hallux duplication, and characteristic facial features including hypertelorism and prominent forehead (115). Novel heterozygous missense mutations have also been identified in KIF7 and shown to be associated with primary cilia defects that lead to ciliopathies, in particular the Bardet-Biedl syndrome (94). Homozygous KIF7 mutations have also been reported in Joubert syndrome; a ciliopathy with phenotype overlapping with the ACL syndrome (116).
HH signaling plays a key role in tissue development and cellular homeostasis, including skeletal patterning, development, and growth; it is highly associated with primary cilia (cell surface organelles involved in regulation of cell polarity and mechanosensing) and implicated in pathobiology of ciliopathies (117). Interestingly, chondrocytes from mice null for KIF3A, a subunit of the Kinesin-2 motor complex required for intraflagellar transport, showed loss of primary cilia, reduced proliferation, defective cell rotation, and accelerated differentiation, resulting in disrupted columnar organization in the growth plate and postnatal dwarfism (118). It has been shown that the mutant KIF7 also results in defects in primary cilia formation and induces abnormal centrosomal duplication and fragmentation of the Golgi network, indicating that KIF7 can potentially participate in microtubule stability and growth direction (119). Analysis of fibroblasts from patients with ACLS showed longer primary cilia without a disruption in the cilia components, suggesting a potential role of KIF7 in cilia length regulation (94). KIF7 was found to localize to the primary cilia tips (the microtubule plus end) in Sonic Hedgehog (SHH)-stimulated cells where it regulates the cilia length by promoting microtubule catastrophe and limiting microtubule polymerization (50). Further research showed that KIF7 mutations cause ciliopathies through dysregulation of the Hedgehog (HH) signaling pathway components and that KIF7 is a key mediator in the HH signaling pathway through the regulation of Gli transcription factor targets (50). Kif7−/− mice, a representative model of ACLS, exhibited most of the features observed in the patients with ACLS and were used to investigate the molecular pathology of the disorder (120). In this model, loss of function of KIF7 resulted in aberrant Gli3 activity and overexpression of SHH signaling, suggesting that KIF7 is essential to regulate Gli3 repressive activity during embryonic development. SHH regulates the anterior-posterior patterning of the limbs, and Indian Hedgehog (IHH) signaling is important for regulation of the chondrocyte proliferation and differentiation in the cartilage growth plate (121). Specifically, IHH is expressed by the prehypertrophic and hypertrophic chondrocytes, regulates the rate of hypertrophic differentiation, and stimulates the expression of parathyroid hormone-related protein (PTHrP) in the resting chondrocytes, which in turn negatively regulates hypertrophy (122). It is also important in the perichondrium where it regulates the osteoblast differentiation and bone formation (123). Disruption of IHH and its associated Gli zinc-finger proteins (Gli1–Gli3) renders proliferative chondrocytes unable to initiate the hypertrophic differentiation process and has a high impact on chondrogenesis and bone formation (123, 124). Deletion of IHH in mice affects osteoblast development during endochondral ossification, reduces chondrocyte proliferation, and results in expanded hypertrophic zone in the growth plate (122). KIF7 has been shown to be a major mediator of IHH signaling, regulating Gli transcription factors together with SUFU (99, 100). During development, KIF7 downregulates SUFU to promote the IHH pathway. In the absence of SUFU, it represses IHH signaling by inhibiting Gli-mediated transcription, reducing HH pathway activity, decreasing chondrocyte proliferation, and expanding the hypertrophic zone. Moreover, studies using several SHH-signaling pathway mutant mouse strains crossed with the Kif7Maki/Maki mouse model developed through ENU mutagenesis confirmed that KIF7 acts downstream of Smoothened (Smo) and upstream of Gli, and its activity is dependent on the presence of the primary cilia (125). Interestingly, differential expression of KIF7 was demonstrated by qRT-PCR on microdissected sections of wild-type mouse growth plate and shown that KIF7 expression is high in the articular and resting chondrocytes and significantly decreased in proliferating, prehypertrophic, and hypertrophic chondrocyte, similar to the SUFU and PTHrP expression (121). In summary, KIF7 is essential for the regulation of chondrocyte proliferation and differentiation, controls cilia length during ciliogenesis by promoting microtubule catastrophe, is a key mediator of HH signaling, and its deletion or disruption results in skeletal defects and ciliopathies.
Compound Heterozygous Mutations in KIF10 Disrupt Cell Division and Are Associated with Primary Microcephalic Osteodysplastic Primordial Dwarfism
Microcephalic osteodysplastic primordial dwarfism (MCPH13) is an autosomal recessive skeletal disorder characterized by limited growth, small hands and metaphyseal sclerosis, mild micrognathia, short metacarpals, and osteopenia (61). Whole genome sequencing has identified two variants of compound heterozygous mutation of KIF10 (c.2797G>A: p.D933N and c.4063A>G: p.K1355E) in individuals with MCPH13 as causative locus of this disorder.
KIF10 (also known as centromere-associated protein E, or CENPE) is a kinetochore-associated kinesin-like motor protein (101). CENPE gene is evolutionary conserved in humans, mice, and zebrafish. During cell division, kinesin proteins participate in the separation of centrosomes to achieve spindle bipolarity and facilitate chromosome congression to the metaphase plate through diverse mechanical activities; transducing regulatory signals, modulating microtubule ends, and directing movements (plus-end, minus-end, or bipolar plus-end motility) (61). Kinesins involved in the kinetochore-microtubule interactions are also essential for chromosome alignment (95). They mediate chromosome attachment and movement along spindle microtubules, generate tension across aligned chromosomes, maintain their alignment, and stabilize spindle poles (95). HeLa cells with depleted CENPE showed chromosome misalignment at metaphase and abnormal spindle microtubules. Interestingly, the ratio of cells in the G2-M phase of the cell cycle was significantly decreased indicating the arrest of the G2-M phase (101, 102). CENPE null mice resulting from a heterozygous cross die in utero and immunofluorescence analysis of in vitro culture of early-stage embryos showed severe abnormal growth, inner cell mass growth retardation, aberrant mitosis, and chromosome misalignment (126). Moreover, examination of CENPE-deficient fibroblasts revealed a significant decrease in the number of kinetochore-associated microtubules, misposition of the chromosome centromeres, and defects or failure in chromosome alignment, highlighting that KIF10 is essential for kinetochore-microtubule interaction stability and for chromosome stability, alignment, attachment to the spindle, and for mitotic check-point activation (126). Furthermore, inhibition of the ATP binding sites of the CENPE motor domain in zebrafish using a small-molecular inhibitor (GSK923295), used as a chemotherapeutic to disrupt mitosis in cancer cells, resulted in severe developmental abnormalities characterized by head malformation, degenerative notochord, smaller lens, and small organs. Analysis of the early embryogenesis stage showed asymmetric cell division of the zygotes and disruption of cell cycle with defects in cell migration during gastrulation, suggesting the role of CENPE in the regulation of cell division and organogenesis (102). In mice, CENPE inhibition using the same molecular inhibitor (GSK923295) significantly reduced the weight of the liver, heart, and kidney, and mutant liver cells showed an abnormal cell morphology and arrest of cell cycle at metaphase, confirming the critical role of CENPE in early development of the organs (102). In contrast, CENPE-deficient liver cells from mice with a conditional disruption of CENPE injured by carbon tetrachloride injection to induce chemical damage manifested abnormal chromosome positioning and unstable microtubules-kinetochores interactions but were able to evade mitotic arrest and consequently regenerate at normal rate and recover their function, suggesting that CENPE plays a critical role in early development but not in late stages of organogenesis (126). Altogether, these studies demonstrate the role of KIF10 in early embryogenesis and early organ development, as key regulator of cell cycle progression and key mediator of cell division, essential for chromosomes alignment and microtubule spindle stability.
Gain of Function Mutations in the Head and Tail Domain of KIF22 Result in Spondyloepimetaphyseal Dysplasia with Joint Laxity
Spondyloepimetaphyseal dysplasia with joint laxity type 2 (SEMDJL-leptodactylic type or SEMDJL2) is an autosomal dominant skeletal disorder characterized by short stature, limb malalignment (genu valgum and/or varum), ligamentous laxity, and mild spinal deformity without cognitive impairment (103). Individuals with SEMDJL2 present with delayed epiphyseal ossification leading to epiphyseal dysplasia, striated metaphyses, and precocious osteoarthritis (127). Other common features include joint laxity, slender metacarpals, constricted femur neck, and progressive scoliosis, all together presenting a premature skeletal aging phenotype (104). The molecular pathogenesis of the SEMDJL-leptodactylic type was thought to be related to ECM abnormalities since the disorder is characterized by highly abnormal tendon collagen fibrils in the affected ligaments and early onset joint degeneration. This hypothesis was encouraged by the observation of phenotypic overlap with skeletal disorders in which either collagen type II or cartilage oligomeric matrix protein (COMP) is affected (spondyloepiphyseal dysplasia congenita and pseudoachondroplasia, respectively) (128). However, the disease-causing mutations in patients with SEMDJL2 remained obscure until the whole genome sequencing of individuals with lepto-SEMDJL in 2011 identified KIF22 mutations as the main cause of the disorder (104).
Analysis of early mouse embryos showed KIF22 attachment to the chromosomes during anaphase/telophase in a chromosome- and microtubule-binding-dependent manner and revealed that KIF22 deficiency results in malformation of nuclear blastomeres and affects cell division. KIF22 deletion affected early embryonic development of pups derived from intercrosses of Kif22+/− with 50% of KIF22 null embryos dying by E9.5 (129). Time-lapse imaging of chromosomes in Kif22−/− zygotes showed less chromosome compaction during anaphase/telophase and 40% of micro- or multinucleation at the 1–8 cell stage. Interestingly, KIF22 deficiency did not severely affect meiosis and the post morula mitosis (129). The Kif22−/− pups that survived and developed to adulthood without an overt phenotype were those that passed the 4- to 8-cell stage without multinucleated blastomeres. Four of the discovered SEMDJL2 mutations affect the KIF22 motor domain and result in the substitution of two highly conserved amino acids in the α2 helix of the KIF22 motor domain near the ATP binding site [Proline 148 (P148S, P148L) and Arginine 149 (R149Q, R149L)] and consequently interfere with the ATP binding (104, 105, 128). The mutations were exclusively identified in individuals with SEMDJL2 and had largely tissue-specific effects particularly in the connective tissues, despite the KIF22 mRNA expression detected in various human somatic tissues (130). An additional SEMDJL2 mutation was recently identified in the KIF22 coiled-coil domain in the kinesin tail, with a highly conserved Valine 475 residue substituted by Glycine, and suggesting that a second microtubule binding site may be present in the tail of KIF22 that is as important as the head motor domain (131). It has been suggested that mutations in either the tail or motor domain of KIF22 can impede the interaction between these domains and subsequently prevent KIF22 inactivation. Moreover, phosphorylation of two residues in KIF22 (T158 and T463) that are in the vicinity of the mutation sites disrupted KIF22 inactivation and hampered chromosome segregation, which highlighted the essential role of T463 and T158 dephosphorylation in promoting the head-tail interaction to inactivate KIF22. Interestingly, KIF22 with SEMDJL2 mutations was able to attach to both chromosome arms and spindle microtubules in Kyoto-Hela cells (131). In normal conditions, KIF22 inactivation during anaphase produces ejection forces to facilitate chromosome movement of the spindle poles (132, 133). KIF22 tail mutations disrupted the chromosome segregation during anaphase due to its hyperactivation and generation of aberrant polar ejection forces, which limit chromosome arm movement, disrupt spindle separation, and lead to chromosome misplacement in the spindle (131). Daughter cells resulting from the HeLa cells overexpressing recombinant mutant KIF22 were multinucleated and exhibited lack of anaphase chromosome mass compaction along the spindle axis (129). Proliferation of cells expressing mutant KIF22 was significantly reduced due to the nuclear deformity of daughter cells and inhibition of cytokinesis. Moreover, activation of the motor domain resulted in the same phenotypes as observed for the KIF22 SEMDJL2 mutations, suggesting that the motor domain potentially regulates KIF22 activity at the metaphase to anaphase transition. Motor domain deletion in NIH 3T3 cells led to KIF22 mislocalization to anaphase chromosomes and KIF22 tail-mutant was strictly localized to the kinetochore microtubules (129). Thus, both the motor domain and the DNA binding tail domain are important for Kinesin-10 spindle localization and function during cell division.
These findings highlight the essential role of KIF22 in chromosome localization and a role in preventing cell multinucleation by mediating anaphase/telophase chromosome compaction. However, further studies are required to explain the mechanism by which KIF22 contributes to chromosome compaction, as well as the exclusively musculoskeletal phenotype of the patients with SEMDJL2 (129, 134). Analysis of KIF22 gene expression in wild-type mouse growth plate revealed its overexpression in the proliferative zone, which supports its involvement in chondrocyte proliferation (128). Interestingly, recent studies in lung carcinoma (cells and tissues from xenografts) have revealed interaction of KIF22 with cell surface receptors such as EGFR and CAR (important for the cell-cell adhesion in various tumors) and suggested its role in membrane receptor trafficking and dynamics. These data highlight the potential new roles of the chromokinesins in intracellular transport of cargo in addition to their fundamental roles in chromosome movement during cell division, which requires further investigation in the context of skeletal dysplasia (135, 136).
SUMMARY
Mutations in KIF5B, KIF7, KIF10, and KIF22 kinesin proteins lead to skeletal dysplasia with various severity of clinical features and overlapping phenotypes. Several mutations in the kinesin motor domain, tail, or coiled-coil domain disrupt skeletal development, causing musculoskeletal complications. Interestingly, defects in kinesins have tissue-specific effects, can present with variable severity, and act through various mechanisms during different stages of embryonic development. One possible explanation as to why mutations in several kinesins manifest with pronounced skeletal phenotypes is that the expression of kinesins varies throughout tissues, indicating their varying relevance during tissue formation and, subsequently, their distinct impacts in case of disruption and dysfunction. In addition, kinesins can be differentially expressed (both spatially and temporally) within the same tissue, for example the cartilage growth plate. Moreover, several kinesins are present in different isoforms, and it is likely that some isoforms can be tissue specific. Several studies highlighted the role of kinesins in the regulation of chondrocyte proliferation and differentiation through a variety of molecular mechanisms, including regulation of signaling pathways and control of cytokinesis. Moreover, motor proteins mediate primary cilia structure and function, and their disruptions can lead to skeletal ciliopathies. Kinesins are also key modulators of cell cycle progression mediating chromosome congression and microtubule-kinetochore stability in addition to their roles in endocytosis and receptor recycling, autophagy, and intracellular trafficking of membrane-bound organelles. Although the coordination and regulation of kinesin expression in skeletal cells and the specific mechanisms in which they are involved in skeletal development and disease are not well understood, the diversity of the microtubule structures and functions together with the complexity of the regulation of the kinesins’ gene expression, splicing, and interaction with other cargo and associated partners encourages further research studies to decipher their role in development.
GRANTS
This work was supported by HORIZON EUROPE Marie Sklodowska-Curie Actions (MSCA) Grant 101072766.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.B. prepared figures; R.B. drafted manuscript; F.M.d.S.B. and K.A.P. edited and revised manuscript; K.A.P. approved final version of manuscript.
REFERENCES
- 1. Warman ML, Cormier‐Daire V, Hall C, Krakow D, Lachman R, Lemerrer M, Mortier G, Mundlos S, Nishimura G, Rimoin DL, Robertson S, Savarirayan R, Sillence D, Spranger J, Unger S, Zabel B, Superti‐Furga A. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A 155A: 943–968, 2011. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Krakow D, Rimoin DL. The skeletal dysplasias. Genet Med 12: 327–341, 2010. doi: 10.1097/GIM.0b013e3181daae9b. [DOI] [PubMed] [Google Scholar]
- 3. Orioli IM, Castilla EE, Barbosa-Neto JG. The birth prevalence rates for the skeletal dysplasias. J Med Genet 23: 328–332, 1986. doi: 10.1136/jmg.23.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kolambage YD, Walpita YN, Liyanage UA, Dayaratne BMKDR, Dissanayake VHW. The burden of hospital admissions for skeletal dysplasias in Sri Lanka: a population-based study. Orphanet J Rare Dis 18: 279, 2023. doi: 10.1186/s13023-023-02884-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Unger S, Ferreira CR, Mortier GR, Ali H, Bertola DR, Calder A, Cohn DH, Cormier-Daire V, Girisha KM, Hall C, Krakow D, Makitie O, Mundlos S, Nishimura G, Robertson SP, Savarirayan R, Sillence D, Simon M, Sutton VR, Warman ML, Superti-Furga A. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet A 191: 1164–1209, 2023. doi: 10.1002/ajmg.a.63132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marini JC, Blissett AR. New genes in bone development: what’s new in osteogenesis imperfecta. J Clin Endocrinol Metab 98: 3095–3103, 2013. doi: 10.1210/jc.2013-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, Nishimura G, Robertson S, Sangiorgi L, Savarirayan R, Sillence D, Superti-Furga A, Unger S, Warman ML. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A 179: 2393–2419, 2019. doi: 10.1002/ajmg.a.61366. [DOI] [PubMed] [Google Scholar]
- 8. Horton WA. Molecular genetic basis of the human chondrodysplasias. Endocrinol Metab Clin North Am 25: 683–697, 1996. doi: 10.1016/s0889-8529(05)70347-9. [DOI] [PubMed] [Google Scholar]
- 9. Tompson SW, Merriman B, Funari VA, Fresquet M, Lachman RS, Rimoin DL, Nelson SF, Briggs MD, Cohn DH, Krakow D. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am J Hum Genet 84: 72–79, 2009. doi: 10.1016/j.ajhg.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Ravenswaaij-Arts CM, Losekoot M. [From gene to disease; achondroplasia and other skeletal dysplasias due to an activating mutation in the fibroblast growth factor]. Ned Tijdschr Geneeskd 145: 1056–1059, 2001. [PubMed] [Google Scholar]
- 11. Guasto A, Cormier-Daire V. Signaling pathways in bone development and their related skeletal dysplasia. Int J Mol Sci 22: 4321, 2021. doi: 10.3390/ijms22094321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nishimura G, Lausch E, Savarirayan R, Shiba M, Spranger J, Zabel B, Ikegawa S, Superti‐Furga A, Unger S. TRPV4‐associated skeletal dysplasias. Am J Med Genet C Semin Med Genet 160: 190–204, 2012. doi: 10.1002/ajmg.c.31335. [DOI] [PubMed] [Google Scholar]
- 13. Shen X, Li Z, Pan X, Yao J, Shen G, Zhang S, Dong M, Fan L. Prenatal diagnosis of recurrent moderate skeletal dysplasias in lamin B receptors. Front Genet 13: 1020475, 2023. doi: 10.3389/fgene.2022.1020475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dodding MP, Way M. Coupling viruses to dynein and kinesin-1. EMBO J 30: 3527–3539, 2011. doi: 10.1038/emboj.2011.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vale R, Reese T, Sheetz M. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42: 39–50, 1985. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 10: 682–696, 2009. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- 17. Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43: 513–525, 2004. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 18. Yokota S, Shah SH, Huie EL, Wen RR, Luo Z, Goldberg JL. Kif5a regulates mitochondrial transport in developing retinal ganglion cells in vitro. Invest Ophthalmol Vis Sci 64: 4, 2023. doi: 10.1167/iovs.64.3.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu M, Pi H, Xi Y, Wang L, Tian L, Chen M, Xie J, Deng P, Zhang T, Zhou C, Liang Y, Zhang L, He M, Lu Y, Chen C, Yu Z, Zhou Z. KIF5A-dependent axonal transport deficiency disrupts autophagic flux in trimethyltin chloride-induced neurotoxicity. Autophagy 17: 903–924, 2021. doi: 10.1080/15548627.2020.1739444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kamata H, Tsukasaki Y, Sakai T, Ikebe R, Wang J, Jeffers A, Boren J, Owens S, Suzuki T, Higashihara M, Idell S, Tucker TA, Ikebe M. KIF5A transports collagen vesicles of myofibroblasts during pleural fibrosis. Sci Rep 7: 4556, 2017. doi: 10.1038/s41598-017-04437-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gan H, Xue W, Gao Y, Zhu G, Chan D, Cheah KSE, Huang J. KIF5B modulates central spindle organization in late-stage cytokinesis in chondrocytes. Cell Biosci 9: 85, 2019. doi: 10.1186/s13578-019-0344-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414: 643–648, 2001. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 23. Kural C, Kim H, Syed S, Goshima G, Gelfand VI, Selvin PR. Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement? Science 308: 1469–1472, 2005. doi: 10.1126/science.1108408. [DOI] [PubMed] [Google Scholar]
- 24. Metzger T, Gache V, Xu M, Cadot B, Folker ES, Richardson BE, Gomes ER, Baylies MK. MAP and kinesin-dependent nuclear positioning is required for skeletal muscle function. Nature 484: 120–124, 2012. doi: 10.1038/nature10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morris RL, Scholey JM. Heterotrimeric kinesin-II is required for the assembly of motile 9+2 ciliary axonemes on sea urchin embryos. J Cell Biol 138: 1009–1022, 1997. doi: 10.1083/jcb.138.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cruz NM, Reddy R, McFaline-Figueroa JL, Tran C, Fu H, Freedman BS. Modelling ciliopathy phenotypes in human tissues derived from pluripotent stem cells with genetically ablated cilia. Nat Biomed Eng 6: 463–475, 2022. [Erratum in Nat Biomed Eng 6: 463–475, 2022]. doi: 10.1038/s41551-022-00880-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA 100: 5286–5291, 2003. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kovacs JJ, Whalen EJ, Liu R, Xiao K, Kim J, Chen M, Wang J, Chen W, Lefkowitz RJ. Beta-arrestin-mediated localization of smoothened to the primary cilium. Science 320: 1777–1781, 2008. doi: 10.1126/science.1157983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yue Y, Engelke MF, Blasius TL, Verhey KJ. Hedgehog-induced ciliary trafficking of kinesin-4 motor KIF7 requires intraflagellar transport but not KIF7's microtubule binding. Mol Biol Cell 33: br1, 2022. doi: 10.1091/mbc.E21-04-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alsabban AH, Morikawa M, Tanaka Y, Takei Y, Hirokawa N. Kinesin Kif3b mutation reduces NMDAR subunit NR2A trafficking and causes schizophrenia-like phenotypes in mice. EMBO J 39: e101090, 2020. doi: 10.15252/embj.2018101090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao C, Omori Y, Brodowska K, Kovach P, Malicki J. Kinesin-2 family in vertebrate ciliogenesis. Proc Natl Acad Sci USA 109: 2388–2393, 2012. doi: 10.1073/pnas.1116035109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gumy LF, Chew DJ, Tortosa E, Katrukha EA, Kapitein LC, Tolkovsky AM, Hoogenraad CC, Fawcett JW. The kinesin-2 family member KIF3C regulates microtubule dynamics and is required for axon growth and regeneration. J Neurosci 33: 11329–11345, 2013. doi: 10.1523/JNEUROSCI.5221-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Setou M, Nakagawa T, Seog DH, Hirokawa N. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science 288: 1796–1802, 2000. doi: 10.1126/science.288.5472.1796. [DOI] [PubMed] [Google Scholar]
- 34. Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 81: 769–780, 1995. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- 35. Yonekawa Y, Harada A, Okada Y, Funakoshi T, Kanai Y, Takei Y, Terada S, Noda T, Hirokawa N. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J Cell Biol 141: 431–441, 1998. doi: 10.1083/jcb.141.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, Hirokawa N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 79: 1209–1220, 1994. doi: 10.1016/0092-8674(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 37. Xu F, Takahashi H, Tanaka Y, Ichinose S, Niwa S, Wicklund MP, Hirokawa N. KIF1Bβ mutations detected in hereditary neuropathy impair IGF1R transport and axon growth. J Cell Biol 217: 3480–3496, 2018. doi: 10.1083/jcb.201801085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dorner C, Ciossek T, Müller S, Møller PH, Ullrich A, Lammers R. Characterization of KIF1C, a new kinesin-like protein involved in vesicle transport from the Golgi apparatus to the endoplasmic reticulum. J Biol Chem 273: 20267–20275, 1998. doi: 10.1074/jbc.273.32.20267. [DOI] [PubMed] [Google Scholar]
- 39. Nakagawa T, Setou M, Seog D, Ogasawara K, Dohmae N, Takio K, Hirokawa N. A novel motor, KIF13A, transports mannose-6-phosphate receptor to plasma membrane through direct interaction with AP-1 complex. Cell 103: 569–581, 2000. doi: 10.1016/s0092-8674(00)00161-6. [DOI] [PubMed] [Google Scholar]
- 40. Yamada KH, Nakajima Y, Geyer M, Wary KK, Ushio-Fukai M, Komarova Y, Malik AB. KIF13B regulates angiogenesis through Golgi to plasma membrane trafficking of VEGFR2. J Cell Sci 127: 4518–4530, 2014. doi: 10.1242/jcs.156109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Serra-Marques A, Martin M, Katrukha EA, Grigoriev I, Peeters CA, Liu Q, Hooikaas PJ, Yao Y, Solianova V, Smal I, Pedersen LB, Meijering E, Kapitein LC, Akhmanova A. Concerted action of kinesins KIF5B and KIF13B promotes efficient secretory vesicle transport to microtubule plus ends. eLife 9: e61302, 2020. doi: 10.7554/eLife.61302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carleton M, Mao M, Biery M, Warrener P, Kim S, Buser C, Marshall CG, Fernandes C, Annis J, Linsley PS. RNA interference-mediated silencing of mitotic kinesin KIF14 disrupts cell cycle progression and induces cytokinesis failure. Mol Cell Biol 26: 3853–3863, 2006. doi: 10.1128/MCB.26.10.3853-3863.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Torres JZ, Summers MK, Peterson D, Brauer MJ, Lee J, Senese S, Gholkar AA, Lo YC, Lei X, Jung K, Anderson DC, Davis DP, Belmont L, Jackson PK. The STARD9/Kif16a kinesin associates with mitotic microtubules and regulates spindle pole assembly. Cell 147: 1309–1323, 2011. doi: 10.1016/j.cell.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blatner NR, Wilson MI, Lei C, Hong W, Murray D, Williams RL, Cho W. The structural basis of novel endosome anchoring activity of KIF16B kinesin. EMBO J 26: 3709–3719, 2007. doi: 10.1038/sj.emboj.7601800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hoepfner S, Severin F, Cabezas A, Habermann B, Runge A, Gillooly D, Stenmark H, Zerial M. Modulation of receptor recycling and degradation by the endosomal kinesin KIF16B. Cell 121: 437–450, 2005. doi: 10.1016/j.cell.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 46. Almeida AC, Maiato H. Chromokinesins. Curr Biol 28: R1131–R1135, 2018. doi: 10.1016/j.cub.2018.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sheng L, Hao SL, Yang WX, Sun Y. The multiple functions of kinesin-4 family motor protein KIF4 and its clinical potential. Gene 678: 90–99, 2018. doi: 10.1016/j.gene.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 48. He M, Agbu S, Anderson KV. Microtubule motors drive hedgehog signaling in primary cilia. Trends Cell Biol 27: 110–125, 2017. doi: 10.1016/j.tcb.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhu C, Zhao J, Bibikova M, Leverson JD, Bossy-Wetzel E, Fan JB, Abraham RT, Jiang W. Functional analysis of human microtubule-based motor proteins, the kinesins and dyneins, in mitosis/cytokinesis using RNA interference. Mol Biol Cell 16: 3187–3199, 2005. doi: 10.1091/mbc.e05-02-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. He M, Subramanian R, Bangs F, Omelchenko T, Liem KF Jr, Kapoor TM, Anderson KV. The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat Cell Biol 16: 663–672, 2014. doi: 10.1038/ncb2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bianchi S, van Riel WE, Kraatz SH, Olieric N, Frey D, Katrukha EA, Jaussi R, Missimer J, Grigoriev I, Olieric V, Benoit RM, Steinmetz MO, Akhmanova A, Kammerer RA. Structural basis for misregulation of kinesin KIF21A autoinhibition by CFEOM1 disease mutations. Sci Rep 6: 30668, 2016. doi: 10.1038/srep30668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hooikaas PJ, Damstra HG, Gros OJ, van Riel WE, Martin M, Smits YT, van Loosdregt J, Kapitein LC, Berger F, Akhmanova A. Kinesin-4 KIF21B limits microtubule growth to allow rapid centrosome polarization in T cells. eLife 9: e62876, 2020. doi: 10.7554/eLife.62876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wilson CW, Nguyen CT, Chen MH, Yang JH, Gacayan R, Huang J, Chen JN, Chuang PT. Fused has evolved divergent roles in vertebrate Hedgehog signalling and motile ciliogenesis. Nature 459: 98–102, 2009. doi: 10.1038/nature07883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Blangy A, Lane HA, d'Hérin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 83: 1159–1169, 1995. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 55. Birtel J, Gliem M, Mangold E, Tebbe L, Spier I, Müller PL, Holz FG, Neuhaus C, Wolfrum U, Bolz HJ, Charbel Issa P. Novel insights into the phenotypical spectrum of KIF11-associated retinopathy, including a new form of retinal ciliopathy. Invest Ophthalmol Vis Sci 58: 3950–3959, 2017. doi: 10.1167/iovs.17-21679. [DOI] [PubMed] [Google Scholar]
- 56. Bartoli KM, Jakovljevic J, Woolford JL Jr, Saunders WS. Kinesin molecular motor Eg5 functions during polypeptide synthesis. Mol Biol Cell 22: 3420–3430, 2011. doi: 10.1091/mbc.E11-03-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nguyen PA, Groen AC, Loose M, Ishihara K, Wühr M, Field CM, Mitchison TJ. Spatial organization of cytokinesis signaling reconstituted in a cell-free system. Science 346: 244–247, 2014. doi: 10.1126/science.1256773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Echard A, Jollivet F, Martinez O, Lacapère JJ, Rousselet A, Janoueix-Lerosey I, Goud B. Interaction of a Golgi-associated kinesin-like protein with Rab6. Science 279: 580–585, 1998. doi: 10.1126/science.279.5350.580. [DOI] [PubMed] [Google Scholar]
- 59. Kamimoto T, Zama T, Aoki R, Muro Y, Hagiwara M. Identification of a novel kinesin-related protein, KRMP1, as a target for mitotic peptidyl-prolyl isomerase Pin1. J Biol Chem 276: 37520–37528, 2001. doi: 10.1074/jbc.M106207200. [DOI] [PubMed] [Google Scholar]
- 60. Liljeholm M, Irvine AF, Vikberg AL, Norberg A, Month S, Sandström H, Wahlin A, Mishima M, Golovleva I. Congenital dyserythropoietic anemia type III (CDA III) is caused by a mutation in kinesin family member, KIF23. Blood 121: 4791–4799, 2013. doi: 10.1182/blood-2012-10-461392. [DOI] [PubMed] [Google Scholar]
- 61. Mirzaa GM, Vitre B, Carpenter G, Abramowicz I, Gleeson JG, Paciorkowski AR, Cleveland DW, Dobyns WB, O'Driscoll M. Mutations in CENPE define a novel kinetochore-centromeric mechanism for microcephalic primordial dwarfism. Hum Genet 133: 1023–1039, 2014. doi: 10.1007/s00439-014-1443-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stumpff J, von Dassow G, Wagenbach M, Asbury C, Wordeman L. The kinesin-8 motor Kif18A suppresses kinetochore movements to control mitotic chromosome alignment. Dev Cell 14: 252–262, 2008. doi: 10.1016/j.devcel.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tanenbaum ME, Macurek L, van der Vaart B, Galli M, Akhmanova A, Medema RH. A complex of Kif18b and MCAK promotes microtubule depolymerization and is negatively regulated by Aurora kinases. Curr Biol 21: 1356–1365, 2011. doi: 10.1016/j.cub.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 64. Niwa S, Nakajima K, Miki H, Minato Y, Wang D, Hirokawa N. KIF19A is a microtubule-depolymerizing kinesin for ciliary length control. Dev Cell 23: 1167–1175, 2012. doi: 10.1016/j.devcel.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 65. Konjikusic MJ, Yeetong P, Boswell CW, Lee C, Roberson EC, Ittiwut R, Suphapeetiporn K, Ciruna B, Gurnett CA, Wallingford JB, Shotelersuk V, Gray RS. Mutations in Kinesin family member 6 reveal specific role in ependymal cell ciliogenesis and human neurological development. PLoS Genet 14: e1007817, 2018. doi: 10.1371/journal.pgen.1007817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Konjikusic MJ, Lee C, Yue Y, Shrestha BD, Nguimtsop AM, Horani A, Brody S, Prakash VN, Gray RS, Verhey KJ, Wallingford JB. Kif9 is an active kinesin motor required for ciliary beating and proximodistal patterning of motile axonemes. J Cell Sci 136: jcs259535, 2023. doi: 10.1242/jcs.259535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cornfine S, Himmel M, Kopp P, El Azzouzi K, Wiesner C, Krüger M, Rudel T, Linder S. The kinesin KIF9 and reggie/flotillin proteins regulate matrix degradation by macrophage podosomes. Mol Biol Cell 22: 202–215, 2011. doi: 10.1091/mbc.E10-05-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou R, Niwa S, Homma N, Takei Y, Hirokawa N. KIF26A is an unconventional kinesin and regulates GDNF-Ret signaling in enteric neuronal development. Cell 139: 802–813, 2009. doi: 10.1016/j.cell.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 69. Uchiyama Y, Sakaguchi M, Terabayashi T, Inenaga T, Inoue S, Kobayashi C, Oshima N, Kiyonari H, Nakagata N, Sato Y, Sekiguchi K, Miki H, Araki E, Fujimura S, Tanaka SS, Nishinakamura R. Kif26b, a kinesin family gene, regulates adhesion of the embryonic kidney mesenchyme. Proc Natl Acad Sci USA 107: 9240–9245, 2010. doi: 10.1073/pnas.0913748107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stalke A, Sgodda M, Cantz T, Skawran B, Lainka E, Hartleben B, Baumann U, Pfister ED. kif12 variants and disturbed hepatocyte polarity in children with a phenotypic spectrum of cholestatic liver disease. J Pediatr 240: 284–291.e9, 2022. doi: 10.1016/j.jpeds.2021.09.019. [DOI] [PubMed] [Google Scholar]
- 71. Brouwers N, Mallol Martinez N, Vernos I. Role of Kif15 and its novel mitotic partner KBP in K-fiber dynamics and chromosome alignment. PLoS One 12: e0174819, 2017. doi: 10.1371/journal.pone.0174819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Eskova A, Knapp B, Matelska D, Reusing S, Arjonen A, Lisauskas T, Pepperkok R, Russell R, Eils R, Ivaska J, Kaderali L, Erfle H, Starkuviene V. An RNAi screen identifies KIF15 as a novel regulator of the endocytic trafficking of integrin. J Cell Sci 127: 2433–2447, 2014. doi: 10.1242/jcs.137281. [DOI] [PubMed] [Google Scholar]
- 73. Kobayashi T, Tsang WY, Li J, Lane W, Dynlacht BD. Centriolar kinesin Kif24 interacts with CP110 to remodel microtubules and regulate ciliogenesis. Cell 145: 914–925, 2011. doi: 10.1016/j.cell.2011.04.028. [DOI] [PubMed] [Google Scholar]
- 74. Decarreau J, Wagenbach M, Lynch E, Halpern AR, Vaughan JC, Kollman J, Wordeman L. The tetrameric kinesin Kif25 suppresses pre-mitotic centrosome separation to establish proper spindle orientation. Nat Cell Biol 19: 384–390, 2017. [Erratum in Nat Cell Biol 19: 740, 2017]. doi: 10.1038/ncb3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Manning AL, Ganem NJ, Bakhoum SF, Wagenbach M, Wordeman L, Compton DA. The kinesin-13 proteins Kif2a, Kif2b, and Kif2c/MCAK have distinct roles during mitosis in human cells. Mol Biol Cell 18: 2970–2979, 2007. doi: 10.1091/mbc.e07-02-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schlager MA, Kapitein LC, Grigoriev I, Burzynski GM, Wulf PS, Keijzer N, de Graaff E, Fukuda M, Shepherd IT, Akhmanova A, Hoogenraad CC. Pericentrosomal targeting of Rab6 secretory vesicles by Bicaudal-D-related protein 1 (BICDR-1) regulates neuritogenesis. EMBO J 29: 1637–1651, 2010. doi: 10.1038/emboj.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Meng W, Mushika Y, Ichii T, Takeichi M. Anchorage of microtubule minus ends to adherens junctions regulates epithelial cell-cell contacts. Cell 135: 948–959, 2008. doi: 10.1016/j.cell.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 78. Miki H, Setou M, Kaneshiro K, Hirokawa N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci USA 98: 7004–7011, 2001. doi: 10.1073/pnas.111145398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yip YY, Pernigo S, Sanger A, Xu M, Parsons M, Steiner RA, Dodding MP. The light chains of kinesin-1 are autoinhibited. Proc Natl Acad Sci USA 113: 2418–2423, 2016. doi: 10.1073/pnas.1520817113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. D'Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem Sci 28: 655–662, 2003. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 81. Konjikusic MJ, Gray RS, Wallingford JB. The developmental biology of kinesins. Dev Biol 469: 26–36, 2021. doi: 10.1016/j.ydbio.2020.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science 279: 519–526, 1998. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- 83. Verhey KJ, Lizotte DL, Abramson T, Barenboim L, Schnapp BJ, Rapoport TA. Light chain-dependent regulation of Kinesin’s interaction with microtubules. J Cell Biol 143: 1053–1066, 1998. doi: 10.1083/jcb.143.4.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Soppina P, Patel N, Shewale DJ, Rai A, Sivaramakrishnan S, Naik PK, Soppina V. Kinesin-3 motors are fine-tuned at the molecular level to endow distinct mechanical outputs. BMC Biol 20: 177, 2022. doi: 10.1186/s12915-022-01370-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci 6: 201–214, 2005. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- 86. Sanger A, Yip YY, Randall TS, Pernigo S, Steiner RA, Dodding MP. SKIP controls lysosome positioning using a composite kinesin-1 heavy and light chain-binding domain. J Cell Sci 130: 1637–1651, 2017. doi: 10.1242/jcs.198267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kawano T, Araseki M, Araki Y, Kinjo M, Yamamoto T, Suzuki T. A small peptide sequence is sufficient for initiating kinesin-1 activation through part of TPR region of KLC1. Traffic 13: 834–848, 2012. doi: 10.1111/j.1600-0854.2012.01350.x. [DOI] [PubMed] [Google Scholar]
- 88. Cason SE, Holzbaur ELF. Selective motor activation in organelle transport along axons. Nat Rev Mol Cell Biol 23: 699–714, 2022. doi: 10.1038/s41580-022-00491-w. [DOI] [PubMed] [Google Scholar]
- 89. Niwa S. Kinesin superfamily proteins and the regulation of microtubule dynamics in morphogenesis. Anat Sci Int 90: 1–6, 2015. doi: 10.1007/s12565-014-0259-5. [DOI] [PubMed] [Google Scholar]
- 90. Ocbina PJ, Eggenschwiler JT, Moskowitz I, Anderson KV. Complex interactions between genes controlling trafficking in primary cilia. Nat Genet 43: 547–553, 2011. doi: 10.1038/ng.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Harada A, Takei Y, Kanai Y, Tanaka Y, Nonaka S, Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J Cell Biol 141: 51–59, 1998. doi: 10.1083/jcb.141.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yao M, Qu H, Han Y, Cheng CY, Xiao X. Kinesins in mammalian spermatogenesis and germ cell transport. Front Cell Dev Biol 10: 837542, 2022. doi: 10.3389/fcell.2022.837542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Putoux A, Thomas S, Coene KL, Davis EE, Alanay Y, Ogur G, et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet 43: 601–606, 2011. doi: 10.1038/ng.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Garcia-Saez I, Yen T, Wade RH, Kozielski F. Crystal structure of the motor domain of the human kinetochore protein CENP-E. J Mol Biol 340: 1107–1116, 2004. doi: 10.1016/j.jmb.2004.05.053. [DOI] [PubMed] [Google Scholar]
- 96. Wiesner C, Faix J, Himmel M, Bentzien F, Linder S. KIF5B and KIF3A/KIF3B kinesins drive MT1-MMP surface exposure, CD44 shedding, and extracellular matrix degradation in primary macrophages. Blood 116: 1559–1569, 2010. doi: 10.1182/blood-2009-12-257089. [DOI] [PubMed] [Google Scholar]
- 97. Cardoso CMP, Groth-Pedersen L, Høyer-Hansen M, Kirkegaard T, Corcelle E, Andersen JS, Jäättelä M, Nylandsted J. Depletion of Kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS One 4: e4424, 2009. doi: 10.1371/journal.pone.0004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Marom R, Zhang B, Washington ME, Song IW, Burrage LC, Rossi VC, Berrier AS, Lindsey A, Lesinski J, Nonet ML, Chen J, Baldridge D, Silverman GA, Sutton VR, Rosenfeld JA, Tran AA, Hicks MJ, Murdock DR, Dai H, Weis M, Jhangiani SN, Muzny DM, Gibbs RA, Caswell R, Pottinger C, Cilliers D, Stals K; Undiagnosed Diseases Network; Eyre D, Krakow D, Schedl T, Pak SC, Lee BH. Dominant negative variants in KIF5B cause osteogenesis imperfecta via down regulation of mTOR signaling. PLOS Genet 19: e1011005, 2023. doi: 10.1371/journal.pgen.1011005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Li ZJ, Nieuwenhuis E, Nien W, Zhang X, Zhang J, Puviindran V, Wainwright BJ, Kim PC, Hui CC. Kif7 regulates Gli2 through Sufu-dependent and -independent functions during skin development and tumorigenesis. Development 139: 4152–4161, 2012. doi: 10.1242/dev.081190. [DOI] [PubMed] [Google Scholar]
- 100. Law KK, Makino S, Mo R, Zhang X, Puviindran V, Hui CC. Antagonistic and cooperative actions of Kif7 and sufu define graded intracellular Gli activities in Hedgehog signaling. PLoS One 7: e50193, 2012. doi: 10.1371/journal.pone.0050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yen TJ, Compton DA, Wise D, Zinkowski RP, Brinkley BR, Earnshaw WC, Cleveland DW. CENP-E, a novel human centromere-associated protein required for progression from metaphase to anaphase. EMBO J 10: 1245–1254, 1991. doi: 10.1002/j.1460-2075.1991.tb08066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yu KW, She ZY, Wei YL, Zhong N. Kinesin-7 CENP-E regulates cell division, gastrulation and organogenesis in development. Eur J Cell Biol 99: 151107, 2020. doi: 10.1016/j.ejcb.2020.151107. [DOI] [PubMed] [Google Scholar]
- 103. Hall CM, Elçioglu NH, Shaw DG. A distinct form of spondyloepimetaphyseal dysplasia with multiple dislocations. J Med Genet 35: 566–572, 1998. doi: 10.1136/jmg.35.7.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Min BJ, Kim N, Chung T, Kim OH, Nishimura G, Chung CY, Song HR, Kim HW, Lee HR, Kim J, Kang TH, Seo ME, Yang SD, Kim DH, Lee SB, Kim JI, Seo JS, Choi JY, Kang D, Kim D, Park WY, Cho TJ. Whole-exome sequencing identifies mutations of KIF22 in spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type. Am J Hum Genet 89: 760–766, 2011. doi: 10.1016/j.ajhg.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tüysüz B, Yılmaz S, Erener-Ercan T, Bilguvar K, Günel M. Spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type: longitudinal observation of radiographic findings in a child heterozygous for a KIF22 mutation. Pediatr Radiol 45: 771–776, 2015. doi: 10.1007/s00247-014-3159-x. [DOI] [PubMed] [Google Scholar]
- 106. Soul J, Hardingham TE, Boot-Handford RP, Schwartz J-M. SkeletalVis: an exploration and meta-analysis data portal of cross-species skeletal transcriptomics data. Bioinformatics 35: 2283–2290, 2019. doi: 10.1093/bioinformatics/bty947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Itai T, Wang Z, Nishimura G, Ohashi H, Guo L, Wakano Y, Sugiura T, Hayakawa H, Okada M, Saisu T, Kitta A, Doi H, Kurosawa K, Hotta Y, Hosono K, Sato M, Shimizu K, Takikawa K, Watanabe S, Ikeda N, Suzuki M, Fujita A, Uchiyama Y, Tsuchida N, Miyatake S, Miyake N, Matsumoto N, Ikegawa S. De novo heterozygous variants in KIF5B cause kyphomelic dysplasia. Clin Genet 102: 3–11, 2022. doi: 10.1111/cge.14133. [DOI] [PubMed] [Google Scholar]
- 108. Hirokawa N, Noda Y. Intracellular transport and kinesin superfamily proteins, KIFs: structure, function, and dynamics. Physiol Rev 88: 1089–1118, 2008. doi: 10.1152/physrev.00023.2007. [DOI] [PubMed] [Google Scholar]
- 109. Xia CH, Roberts EA, Her LS, Liu X, Williams DS, Cleveland DW, Goldstein LSB. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol 161: 55–66, 2003. doi: 10.1083/jcb.200301026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Santos-Ledo A, Garcia-Macia M, Campbell PD, Gronska M, Marlow FL. Kinesin-1 promotes chondrocyte maintenance during skeletal morphogenesis. PLoS Genet 13: e1006918, 2017. [Erratum in PLoS Genet 13: e1007099, 2017]. doi: 10.1371/journal.pgen.1006918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 93: 1147–1158, 1998. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- 112. Marchesin V, Castro-Castro A, Lodillinsky C, Castagnino A, Cyrta J, Bonsang-Kitzis H, Fuhrmann L, Irondelle M, Infante E, Montagnac G, Reyal F, Vincent-Salomon A, Chavrier P. ARF6-JIP3/4 regulate endosomal tubules for MT1-MMP exocytosis in cancer invasion. J Cell Biol 211: 339–358, 2015. doi: 10.1083/jcb.201506002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Linklater E, Jewett CE, Prekeris R. Chapter 5 - polarized membrane trafficking in development and disease: from epithelia polarization to cancer cell invasion. In: Cell Polarity in Development and Disease, edited by Michael Conn P. Boston, MA: Academic Press, 2018, p. 121–146. [Google Scholar]
- 114. Ali BR, Silhavy JL, Akawi NA, Gleeson JG, Al-Gazali L. A mutation in KIF7 is responsible for the autosomal recessive syndrome of macrocephaly, multiple epiphyseal dysplasia and distinctive facial appearance. Orphanet J Rare Dis 7: 27, 2012. doi: 10.1186/1750-1172-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Schinzel A, Schmid W. Hallux duplication, postaxial polydactyly, absence of the corpus callosum, severe mental retardation, and additional anomalies in two unrelated patients: a new syndrome. Am J Med Genet 6: 241–249, 1980. doi: 10.1002/ajmg.1320060308. [DOI] [PubMed] [Google Scholar]
- 116. Nelson MM, Thomson AJ. The acrocallosal syndrome. Am J Med Genet 12: 195–199, 1982. doi: 10.1002/ajmg.1320120209. [DOI] [PubMed] [Google Scholar]
- 117. Lai B, Jiang H, Gao Y, Zhou X. Skeletal ciliopathy: pathogenesis and related signaling pathways. Mol Cell Biochem 479: 811–823, 2024. doi: 10.1007/s11010-023-04765-5. [DOI] [PubMed] [Google Scholar]
- 118. Song B, Haycraft CJ, Seo H-S, Yoder BK, Serra R. Development of the post-natal growth plate requires intraflagellar transport proteins. Dev Biol 305: 202–216, 2007. doi: 10.1016/j.ydbio.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Dafinger C, Liebau MC, Elsayed SM, Hellenbroich Y, Boltshauser E, Korenke GC, Fabretti F, Janecke AR, Ebermann I, Nürnberg G, Nürnberg P, Zentgraf H, Koerber F, Addicks K, Elsobky E, Benzing T, Schermer B, Bolz HJ. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J Clin Invest 121: 2662–2667, 2011. doi: 10.1172/JCI43639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Putoux A, Baas D, Paschaki M, Morlé L, Maire C, Attié-Bitach T, Thomas S, Durand B. Altered GLI3 and FGF8 signaling underlies acrocallosal syndrome phenotypes in Kif7 depleted mice. Hum Mol Genet 28: 877–887, 2019. doi: 10.1093/hmg/ddy392. [DOI] [PubMed] [Google Scholar]
- 121. Hsu SH, Zhang X, Yu C, Li ZJ, Wunder JS, Hui CC, Alman BA. Kif7 promotes hedgehog signaling in growth plate chondrocytes by restricting the inhibitory function of Sufu. Development 138: 3791–3801, 2011. doi: 10.1242/dev.069492. [DOI] [PubMed] [Google Scholar]
- 122. St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev 13: 2072–2086, 1999. [Erratum in Genes Dev 13: 2617, 1999]. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian Hedgehog and PTH-Related protein. Science 273: 613–622, 1996. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 124. Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell 15: 801–812, 2008. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Liem KF, He M, Ocbina PJR, Anderson KV. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci USA 106: 13377–13382, 2009. doi: 10.1073/pnas.0906944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Putkey FR, Cramer T, Morphew MK, Silk AD, Johnson RS, McIntosh JR, Cleveland DW. Unstable kinetochore-microtubule capture and chromosomal instability following deletion of CENP-E. Dev Cell 3: 351–365, 2002. doi: 10.1016/s1534-5807(02)00255-1. [DOI] [PubMed] [Google Scholar]
- 127. Mégarbané A, Ghanem I, Le Merrer M. Spondyloepimetaphyseal dysplasia with multiple dislocations, leptodactylic type: report of a new patient and review of the literature. Am J Med Genet A 122A: 252–256, 2003. doi: 10.1002/ajmg.a.20262. [DOI] [PubMed] [Google Scholar]
- 128. Boyden ED, Campos-Xavier AB, Kalamajski S, Cameron TL, Suarez P, Tanackovic G, Andria G, Ballhausen D, Briggs MD, Hartley C, Cohn DH, Davidson HR, Hall C, Ikegawa S, Jouk PS, König R, Megarbané A, Nishimura G, Lachman RS, Mortier G, Rimoin DL, Rogers RC, Rossi M, Sawada H, Scott R, Unger S, Valadares ER, Bateman JF, Warman ML, Superti-Furga A, Bonafé L. Recurrent dominant mutations affecting two adjacent residues in the motor domain of the monomeric kinesin KIF22 result in skeletal dysplasia and joint laxity. Am J Hum Genet 89: 767–772, 2011. [Erratum in Am J Hum Genet 90: 170, 2012]. doi: 10.1016/j.ajhg.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ohsugi M, Adachi K, Horai R, Kakuta S, Sudo K, Kotaki H, Tokai-Nishizumi N, Sagara H, Iwakura Y, Yamamoto T. Kid-mediated chromosome compaction ensures proper nuclear envelope formation. Cell 132: 771–782, 2008. doi: 10.1016/j.cell.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 130. Tokai N, Fujimoto-Nishiyama A, Toyoshima Y, Yonemura S, Tsukita S, Inoue J, Yamamota T. Kid, a novel kinesin-like DNA binding protein, is localized to chromosomes and the mitotic spindle. EMBO J 15: 457–467, 1996. doi: 10.1002/j.1460-2075.1996.tb00378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Thompson AF, Blackburn PR, Arons NS, Stevens SN, Babovic-Vuksanovic D, Lian JB, Klee EW, Stumpff J. Pathogenic mutations in the chromokinesin KIF22 disrupt anaphase chromosome segregation. eLife 11: e78653, 2022. doi: 10.7554/eLife.78653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Soeda S, Yamada-Nomoto K, Ohsugi M. The microtubule-binding and coiled-coil domains of Kid are required for turn off the polar ejection force at anaphase. J Cell Sci 129: 3609–3619, 2016. doi: 10.1242/jcs.189969. [DOI] [PubMed] [Google Scholar]
- 133. Wolf F, Wandke C, Isenberg N, Geley S. Dose-dependent effects of stable cyclin B1 on progression through mitosis in human cells. EMBO J 25: 2802–2813, 2006. doi: 10.1038/sj.emboj.7601163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ohsugi M, Tokai-Nishizumi N, Shiroguchi K, Toyoshima YY, Inoue JI, Yamamoto T. Cdc2-mediated phosphorylation of Kid controls its distribution to spindle and chromosomes. EMBO J 22: 2091–2103, 2003. doi: 10.1093/emboj/cdg208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pike R, Ortiz-Zapater E, Lumicisi B, Santis G, Parsons M. KIF22 coordinates CAR and EGFR dynamics to promote cancer cell proliferation. Sci Signal 11: eaaq1060, 2018. doi: 10.1126/scisignal.aaq1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ren Y, Yang B, Guo G, Zhang J, Sun Y, Liu D, Guo S, Wu Y, Wang X, Wang S, Zhang W, Guo X, Li X, Li R, He J, Zhou Z. GBP2 facilitates the progression of glioma via regulation of KIF22/EGFR signaling. Cell Death Discov 8: 208, 2022. doi: 10.1038/s41420-022-01018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]